

Abstract
Increasing the success rate and throughput of drug discovery will require efficiency improvements throughout the process that is currently used in the pharmaceutical community, including the crucial step of identifying hit compounds to act as drivers for subsequent optimization. Hit identification can be carried out through large compound collection screening and often involves the generation and testing of many hypotheses based on available knowledge. In practice, hypothesis generation can involve the selection of promising chemical structures from compound collections using predictive models built from previous screening/assay results. Available physical collections, typically used during hit identification, are of the order of 106 compounds but represent only a small fraction of the small molecule drug-like chemical space. In an effort to survey a larger portion of chemical space and eliminate inefficiencies during hit identification, we introduce a new process, termed Idea2Data (I2D) that tightly integrates computational and experimental components of the drug discovery process. I2D provides the ability to connect a vast virtual collection of compounds readily synthesizable on automated synthesis systems with computational predictive models for the identification of promising structures. This new paradigm enables researchers to process billions of virtual molecules and select structures that can be prepared on automated systems and made available for biological testing, allowing for timely hypothesis testing and follow-up. Since its introduction, I2D has positively impacted several portfolio efforts through identification of new chemical scaffolds and functionalization of existing scaffolds. In this Innovations paper, we describe the I2D process and present an application for the discovery of new ULK inhibitors.
Keywords: Hit identification, virtual screening, automated synthesis, ULK1 serine/threonine protein kinase
A key step in the small molecule drug discovery process involves the identification of so-called hit compounds, chemical structures with measurable although typically weak activity that can serve as tools for subsequent target specific exploration and optimization. The success of hit identification (HI) often relies on the availability of large, diverse, drug-like compound collections intensively searched against targets of interest. In this setting, it is only natural to conclude that the larger and more diverse the collection, the larger the coverage of the chemical space and therefore the higher the possibility of identifying a new, promising hit structure.
Drug discovery organizations maintain physical collections on the order of 105–106 compounds that are routinely used for primary screening campaigns and structure–activity relationship (SAR) elucidation. Maintaining such collections comes at significant cost (compound synthesis/acquisition, storage, distribution, plating, replenishment, etc.). Putting such collections to use in the form of physical screening requires substantial resources, time, and cost.1 Accordingly, the time required to initiate a screening campaign and obtain results can range from a few days for small sets of e.g. 104 compounds to three months for compound collections of 106.2 Time and cost requirements naturally limit the applicability of such methodologies to large, well-resourced organizations. In an effort to manage costs but also expedite the identification of hits from such collections, virtual screening (VS) approaches, the computational counterpart of experimental screening, may be used provided that enough knowledge and appropriate computational predictive models exist to guide compound selection.3 VS may also be used to process virtual compound collections consisting of chemical structures that can be purchased from vendors or virtual compounds that are believed synthesizable using current means and synthetic knowledge. Such virtual collections are not only intended to lower the cost and speed-up the HI process but also provide access to larger sections of chemical space. Once virtual hits are identified and determined to be of interest, follow-up involves compound acquisition or synthesis and experimental verification.
Recently we introduced the Proximal Lilly Collection (PLC), Eli Lilly’s version of a virtual, synthesizable compound collection.4 In this work we lay out the Idea2Data (I2D) paradigm, a new approach to early discovery chemistry designed to provide holistic support spanning from chemical structure design to synthesis, purification, and biological testing. I2D aspires to bridge the chemical synthesis design and experience in our organization, largely captured by the PLC, with automated synthesis capabilities and quantitative biology to meet the needs of ongoing discovery chemistry projects and thereby enable the exploitation of a larger chemical space. This new drug discovery paradigm is founded on the tight integration of computational methods for virtual hit identification, with experimental processes such as automated synthesis, purification, and testing. Emphasis is placed on the integration of research efforts from distinct units of a discovery chemistry organization, which render I2D implementation and use possible. We also present an application example of I2D to identify ULK1 kinase inhibitors.
Hit Identification
Hit identification may be initiated in a number of ways depending on the type and availability of project related information. If known ligands exist, analog-based methods, e.g., similarity search, may be used to select compounds.5 Similarly, if a high quality target structure is available, docking techniques can be used to predict the binding affinity of chemical structures.6 In the case where no adequate information is available, experimental high-throughput screening (HTS) is often used to probe the target receptor and provide ideas for follow up.2 Subsequent rounds of HI take advantage of data acquired during the process. In a typical scenario, following initial hit identification attempts through virtual and/or experimental screening, a project team reviews all available information and proposes one or more hypotheses with the support of computational techniques, expert knowledge, and intuition. Synthetic chemists plan and execute the route to chemical structure synthesis. The resulting material is delivered to the analytical group for quality assessment and purification. If the reaction has been successful, the purified sample is sent for experimental screening. Upon completion of this process, the screening results are provided to the project team for consideration in the next round of hypothesis generation. Data is thoroughly recorded during each step of this learning cycle and is frequently used to optimize future exploration. A significant number of learning cycles is often required before molecular design has matured to a stage where the compound characteristics meet project requirements for biological activity and selectivity together with favorable toxicology, formulation, and pharmacokinetics. Due to the number of cycles required and the length of time for each cycle, the process can be both time and resource expensive.
The above learning cycle requires contributions from numerous highly specialized groups and involves multiple creativity and process-driven operations and data handoffs.7 Each participating group has well established practices guiding its operations and ensuring high productivity and quality of work. The presence of many groups each with its own objectives, the diverse background of the scientists involved and, more often than not, the geographical isolation between groups impede collaboration and information flow. Analysis of the steps of this process suggests that a sizable fraction of time to go from compound design to data generation is of no value attributed to sample handoffs and the manipulation of compounds for the next step.8 As well as increasing cycle time, typically ranging between 2 and 5 weeks, inefficient sample handoffs also lead to greater sample consumption and higher material cost. The task of a process owner is to monitor progress, communicate information, and ensure that obstacles are overcome timely. However, the complexity and scale of the process make efficient management challenging. New approaches are needed to streamline HI while enabling investigation of larger portions of chemical space especially given the interest in novel pharmaceutical targets for which, in all likelihood, current physical collections are not well suited.4 A path forward, explored in this Innovations, is to integrate the various discovery tasks and expand technological frontiers as needed. Alternative promising approaches with similar goal have been reported in the literature. Interested readers are referred to refs (9−12).
