

Abstract

Herein, we report the discovery of a novel potent, selective, CNS penetrant, and orally bioavailable mGlu4 PAM, VU0652957 (VU2957, Valiglurax). VU2957 possessed attractive in vitro and in vivo pharmacological and DMPK properties across species. To advance toward the clinic, a spray-dried dispersion (SDD) formulation of VU2957 was developed to support IND-enabling toxicology studies. Based on its overall profile, VU2957 was evaluated as a preclinical development candidate for the treatment of Parkinson’s disease.
Keywords: Positive allosteric modulator (PAM), metabotropic glutamate receptor 4 (mGlu4), VU2957, Parkinson’s disease
Selective activation of the metabotropic glutamate receptor subtype 4 (mGu4), via either subtype selective agonists or positive allosteric modulators (PAMs), has been shown to decrease output of the indirect pathway in the basal ganglia and significantly reduce or eliminate motor symptoms in preclinical models of Parkinson’s disease (PD).1−25 Other preclinical data with mGlu4 PAMs suggest disease modification potential and that potentiation of mGlu4 can be considered a “pharmacological mimic” of deep brain stimulation (DBS).16−25 Recent data in DBS patients further strengthens the argument for mGlu4 PAMs as a disease modifying, as well as a symptomatic, therapeutic approach.26−28 A diverse array of mGlu4 PAM chemotypes 1–5 have demonstrated preclinical efficacy in PD models (Figure 1), but only recently has an mGlu4 PAM (Prexton Therapeutics Foliglurax, 6) entered clinical development.29,30 However, due to concerns with 6 (an α,β-unsaturated chromene oxime),30 efforts in the field continued to advance alternative mGlu4 PAMs. In this Letter, we detail the discovery of a potent, selective (versus the other seven mGlu receptor subtypes, mGlu1–3,5–8), and orally bioavailable mGlu4 PAM (VU2957) from a novel scaffold that was evaluated as a preclinical candidate.
Figure 1.

Structures of reported mGlu4 PAMs 1–5 with efficacy in preclinical rodent and/or nonhuman primate models of PD and 6, the first mGlu4 PAM to advance into human clinical studies.
PAM 4 was our first compound to advance as a potential development candidate, but a CYP1A2 autoinduction liability prevented it from advancing into IND-enabling studies due to an inability to chronically administer the compound in vivo.21,22 Further optimization of 4 led to several subseries of indazoles, benzo[d]isoxazoles, and benzo[d]isothiazoles (e.g., 3) as 5,6-heterobicyclic replacements for the halogenated phenyl ring of 4, resulting in potent, CNS penetrant mGlu4 PAMs wherein the CYP1A2 autoinduction liability was resolved by engendering a robust CYP metabolism phenotype and/or mitigating induction of CYP1A2 (Figure 2).23,24 However, these analogs, while displaying robust efficacy in preclinical PD models, did not possess profiles suitable as preclinical candidates due to solubility limited absorption, high projected human dose, and a small therapeutic window. Thus, we focused on 6,6-heterobicyclic systems and a 1-trifluormethyl isoquinoline-based PAM 7 (VU0652957, VU2957) emerged from the optimization effort as an mGlu4 PAM worthy of in-depth profiling as a preclinical development candidate.
Figure 2.
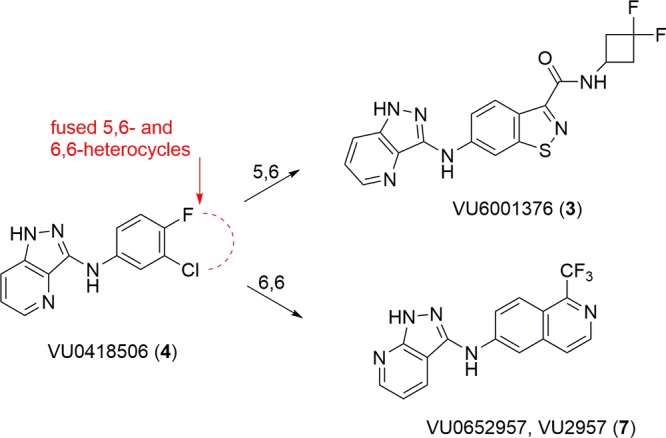
Optimization strategy for 4, which involved replacement of the halogenated phenyl moiety with diverse 5,6- and 6,6-heterobicyclic ring systems. Representative 5,6-based PAM, 3, ablated the CYP1A2 liabilities, and 6,6-based PAM 7 (VU2957) possessed a strong profile for advancement.
