

Abstract

The four members of the Janus family of nonreceptor tyrosine kinases play a significant role in immune function. The JAK family kinase inhibitor, tofacitinib 1, has been approved in the United States for use in rheumatoid arthritis (RA) patients. A number of JAK inhibitors with a variety of JAK family selectivity profiles are currently in clinical trials. Our goal was to identify inhibitors that were functionally selective for JAK1 and JAK3. Compound 22 was prepared with the desired functional selectivity profile, but it suffered from poor absorption related to physical properties. Use of the phosphate prodrug 32 enabled progression to a murine collagen induced arthritis (CIA) model. The demonstration of a robust efficacy in the CIA model suggests that use of phosphate prodrugs may resolve issues with progressing this chemotype for the treatment of autoimmune diseases such as RA.
Keywords: JAK1, JAK2, JAK3, TYK2, prodrug, mouse CIA model
The Janus Kinase (JAK) family is made up of four structurally related kinases, JAK1, JAK2, JAK3, and TYK2 (Tyrosine Kinase-2). The JAK family kinases, through the actions of specific cytokines binding to their receptors, are key drivers of immune system and inflammatory responses.1 Once these receptors are activated, the kinases phosphorylate cytokine receptors and subsequently activate signal transducers and activators of transcription (STATs). The STATs dimerize and are translocated to the nucleus where they bind to regions of genes that promote cytokine production leading to a propagation of inflammatory and immune signals. As a result of their function, the JAK family kinases have been of significant interest to the pharmaceutical industry, and various inhibitor profiles have been explored as possible treatments for autoimmune disease. Tofacitinib is a pan-JAK family inhibitor that has been approved for the treatment of rheumatoid arthritis (RA),2 psoriatic arthritis,3 and ulcerative colitis.4 Baricitinib is a selective JAK1/2 inhibitor that has been recently approved for the treatment of RA.5 Selectively targeting JAK1 would be expected to not only inhibit γc chain cytokine signaling (IL-2, IL-4, IL-7, IL-9, IL-15, IL-21), but also block signaling from other pro-inflammatory cytokines such as IL-6 and Type I interferon. Selective inhibitors of JAK1 such as figlotinib,6 upadicitnib,7 and abrocitinib8 are currently in clinical trials for the treatment of RA and other autoimmune or inflammatory diseases. Early attempts to identify JAK3 selective inhibitors suggested that JAK1 inhibition was also required to obtain potent inhibitors of γc chain cytokine signaling.9 A detailed analysis of the JAK family members Km for ATP helped explain this observation.10 This limitation toward selectively targeting JAK3 has recently been overcome by pursuing a covalent inhibition strategy, which has identified inhibitors that selectively block γc chain cytokine signaling.11−16 PF-06651600 is one such covalent inhibitor that has advanced to clinical trials. As a result of these considerations and our own difficulty in identifying potent selective noncovalent JAK3 inhibitors, we chose to pursue a JAK1/3 selective strategy in the hope of accessing a ‘JAK1 selective profile’.
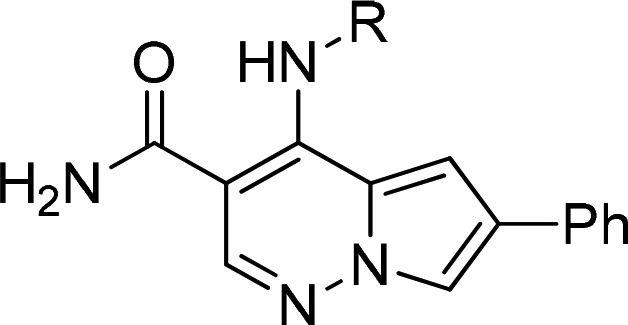
Our initial report described the discovery of pyrrolopyridazine-3-carboxamides (PPZ) as potent inhibitors of the JAK kinase family.17 Subsequently, we discovered that addition of an aryl or heteroaryl group at the C-6 position significantly increased JAK family potency. Compound 2 (Figure 1) demonstrated efficacy in a pseudoestablished mouse collagen-induced arthritis (mouse CIA) model; however, a dose response was not obtained.18 Further progression of 2 was also hampered by multiple cardiovascular liabilities, which included hemodynamic effects attributed to inhibition of Rho kinase (ROCK1 IC50 = 50 nM) and the potential for QTc prolongation due to significant inhibition of the hERG ion channel.