Proximal Lilly Collection Implementation
Efforts to explore the diversity of the drug-like chemical space and identify structurally novel, potentially promising regions for pharmaceutical development have been hampered by the sheer number of theoretically feasible compounds and practical concerns on the synthesizability of the chemical structures proposed. In a typical setting, such virtual compounds are conceived through the enumeration of the products of chemical reactions when supplied building blocks appropriate to the specific reaction. Alternatively, virtual compounds may be proposed simply through the permutation of a set of atoms or fragments abiding to some rudimentary chemical structure rules. In either case the result is a large virtual collection of chemical structures of questionable synthesizability and, therefore, practical use. To address this problem we have implemented the Proximal Lilly Collection designed to bridge the chemical synthesis knowhow at Eli Lilly with the needs of ongoing discovery chemistry projects.4 The PLC exploits the capabilities of our Automated Synthesis Lab (ASL) system,13 which served as the main motivation for this work. In practical terms, the availability of PLC empowers Lilly Discovery Chemistry scientists to routinely access and investigate a chemical space in the order of 1011 compounds for the purposes of their daily pharmaceutical endeavors. The PLC design guarantees that synthesis can be attempted with high probability of success on any structure identified on internal robotic systems using available reactants. The confidence is crucial to user acceptance of the system and the success of the I2D initiative. A detailed description of the technology enabling PLC search and exploitation is provided in ref (4). An application of PLC for the discovery of RIO2 inhibitors can be found in ref (14).
Idea2Data Process
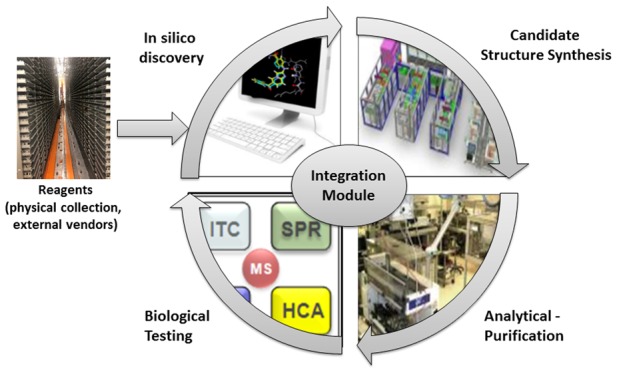
The Idea2Data process was developed by considering every interface between steps from molecular design to data generation. These steps include in silico design, chemical synthesis, purification/characterization, compound management, and biological testing (see Figure 1). In order to reduce the time and material transitioning from idea to data, innovative automation and process optimization is utilized. Critical to this optimization is matching the required input sample format for each step to the output format of the previous step, thus avoiding additional sample reformatting. Another key activity is sample concentration determination at the time of characterization using quantitative NMR; aliquots from this characterized and quantified solution are subsequently used for all future testing, thus removing time-consuming steps of sample drying, manual weighing, and reformatting.
Figure 1.
Schematic representation of the I2D process.
In addition to physical sample considerations, management of sample metadata is also important. The process is optimized for seamless data flow between each step avoiding manual reformatting of information and multiple submission, tracking, and result systems. Unlike several reported systems that fully integrate key steps of the learning cycle into one single closed platform, Idea2Data is designed to utilize the wide range of existing assays available to project teams within Lilly. Rapid access to a large number of assays, coupled with a wide range of chemistries available on the ASL and encoded in the PLC, provides scientists with flexibility to test hypotheses retrieved from a large, diverse chemical space. Overall, the process provides a project team with tools to perform a much higher number of design loops in a given time frame, thereby increasing the quality and efficiency of hypothesis generation.
The first step of an I2D project involves the use of computational techniques to identify promising PLC compounds for synthesis. Utilities for near neighbor chemical structure search may be used if the project needs require the identification of similar compounds to a known ligand. If the goal is to explore the structure–activity relationship landscape of a compound or scaffold, tools to enumerate focused libraries based on the input structure may be used. More traditional virtual screening methods can be used when appropriate pharmacophore or docking models exist, to select structures from diverse PLC subsets. Once a candidate set of PLC compounds has been identified, synthetic profiling is performed to retrieve detailed information on the reaction needed to prepare each compound as well the reactants involved including source and structure.
Research scientists can interact with PLC via multiple interfaces including custom command line tools, Knime15 nodes and MD316 utilities to “grow” a scaffold with PLC reactions and building blocks. Exploiting PLC through traditional virtual screening techniques uses enumerated PLC structure sets prepared specifically for this purpose. The process involves the use of computational models as filters to search through large compound collections to identify virtual hits.5,6 If properly designed, VS workflows can process very large compound collections in little time and, virtually, no cost. The PLC sets used for VS vary in size from a few million to several billion and may be sampled quasi-randomly from the entire PLC space or custom designed to satisfy certain user defined objectives.4 While virtual screening is common-place in modern drug discovery efforts, VS workflows for PLC libraries typically need to process much larger numbers of compounds. For this purpose, custom computational infrastructure has been developed that exploits modern hardware to cope with very large databases in order to complete virtual screens in a reasonable amount of time. For instance, the PLC infrastructure has been used to process data sets in the order 109 and successfully identify promising structures (vide infra).