Two chemical routes were developed to prepare VU2957 and related analogs (Scheme 1).31 The original medicinal chemistry route (up to 1 g scales) focused on a Bischler–Napieralski approach starting from commercial phenethylamine 8. Acylation afforded 9, which was then subjected to a microwave-assisted Bischler–Napieralski reaction to afford trifluoromethyl imine 10. Oxidation/aromatization with MnO2 in refluxing xylene gave the desired bromo isoquinoline 13; however, under these conditions, 13 was advanced crude into the next step due to isolation challenges, and yields dropped precipitously as scale increased. Thus, an alternative, scalable route was developed. Based on the precedent of Kuninobu,32 isoquinoline 11 was converted into the corresponding N-oxide 12, and upon treatment with trifluoromethyl trimethylsilane, 13 was readily purified and obtained in good yield (scalable to >100 g batches). A Buchwald coupling with protected 1-(4-methoxybenzyl)-1H-pyrazolo[3,4-b]pyridine-3-amine provided 14 was then deprotected with TFA to deliver VU2957 (7).31
Scheme 1. Synthesis of VU2957 (7).

Reagents and conditions: (a) (CF3CO)2O, CH2Cl2, Et3N, rt, 1 h, 87%; (b) (i) 2-chloropyridine, CH2Cl2, −78 °C, then Tf2O, (ii) μwave, 160 °C, 5 min, 56%; (c) MnO2, xylene, 130 °C, 18 h, 44%; (d) 70% mCPBA, CH2Cl2, 5–10 °C, then warm to rt for 18 h, 80%; (e) F3C-SiMe3, THF, molecular sieves, K–OtBu, −20 °C, 46–54%; (f) 1-(4-methoxybenzyl)-1H-pyrazolo[3,4-b]pyridine-3-amine, 5 mol % Pd2(dba)3, 5 mol % XantPhos, Cs2CO3, 1,4-dioxane, μwave, 140 °C, 30 min, 65%; (g) TFA, toluene, 100 °C, 30 min, 80%.
VU2957 (7) was a potent mGlu4 PAM (Figure 3) at both human (hmGlu4/Gqi5 EC50 = 64.6 nM, pEC50 = 7.19 ± 0.14, 92.6 ± 5.0% Glu Max) and rat (rmGlu4 GIRK EC50 = 197 nM, pEC50 6.71 ± 0.17, 132% ± 1.1% Glu Max) receptors with a predicted affinity of 229 nM (log Kb = −6.64 ± 0.07) and a favorable cooperativity of 21.8 (Log β = 1.34 ± 0.05). Not only was 7 selective versus the other seven mGlu receptors (>10 μM vs mGlu1–3,5–8) and MAO-A and MAO-B (>30 μM) but it also was devoid of ancillary pharmacology (<50% inhibition @ 10 μM) in an internal Bristol-Myers Squibb (BMS) functional selectivity panel of diverse molecular targets.31 In a Eurofins Cardiac Profiler functional EP panel, 7 was unremarkable (<50% inhibition @ 10 μM) against hERG and a range of other potassium, calcium, and sodium cardiac ion channels.31,33 Furthermore, 7 was negative in both an AMES assay (TA98 and TA99, with and without S9) as well as a standard cytotoxicity assay.
Figure 3.

Molecular pharmacology profile of VU2957 (7). (A) PAM concentration–response curve for 7, affording a human EC50 of 64.6 nM with 92.6% Glu Max. (B) Progressive-fold shift assay and operational modeling to derive at a predicted affinity, Kb, of 233 nM with a cooperativity of 22.1.
