

Abstract

We encountered a dilemma in the course of studying a series of antagonists of the G-protein coupled receptor CC chemokine receptor-2 (CCR2): compounds with polar C3 side chains exhibited good ion channel selectivity but poor oral bioavailability, whereas compounds with lipophilic C3 side chains exhibited good oral bioavailability in preclinical species but poor ion channel selectivity. Attempts to solve this through the direct modulation of physicochemical properties failed. However, the installation of a protonation-dependent conformational switching mechanism resolved the problem because it enabled a highly selective and relatively polar molecule to access a small population of a conformer with lower polar surface area and higher membrane permeability. Optimization of the overall properties in this series yielded the CCR2 antagonist BMS-741672 (7), which embodied properties suitable for study in human clinical trials.
Keywords: Chemokine, G protein-coupled receptor, conformational analysis, hydrogen bonding, oral bioavailability, ion channels
Drug-discovery efforts are frequently characterized by challenges in obtaining compounds with the right combination of biological activity, off-target selectivity, and oral bioavailability. Our studies on a series of antagonists of the G-protein coupled receptor CCR2 have uncovered similar issues, as have many other programs at different institutions.1−3 Although each research program is unique, general correlations between physicochemical properties and drug candidate attrition have now been posited.4 Herein, we describe the discovery of a protonation-dependent conformational switching mechanism that enabled the discovery of small molecules with both sufficient polarity to engender selectivity and the ability to adopt a more hydrophobic conformation to facilitate oral bioavailability. This approach enabled us to identify a CCR2 antagonist suitable for clinical studies and may be valuable for other scientists engaged in drug design.
CCR2 is a G protein-coupled receptor that plays a central role in the biology of classical “inflammatory” monocytes,5 mediating their emigration from the bone marrow and extravasation into inflamed tissues,6 wherein they differentiate into either monocyte-derived macrophages or monocyte-derived dendritic cells.7 Unlike their tissue-resident counterparts, CCR2+ macrophages promote inflammation through the secretion of cytokines and other inflammatory mediators.8 Consistent with this paradigm, gene knockout or knockdown studies have implicated CCR2 as playing a contributory role in numerous preclinical analogs of human diseases, including models of diabetes, atherosclerosis, autoimmune disease, and cancer.9−12 Our own efforts focused on the development of small molecule CCR2 antagonists, as highlighted by the two representative molecules below (Figure 1).
Figure 1.

Compounds with similar CCR2 binding affinity but strikingly different overall profiles.
Compound 1, a potent CCR2/CCR5 dual antagonist, exhibited good oral bioavailability in the rat, as well as mouse, monkey, and dog.13 Although it was selective over hERG, as assessed by a >2000-fold difference in CCR2 binding IC50 (0.7 nM) and its hERG patch clamp IC50 (1,800 nM), our subsequent detailed in vivo electrophysiology studies suggested that the molecule did not have a sufficient safety margin for advancement. In contrast, the CCR2-selective antagonist 2 exhibited high selectivity over hERG, which was confirmed in vivo but did not have good oral bioavailability in the rat, mouse, or monkey.14 To improve oral bioavailability and ion channel selectivity in our trisubstituted cyclohexyl series, we engaged in extensive structure–activity relationship (SAR) studies of the exocyclic amine, cyclohexyl C3-side chain, and benzamide region.15 Generally speaking, our efforts were aimed at introducing polarity into analogs of 1 and hydrophobicity into analogs of 2, in an attempt to find a compound with more-balanced properties.16 A particularly noteworthy result emerged from the replacement of the proximal methylene of sulfone 2 with an amine to give sulfonamide 3 (Table 1). Although the sulfonamide exhibited similar CCR2 and CCR5 binding affinities to the sulfone, it had lower hERG selectivity and, to our surprise, higher PAMPA permeability, making it closer to the n-propyl series (cf. 1–3, Table 1). In contrast, the N-methyl sulfonamide 4, which was ostensibly more lipophilic than 3, had good hERG selectivity and poor PAMPA permeability, like the polar sulfone 2. The differences in permeability and metabolism between compounds 2 and 4 were confirmed in vivo. Relative to sulfone 2, sulfonamide 3 exhibited higher oral bioavailability in mouse (35 versus 16 F%) despite having higher clearance (258 versus 70 mL/min/kg). In contrast, N-Me sulfonamide 4 exhibited poor mouse pharmacokinetics, like 2 (IV Cl: 67 versus 70 mL/min/kg; PO F% 11 versus 16 mL/min/kg).