The list of virtual PLC hits identified, with synthetic profiling information included, is forwarded to the I2D chemical synthesis team. This team leads the effort to review, select, and synthesize PLC structures using available in-house resources, primarily the Automated Synthesis Lab. The ASL is a state-of-the-art innovative central synthesis suite, which integrates synthesis, analysis, isolation, evaporation, information management, and automation technologies into one system.13 The ASL has proven utility in minimizing the burden of repetitive, routine, rule-based operations, and delivering solutions in versatile synthetic transformations commonly used in drug discovery efforts, such as C–C bond cross coupling, C–N bond formation, oxidation, reduction, and heterocyclic formation. As the suite permits the synthesis to be designed, submitted, and manipulated under the direction of a remote user at his or her desktop in real-time, the ASL has become a valuable tool to address synthetic needs in the discovery value chain regardless of geographic location. With demonstrated synthetic capabilities and workflow processes, the ASL is an integral part of the PLC initiative. Conversely, PLC projects drive the enhancement of synthetic technologies and capabilities amenable to the ASL to augment applicability and user experiences.
Prior to synthesis, library size and quantity of materials to be prepared must be determined. In a typical I2D run, 50–150 chemical structures are selected for synthesis on the ASL from 100 to 300 PLC hits based on an overall desirability assessment. Based on this, chemists select the specific chemical reactions and conditions (solvent, concentration, temperature, duration of the reaction) to be used during synthesis. If multiple synthetic steps are required, development of a one-pot synthetic sequence to increase the synthesis efficiency is often considered. In addition, selection of a water miscible solvent for the reaction is desirable to facilitate purification with reverse phase chromatography without any additional workup. Targets to be prepared with the same reaction type are grouped together to achieve efficiencies in synthesis and purification.
Purification of products can be carried out using the Automated Purification Laboratory (APL). Samples from the ASL are typically received predissolved, and aliquots are dispensed to microtiter plates and prepared appropriately for the execution of purity analysis by, e.g., LCMS and NMR. Preparative chromatography is also performed on each sample using the solvent system selected by the analyst. All run parameters as well as analysis of results at each step are recorded and used for sample assessment. Upon completion of the process, the remainder of each sufficiently pure sample is plated in assay-expected format and forwarded for experimental testing.
Successfully synthesized and purified structures are sent to the collaborating quantitative biology team. The compounds may be subjected to any available biochemical assay to assess potency to the target of interest. Biophysical methods can also be used. The latter are particularly useful to measure weak potency or verify binding affinity to the appropriate receptor site, especially when the biochemical assay resolution is low or the I2D design relies primarily on the use of virtual screening.
Integration Challenges
The pharmaceutical industry is organized similarly to most other modern industries involved in the research and development of complex, knowledge intensive products: clusters of highly specialized expert teams with clear objectives and priorities focus on specific tasks of the process under the supervision of a group head. As new requests for work are received, they are assigned to the team queue and processed based on priority. Each of these expert teams plays a crucial role in the process. Together they form a drug discovery production line coordinated by a pharmaceutical project manager, typically a senior medicinal chemist with good understanding of the work performed by each group but little, if any, involvement in the management of any group.
The I2D approach promotes a radically different approach focusing on the tight integration of all teams to form a coherent project team. Recognizing that the teams involved have been functioning independently with limited coordination of operations at a managerial level, we placed emphasis on eliminating communication barriers and facilitating information flow. Central to these efforts has been the implementation of a management software tool to act as a central repository of project related information and a progress monitoring and management instrument. The tool captures the process from the in silico hypothesis generation step through to compound registration and has been accessible to all team members to ensure real time information flow. Figure 2 provides an overview of the tool components and information flow.
Figure 2.

Schematic representation of the I2D management tool. The purpose of the tool is to capture the I2D process and facilitate progress monitoring and exchange of information among stakeholders.
The idea generation step of the process includes the identification of virtual hits using the in silico discovery approach described above and the loading of those hits into the tool. A cross functional team meets to discuss the list of virtual hits and select which will move forward for synthesis. It is common to have more than one synthetic route to several virtual hits and repeated use of a specific building block. It is also common to have multiple choices for each building block, and therefore, the team needs to carefully select not only virtual hits but also synthetic routes to maximize the number of synthesis experiments and provide for all reactions from the best possible source. To support the selection process the tool provides information on the available synthetic routes for a given structure and current available reactant inventory. Reactants necessary for the candidates selected for synthesis are examined in detail, and an automated vial selection process is used to order the optimal samples. For the purposes of this task, attributes such as creation date, available analytical data, and QC score (i.e., structure and purity confirmations) are collected and considered. ASL experts implement the appropriate synthesis workflow for each candidate and, upon reactant delivery, initiate synthesis. The products are forwarded to the APL laboratory for purification and characterization. Successfully synthesized candidates are registered and forwarded to the quantitative biology team for experimental testing. An attrition analysis component prepares a visual representation of progression with statistics and notes on the outcome at each step of the process. The use of the tool allows the tracking of time for each phase of a given project and, thus, helps identify issues and inefficiencies in the process.
Idea2Data in Practice
The I2D platform provides access to the PLC space in numerous ways. In the remainder of this section, we describe and discuss a virtual screening application using a large, diverse PLC subset. An analog search-based application utilizing PLC has been described in ref (14).
Case Study
ULK1 Inhibitors. The PLC provides an excellent opportunity to access a large chemical space for biological active exploration. It also represents several challenges due to the sheer size of the chemical space available, currently upward of 350 billion compounds. The size of this virtual library requires a well thought out virtual screening strategy in order for it to yield interesting active compounds. Considerations of the virtual screening methods must include elements of speed and accuracy to make use of the large virtual space. For example, in typical high throughput screening campaigns, the hit rate is 1 per 100 000 compounds screened for “easy” targets.17 This level of hit rate is acceptable for screening large libraries, but significant enrichment is required for design of synthetic libraries on the order of hundreds of compounds. Thus, a hit rate enrichment on the order of 1000-fold or higher is required to find hits in 100-member compound libraries.
