

Abstract
An international advisory group met at the National Institutes of Health in Bethesda, Maryland in 2017, to discuss a new classification system for the ectodermal dysplasias that would integrate both clinical and molecular information. We propose the following, a working definition of the ectodermal dysplasias building on previous classification systems and incorporating current approaches to diagnosis: ectodermal dysplasias are genetic conditions affecting the development and/or homeostasis of two or more ectodermal derivatives, including hair, teeth, nails, and certain glands. Genetic variations in genes known to be associated with ectodermal dysplasias that affect only one derivative of the ectoderm (attenuated phenotype) will be grouped as non-syndromic traits of the causative gene (e.g. non-syndromic hypodontia or missing teeth associated with pathogenic variants of EDA “ectodysplasin” ). Information for categorization and cataloging includes the phenotypic features, Online Mendelian Inheritance in Man number, mode of inheritance, genetic alteration, major developmental pathways involved (e.g. EDA, WNT “wingless-type”, TP63 “tumor protein p63”) or the components of complex molecular structures ( e.g. connexins, keratins, cadherins).
Keywords: ectodermal, dysplasia, classification, molecular, genetic, signaling pathway
Introduction
Ectoderm gives rise to the epidermis, the central and peripheral nervous system, the placodes (including cranial placodes), and neural crest cells. Cell fate decisions that lead to ectodermal lineage specification are regulated by highly conserved signaling pathways such as the WNT, BMP “bone morphogenic protein, and FGF “fibroblast growth factor” pathways.[Patthey and Gunhaga 2014] Ectodermal appendages (e.g. hair, teeth, nails) arise from signaling cross-talk interactions between ectodermal epithelium and mesenchyme.[Naveau and others 2014] Numerous hereditary conditions are characterized by abnormal development of ectodermal tissues. Classification and nosology for these conditions exist to guide clinicians in deriving a correct diagnosis and aid in communicating with patients and investigators. Ideally, these classification systems and approaches should be useful for diverse stakeholders including students, patients, researchers, and clinicians.
Hereditary conditions historically classified as an ectodermal dysplasia (ED) are heterogeneous in their genetic causes and clinical phenotypes. A unifying feature is that all of the ED conditions are genetically determined developmental defects of tissues of ectodermal origin. Initial classification systems for the EDs predate molecular genetics and were categorized and grouped according to phenotypic features and mode of inheritance. The most widely known such nosology was developed by Dr. Newton Freire-Maia in the 1970s and included conditions with “classical signs” involving hair, teeth, nails, and/or sweat glands.[Freire-Maia 1971; Freire-Maia 1977; Freire-Maia and Pinheiro 1984; Freire-Maia and Pinheiro 1988] The disorders were subdivided into Group A – those having at least two of these tissues affected – and Group B – conditions affecting one of the aforementioned tissues and at least one other tissue of ectodermal origin (e.g. mammary gland).[Freire-Maia and Pinheiro 1984; Nguyen-Nielsen and others 2013] The Freire-Maia classification system serves as a strong foundation that has advanced our understanding of these diverse conditions and provided valuable information on how best to manage affected individuals.
While the ectodermal dysplasias are considered rare conditions, there are contradictory reports on their prevalence. A nationwide cross-sectional study in Denmark that leveraged data from national registries and clinical departments suggests that X-linked hypohidrotic ED (XLHED; MIM #305100) may be more common than previously thought.[Nguyen-Nielsen and others 2013] The authors reported a possible 1224 HED cases between 1995 and 2010 with 90 of these confirmed at the molecular level, 146 clinically diagnosed, and 988 possible HED cases. The prevalence of molecularly confirmed XLHED was 1.6:100,000.[Nguyen-Nielsen and others 2013]
Although the historical nosological grouping of the EDs was predicated on phenotype, our understanding of the human genome and its role in development and disease has advanced providing new opportunities to recognize how some conditions are related at the molecular level. [Freire-Maia 1977]; [Itin 2014; Wright and others 2009] The first genetic alteration identified as causative for ED was a loss-of-function variant of the gene EDA.[Kere and others 1996] Following this discovery, the causes of other hypohidrotic ED conditions with similar phenotypes were discovered in defects of the EDA receptor (EDAR) and the adaptor proteins EDARADD “EDAR-associated death domain” [Headon and others 2001] and TRAF6 “TNF receptor-associated factor 6”. [Wisniewski and Trzeciak 2012] [Chassaing and others 2006] Pathogenic alterations in any of these genes can result in a clinical phenotype similar to XLHED, and both heterozygosity and compound heterozygosity or homozygosity for these can cause the disorders. Subsequently, genetic alterations in genes associated with other ED types were discovered in the NFkB pathway that plays a variety of roles in normal ectodermal tissue development. The genetic basis for nearly 50% of conditions historically classified as EDs and the causative genetic alterations underlying most of the more prevalent ED conditions are known.[Pagnan and Visinoni 2014; Wright and others 2009] Further, it is now clear that many of the genes affected in EDs function in common molecular pathways that are known to be of importance in development of the ectodermal derivatives (e.g. NFkB “nuclear factor kappa-B”, WNT, TP63 pathways).[Cluzeau and others 2011; Kantaputra and Carlson 2018; Koster 2010]
Developing a classification system that incorporates the molecular etiology and the molecular pathway will help clinicians about the diagnosis of the diverse ED conditions at both the clinical and genetics levels.[Itin 2014] Understanding the molecular pathogenesis of the EDs will better inform researchers as to phenotypic features often associated with specific pathways thereby illuminating potential causative candidate genes for ED conditions undiagnosed at the molecular level (e.g. TP63 pathway-associated phenotypes include features such as cleft lip/palate and hand/foot malformations).[Koster 2010; Vera-Carbonell and others 2012]
The National Foundation for Ectodermal Dysplasias (NFED) embarked on developing a new classification system through a series of conferences that brought together stakeholders and international experts. Dr. Carlos Salinas chaired two conferences in Charleston, South Carolina, in 2008 and 2012; with the proceedings published in the American Journal of Medical Genetics.[Salinas and others 2014; Salinas and others 2009] In 2017, an international working group of individuals, many of whom had participated in the previous conferences, met on the National Institutes of Health campus in Bethesda, Maryland. The outcome of these meetings was a refined definition of what comprises an ectodermal dysplasia for developing a classification approach that would incorporate phenotype, inheritance, and molecular etiology including developmental pathway or structural assembly to organize and cluster the ED conditions.