PAM 7 is a low molecular weight compound (329.3) with an acceptable logP (3.78) for an mGlu allosteric modulator, but possesses poor inherent solubility (<5 μg/mL in FaSSIF and 9 μg/mL in FaSSGF). In terms of the in vitro drug metabolism profile, 7 displayed a modest fraction unbound in plasma across multiple species (fu mouse, rat, dog, cyno, and human = 0.015, 0.010, 0.014. 0.016, and 0.016, respectively) and exhibited predicted hepatic clearance values obtained from hepatic microsomal intrinsic clearance assays in human and cyno in the moderate range (9.1 and 8.8 mL/min/kg, respectively), moderate/high range in dog (18.1 mL/min/kg), and low/stable in rat (0 mL/min/kg). Both CYP450 inhibition profiles and/or CYP450 induction or autoinduction of metabolism have been long-standing issues with multiple mGlu4 PAM chemotypes; however, 7 displayed an acceptable CYP450 inhibition profile (>30 μM vs 3A4, 2D6, and 2C9, 12.5 μM vs 2C19, and 1.5 μM vs 1A2) with no upstream receptor activation (human or rat AhR or PXR) or CYP induction (human hepatocytes for CYPs 1A2, 2B6, or 3A4) noted. The lack of induction potential was confirmed in vivo in a four-day rat subchronic multiday dosing paradigm, where levels of 7 remained constant from day 1 to day 4 as measured by full plasma AUCs.31 PAM 7 was also CNS penetrant in both rat (brain-to-plasma partition coefficient, Kp, = 1.51) and cyno (Kp = 1.24), but with lower unbound partition coefficients, Kp,uus, in these species (0.09 and 0.07, respectively) due to high brain homogenate binding (fu’s ≥ 0.001). However, we have noted that in vivo efficacy in rodent haloperidol-induced catalepsy (HIC) models, with multiple lipophilic mGlu4 PAMs, correlates better with CSF levels rather than with total or unbound plasma and brain concentrations.21,22 In addition, PAM 7 was not a human P-gp substrate (ER = 1.3, Papp = 8 × 10–6 cm/s).
In vivo pharmacokinetic parameters for 7 across species were favorable for further advancement (Table 2). Importantly, 7 showed good oral bioavailability in rat (100%), mouse (79%), dog (37.5%), and cyno (31.6%) as parent API. Clearance was moderate in rat (CLp = 37.7 mL/min/kg) and cyno (CLp = 17.7 mL/min/kg), but high in dog (CLp = 31.6 mL/min/kg) and mouse (CLp = 78.3 mL/min/kg) (with acceptable elimination half-lives (t1/2 ≈ 1–4 h) and moderate volume of distribution at steady state (Vss ≈ 3–4 L/kg). The in vivo PK, coupled with metabolite identification studies across species (Figure 4), indicated that rat and cyno provided excellent coverage of human metabolites and would be the most appropriate preclinical safety species.31
Table 2. In Vivo Pharmacokinetic Parameters of VU2957 (7).
| parameter | mousea | rata (SD) | doga (beagle) | NHPa (cyno) |
|---|---|---|---|---|
| dose (mg/kg) iv/po | 1/3 | 0.2/3 | 0.5/3 | 0.2/3 |
| CLp (mL/min/kg) | 78.3 | 37.7 | 31.6 | 17.1 |
| Vss (L/kg) | 9.93 | 3.0 | 4.3 | 3.2 |
| elimination t1/2 (h) | 2.98 | 1.05 | 2.95 | 3.98 |
| Cmax (μM) po | 1.10 | 1.44 | 0.49 | 0.26 |
| Tmax (h) po | 0.42 | 2.0 | 1.0 | 6.0 |
| AUCo-inf (μM·h) po | 1.55 | 9.66 | 1.82 | 2.81 |
| F (%) po | 79 | 100 | 37.5 | 31.6 |
| total brain/total plasma (Kp) | 0.78 | 1.51 | ND | 1.24 |
| unbound brain/unbound plasma (Kp,uu) | 0.16 | 0.09 | ND | 0.07 |
Values represent means from two to three animals; ND = not determined.
Figure 4.

In vivo behavioral profile of 7. (A) VU2957 (7) dose-dependently (0.3–30 mg/kg, po) reverses haloperidol-induced catalepsy (HIC) in rats (haloperidol, 1.5 mg/kg, i.p., *p < 0.05 vs vehicle + haloperidol, N = 8–10 rats/group. At the MED of 1 mg/kg, CSF levels of 7 are 148 nM, similar to the rat in vitro EC50. (B) PAM 7 exhibits efficacy up to 6 h after 30 mg/kg p.o. administration. (C) Dose-dependent increases in CSF exposures result in dose-dependent reversals in HIC for 7. (D) PK of 7 in satellite rats mirroring the HIC study in panel B.