Figure 1.

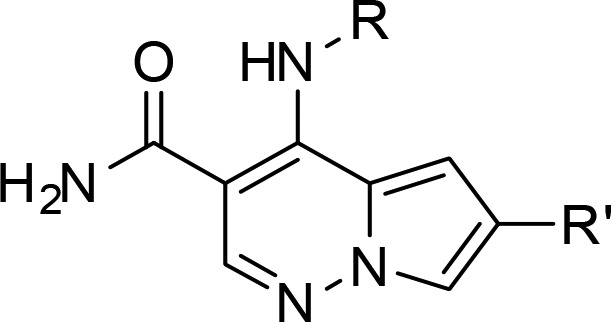
Our chemistry library plan centered on utilizing intermediate 6, 6-bromo-4-chloro-pyrrolopyridazine-3-carboxamide, to rapidly expand SAR at the 4 and 6 position in this series (Scheme 1). Reaction of 6 with amines provided 7, which could be further elaborated at C-6 via a Suzuki-Miyaura coupling procedure. However, when we attempted to conduct the Suzuki-Miyaura coupling as the first step on 6, we obtained undesired 8 as the major product. To circumvent this regiochemical outcome, 6 was reacted with ethanethiol to provide the more versatile C-6 ethylsulfide intermediate, which enabled regio-controlled installation of the aryl group at C-6 followed by oxidation of the sulfide with Oxone to afford 9a and 9b. Reaction of 9a and 9b with amines then provided the desired analog 10.
Scheme 1. Synthetic Strategy for C-4 Diversity.

Experimental details described in the Supporting Information. Scheme conditions: (a) (i) NaH, DMF; (ii) Chloramine solution in MTBE (90%); (b) (i) 3,3-diethyoxypropionitrile, IPA, 85 °C; (ii) DBU, DCE, 85 °C (c) POCl3, 75 °C. 29%, 2 steps); (d) Conc. H2SO4, 55 °C (89%); (e) R-NH2; (f) Ar–B(OH)2/Suzuki conditions; (g) EtSH, K2CO3, DMF, rt, 18 h (65%); (h) ArB(OH)2, X-Phos, Pd(OAc)2, 2 M K3PO4, dioxane 125 °C ; (i) oxone, acetone, water; (j) R-NH2, DMF, 85 °C.
Compounds were evaluated in JAK1, JAK2, JAK3, and TYK2 enzymatic assays and an IL-2 dependent T-cell proliferation assay.19 Compounds of interest were also evaluated in a human whole blood IL-2 driven IFNγ production assay (IL-2 IFNγ hWB), which provided an indication of the level of protein binding and suggested a minimal concentration to target for PK/PD and in vivo efficacy studies. Selected compounds were further profiled for cellular JAK2 activity (SET2 proliferation assay20 and an EPO phosphoSTAT5 assay) and cellular TYK2 activity (IL23 KIT225 STAT reporter assay).
Initial efforts involved fixing the C-6 substituent as phenyl while varying the C-4 position using aliphatic amines as represented by compounds 11–15 (Table 1). These analogs were potent nanomolar inhibitors of the JAK family, particularly JAK3. However, 11–15 showed poor inhibition in the IL2 IFNγ hWB assay (IC50 > 4 μM), poor metabolic stability (<50% remaining after 10 min incubation with mouse microsomes), and poor aqueous solubility (<1 μg/mL). The overall lipophilicity of the molecules (cLogP 2.9–3.7, LogP 12 = 3.94, 13 = 5.08) was suspected to be a contributing factor to the observed liabilities.
Table 1. Secondary Alkyl Amines 11–15a,b.

Assay protocols are described in the Supporting Information.
Values in table represent n = 1.
Synthesized in library format.
Subsequent efforts focused on reducing lipophilicity and improving solubility within the series,21,22 with the hope of improving whole blood potency, by incorporating a more polar 4-methoxy-3-pyridyl group at C-6 and exploring amino alcohol side chains at C-4 (Table 2). Given the potent hERG activity of 2, we avoided incorporation of basic amines as part of this SAR effort. Compounds 16, 17, 19, 21, and 22 were potent inhibitors of JAK3 and gratifyingly showed submicromolar potency in the IL-2 driven T-cell proliferation assay (IL-2 T-cell) with the more potent c-propyldiol diastereomer 19 displaying submicromolar potency in IL2 INFγ hWB (IC50 = 780 nM). Truncating the alkyl side chain in 20 resulted in a significant decrease in JAK family potency. We postulated that the cyclopropyl group might be projecting into the tofacitinib “methyl pocket”.23
Table 2. C-4 Amino Alcohol Derivativesa,c.