In order to prospectively examine the use of I2D for hit identification, we selected a target and modeling method that met the speed and accuracy requirements needed for PLC size space. To this end we chose a target with some existing screening data from which accurate models could be constructed, but also could benefit from identification of novel actives. ULK1, an oncology target involved in autophagy, fit all of these criteria. ULK1 and ULK2 or (unc)-51-like kinase 1 and 2, have been shown to play a role in autophagy which is a critical process for cell survival mechanism in conditions of external nutrient deprivation.18,19 Cancer cells are often operating in such stress conditions especially under treatment with other chemotherapeutic agents. As such, inhibitors of ULK1 are of interest for oncology applications.20 There were very few reports of ULK1/2 inhibitors, thus strategies for inhibitor discovery must include screening of compound collections. Fortunately, Lilly has been engaged in routine compound profiling of kinases in order to identify inhibitors as well as for the generation of selectivity data.21,22 Utilization of this data to build models has been shown previously to be highly effective in not only designing potent kinase inhibitors but also showing very high accuracy in the prediction of active compounds.23,24 The I2D prospective design strategy for ULK1 inhibitors is shown in Figure 3. The outlined strategy begins with a haystack of compounds, in this case, generated by PLC subset enumeration (vida supra). Specifically, the PLC annotated reactions were used to create a library with available reactants using a sampling approach taking into account reactant space.4 In total, 950 million compounds were enumerated and served as the starting collection for the ULK1 library design.
Figure 3.

I2D flow scheme used for ULK1 library design.
Library Design
Iteration 1
In order to focus the PLC haystack to ULK1 actives, a staged protocol using various models was undertaken. Initial filtering was carried out to remove molecules with more than 40 non-hydrogen atoms and those more likely to be false positives according to a set of heuristics developed to identify promiscuous and reactive compounds.25 Given the difficulty in identifying actives in such a large space, top priority was given to ULK1 binding. Additionally, solubility was considered a top criterion given its importance in synthesis and testing. For both ULK1 and solubility, support-vector machine (SVM) predictive models were chosen due to their speed and accuracy. This was demonstrated previously in prospective virtual screening and library design efforts that yielded hit rates up to 84 and 92%, respectively.24 In the current application, biochemical ULK1 enzyme inhibition data26 gathered from a general profiling effort was used to train SVM models for use in selection of compounds. Specifically, the ULK1 activity model was trained on % inhibition data at 20 μM for 5620 compounds. Ten-fold cross validation studies using a random 25% of the set for training and the remainder as a test set yielded an average cross-validated r2 and q2 of ca. 0.5. A solubility model was also used to filter the PLC haystack. The latter model, also using the SVM methodology, was constructed on 20 378 internally measured data points to classify compounds into two classes, soluble and insoluble. A total of 6881 compounds in the training set were classified as soluble, at least 0.1 mg per solvent mL at pH 6, with the remaining 13 497 compounds forming the insoluble class. A training procedure similar to that of the inhibition model described above was followed with the final model yielding 79% accuracy, specificity of 89%, and selectivity of 74%. These two SVM predictive models and the heavy atom count filter reduced the PLC set to roughly 145K compounds. Additional filters for novelty and physical properties were also used (see Figure 3). In particular, it was noticed that a number of compounds contained the familiar donor–acceptor of the amino-aromatic nitrogen hinge binding motif common to many known kinase inhibitors. It was decided to remove these molecules for increased novelty; therefore, a substructure filter was applied reducing the PLC set to ∼50K. A further restriction of the property space on size (35 heavy atoms), lipophilicity (clogP < 5), flexibility (# of rotatable bonds < 5), and the degree of aromaticity (percentage of sp3 carbon atoms >25%) was applied leaving ∼10K compounds.
Selection from this final set included maximizing the number of chemotypes. This was accomplished by clustering the compounds into 249 groups based on chemical structure similarity.27 Since the ultimate goal was to synthesize approximately 100 total compounds, an expert driven cluster-based selection of 285 was first made to maximize the number of clusters while balancing predicted activity and similarity to compounds already existing in the Lilly collection. A plot of the final selection of compounds with their respective cluster number versus similarity distance to the nearest neighbor in the Lilly collection and colored by predicted activity is shown in SI1(a). A total of 179 out of 249 clusters making up the final focused 10K library were represented. No more than four compounds were selected from any one cluster, and near neighbor (NN) distances from existing compounds ranged from 0.12 to 0.34.
With the assurance of a spread in chemotype, the final set of 76 compounds was selected from the 285 based on synthetic considerations, specifically predicted activity, reactivity, selectivity, and available inventory of input reactants. Structures resulting from various PLC reaction types are represented including amide coupling, SnAr, reductive amination, and Buchwald cross coupling with products from the latter in the majority. The building blocks for each of the 76 selected target compounds were retrieved from storage, and each of the reactions was performed on the ASL. LCMS analysis was also performed on the ASL. Successful reaction mixtures were sent to APL for purification where samples were subjected to both LCMS and NMR analysis (see SI2 section). Overall, of the 76 compounds targeted for synthesis, 23 were successfully made, purified, and assayed in biochemical and NMR assays.
In order to assess the compounds for ULK1 inhibition, a combination of biophysical and biochemical methods was employed. Specifically, the ULK1 enzyme inhibition assay, i.e., the transcreener ADP-FP (fluorescent polarization) assay26 and an NMR assay (see SI2) were used. The results from the ULK1 biochemical FP assay showed activity for two of the 23 compounds (17.6 and 18.2 μM IC50). In addition, another three compounds showed evidence of binding in the NMR study. These results, a hit rate greater than 20%, are quite promising given the daunting size of the initial PLC database. The 23 compounds made are quite diverse, representing 20 of the 179 clusters, and show a range of similarity within the 10K initial haystack. The five actives, although somewhat weak binders, come from five different clusters. This is important to expand the range of ULK1 inhibitor scaffolds available for consideration in lead optimization, the main goal of this experiment. The position of the PLC compounds assayed with respect to the distance to the nearest compound in the Lilly collection and the cluster membership space is shown in SI1(b).