PAM 7 also proved to be efficacious in our standard rat HIC model (Figure 4) from which we assess the minimum effective dose (MED) required for efficacy as well as the duration of efficacy with regards to exposure.31 As shown in Figure 4A, PAM 7 provides a dose-dependent reversal (0.3 mg/kg to 30 mg/kg) in HIC in rats with a MED of 1 mg/kg p.o., correlating with a total plasma concentration of 322 nM and a CSF concentration of 148 nM (in good agreement with the rat mGlu4 EC50 of 136 nM). Moreover, PAM 7 exhibits efficacy in the HIC model for up to 6 h after a single 30 mg/kg dose (Figure 4B). As mentioned previously, efficacy in HIC tracks with CSF concentrations of 7 (Figure 4C,D).
Based on all of these data, we were able to generate preliminary human PK predictions (Table 3) using complementary methods of single-species scaling (SSS) as well as full multispecies allometry. Regardless of the method, PAM 7 was predicted to be a low-to-moderately cleared compound in human (human CLps 5.59 to 9.03 mL/min/kg) with a low/moderate predicted volume (Vds 1.04 to 2.09 L/kg) and with half-lives ranging from 2.5 to 4.5 h to support BID dosing in man (assuming stand-alone therapy).
Table 3. Predicted Human PK Parameters for VU2957 (7).
| methoda | predicted human CLp (mL/min/kg) | predicted human Vd (L/kg) |
|---|---|---|
| SSS rat | 6.87 | 2.09 |
| SSS cyno | 5.71 | 1.72 |
| allometry (R, C, M) | 9.03 | 1.68 |
| allometry (R, C, M) fu correction | 5.59 | 1.04 |
Single species scaling (SSS) of human pharmacokinetic parameters from rat or cyno monkey in vivo PK data with corrections for species differences in unbound fraction in plasma and multispecies allometric scaling of human PK with and without correction for species differences in fu. Half-life ranges from 2.5 to 4.5 h. R, rat; C, cyno monkey; M, mouse. Dog was considered an outlier with regards to clearance due to CLp ≈ QH and was not included in the multispecies allometry.
Dose escalation studies were initiated to support a maximum tolerated dose (MTD) prior to 28-day toxicology. PAM 7 is not basic, and no stable salts could be formed; therefore, dose escalation studies progressed with the free base API. In both rat and cyno, across a broad range of vehicles, plasma exposure increased in the dose range of 3–30 mg/kg p.o., but not linearly and not sufficiently to initiate 28 day toxicology with acceptable exposure margins (data not shown). Large vehicle screens and milling exercises were undertaken but proved unsuccessful. Ultimately, we identified a spray-dried dispersion (SDD) formulation of VU2957 (7) that allowed an approximately 40-fold increase in the dissolution of 7 in intestinal fluids.31,34 As can be seen in Figure 5, PAM 7 API, in an amorphous form, demonstrated an AUC after dissolution of 1516 μg/mL·min. Various suspension formulations were prepared using different polymers; one of these, HPMCP-HP55, resulted in increased solubility in suspension form (AUC of 25079 μg/mL·min, gray bar in Figure 5). This formulation was then used to create an SDD batch of VU2957, resulting in a further boost in solubility to 47413 μg/mL·min, a >40-fold increase. These data translated into a nonsink dissolution gastric transfer experiment, where the 25% VU2957/HPMCP-HP55 SDD again displayed a significant increase in solubility relative to the API.31,34
Figure 5.