Assay protocols are described in the Supporting Information.
Synthesized in library format.
Values in table represent n = 1 unless otherwise noted.
T-cell proliferation assay.
Octanol–water (pH 6.5) HPLC determination.
Proceeding on this assumption we decided to explore a change of the cyclopropyl group to a methyl group to provide 21 and 22, both of which were in the same potency range as the cyclopropyl analog 19, and significantly more potent than 20. All four possible diol isomers were prepared and 22, derived from the natural amino acid, threonine, proved to be the most potent against JAK1/3, and more importantly the most potent in the human whole blood assay. On examination of the JAK3 inhibitor complex with 22, we were somewhat surprised to see that the methyl group of the secondary alcohol did not fully occupy the “methyl pocket” observed in the tofacitinib JAK3 crystal structure and the pocket was altered due to a change in the orientation of Leu956 (see Figure 2). Additionally we noticed the primary alcohol formed a hydrogen bond with the glycine rich P-loop (Leu828). It is interesting to note that diasterioisomers 23 and 25 are significantly less potent than 22. The secondary alcohol of 22 did not appear to make any specific interaction with the protein; however, other crystal structures of JAK3 (e.g., 3LXK) showed the existence of a water network in this region of the protein.
Figure 2.
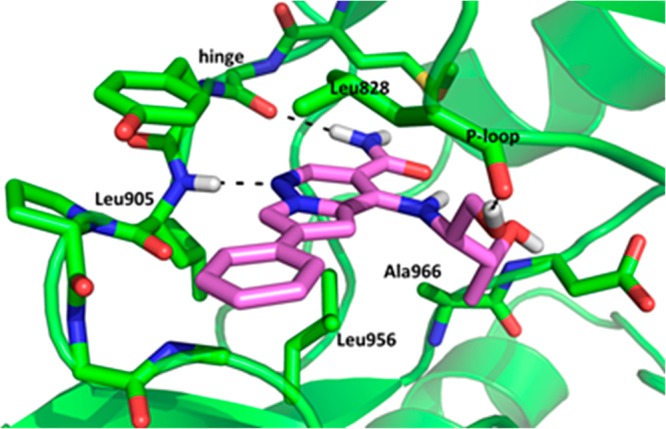
Crystal structure of compound 22 bound to the kinase catalytic domain of JAK3. The carbon atoms are colored green/magenta, oxygens are colored red, and nitrogens are colored blue. Hydrogen bonds are indicated with dashed lines.24
Compound 22 was chosen for additional profiling. 22 demonstrated reduced hERG channel activity compared to 2 (>10 fold improvement) and was deemed to have an acceptable kinome profile. Additionally, 22 was stable in human, mouse, and rat liver microsomes and was permeable in the PAMPA assay. On the basis of this profile, 22 was advanced to an oral mouse PK study. A 10 mg/kg oral dose of 22 (formulated with 5% EtOH, 5% TPGS, 90% PEG 300) provided a plasma Cmax of >3 μM and a time above the IL2-IFNγ mouseWB IC50 of about 3 h. This appeared to be a promising result, although we wanted to explore higher dose PK to allow for greater exposure during the dosing period.
After resynthesis of 22, we planned a PK study at doses of 25 and 50 mg/kg. This new lot failed to dissolve in the original formulation (even at 10 mg/kg dosing concentration). A number of formulation options were explored, but none was found to be satisfactory. The new lot was found to be crystalline and exhibited poor solubility (<1 μg/mL in 50 mM phosphate buffer). The original amorphous lot was equally insoluble in water. On the basis of these observations, we explored spray dried dispersion but were unable to prevent crystallization at higher drug loads in the matrix. Ultimately we resorted to a microsuspension formulation at a 50 mg/kg oral dose; however, the plasma Cmax for this study was only 0.1 μM. A single crystal X-ray structure of 22 shows that the compound forms a stable crystal lattice due to an extensive intermolecular hydrogen bonding matrix between neighbor molecules within the unit cell.25 This observation is also supported by a higher melting point for the dihydroxy analog 22 (280 °C dec) versus the t-butylmethyl analog 14 (204 °C dec).