Table 1. Key in Vitro Data for Analogs of 1 and 2a.

| compound | X | CCR2 binding (IC50, nM) | CCR5 binding (IC50, nM) | hERG patch clamp (percent inhibition) | PAMPA permeability (nanometers per second) | HLM (rate, percent remaining) |
|---|---|---|---|---|---|---|
| 2 | CH2SO2CH3 | 1.8 ± 0.8 (12) | 1636 ± 760 (6) | 4%, 10 μM | 94 ± 58 (10) | 0.00, 100% |
| 3 | NHSO2CH3 | 1.9 ± 0.8 (10) | 3469 ± 906 (3) | 45%, 10 μM | 593 (1) | 0.05, 85% |
| 4 | N(Me)SO2CH3 | 1.4 ± 0.6 (8) | 1570 (1) | 7%, 10 μM | 65 ± 43 (3) | 0.00, 100% |
| 1 | CH2CH2CH3 | 0.7 ± 0.3 (23) | 2.4 ± 1.0 (200) | 83%, 10 μM | 529 ± 157 (9) | 0.00, 100% |
| 5 | CH2SO2CH3 | 0.9 ± 0.3 (5) | 190 (1) | 16%, 30 μM | 14 (1) | 0.00, 99% |
| 6 | NHSO2CH3 | 0.4 ± 0.20 (5) | 287 ± 196 (2) | 74%, 10 μM | 394 ± 340 (2) | 0.13, 58% |
| 7 | NHC(O)CH3 | 1.1 ± 0.7 (18) | 780b | 33%, 10 μM | 443 ± 114 (8) | 0.01, 95% |
Both binding values are reported as mean plus or minus the standard deviation (n). CCR2 binding was measured through blockade of MCP-1 (CCL2) binding to human peripheral blood mononuclear cells. CCR5 binding was assessed through antagonism of MIP-1β binding to HT1080 cells stably expressing CCR5. Inhibition of hERG was assessed in a manual patch clamp assay and is expressed as percent inhibition at a single test concentration. PAMPA permeability (reported in nanometers per second) was assessed at room temperature and a pH of 7.4; data are reported as the average of a single experiment performed in triplicate (standard deviation of <10% mean) or as the average of multiple runs (with standard deviation across those experiments). The metabolism in human liver microsomes at a substrate concentration of 3 μM is shown as a rate of disappearance (nanomoles per minus per milligram) and percent parent remaining (10 min).
The CCR5 binding for this lead compound 7 was also assessed in native T-cells, wherein it exhibited a binding value of 23.6 ± 12 nM.
The replacement of the sulfone side chain with a sulfonamide was also studied in the context of the quinazoline capping group. The sulfonamide 6 retained the CCR2 selectivity of the sulfone 5 but exhibited improved PAMPA permeability (Table 1). It was surprising to us that sulfonamide 6 had a PAMPA Pc that was similar to that of the n-propyl analog 1, despite the marked difference in their polarity (cLog P of 1.0 versus 4.7). We synthesized a range of compounds that contained different side chain motifs and studied the PAMPA permeability of those that exhibited appropriate CCR2 affinity. A selection of these compounds is shown in Figure 2. A clear structure–property relationship was evident: those compounds that contained an −N(H)R motif as the linking unit on the C3 side chain (Figure 2, blue data series) exhibited higher PAMPA values than those that did not (Figure 2, red data series). This trend held largely independent of cLog P, although there were exceptions at the high and the low ends of the cLog P range (as expected).
Figure 2.