Iteration 2
In order to leverage the five ULK1 actives, a second iteration was performed. The PLC experiment is uniquely equipped for iterating on actives given the reactants and coupling reactions leading to the active compounds are already set up and available. In this case, the reactants making up the three NMR actives along with their near neighbors were used for PLC enumeration, yielding a total of 259 compounds (see ExpandSearch PLC search method in ref (4)). Due to the low synthetic success rate in the first round, reaction workflows were further scrutinized. A total of 116 compounds of the total 259 were selected based on synthetic feasibility, chemotype (compounds from four clusters) and predicted activity (>50% Inh). Of the 116, 71 (61%) were successfully made, purified, and tested.
Due to the weak binding of the novel compounds found in the first round, an initial biochemical screen was run at 100 μM. This high concentration biochemical data plus chemical diversity was used to select a subset of 24 compounds to be examined in the NMR binding assay. Of the chosen 24, 12 showed binding in NMR assay. Figure 4 plots show that the actives arose from three scaffolds and represented novel and diverse chemical space compared to known ULK1 actives. Figure 4a shows that the closest near neighbor distance to a known ULK1 active reported in the Chembl database28 of curated literature bioactive compounds is 0.34, well beyond typical high similarity cut-offs. Even compared to the in-house actives from screening, as shown in Figure 4b, most of these compounds represent new chemical space. One of the compounds made, however, was similar to previous actives with a near neighbor distance of 0.1. These expanded scaffolds along with those identified in the first PLC iteration were added to the compounds from general screening for consideration for lead optimization. An example of one of the scaffolds is a benzotriazolecarboxamide (BTC), which is very unique for a kinase hinge-binding motif. In order to verify the binding mode, the crystal structure of ULK1 in complex with BTC was determined and refined at 1.73 Å resolution (see Figure 5, SI3). The high resolution permitted the unambiguous placement of all non-hydrogen ligand atoms further confirmed by calculating the final omit electron density map with only the ligand molecule excluded from the map calculation. All non-hydrogen ligand atoms with hydrogen bond donor or acceptor potential are involved in hydrogen bonds with good geometry (see SI3).
Figure 4.

Plot of scaffold type vs the near neighbor distance to ULK1 actives reported in (a) the Chembl database and (b) the Lilly collection. ULK1 active compounds are in green, inactive in red.
Figure 5.
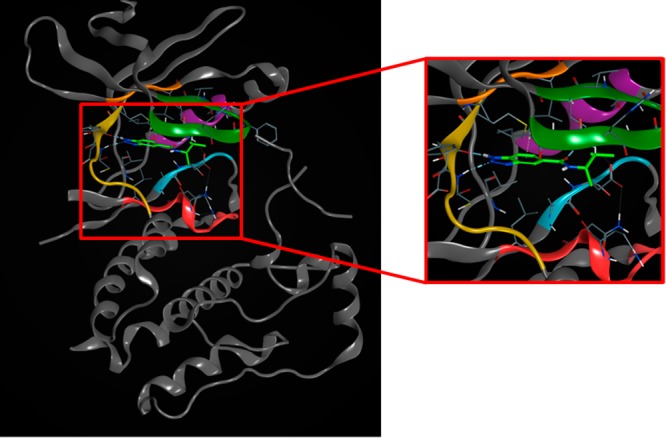
X-ray crystal structure of BTC bound to ULK1.
The triazole moiety of BTC engages the ULK1 hinge binding site with two typical hydrogen bonds observed in many crystal structures from kinase inhibitors containing five-membered heterocycles, e.g., pyrazole. However, the orientation of the fused ring system is very unusual. While typical fused heterocycles binding to the kinase hinge do so alongside the hinge in a parallel fashion engaging the hinge with both rings, the benzotriazole binds perpendicular to the hinge. As a consequence, the edge of the benzene ring packs tightly against the sulfur atom of the gate keeper methionine thereby excluding water from the hydrophobic pocket between the hinge and the gatekeeper. The amide moiety attached to the benzene ring is positioned to accept the hydrogen bond from the catalytic lysine, which in turn is engaged to the conserved glutamic acid of the so-called C-helix, locking the kinase domain in an active conformation. A nearest neighbor found in the public domain, the CDK2 inhibitor structure PDB ID: 3R8M, contains an indazole superimposing very well with the benzotriazole of BTC. However, the hydrazine amide attached to the pyrazole ring of 3R8M inhibitor engages the CDK2 hinge in a third hydrogen bond resulting in the appearance of a classical hinge binder.29
A member of scaffold 2, Figure 4, shows that this class of ULK1 inhibitors is very unique compared to both literature and in-house scaffolds. This structure is also fairly unique within the manifold of kinase inhibitors with available bound X-ray structures. This is further highlighted by the near-neighbor distance plot to kinase ligands from the PDB shown in Figure 6.
Figure 6.
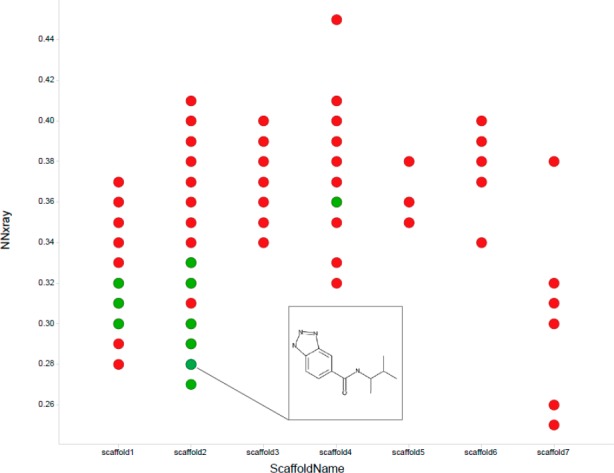
Plot of scaffold type vs the near neighbor distance to compounds with kinase bound X-ray structures in the PDB. ULK1 active compounds are in green, inactive in red.