Next we explored the use of a solubilizing prodrug of 22 by preparing and evaluating a phosphate of the primary alcohol (Scheme 2). Benzyl protected Boc-l-threonine (26) was reduced to the corresponding alcohol, 27, with LAH in 90% yield. Treatment with anhydrous HCl afforded the amino-alcohol hydrochloride, 28, which was treated with 6 and Hunig’s base at elevated temperature in DMA to afford 29 in a 95% yield over two steps. Suzuki-Miyaura reaction of 29 with phenylboronic acid afforded 30 in 73% yield. Phosphorylation with dibenzyl diisopropylphosphoramidite and hydrogen peroxide, followed by deprotection and salt formation, afforded the disodium salt of the phosphate 32 in good yield over three steps.
Scheme 2. Synthesis of Phosphate Prodrug (32).

Conditions: (a) LAH, Et2O, 0 °C (90%); (b) HCl, dioxane, rt (99%); (c) 6, Hunig’s base, DMA, 100 °C (95%); (d) PhB(OH)2, X-Phos, Pd(OAc)2, 2 M K3PO4, 100 °C (73%.); (e) (i) dibenzyl diisopropylphosphoramidite, tetrazole, DCM/THF, 0 °C; (ii) 30% H2O2, 0 °C (73%); (f) (i) H2, Pd/C, EtOH (74%); (ii) NaHCO3, MeOH/H2O (99%).
The phosphate prodrug (32) proved to be freely soluble in water (>1 mg/mL). We progressed the prodrug to PK and found that exposure of the parent was dose proportional (10 mg/kg, AUC ∼1 μM h; 40 mg/kg, AUC ∼6 μM h). On the basis of this promising result, we designed a mouse collagen induced arthritis study where the low dose was selected to cover the whole blood IC50 at trough for 67% of a 24 h period, and the higher dose was selected to cover the whole blood IC50 at trough over a 24 h period (mouse IL-15 WB IC50= 810 nM).26 In addition, 1 was used as a positive control and was dosed to cover the whole blood IC50 for 50% of the time. This level of coverage was targeted to approach the exposure observed at the clinical dose of 10 mg BID in humans.27 The lower dose of 32 demonstrated comparable efficacy to 1, and we were gratified to observe increased efficacy at the high dose of 32, which demonstrated that the phosphate prodrug strategy successfully enabled dose escalation (see Figure 3).
Figure 3.
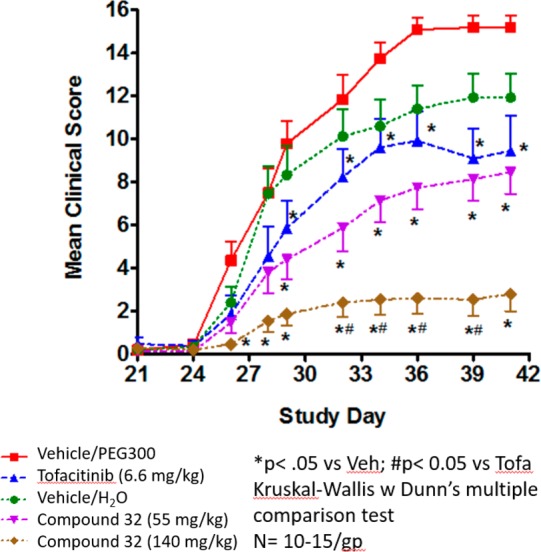
Pseudo-Established Mouse Collagen-Induced Arthritis Study with 32.* All compounds were dosed orally BID.
Drug levels of the parent 22 after dosing of 32 were close to the initial projections discussed above with the high dose achieving greater than 90% coverage of the whole blood IC50 over 24 h. This result clearly demonstrated the utility of a phosphate prodrug strategy to overcome solubility issues associated with the highly crystalline form of the parent.
In summary, we were able to identify novel pyrrolopyridazine diol 22 as a potent inhibitor of JAK1 and JAK3. The compound displayed poor PK properties, which were largely attributed to poor kinetic solubility of a highly crystalline form. However, this limitation was overcome by use of a phosphate prodrug. Prodrug 32 displayed robust dose dependent efficacy in a mouse CIA model, which demonstrated preclinically that a JAK1/3 inhibitor profile may have utility in the treatment of autoimmune disease.