Shown is a plot of cLog P vs PAMPA (normalized to reference standard 1) for a series of analogs from the trisubstituted cyclohexyl series. All of these compounds are analogs of 1, 5, and 6 in that they contain the N-isopropyl, N-methyl-amine motif, the γ-lactam linker, and the quinazoline capping group.
Relative to their sulfone counterparts, both sulfonamides exhibited worse metabolic stability (Table 1, cf. 3 and 2 as well as 6 and 5). Intriguingly, the N-Me analog 4, which had a new metabolic soft spot, exhibited superior metabolic stability to the parent 3 in both human and rodent liver microsomes. Analysis of the in vitro metabolites indicated that the dominant metabolic pathway for 2 and 3 was identical: oxidative demethylation of the tertiary amine. Quite remarkably, the simple acetamide did not exhibit this metabolic liability and proved to be one of the most effective replacements of the sulfonamide that we identified in this survey. Indeed, acetamide 7 retained potent CCR2 binding activity while exhibiting high hERG selectivity, PAMPA permeability, and in vitro metabolic stability (Table 1).17 Furthermore, compound 7 (BMS-741672) had improved mouse oral bioavailability (28 F%) and IV half-life (38 mL/min/kg) relative to 2–4, so we advanced it into additional studies (see below and ref (17)).
The optimized first-generation synthesis of compound 7 is shown below in Scheme 1. Our previously described18 key intermediate S.1 was subjected to a four-step sequence19 to install the γ-lactam. The hydrolytic opening of S.2 to give acid S.3 was followed by coupling with ammonia to yield the carboxamide, which was subjected to Hoffman rearrangement (including trapping with acetic anhydride) to yield acetamide S.4. Conversion of S.4 to the key compound BMS-741672 (7) followed a simple sequence consisting of acid-mediated Boc removal, double-reductive amination, hydrogenolysis of the Cbz, and coupling with 4-chloro-6-trifluoromethylquinazoline. This route served to provide material for all preclinical studies and was an important precursor to our recently reported second-generation route.20
Scheme 1. Synthesis of BMS-741672 (7).

Reagents and conditions: (a) 1 atm H2, Pd/C, EtOAc; (b) Cbz-Met-OH, TBTU, iPr2NEt, MeCN; (c) MeI, 48 h; (d) Cs2CO3, DMSO; (e) LiOH, THF/H2O; (f) EDC, HOBt, CH2Cl2, then NH3; (g) PhI(OAc)2, MeCN/H2O, then Ac2O, iPr2NEt; (h) TFA, CH2Cl2; (i) acetone, NaBH3CN, MeOH; (j) CH2O, NaB(OAc)3H, CH2Cl2; (k) 1 atm H2, Pd/C, MeOH; (l) 4-Cl,6-CF3-quinazoline, iPr2NEt, iPrOH.
In the context of investigating the optimal formulation of compound 7 to support clinical trials, we conducted a number of small molecule crystal studies with various salt forms. The conformation of the molecule depended on whether the compound was studied as a salt or free base. Specifically, when studied as an amine salt, we found that the lactam of 7 was axial, while the acetamide and amine moieties were equatorially disposed (Figure 3A, orange, Lactam Axial DiEquatorial, “LADE”). This conformation matched that of the receptor-bound,21 protonated 1 (Figure 3C, blue) as well as the TFA salt of compound 6 (Figure 3C, orange; see also Figure 3D). The free base of compound 1 was also shown to exhibit this LADE conformation (Figure 3C, red). However, the crystalline form of the free base of 7 displayed a very different conformation, in which the cyclohexyl chair flipped such that the lactam was equatorial, while the acetamide and amino moieties were diaxially disposed (Figure 3A, blue, lactam equatorial diaxial, “LEDA”). Note that the acetamide bridges the tertiary amine and quinazoline NH via intramolecular H-bonds in this LEDA conformation (Figure 3B). The pH-dependent conformational switching of 7 that was observed in the solid state was further confirmed in solution studies via 1H NMR spectroscopy (see the Supporting Information).
Figure 3.