It is worth noting the increase in synthesis success rate from 30% in iteration 1 (23 made from 76 attempted) to 61% in iteration 2 (71 from 116). An attrition analysis following iteration 1 indicated that problems with reactant purity explained over 20% of the reaction failures. Lessons from the remaining failures were used to update PLC annotation rules to improve future synthesis attempts. The increase in iteration 2 is attributed to the quality control of reactants and the use of combinations of reactants and reaction conditions similar to those successful in round 1. Similar attrition analyses are regularly performed to summarize learnings and improve the process. Iteration 2 also required noticeably less time since the experimental and computational infrastructure was already in place.
The goal of this proof of concept experiment was to investigate the feasibility of Idea2Data, a new, paradigm-shifting process for early drug discovery process. The process combined cutting edge computational and automation technologies to answer the question: “Can novel biologically active molecules be designed, synthesized, tested, and optimized in rapid development cycles using reaction informatics, virtual screening, and automated synthesis platforms?” The application on ULK1 took approximately 3 months to complete including experiment design, computational model preparation, virtual screening, compound synthesis, purification, and testing for both iterations performed. This pilot study indicates the ability to both, identify novel hit compounds and significantly expedite the hypothesis generation to testing cycle.
Conclusions
The last decade of the 20th century has marked a turn for the science of drug discovery. The traditional approach relying on leads from nature in the search for new medicinal agents,30 complemented with human intuition and expert labor has been gradually transforming into a rationalized, data driven process following the introduction of several disruptive technologies. For the first time, unprecedented numbers of chemical structures could be delivered by combinatorial chemistry. High-throughput screening, enabled by robotic systems, provided the means to test such large collections to identify compounds of pharmaceutical interest. This was just the beginning as a revolution in biology and genomics was starting to elucidate the human genome fueling hopes for the identification of new targets for drug discovery. In such an environment, computational chemistry and bio- and cheminformatics thrived, and algorithms intended to address the needs of the evolving drug discovery process, including virtual screening, were introduced.
The transformation has been gradual and not without setbacks. Once the initial enthusiasm for combinatorial chemistry settled researchers realized that the quality and diversity of the libraries produced was often not up to required standards for use.30 Early HTS systems, although capable of handling very large collections of compounds, produced results with considerable noise in the form of false positives and negatives.31 Similarly, the conclusion of the human genome project did not immediately lead to the identification of multiple, readily druggable pharmaceutical targets; rather, as concluded early on due to the multitude of evidence, mere presence of a biological marker in a disease state (correlation) does not imply causation.32 Such setbacks have proven temporary. Combinatorial chemistry concepts are resurfacing in, e.g., DNA encoded libraries, now incorporating DNA tags to facilitate hit follow-up efforts coupled with affinity-based screening for rapid identification of promising hit structures.33 Investments in automation are resulting in much improved data quality of HTS2, automated synthesis systems such as the ASL13 and the Abbvie platform,10 and lab-on-chip platforms for chemical synthesis and purification.34 These technologies, combined with advancements in synthetic knowledge, are providing practical access to sections of the chemical space much larger and more diverse than ever before.4,35 Custom cheminformatics systems designed to manage and search chemical spaces of such dimensions enable in silico screening for the rapid identification of promising structures.36,37 Overall, the technologies introduced seem to have reached a level of maturity to become indispensable tools in the search for new drugs. Alas, the drug discovery process has largely remained the same, centered on a sequential paradigm that progresses chemical structures from conception to assessment in steady, slow steps, and as a probable consequence, the average number of NMEs per year has not seen an analogous increase, an indication that may simply reflect the innovative capacity of the current R&D model.38,39
Idea2Data is an initiative aiming to disrupt the existing paradigm and increase the throughput of drug discovery efforts by exploiting technological advancements holistically. The early application presented herein has convinced us that such initiative is now well within our reach. The interdisciplinary team formed to support I2D completed two rounds of in silico design to compound preparation and testing resulting in novel hits for ULK in a timely manner. We expect that as I2D enters production the time required per cycle will be significantly reduced. The task is both complex and challenging as it requires the redesign of established discovery workflows (i) to allow close cross-department cooperation by stakeholders from the design, synthesis, purification, and quantitative biology groups and (ii) to incorporate the use of advanced automation tools to streamline process-driven steps of the HI effort. This new paradigm, heavily relying on establishing communication channels among researchers that often have few opportunities for interaction, can contribute to better coordination of activities and enable exploitation of modern, higher performance tools. It is the opinion of the authors that such steps are necessary to fully exploit the capabilities of modern tools and drastically improve the productivity of modern drug discovery.
Acknowledgments
The authors would like to acknowledge Dr. Sheehan for his strong encouragement during the implementation of the project and his contribution to the Idea2Data initiative vision. We would also like to thank Drs. Beck, Weidner, Godfrey, and McLoughlin, as well as numerous colleagues from the Computational Chemistry and Cheminformatics, Synthesis, Analytical and Purification groups at Eli Lilly and Co. for their work that led to the meticulous planning and successful implementation of the ULK1 I2D pilot project. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357 (https://www.aps.anl.gov/Science/Publications/Acknowledgment-Statement-for-Publications). Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility http://lrlcat.lilly.com/
Glossary
ABBREVIATIONS
- APL
Automated Purification Laboratory
- ASL
Automated Synthesis Laboratory
- BTC
benzotriazolecarboxamide
- HI
hit identification
- HTS
high throughput screening
- I2D
Idea2Data
- NN
near neighbor
- PLC
Proximal Lilly Collection
- SVM
support vector machines
- ULK1
serine/threonine protein kinase
- VS
virtual screening
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00488.
Plots of the selected PLC set of 285 compounds and the compounds prepared; detailed description of the methods; crystal structure of the complex of ULK1 and BTC, 6MNH (PDF)
Author Present Address
† Lilly Biotechnology Center, 10290 Campus Point Drive, San Diego, CA 92121.