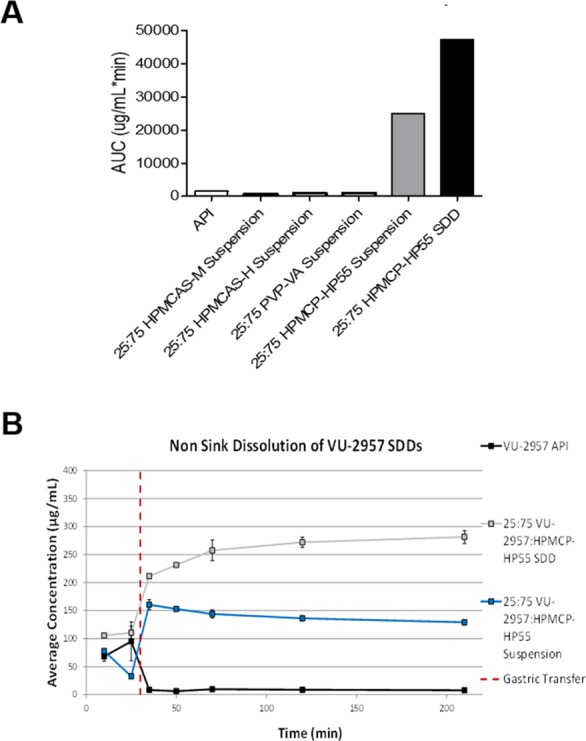
Spray-Dried Dispersion of VU2957 (7). (A) Increased solubility of the mGlu4 PAM VU2957 is achieved with an HPMCP-HP55 polymer and spray-dried dispersion (SDD) formulation. Various polymers were complexed with VU2957, and solubility in intestinal fluids, after gastric transfer, was measured. Compared to the active pharmaceutical ingredient (API) formulation, use of HPMCP-HP55 and a spray drying procedure resulted in an approximately 40-fold increase in solubility. (B) Increased solubility of the mGlu4 PAM VU2957 is achieved with a 25%/75% ratio of VU2957 to HPMCP-HP55 polymer and spray dried dispersion (SDD) formulation.
With the SDD formulation of VU2957 in hand, we revisited rat and cyno oral dose escalation studies. In both species, the SDD formulation of 7 was superior (Figure 6), providing linear dose escalation in rat (3 to 30 mg/kg in 30% Dexolve) and increasing oral bioavailability in cyno (58%F). Based on this data, we conducted “best case scenario” preliminary human dose projections by targeting total plasma concentrations that would yield a projected CSF concentration, which elicits efficacy in the rodent HIC (MED), or that would cover the in vitro EC50 in rodent and human. In doing these preliminary calculations, we found that doses between 100 and 300 mg BID would only provide a 2–4-fold margin at the projected human plasma Cmax at steady state.
Figure 6.
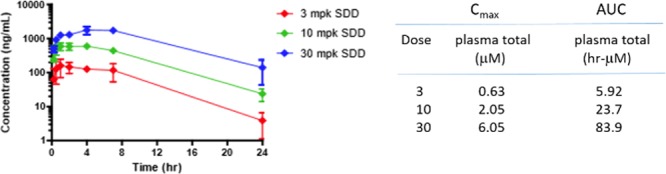
Spray-dried dispersion of VU2957 (7) in rat. The SDD formulation afforded linear dose-dependent increases in Cmax (3–30 mg/kg p.o.) and supra-linear dose escalation in AUC (total plasma) in the same dose range. N = 3.
Other scenarios, including targeting coverage of the in vivo EC50 from the rodent HIC study (which is right-shifted from the MED) using either total or unbound plasma Cmin or projected CSF concentration coverage would increase the dose of 7 dramatically (>700 mg BID). Thus, despite the formulation efforts, it became clear that PAM 7, in its current formulation, was not an appropriate choice for advancement into IND-enabling studies based on the projected inability to establish a no adverse effect level (NOAEL) at a relevant plasma concentration or reasonable human dose. Thus, VU2957 (7), a novel mGlu4 PAM, could not advance toward human clinical testing in its current formulation.
Still, 7 represents a major advance in the field, as a novel mGlu4 PAM tool compound with preclinical PK to support in vivo behavioral pharmacology studies. Moreover, VU2957 demonstrated that efficacy in HIC is sustained for up to 6 h after a single dose and that CSF exposure correlates well with in vivo efficacy. VU2957 also was a pivotal compound in demonstrating how SDD formulation is a viable solution for lipophilic GPCR PAMs to greatly enhance solubility and PK and enable dose escalation. Work with additional formulations of VU2957, as well as with close analogs of VU2957, toward clinical development are in progress and will be reported in due course.
Acknowledgments
The authors would like to thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
Glossary
ABBREVIATIONS
- mGlu
metabotropic glutamate receptor
- HIC
haloperidol-induced catalepsy
- DMPK
drug metabolism and pharmacokinetics
- NOAEL
no adverse effect level
- MED
minimum effective dose
- BID
“bis in die” which in Latin means twice a day
- PAM
positive allosteric modulator
- CSF
cerebrospinal fluid
- HPMCP-HP55
hydroxypropyl methylcellulose phthalate
- SDD
spray-dried dispersion
- API
active pharmaceutical ingredient
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00426.