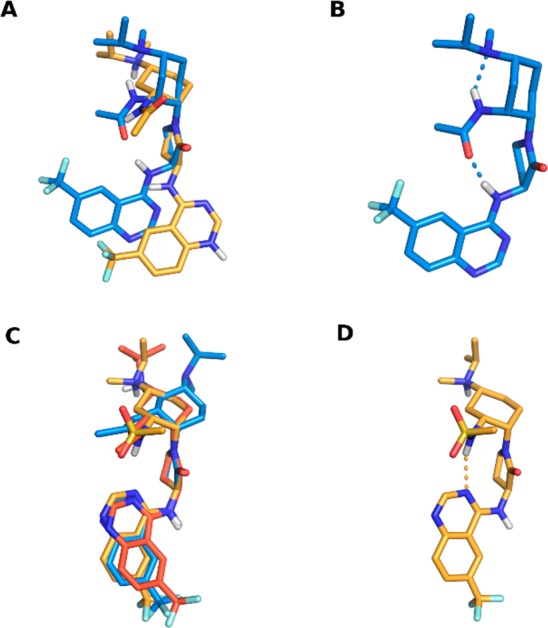
X-ray crystal structures of compounds 1, 6, and 7. (A) The bis-benzenesulfonic acid (omitted for clarity) salt of 7 (orange) exists in the chair configuration with γ-lactam disposed axially. The free base of 7 (blue) exists in the chair configuration with the γ-lactam disposed equatorially. (B) Hydrogen-bonding network in the 7 free-base crystal structure. (C) The X-ray structures of the free-base of 1 (red) and TFA (omitted for clarity) salt of 6 (orange) overlaid with the CCR2-bound conformation of the protonated form of 1 (blue) extracted from the 1/CCR2 complex crystal structure (PDB ID: 5T1A). All exist in the LADE chair conformation. (D) Hydrogen-bonding network in the 6 TFA salt crystal structure.
The aforementioned crystal structures were coupled with ab initio calculations to enable a detailed understanding of the protonation-dependent conformational biases within these systems. When the tertiary amine is protonated, the LADE chair conformation is lower in energy for every substituent in the third position (Table 2, protonated; see the Supporting Information for additional details). However, in the free-base form, the conformational preferences vary depending on the nature of the C3 substituent. The polar moieties in compounds 6 (sulfonamide) and 7 (acetamide) are able to engage the hydrogen bonding network observed in the compound 7 free-base crystal structure (Figure 3B). These hydrogen bonds stabilize the LEDA conformation and overcome the unfavorable steric clashes inherent to the diaxial disposition of the two substituents. Indeed, calculations predict the LEDA conformation to be favored by 4.8 and 5.4 kcal/mol for compounds 6 and 7, respectively (Table 2, free base). Compound 5 shows a smaller calculated bias (0.6 kcal/mol) toward the LEDA conformation because, while the sulfone still hydrogen bonds the aminoquinazoline NH, the hydrogen bond to the tertiary amine is not present in the structure. The n-propyl group in compound 1 is unable to stabilize the LEDA conformation through any hydrogen bonding, and the calculated conformational preference is LADE in both the protonated and the free-base species.
Table 2. Ab Initio Calculated Relative Energies and Polar Surface Areas.

| protonated |
free
base |
|||||
|---|---|---|---|---|---|---|
| LEDA |
LADE | LEDA |
LADE | |||
| compound | ΔEa | PSAb | PSAb | ΔEa | PSAb | PSAb |
| 1 | 1.7 | 267 | 266 | 3.6 | 247 | 242 |
| 5 | 0.9 | 270 | 310 | –0.6 | 252 | 288 |
| 6 | 1.5 | 270 | 299 | –4.8 | 255 | 282 |
| 7 | 1.9 | 272 | 282 | –5.4 | 234 | 264 |
ΔE = ELEDA – ELADE (kilocalories per mole). Representative LEDA and LADE conformations for compound 1 are illustrated above the table. The minimized conformations for compounds 1, 5, 6, and 7 are shown in Figure 1 in the Supporting Information.
Polar surface area, PSA (square angstroms), is computed from the 3D conformations.