Author Present Address
‡ Centro de Investigación Lilly, SA, Avenida de la Industria 30, 28108 Alcobendas, Spain.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. * These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Dahlin J. L.; Walters M. A. The Essential Roles of Chemistry in High-Throughput Screening Triage. Future Med. Chem. 2014, 6, 1265–1290. 10.4155/fmc.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarron R.; Banks M. N.; Bojanic D.; Burns D. J.; Cirovic D. A.; Garyantes T.; Green D. V.; Hertzberg R. P.; Janzen W. P.; Paslay J. W.; Schopfer U.; Sittampalam G. S. Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discovery 2011, 10, 188–195. 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- Glick M.; Jacoby E. The Role of Computational Methods in the Identification of Bioactive Compounds. Curr. Opin. Chem. Biol. 2011, 15, 540–546. 10.1016/j.cbpa.2011.02.021. [DOI] [PubMed] [Google Scholar]
- Nicolaou C. A.; Watson I. A.; Hu H.; Wang J. The Proximal Lilly Collection: Mapping, Exploring and Exploiting Feasible Chemical Space. J. Chem. Inf. Model. 2016, 56, 1253–1266. 10.1021/acs.jcim.6b00173. [DOI] [PubMed] [Google Scholar]
- Riniker S.; Fechner N.; Landrum G. A. Heterogeneous Classifier Fusion for Ligand-Based Virtual Screening: Or, How Decision Making by Committee Can Be a Good Thing. J. Chem. Inf. Model. 2013, 53, 2829–2836. 10.1021/ci400466r. [DOI] [PubMed] [Google Scholar]
- Spyrakis F.; Cavasotto C. N. Open Challenges in Structure-Based Virtual Screening: Receptor Modeling, Target Flexibility Consideration and Active Site Water Molecules Description. Arch. Biochem. Biophys. 2015, 583, 105–119. 10.1016/j.abb.2015.08.002. [DOI] [PubMed] [Google Scholar]
- Ullman F.; Boutellier R. A Case Study of Lean Drug Discovery: From Project Driven Research to Innovation Studios and Process Factories. Drug Discovery Today 2008, 13, 543–550. 10.1016/j.drudis.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Weller H. N.; Nirschl D. S.; Paulson J. L.; Hoffman S. L.; Bullock W. H. Addressing the Medicinal Chemistry Bottleneck: A Lean Approach to Centralized Purification. ACS Comb. Sci. 2012, 14, 520–526. 10.1021/co300075g. [DOI] [PubMed] [Google Scholar]
- Gesmundo N. J.; Sauvagnat B.; Curran P. J.; Richards M. P.; Andrews C. L.; Dandliker P. J.; Cernak T. Nanoscale Synthesis and Affinity Ranking. Nature 2018, 557, 228–232. 10.1038/s41586-018-0056-8. [DOI] [PubMed] [Google Scholar]
- Baranczak A.; Tu N. P.; Marjanovic J.; Searle P. A.; Vasudevan A.; Djuric S. W. Integrated Platform for Expedited Synthesis-Purification-Testing of Small Molecule Libraries. ACS Med. Chem. Lett. 2017, 8, 461–465. 10.1021/acsmedchemlett.7b00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitrago Santanilla A.; Regalado E. L.; Pereira T.; Shevlin M.; Bateman K.; Campeau L. C.; Schneeweis J.; Berritt S.; Shi Z. C.; Nantermet P.; Liu Y.; Helmy R.; Welch C. J.; Vachal P.; Davies I. W.; Cernak T.; Dreher S. D. Organic Chemistry. Nanomole-Scale High-Throughput Chemistry for the Synthesis of Complex Molecules. Science 2015, 347, 49–53. 10.1126/science.1259203. [DOI] [PubMed] [Google Scholar]
- Desai B.; Dixon K.; Farrant E.; Feng Q.; Gibson K. R.; van Hoorn W. P.; Mills J.; Morgan T.; Parry D. M.; Ramjee M. K.; Selway C. N.; Tarver G. J.; Whitlock G.; Wright A. G. Rapid Discovery of a Novel Series of Abl Kinase Inhibitors by Application of an Integrated Microfluidic Synthesis and Screening Platform. J. Med. Chem. 2013, 56, 3033–3047. 10.1021/jm400099d. [DOI] [PubMed] [Google Scholar]
- Godfrey A. G.; Masquelin T.; Hemmerle H. A Remote-Controlled Adaptive Medchem Lab: An Innovative Approach to Enable Drug Discovery in the 21st Century. Drug Discovery Today 2013, 18, 795–802. 10.1016/j.drudis.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Varin T.; Godfrey A. G.; Masquelin T.; Nicolaou C. A.; Evans D. A.; Vieth M. Discovery of Selective Rio2 Kinase Small Molecule Ligand. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 1630–1636. 10.1016/j.bbapap.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Berthold M. R.; Cebron N.; Dill F.; Gabriel T. R.; Kötter T.; Meinl T.; Ohl P.; Sieb C.; Thiel K.; Wiswedel B.. Knime: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications: Proceedings of the 31st Annual Conference of the Gesellschaft Für Klassifikation E.V., Albert-Ludwigs-Universität Freiburg, March 7–9, 2007; Preisach C., Burkhardt H., Schmidt-Thieme L., Decker R., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2008; pp 319–326.