General methods for the synthesis and characterization of all compounds, methods for the in vitro and in vivo DMPK protocols, and supplemental figures (PDF)
Author Contributions
C.W.L., A.L.B., C.R.H., P.J.C., C.M.N., and C.J.K. drafted/corrected the manuscript. J.D.P., D.W.E., Y.J.W., A.C., A.S.F., and J.L.E. performed the chemical synthesis. C.W.L., C.R.H., J.J.B., J.E.M., K.A.E., P.J.C., C.M.N., C.J.K., A.L.B., and A.L.R. oversaw the target selection and interpreted the biological data. A.L.R. and C.M.N. performed the in vitro molecular pharmacology studies. A.L.B. oversaw the in vitro and in vivo DMPK studies. C.K.J. performed the in vivo experiments. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Authors hold patents on mGlu4 PAMs and are actively developing mGlu4 PAMs for PD patients.
Supplementary Material
References
- Tysnes O.-B.; Storstein A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. 10.1007/s00702-017-1686-y. [DOI] [PubMed] [Google Scholar]
- Bedard P. J.; Blanchet P. J.; Levesque D.; Soghomonian J. J.; Grondin R.; Morissette M.; Goulet M.; Calon F.; Falardeau P.; Gomez-Mancilla B.; Doucet J. P.; Robertson G. S.; Di Paolo T. Pathophysiology of L-dopa-induced dyskinesias. Mov. Dis. 1999, 14, 4–8. [PubMed] [Google Scholar]
- Jenner P. Preventing and controlling dyskinesia in Parkinson’s disease-a view of current knowledge and future opportunities. Mov. Disord. 2008, 23, S585–S598. 10.1002/mds.22022. [DOI] [PubMed] [Google Scholar]
- Calabresi P.; Picconi B.; Tozzi A.; Ghiglieri V.; Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci. 2014, 17, 1022–1030. 10.1038/nn.3743. [DOI] [PubMed] [Google Scholar]
- Obeso J. A.; Rodríguez-Oroz M. C.; Benitez-Temino B.; Blesa F. J.; Guridi J.; Marin C.; Rodriguez M. Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov. Disord. 2008, 23, S548–S559. 10.1002/mds.22062. [DOI] [PubMed] [Google Scholar]
- Marino M. J.; Hess J. F.; Liverton N. Targeting the metabotropic glutamate receptor mGluR4 for the treatment of diseases of the central nervous system. Curr. Top. Med. Chem. 2005, 5, 885–895. 10.2174/1568026054750263. [DOI] [PubMed] [Google Scholar]
- Corti C.; Aldegheri L.; Somogyi P.; Ferraguti F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience 2002, 110, 403–420. 10.1016/S0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- Battaglia G.; Busceti C. L.; Molinaro G.; Giagioni F.; Traficante A.; Nicoletti F.; Bruno V. Pharmacological activation of mGluR4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Neurosci. 2006, 26, 7222–7229. 10.1523/JNEUROSCI.1595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno V.; Battaglia G.; Copani A.; D’Onofrio M.; Di Iorio P.; De Blasi A.; Melchiorri D.; Flor J. P.; Nicoletti F. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J. Cereb. Blood Flow Metab. 2001, 21, 1013–1033. 10.1097/00004647-200109000-00001. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Pin J.-P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205–237. 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Niswender C. M.; Engers D. W.; Hopkins C. R. Recent progress in the development of mGluR4 positive allosteric modulators for the treatment of Parkinson’s disease. Curr. Top Med. Chem. 2009, 9, 949–963. [PubMed] [Google Scholar]