The above observations led us to propose a model of pH-dependent conformational flipping to enable oral bioavailability, as shown in Figure 4. The progression begins with the tertiary amine predominately in the protonated form within the slightly acidic to neutral environment of the intestinal lumen (measured pKa = 9.5, calculated population of 98%). In aqueous solution, BMS-741672 exists in equilibrium between its diprotonated, monoprotonated, and unprotonated states. Potentiometric titration reveals pKa values of 9.5 (tertiary amine) and 4.5 (quinazoline). Figure 4 considers only the equilibrium between monoprotonated and unprotonated states, in which the monoprotonated form (i.e., protonated tertiary amine) will dominate at physiological pH. In this state, compound 7 is predicted to be almost entirely LADE as it is calculated to be 1.9 kcal/mol lower in energy than LEDA (see the Supporting Information for detailed discussion of the ab initio calculations). However, within the small non-protonated subpopulation, the conformational stabilities are reversed with the LEDA conformation being 5.4 kcal/mol lower in energy. Its relative stability arises from two intramolecular hydrogen bonds, which masks two hydrogen bond donors, lowers PSA, and desolvates the acetamide moiety. The small relative concentrations of the nonprotonated, LEDA form can readily enter the lipid environment, driving the equilibrium toward this species. The compound then exits the lipid environment to enter either the aqueous environment of the lumen or the bloodstream. Once the compound exits the membrane into the circulation, oral bioavailability has been secured. In this environment, the equilibrium population will again favor the protonated LADE conformation, which is the conformation required for CCR2 binding.21 In this conformation, the polar side chain is exposed, and the selectivity for the molecule relative to ion channels is therefore high. Thus, the model suggests that compound 7 largely exists in protonated LADE in aqueous environments but that a small equilibrium population of unprotonated LEDA is sufficient to act as a shuttle between the intestine and the bloodstream by enabling 7 to enter the lipid environment and thereby traverse the intestinal wall.
Figure 4.
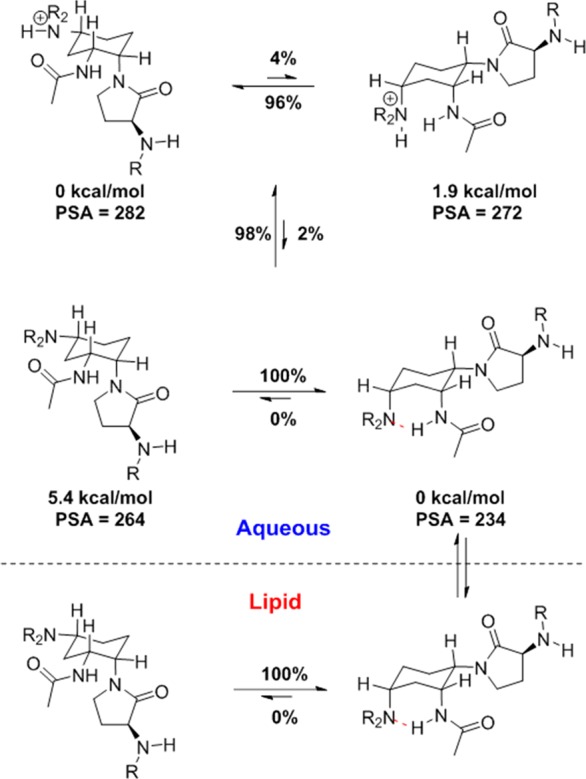
Working model for the equilibrium populations of acetamide 7 in both high-dielectric aqueous and low-dielectric lipid environments. Percentages on the horizontal arrows are Boltzmann populations calculated from the relative ab initio conformational energies, denoted beneath each structure. The Boltzmann populations on the vertical arrows are calculated using the Henderson–Hasselbalch equation using an amine pKa of 9.5 and buffer pH of 7.3.