Acknowledgments
We gratefully acknowledge our colleagues at BBRC and Richard A. Rampulla for the scale-up synthesis of 6-bromo-4-chloropyrrolo[1,2-b]pyridazine-3-carboxamide and our colleagues in the Lead Evaluation group for their assistance during this SAR study.
Glossary
Abbreviations
- JAK1
Janus family kinase 1
- JAK2
Janus family kinase 2
- JAK3
Janus family kinase 3
- TYK2
tyrosine kinase 2
- IL-2
interleukin 2
- IL-4
interleukin 4
- IL-6
interleukin 6
- IL-7
interleukin 7
- IL-9
interleukin 9
- IL-15
interleukin 15
- IL-21
interleukin 21
- ATP
adenosine triphosphate
- PK
pharmacokinetics
- hERG
human ether-a-go-go-related gene
- PAMPA
parallel artificial membrane permeability assay
- SAR
structure activity relationship
- GCK
germinal center kinase
- GLK
germinal center kinase like kinase
- ROCK
rho associated protein kinase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00508.
Synthetic procedures and characterization data for compounds 4–32, in vitro profile of 22, kinome selectivity, biological methods, efficacy study terminal bleed data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Schwartz D. M.; Kanno Y.; Villarino A.; Ward M.; Gadina M.; O’Shea J. J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases.. Nat. Rev. Drug Discovery 2017, 16, 843. 10.1038/nrd.2017.201. [DOI] [PubMed] [Google Scholar]
- Dhillon S. Tofacitinib: A review in rheumatoid arthritis.. Drugs 2017, 77, 1987. 10.1007/s40265-017-0835-9. [DOI] [PubMed] [Google Scholar]
- Berekmeri A.; Mahmood F.; Wittmann M.; Helliwell P. Tofacitinib for the treatment of psoriasis and psoriatic arthritis.. Expert Rev. Clin. Immunol. 2018, 14, 719. 10.1080/1744666X.2018.1512404. [DOI] [PubMed] [Google Scholar]
- Fernandez-Clotet A.; Castro-Poceiro J.; Panes J. Tofacitinib for the treatment of ulcerative colitis.. Expert Rev. Clin. Immunol. 2018, 14 (11), 881. 10.1080/1744666X.2018.1532291. [DOI] [PubMed] [Google Scholar]
- Al-Salama Z. T.; Scott L. J. Baracitinib: A review in rheumatoid arthritis.. Drugs 2018, 78, 761. 10.1007/s40265-018-0908-4. [DOI] [PubMed] [Google Scholar]
- Menet C. J.; Fletcher S. R.; Van Lommen G.; Geney R.; Blanc J.; Smits K.; Jouannigot N.; Deprez P.; van der Aar R. M.; Clement-Lacroix P.; Lepescheux L.; Galien R.; Vayssiere B.; Nelles L.; Christophe T.; Brys R.; Uhring M.; Ciesielski F.; Van Rompaey L. Triazolopyridines as selective JAK1 inhibitors: From hit identification to GLP-634.. J. Med. Chem. 2014, 57, 9323. 10.1021/jm501262q. [DOI] [PubMed] [Google Scholar]
- Serhal L.; Edwards C. J. Upadacitinib for the treatment of rheumatoid arthritis.. Expert Rev. Clin. Immunol. 2019, 15, 13. 10.1080/1744666X.2019.1544892. [DOI] [PubMed] [Google Scholar]
- Vazquez M. L.; Kaila N.; Strohbach J. W.; Trzupek J. D.; Brown M. F.; Flanagan M. E.; Mitton-Fry M. J.; Johnson T. A.; TenBrink R. E.; Arnold E. P.; Basak A.; Heasley S. E.; Kwon S.; Langille J.; Parikh M. D.; Griffin S. H.; Casavant J. M.; Duclos B. A.; Fenwick A. E.; Harris T. M.; Han S.; Caspers N.; Dowty M. E.; Yang X.; Banker M. E.; Hegen M.; Symanowicz P. T.; Li L.; Wang L.; Lin T. H.; Jussif J.; Clark J. D.; Telliez J.-B.; Robinson R. P.; Unwalla R. Identification of N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (PF-04965842): A Selective JAK1 Clinical Candidate for the Treatment of Autoimmune Diseases.. J. Med. Chem. 2018, 61, 1130. 10.1021/acs.jmedchem.7b01598. [DOI] [PubMed] [Google Scholar]