- Hopkins C. R.; Lindsley C. W.; Niswender C. M. mGluR4-positive allosteric modulation as potential treatment for Parkinson’s disease. Future Med. Chem. 2009, 1, 501–513. 10.4155/fmc.09.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud A. J.; Engers D. W.; Lindsley C. W.; Hopkins C. R. Recent progress on the identification of metabotropic glutamate 4 receptor ligands and their potential utility as CNS therapeutics. ACS Chem. Neurosci. 2011, 2, 433–449. 10.1021/cn200043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley C. W.; Emmitte K. A.; Hopkins C. R.; Bridges T. M.; Gregory K. A.; Niswender C. M.; Conn P. J. Practical strategies and concepts in GPCR allosteric modulator discovery: Recent advances with metabotropic glutamate receptors. Chem. Rev. 2016, 116, 6707–6741. 10.1021/acs.chemrev.5b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieronska J. M.; Stachowicz K.; Palucha-Poniewiera A.; Acher F.; Branski P.; Pilc A. Metabotropic glutamate receptor 4 novel agonist LSP1–2111 with anxiolytic, but not antidepressant-like activity, mediated by serotonergic and GABAergic systems. Neuropharmacology 2010, 59, 627–634. 10.1016/j.neuropharm.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Selvam C.; Oueslati N.; Lemasson I. A.; Brabet I.; Rigault D.; Courtiol T.; Cesarini S.; Triballeau N.; Bertrand H.-O.; Goudet C.; Pin J.-P.; Acher F. C. A virtual screening hit reveals new possibilities for developing group III metabotropic glutamate receptor agonists. J. Med. Chem. 2010, 53, 2797–2813. 10.1021/jm901523t. [DOI] [PubMed] [Google Scholar]
- Selvam C.; Lemasson I. A.; Brabet I.; Oueslati N.; Karaman B.; Cabaye A.; Tora A. S.; Commare B.; Courtiol T.; Cesarini S.; McCort-Tranchepain I.; Rigault D.; Mony L.; Bessiron T.; McLean H.; Leroux F. R.; Colobert F.; Daniel H.; Goupil-Lamy A.; Bertrand H.-O.; Goudet C.; Pin J.-P.; Acher F. C. Increased potency and selectivity for group III metabotropic glutamate receptor agonists binding at dual sites. J. Med. Chem. 2018, 61, 1969–1989. 10.1021/acs.jmedchem.7b01438. [DOI] [PubMed] [Google Scholar]
- Kalinichev M.; Le Poul E.; Bolea C.; Girard F.; Campo B.; Fonsi M.; Royer-Urios I.; Browne S. E.; Uslaner J. M.; Davis M. J.; Raber J.; Duvoisin R.; Bate S. T.; Reynolds I. J.; Poli S.; Celanire S. Characterization of the novel positive allosteric modulator of the metabotropic glutamate receptor 4 ADX88178 in rodent models of neuropsychiatric disorders. J. Pharmacol. Exp. Ther. 2014, 350, 495–505. 10.1124/jpet.114.214437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennouar K.-E.; Uberti M. A.; Melon C.; Bacolod M. D.; Jimenez H. N.; Cajina M.; Kierkerian-Le Goff L.; Doller D.; Gubellini P. Synergy between L-DOPA and a novel positive allosteric modulator of metabotropic glutamate receptor 4: implications for Parkinson’s disease treatment and dyskinesisa. Neuropharmacology 2013, 66, 158–169. 10.1016/j.neuropharm.2012.03.022. [DOI] [PubMed] [Google Scholar]
- Engers D. W.; Niswender C. M.; Weaver C. D.; Jadhav S.; Menon U. N.; Zamorano R.; Conn P. J.; Lindsley C. W.; Hopkins C. R. Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs). J. Med. Chem. 2009, 52, 4115–4118. 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engers D. W.; Blobaum A. L.; Gogliotti R. D.; Cheung Y.-Y.; Salovich J. M.; Garcia-Barrantes P. M.; Daniels J. S.; Morrison R. D.; Jones C. K.; Soars M. G.; Zhuo X.; Hurley J.; Macor J. E.; Bronson J. J.; Conn P. J.; Lindsley C. W.; Niswender C. M.; Hopkins C. R. Discovery, synthesis and pre-clinical characterization of N-(3-chloro-4-fluorophenyl)-1H-pyrazolo[4,3-b]pyridin-3-amine (VU0418506), a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu4). ACS Chem. Neurosci. 2016, 7, 1192–1200. 10.1021/acschemneuro.6b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Jones C. K.; Lin X.; Bubser M.; Gray A. T.; Blobaum A. L.; Engers D. W.; Rodriguez A. L.; Loch M. T.; Daniels J. S.; Lindsley C. W.; Hopkins C. R.; Javitch J. A.; Conn P. J. Development and antiparkinsonian activity of VU0418506, a selective positive allosteric modulator of metabotropic glutamate receptor 4 homomers without activity at mGlu2/4 heteromers. ACS Chem. Neurosci. 