Changes in pH and ion content have previously been shown to enable “molecular switching” in certain predisposed systems, including six-membered carbo- and heterocycles.23,24 Furthermore, intramolecular hydrogen-bonding is known to mask exposed polarity in peptides and can sometimes be correlated with enhanced membrane permeability and even oral bioavailability.25−29 The solution to our problem required that this masking was context-dependent so that the polar form of the molecule was the one predominantly available. Based on the structure–activity relationships relative to CCR2 binding, hERG functional activity, and membrane permeability, the conformational switching mechanism (Figure 4) appeared to remain intact within this trisubstituted cyclohexane series so long as three elements remained in place: a secondary or tertiary amine at C1, an amide or sulfonamide directly linked to C3, and the Friedinger lactam at C4. Modification of these groups (e.g., homologation of the acetamide or replacement of the lactam with a glycinamide) disrupted the SAR trends. However, with these groups in place, optimized molecules for the inhibition of CCR2 could be obtained. Acetamide 7 (BMS-741672) was one such molecule. Specifically, BMS-741672 was found to have 51% oral bioavailability in a rat with a t1/2 value of 5.1 h (IV), and there was 46% oral bioavailability in a cynomolgus monkey with a t1/2 of 3.2 h (IV). It exhibited an IC50 of 0.67 nM for the inhibition of monocyte chemotaxis in vitro and was fully active in both monkey and hCCR2 knock-in mouse models of monocyte chemotaxis (see ref (17)). The compound proved to have a suitable safety margin for advancement based on single-day tolerability studies (rat and monkey) and a 14-day toxicology study in the rat. Relative to our general concerns over hERG selectivity, it was notable that 7 did not show any effects on the QTc interval in telemetrized monkeys at a dose of 30 mg/kg. Taken together, the preclinical studies with BMS-741672 enabled its advancement into human clinical trials.
Acknowledgments
We acknowledge our many colleagues in the CCR2 Discovery Working Group as well as the Department of Discovery Synthesis at the Biocon-BMS R&D Center (Bangalore, India). We also thank Dr. Nicholas A. Meanwell for helpful comments on the manuscript.
Glossary
Abbreviations
- CCR2
CC chemokine receptor 2
- CCR5
CC chemokine receptor 5
- LADE
lactam axial diequatorial
- LEDA
lactam equatorial diaxial
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00439.
This work was funded by Bristol-Myers Squibb Company.
The authors declare no competing financial interest.
Dedication
This paper is dedicated to the memory of Joseph B. Santella, III.
Supplementary Material
References
- Carter P. H. Progress in the discovery of CC chemokine receptor 2 antagonists, 2009 - 2012. Expert Opin. Ther. Pat. 2013, 23 (5), 549–568. 10.1517/13543776.2013.771168. [DOI] [PubMed] [Google Scholar]
- Struthers M.; Pasternak A. CCR2 antagonists. Curr. Top. Med. Chem. 2010, 10 (13), 1278–1298. 10.2174/156802610791561255. [DOI] [PubMed] [Google Scholar]