- Thoma G.; Druckes P.; Zerwes H.-G. Selective inhibitors of the Janus kinase JAK3-Are they effective?. Bioorg. Med. Chem. Lett. 2014, 24, 4617. 10.1016/j.bmcl.2014.08.046. [DOI] [PubMed] [Google Scholar]
- Thorarensen A.; Banker M. E.; Fensome A.; Telliez J.-B.; Juba B.; Vincent F.; Czerwinski R. M.; Casimiro-Garcia A. ATP-Mediated kinome selectivity: The missing link in understanding the contribution of individual JAK kinase isoforms to cellular signaling.. ACS Chem. Biol. 2014, 9, 1552. 10.1021/cb5002125. [DOI] [PubMed] [Google Scholar]
- Tan L.; Akahane K.; McNally R.; Reyskens K. M. S. E.; Ficarro S. B.; Liu S.; Herter-Sprie G. S.; Koyama S.; Pattison M. J.; Labella K.; Johannessen L.; Akbay E. A.; Wong K.-K.; Frank D. A.; Marto J. A.; Look T. A.; Arthur S. C.; Eck M. J.; Gray N. S. Development of selective covalent Janus Kinase 3 inhibitors.. J. Med. Chem. 2015, 58, 6589. 10.1021/acs.jmedchem.5b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedken E. R.; Argiriadi M. A.; Banach D. L.; Fiamengo B. A.; Foley S. E.; Frank K. E.; George J. S.; Harris C. M.; Hobson A. D.; Ihle D. C.; Marcotte D.; Merta P. J.; Michalak M. E.; Murdock S. E.; Tomlinson M. J.; Voss J. W. Tricyclic covalent inhibitors selectivity target Jak3 through an active site thiol.. J. Biol. Chem. 2015, 290, 4573. 10.1074/jbc.M114.595181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telliez J.-B.; Dowty M. E.; Wang L.; Jussif J.; Lin T.; Moy E.; Balbo P.; Li W.; Zhao Y.; Crouse K.; Dickinson C.; Symanowicz P.; Hegen M.; Banker M. E.; Vincent F.; Unwalla R.; Liang S.; Gilbert A. M.; Brown M. F.; Hayward M.; Montgomery J.; Yang X.; Bauman J.; Trujillo J. I.; Casimiro-Garcia A.; Vajdos F. F.; Leung L.; Geoghegan K. F.; Quazi A.; Xuan D.; Jones L.; Hett E.; Wright K.; Clark J. D.; Thorarensen A.; Li L. Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem. Biol. 2016, 11, 3442–3451. 10.1021/acschembio.6b00677. [DOI] [PubMed] [Google Scholar]
- Thorarensen A.; Dowty M. E.; Banker M. E.; Juba B. M.; Jussif J.; Lin T.; Vincent F.; Czerwinski R. M.; Casimiro-Garcia A.; Unwalla R.; Trujillo J. I.; Liang S.; Balbo P.; Che Y.; Gilbert A. M.; Brown M. F.; Hayward M.; Montgomery J.; Yang X.; Soucy S.; Hegen M.; Wadsworth C.; Langille J.; Vajdos F. F.; Chrencik J. E.; Telliez J.-B. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J. Med. Chem. 2017, 60, 1971–1993. 10.1021/acs.jmedchem.6b01694. [DOI] [PubMed] [Google Scholar]
- Kempson J.; Ovalle D.; Guo J.; Wrobleski S. T.; Lin S.; Spergel S. H.; Duan J. J-W.; Jiang B.; Lu Z.; Das J.; Yang B. V.; Hynes J.; Wu H.; Tokarski J.; Sack J. S.; Khan J.; Schieven G.; Bladd Y.; Chaudhry C.; Salter-Cid L. M.; fura A.; Barrish J. C.; Carter P. H.; Pitts W. J. Discovery of highly potent, selective covalent inhibitors of JAK3.. Bioorg. Med. Chem. Lett. 2017, 27, 4622. 10.1016/j.bmcl.2017.09.023. [DOI] [PubMed] [Google Scholar]