- Zhang H.; Wang J.; Gao C.; Nicolaou C.; Humblet C. Md3: A Computational Application to Support Drug Discovery Process Natl. Meet.—Am. Chem. Soc., Div. Comp. Chem., COMP 2013, 216. [Google Scholar]
- Spencer R. W. High-Throughput Screening of Historic Collections: Observations on File Size, Biological Targets, and File Diversity. Biotechnol. Bioeng. 1998, 61, 61–67. 10.1002/(SICI)1097-0290(199824)61:1<61::AID-BIT11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Lum J. J.; DeBerardinis R. J.; Thompson C. B. Autophagy in Metazoans: Cell Survival in the Land of Plenty. Nat. Rev. Mol. Cell Biol. 2005, 6, 439–448. 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- Cheong H.; Lindsten T.; Wu J.; Lu C.; Thompson C. B. Ammonia-Induced Autophagy Is Independent of Ulk1/Ulk2 Kinases. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 11121. 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Ouyang L.; Guo Y.; Zhang J.; Liu B. Unc-51-Like Kinase 1: From an Autophagic Initiator to Multifunctional Drug Target. J. Med. Chem. 2018, 61, 6491–6500. 10.1021/acs.jmedchem.7b01684. [DOI] [PubMed] [Google Scholar]
- Vieth M.; Sutherland J. J.; Robertson D. H.; Campbell R. M. Kinomics: Characterizing the Therapeutically Validated Kinase Space. Drug Discovery Today 2005, 10, 839–846. 10.1016/S1359-6446(05)03477-X. [DOI] [PubMed] [Google Scholar]
- Gao C.; Cahya S.; Nicolaou C. A.; Wang J.; Watson I. A.; Cummins D. J.; Iversen P. W.; Vieth M. Selectivity Data: Assessment, Predictions, Concordance, and Implications. J. Med. Chem. 2013, 56, 6991–7002. 10.1021/jm400798j. [DOI] [PubMed] [Google Scholar]
- Erickson J. A.; Mader M. M.; Watson I. A.; Webster Y. W.; Higgs R. E.; Bell M. A.; Vieth M. Structure-Guided Expansion of Kinase Fragment Libraries Driven by Support Vector Machine Models. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 642–652. 10.1016/j.bbapap.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Vieth M.; Erickson J.; Wang J.; Webster Y.; Mader M.; Higgs R.; Watson I. Kinase Inhibitor Data Modeling and De Novo Inhibitor Design with Fragment Approaches. J. Med. Chem. 2009, 52, 6456–6466. 10.1021/jm901147e. [DOI] [PubMed] [Google Scholar]
- Bruns R. F.; Watson I. A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zalameda L.; Kim K. W.; Wang M.; McCarter J. D. Discovery of Acetyl-Coenzyme a Carboxylase 2 Inhibitors: Comparison of a Fluorescence Intensity-Based Phosphate Assay and a Fluorescence Polarization-Based Adp Assay for High-Throughput Screening. Assay Drug Dev. Technol. 2007, 5, 225–235. 10.1089/adt.2006.045. [DOI] [PubMed] [Google Scholar]
- MacCuish J. D.; MacCuish N. E.. Clustering in Bioinformatics and Drug Discovery; CRC Press, 2010. [Google Scholar]
- Bento A. P.; Gaulton A.; Hersey A.; Bellis L. J.; Chambers J.; Davies M.; Kruger F. A.; Light Y.; Mak L.; McGlinchey S.; Nowotka M.; Papadatos G.; Santos R.; Overington J. P. The Chembl Bioactivity Database: An Update. Nucleic Acids Res. 2014, 42, D1083–1090. 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzi S.; Alam R.; Han H.; Becker A.; Schonbrunn E. PDB Id: 3r8m, Cdk2 in complex with inhibitor L3–3.
- Gershell L. J.; Atkins J. H. A Brief History of Novel Drug Discovery Technologies. Nat. Rev. Drug Discovery 2003, 2, 321–327. 10.1038/nrd1064. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. Virtual Screening of Chemical Libraries. Nature 2004, 432, 862–865. 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol A.; Mosca R.; Farre J.; Aloy P. Unveiling the Role of Network and Systems Biology in Drug Discovery. Trends Pharmacol. Sci. 2010, 31, 115–123. 10.1016/j.tips.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Goodnow R. A.; Dumelin C. E.; Keefe A. D. DNA-Encoded Chemistry: Enabling the Deeper Sampling of Chemical Space. Nat. Rev. Drug Discovery 2017, 16, 131. 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Keng P. Y.; Chen S.; Ding H.; Sadeghi S.; Shah G. J.; Dooraghi A.; Phelps M. E.; Satyamurthy N.; Chatziioannou A. F.; Kim C. J.; van Dam R. M. Micro-Chemical Synthesis of Molecular Probes on an Electronic Microfluidic Device. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 690–695. 10.1073/pnas.1117566109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q.; Peng Z.; Kostrowicki J.; Kuki A. Leap into the Pfizer Global Virtual Library (Pgvl) Space: Creation of Readily Synthesizable Design Ideas Automatically. Methods Mol. Biol. 2011, 685, 253–276. 10.1007/978-1-60761-931-4_13. [DOI] [PubMed] [Google Scholar]
- Peng Z.; Yang B.; Mattaparti S.; Shulok T.; Thacher T.; Kong J.; Kostrowicki J.; Hu Q.; Na J.; Zhou J. Z.; Klatte D.; Chao B.; Ito S.; Clark J.; Sciammetta N.; Coner B.; Waller C.; Kuki A. Pgvl Hub: An Integrated Desktop Tool for Medicinal Chemists to Streamline Design and Synthesis of Chemical Libraries and Singleton Compounds. Methods Mol. Biol. 2011, 685, 295–320. 10.1007/978-1-60761-931-4_15. [DOI] [PubMed] [Google Scholar]
- Virshup A. M.; Contreras-Garcia J.; Wipf P.; Yang W.; Beratan D. N. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. J. Am. Chem. Soc. 2013, 135, 7296–7303. 10.1021/ja401184g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munos B. Lessons from 60 Years of Pharmaceutical Innovation. Nat. Rev. Drug Discovery 2009, 8, 959–968. 10.1038/nrd2961. [DOI] [PubMed] [Google Scholar]
- Cheshire D. R. How Well Do Medicinal Chemists Learn from Experience?. Drug Discovery Today 2011, 16, 817–821. 10.1016/j.drudis.2011.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.