- Solari R.; Pease J. E.; Begg M. Chemokine receptors as therapeutic targets: why aren’t there more drugs?. Eur. J. Pharmacol. 2015, 746, 363–367. 10.1016/j.ejphar.2014.06.060. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Arrowsmith J.; Leach A. R.; Leeson P. D.; Mandrell S.; Owen R. M.; Pairaudeau G.; Pennie W. D.; Pickett S. D.; Wang J.; Wallace O.; Weir A. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discovery 2015, 14 (7), 475–486. 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- Gordon S.; Taylor P. R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5 (12), 953–964. 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Ingersoll M. A.; Platt A. M.; Potteaux S.; Randolph G. J. Monocyte trafficking in acute and chronic inflammation. Trends Immunol. 2011, 32 (10), 470–477. 10.1016/j.it.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F.; Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14 (6), 392–404. 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- Wynn T. A.; Chawla A.; Pollard J. W. Macrophage biology in development, homeostasis, and disease. Nature 2013, 496, 445–455. 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y. S.; Cha J. Jo.; Hyun Y. Y.; Cha D. R. Novel C-C chemokine receptor 2 antagonists in metabolic disease: a review of recent developments. Expert Opin. Invest. Drugs 2011, 20 (6), 745–756. 10.1517/13543784.2011.575359. [DOI] [PubMed] [Google Scholar]
- Charo I. F.; Ransohoff R. M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354 (6), 610–621. 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Feria M.; Díaz-González F. The CCR2 receptor as a therapeutic target. Expert Opin. Ther. Pat. 2006, 16 (1), 49–57. 10.1517/13543776.16.1.49. [DOI] [Google Scholar]
- Leuschner F.; Dutta P.; Gorbatov R.; Novobrantseva T. I.; Donahoe J. S.; Courties G.; Lee K. M.; Kim J. I.; Markmann J. F.; Marinelli B.; Panizzi P.; Lee W. W.; Iwamoto Y.; Milstein S.; Epstein-Barash H.; Cantley W.; Wong J.; Cortez-Retamozo V.; Newton A.; Love K.; Libby P.; Pittet M. J.; Swirski F. K.; Koteliansky V.; Langer R.; Weissleder R.; Anderson D. G.; Nahrendorf M. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29 (11), 1005–1010. 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter P. H.; Brown G. D.; Cherney R. J.; Batt D. G.; Chen J.; Clark C. M.; Cvijic M. E.; Duncia J. V.; Ko S. S.; Mandlekar S.; Mo R.; Nelson D. J.; Pang J.; Rose A. V.; Santella J. B. 3rd; Tebben A. J.; Traeger S. C.; Xu S.; Zhao Q.; Barrish J. C. Discovery of a Potent and Orally Bioavailable Dual Antagonist of CC Chemokine Receptors 2 and 5. ACS Med. Chem. Lett. 2015, 6 (4), 439–44. 10.1021/ml500505q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherney R. J.; Mo R.; Yang M. G.; Xiao Z.; Zhao Q.; Mandlekar S.; Cvijic M. E.; Charo I. F.; Barrish J. C.; Decicco C. P.; Carter P. H. Alkylsulfone-containing trisubstituted cyclohexanes as potent and bioavailable chemokine receptor 2 (CCR2) antagonists. Bioorg. Med. Chem. Lett. 2014, 24 (7), 1843–1845. 10.1016/j.bmcl.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter P. H.; Cherney R. J.; Batt D. G.; Duncia John V.; Gardner D. S.; Ko S. S.; Srivastava A. S.; Yang M. G.. Cyclic derivatives as modulators of chemokine receptor activity. Patent no. WO 2005/021500; March 10, 2005.
- Meanwell N. A. Improving Drug Design: An Update on Recent Applications of Efficiency Metrics, Strategies for Replacing Problematic Elements, and Compounds in Nontraditional Drug Space. Chem. Res. Toxicol. 2016, 29 (4), 564–616. 10.1021/acs.chemrestox.6b00043. [DOI] [PubMed] [Google Scholar]
- Yang M. G.; Cherney R. J.; Eastgate M. G.; Muslehiddinoglu J.; Prasad S. J.; Xiao Z.. Modulators of chemokine receptor activity, crystalline forms and process. Patent no. WO 2008/014381; January 31, 2008.