- Casimiro-Garcia A.; Trujillo J. I.; Vajdos F.; Juba B.; Banker M. E.; Aulabaugh A.; Balbo P.; Bauman J.; Chrencik J.; Coe J. W.; Czerwinski R.; Dowty M.; Knafels J. D.; Kwon S.; Leung L.; Liang S.; Robinson R. P.; Telliez J.-B.; Unwalla R.; Yang X.; Thorarensen A. Identification of cyanamide-based Janus kinase 3 (JAK3) covalent inhibitors.. J. Med. Chem. 2018, 61, 10665. 10.1021/acs.jmedchem.8b01308. [DOI] [PubMed] [Google Scholar]
- Duan J. J.-W.; Lu Z.; Jiang B.; Yang B. V.; Doweyko L. M.; Nirschl D. S.; Haque L. E.; Lin S.; Brown G.; Hynes J.; Tokarski J. S.; Sack J. S.; Khan J.; Lippy J. S.; Zhang R. F.; Pitt S.; Shen G.; Pitts W. J.; Carter P. H.; Barrish J. C.; Nadler S. G.; Salter-Cid L. M.; McKinnon M.; Fura A.; Schieven G. L.; Wrobleski S. T. Discovery of Pyrrolo[1,2-b]pyridazine-3-carboxamides as Janus Kinase (JAK) inhibitors.. Bioorg. Med. Chem. Lett. 2014, 24, 5721. 10.1016/j.bmcl.2014.10.061. [DOI] [PubMed] [Google Scholar]
- Hynes J.; Wu H.; Kempson J.; Duan J. J.W.; Lu Z.; Jiang B.; Tokarski J. S.; Sack J. S.; Khan J.; Lippy J. S.; Zhang R. F.; Pitt S.; Shen G.; Gillooly K.; McIntyre K.; Carter P.; Barrish J. C.; Salter-Cid L. M.; Fura A.; Schieven G. L.; Pitts W. J.; Wrobleski S. T. Discovery of potent and efficacious pyrrolpyridazines as dual JAK1/3 inhibitors.. Bioorg. Med. Chem. Lett. 2017, 27, 3101. 10.1016/j.bmcl.2017.05.043. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for in vitro assay descriptions and profiling data and in vivo model description for compound 22.
- Quentmeier H.; MacLeod R. A. F.; Zaborski M.; Drexler H. G. JAK2 V617F tyrosine kinase mutation in cell lines derived from myeloproliferative disorders.. Leukemia 2006, 20, 471. 10.1038/sj.leu.2404081. [DOI] [PubMed] [Google Scholar]
- O’Brien Z.; Fallah Moghaddam M. Small molecule kinase inhibitors approved by the FDA from 2000 to 2011: a systematic review of preclinical ADME data.. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1597. 10.1517/17425255.2013.834046. [DOI] [PubMed] [Google Scholar]
- Balimane P. V.; Han Y.-H.; Shong S. Permeability and transporter models in drug discovery and development: PAMPA Caco-2, Pgp and beyond.. Int. Drug Dis. 2010, 5, 83. [Google Scholar]
- Chrencik J. E.; Patny A.; Leung I. K.; Korinski B.; Emmons T. L.; Hall T.; Weinberg R. A.; Gormley J. M.; Day J. E.; Hirsch J. L.; Kiefer J. R.; Leone J. W.; Fischer H. D.; Sommers C. D.; Huang H.-C.; Jacobsen E. J.; Tenbrink R. E.; Tomasselli A. G.; Benson T. E. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6.. J. Mol. Biol. 2010, 400, 413. 10.1016/j.jmb.2010.05.020. [DOI] [PubMed] [Google Scholar]
- PDB ID = 6NY4
- See Supporting Information for compound 22 single crystal X-ray information. CCDC ID = 1896615
- Fereshteh M. P.; Li X.; Li S.; Yi F.; Zhang R.; Farr G. A.; Kolodin G.; Lippy J.; Naglich J. G.; Schieven G.; Schweizer L.; Zhang L. Development of a human whole blood screening platform to monitor JAK/STAT signaling using high-throughput flow cytometry.. J. Biomol. Screening 2016, 21, 866. 10.1177/1087057116645095. [DOI] [PubMed] [Google Scholar]
- Krishnaswami S.; Boy M.; Chow V.; Chan G. Safety, tolerability, and pharmacokinetics of single oral doses of tofacitinib, a Janus kinase inhibitor, in healthy volunteers.. Clin. Pharmacol. Drug Dev. 2015, 4, 83. 10.1002/cpdd.171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.