2016, 7, 1201–1211. 10.1021/acschemneuro.6b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engers D. W.; Bollinger S. R.; Engers J. L.; Panarese J. D.; Breiner M. M.; Gregro A.; Blobaum A. L.; Bronson J. J.; Wu Y.-J.; Macor J. E.; Rodriguez A. L.; Zarmorano R.; Liang S.; Venable D.; Conn P. J.; Liindsley C. W.; Niswender C. M.; Hopkins C. R. Discovery and characterization of N-(1,3-dialkyl-1H-indazol-6-yl)-1H-pyrazolo[4,3-b]pyridin-3-amine scaffold as mGlu4 positive allosteric modulators that mitigate CYP1A2 induction liability. Bioorg. Med. Chem. Lett. 2018, 28, 2641–2646. 10.1016/j.bmcl.2018.06.034. [DOI] [PubMed] [Google Scholar]
- Bollinger S. R.; Engers D. W.; Panarese J. D.; West M.; Engers J. L.; Loch M. T.; Rodriguez A. L.; Blobaum A. L.; Jones C. K.; Thompson A. D.; Conn P. J.; Lindsley C. W.; Niswender C. M.; Hopkins C. R. The discovery, SAR and biological characterization of a novel series of 6-((1H-pyrazolo[4,3-b]pyridin-3-yl)amino)-benzo[d]isothiazole-3-carboxamides as mGlu4 positive allosteric modulators. J. Med. Chem. 2018, 10.1021/acs.jmedchem.8b00994. [DOI] [PubMed] [Google Scholar]
- Jones C. K.; Bubser M.; Thompson A. D.; Dickerson J. W.; Turle-Lorenzo N.; Amalric M.; Blobaum A. L.; Bridges T. M.; Morrison R. D.; Jadha S.; Engers D. W.; Italiano K.; Bode J.; Daniels J. S.; Lindsley C. W.; Hopkins C. R.; Conn P. J.; Niswender C. M. The metabotropic glutamate receptor 4-positive allosteric modulator VU0364770 produces efficacy alone and in combination with L-DOPA or an adenosine 2A antagonist in preclinical rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2012, 340, 404–21. 10.1124/jpet.111.187443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker M. L.; DeLong M. R.; Turchan M.; Heusinkveld L. E.; Ostrem J. L.; Molinari A. L.; Currie A. D.; Konrad P. E.; Davis T. L.; Phibbs F. T.; Hedera P.; Cannard K. R.; Drye L. T.; Sternberg A. L.; Shade D. M.; Tonascia J.; Charles D. Effects of deep brain stimulation on rest tremor progression in early stage Parkinson disease. Neurology 2018, 91, e463–e471. 10.1212/WNL.0000000000005903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace Cannard K.; Nacker M. L.; Molinari A. L.; Heusinkveld L. E.; Currie A. D.; Charles D. Recruitment and Retention in Clinical Trials of Deep Brain Stimulation in Early-Stage Parkinson’s Disease: Past Experiences and Future Considerations. J. Parkinson's Dis. 2018, 8, 421–428. 10.3233/JPD-181381. [DOI] [PubMed] [Google Scholar]
- Hacker M.; Charles D.; Finder S. Deep brain stimulation in early stage Parkinson’s disease may reduce the relative risk of symptom worsening. Parkinsonism Relat. Disord. 2016, 22, 112–113. 10.1016/j.parkreldis.2015.11.023. [DOI] [PubMed] [Google Scholar]
- https://parkinsonsnewstoday.com/2017/07/28/prexton-initiates-new-phase-2-trial-foliglurax-parkinsons-disease/.
- Charvin D.; Pomel V.; Ortiz M.; Frauli M.; Scheffler S.; Steinberg E.; Barin L.; Deshons L.; Rudigier R.; Thiarc D.; Morice C.; Manteau B.; Mayer S.; Graham D.; Giethlen B.; Brugger N.; Hedou G.; COnquet F.; Schann S. Discovery, structure-activity-relationship, and anti-parkinsonian effect of a potent and brain-penetrant chemical series of positive allosteric modulators of metabotropic glutamate receptor 4. J. Med. Chem. 2017, 60, 8515–8537. 10.1021/acs.jmedchem.7b00991. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for full details.
- Shirai T.; Kanai M.; Kuninobu Y. 2-Position-selective C-H perfluoroalkylation of quinoline derivatives. Org. Lett. 2018, 20, 1593–1596. 10.1021/acs.orglett.8b00339. [DOI] [PubMed] [Google Scholar]
- https://www.eurofinsdiscoveryservices.com.
- https://www.patheon.com/en-us.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.