- Campbell C. L.; Hassler C.; Ko S. S.; Voss M. E.; Guaciaro M. A.; Carter P. H.; Cherney R. J. Enantioselective synthesis of benzyl (1S,2R,4R)-4-(tert-butoxycarbonylamino)-2-(hydroxyl-methyl)cyclohexylcarbamate using an iodolactamization as the key step. J. Org. Chem. 2009, 74 (16), 6368–6370. 10.1021/jo9011249. [DOI] [PubMed] [Google Scholar]
- Cherney R. J.; Mo R.; Meyer D. T.; Voss M. E.; Yang M. G.; Santella J. B. 3rd; Duncia J. V.; Lo Y. C.; Yang G.; Miller P. B.; Scherle P. A.; Zhao Q.; Mandlekar S.; Cvijic M. E.; Barrish J. C.; Decicco C. P.; Carter P. H. gamma-Lactams as glycinamide replacements in cyclohexane-based CC chemokine receptor 2 (CCR2) antagonists. Bioorg. Med. Chem. Lett. 2010, 20 (8), 2425–2430. 10.1016/j.bmcl.2010.03.035. [DOI] [PubMed] [Google Scholar]
- Deerberg J.; Prasad S. J.; Sfouggatakis C.; Eastgate M. D.; Fan Y.; Chidambaram R.; Sharma P.; Li L.; Schild R.; Müslehiddinoğlu J.; Chung H.-J.; Leung S.; Rosso V. Stereoselective Bulk Synthesis of CCR2 Antagonist BMS-741672: Assembly of an All-cis (S,R,R)-1,2,4-Triaminocyclohexane (TACH) Core via Sequential Heterogeneous Asymmetric Hydrogenations. Org. Process Res. Dev. 2016, 20 (11), 1949–1966. 10.1021/acs.oprd.6b00282. [DOI] [Google Scholar]
- Zheng Y.; Qin L.; Zacarías N. V.; de Vries H.; Han G. W.; Gustavsson M.; Dabros M.; Zhao C.; Cherney R. J.; Carter P.; Stamos D.; Abagyan R.; Cherezov V.; Stevens R. C.; IJzerman A. P.; Heitman L. H.; Tebben A.; Kufareva I.; Handel T. M. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540, 458–461. 10.1038/nature20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Liu X.; Samoshina N. M.; Samoshin V. V.; Franz A. H.; Guo X. Fliposomes: trans-2-aminocyclohexanol-based amphiphiles as pH-sensitive conformational switches of liposome membrane - a structure-activity relationship study. Chem. Phys. Lipids 2018, 210, 129–141. 10.1016/j.chemphyslip.2017.10.004. [DOI] [PubMed] [Google Scholar]
- Knipe P. C.; Thompson S.; Hamilton A. D. Ion-mediated conformational switches. Chem. Sci. 2015, 6 (3), 1630–1639. 10.1039/C4SC03525A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thansandote P.; Harris R. M.; Dexter H. L.; Simpson G. L.; Pal S.; Upton R. J.; Valko K. Improving the passive permeability of macrocyclic peptides: Balancing permeability with other physicochemical properties. Bioorg. Med. Chem. 2015, 23 (2), 322–327. 10.1016/j.bmc.2014.11.034. [DOI] [PubMed] [Google Scholar]
- Hewitt W. M.; Leung S. S.; Pye C. R.; Ponkey A. R.; Bednarek M.; Jacobson M. P.; Lokey R. S. Cell-permeable cyclic peptides from synthetic libraries inspired by natural products. J. Am. Chem. Soc. 2015, 137 (2), 715–721. 10.1021/ja508766b. [DOI] [PubMed] [Google Scholar]
- Shalaeva M.; Caron G.; Abramov Y. A.; O’Connell T. N.; Plummer M. S.; Yalamanchi G.; Farley K. A.; Goetz G. H.; Philippe L.; Shapiro M. J. Integrating intramolecular hydrogen bonding (IMHB) considerations in drug discovery using ΔlogP as a tool. J. Med. Chem. 2013, 56 (12), 4870–4879. 10.1021/jm301850m. [DOI] [PubMed] [Google Scholar]
- Rossi Sebastiano M.; Doak B. C.; Backlund M.; Poongavanam V.; Over B.; Ermondi G.; Caron G.; Matsson P.; Kihlberg J. Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs Beyond the Rule of 5. J. Med. Chem. 2018, 61 (9), 4189–4202. 10.1021/acs.jmedchem.8b00347. [DOI] [PubMed] [Google Scholar]
- Nielsen D. S.; Hoang H. N.; Lohman R. J.; Hill T. A.; Lucke A. J.; Craik D. J.; Edmonds D. J.; Griffith D. A.; Rotter C. J.; Ruggeri R. B.; Price D. A.; Liras S.; Fairlie D. P. Improving on nature: making a cyclic heptapeptide orally bioavailable. Angew. Chem., Int. Ed. 2014, 53 (45), 12059–12063. 10.1002/anie.201405364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.