Definition of Ectodermal Dysplasias
A broad variety of tissues such as the central and peripheral nervous system, adenohypophysis, lens, olfactory epithelium, parts of the pharyngeal arches, pigmented cells, epidermis, and mucosal epithelium are of ectodermal origin. The definition for inclusion as an ectodermal dysplasia does not extend to all derivatives of ectoderm and is most often limited to conditions affecting the skin and mucosa and/or their appendages.[Freire-Maia 1977; Itin 2013; Pagnan and Visinoni 2014] Based on recommendations from the previous classification conferences, the group developed consensus on the following working definition of ectodermal dysplasia for the purposes of this classification system: Ectodermal dysplasias are genetic conditions affecting the development and/or homeostasis of two or more ectodermal derivatives, including hair, teeth, nails, and certain glands. The molecular causes of these diverse conditions involve many genes and multiple developmental pathways and components of complex molecular structures that are necessary for normal formation, structure, and function of the ectodermal derivatives. It should be noted that in some ectodermal dysplasias there are disturbances of epithelial-mesenchymal interaction affecting endodermal as well as ectodermal structures, such as the defects of mucous gland formation in the lung and the colon in XLHED.
Numerous genes may have alterations that are associated with syndromes while in other instances they are associated with changes in only one ectodermal tissue. Examples of this include EDA and WNT10A “wingless-type 10A” variants that result in missing teeth but no other phenotypic features of ED.[van den Boogaard and others 2012; Yang and others 2013] Genetic alterations of ED-associated genes that only affect one derivative of ectoderm (e.g. hair, teeth, nails, sweat glands) should be grouped as non-syndromic traits of the causative gene (e.g. non-syndromic hypodontia or missing teeth associated with pathogenic EDA variants). It is further noted that not all pathogenic mutations in a given gene may cause an ectodermal dysplasia. For example, mutations in GJB2 “gap junction protein beta-2”, a gene coding for connexin, can give rise to isolated deafness, palmoplantar keratoderma, and ichthyosis, as well as K-I-D syndrome (kearatitis-icthyosis-deafness syndrome).
Inclusion/Exclusion
The development of a useful nosology based on the above definition of ED involved establishing inclusion and exclusion criteria. Conditions were included if they met the adopted definition of an ectodermal dysplasia. Conditions already included as part of other classifications or groups of diseases and/or are presented in different chapters in textbooks (e.g. palmoplantar keratodermas such as Papillon-Lefèvre syndrome (OMIM #245000), disorders of DNA repair such as trichothiodystrophy, vesiculobullous disorders) were not included, although they may be associated with alterations in ectodermal structures.[Fine and others 2014; Lucker and others 1994] Complex syndromes that have ED signs but also major non-ED signs (e.g. affecting bone, brain) were also excluded (e.g. trisomy 21). Finally, the group agreed to exclude conditions listed in OMIM with only one case report and no known molecular etiology.
Classification Scheme and Clustering
The proposed ED classification system comprises information from multiple domains including OMIM #, phenotype, mode of inheritance, causal gene, and molecular pathway or structure. Conditions are grouped based on genotype, molecular pathway and phenotype. The clinician is likely to assort these disorders based on the physical features while the molecular geneticist may think in terms of pathways. A classification system should be a useful tool irrespective of the user’s entry point. Knowledge of developmental pathways and molecular structures, and the relationship of different gene products within these domains, show that many EDs result from genes that co-participate in critical developmental processes and structural assemblages of the ectodermal derivatives (Figures 1, 2, 3). In these figures the EDA associated genes are presented in orange ovals, WNT associated genes are presented in purple ovals, and TP63 associated genes are presented in blue ovals. These pathway figures also illustrate how different pathways can be interconnected (Figure 1 – TP63 genes interacting with EDA pathway genes). Other genes and their genetic variants associated with EDs code for proteins important for the structure and/or function of cells. Table 1 illustrates this organizational system showing how ED conditions are clustered based on the gene, molecular pathway, and/or protein function and how these different domains are ordered to provide relevant information. The full list of the known ED conditions included is available in the electronic supplement (Table 2e). The conditions are ordered within clusters based on the most proximal or up-stream gene involved with down-stream genes in the pathway following (e.g. EDA, EDAR, IKBKG “inhibitor of kappa light plypeptide gene enhance in B cells”). In the case of EDA, this also happens to coincide with XLHED being the most prevalent form of HED. As ED prevalence remains poorly characterized, meaningful ordering or reporting based on frequency of occurrence in the population is difficult. Conditions meeting the definition of an ED but of unknown etiology are grouped with other EDs that share the most similar phenotype. Identifying the molecular basis of these conditions will allow classification with existing ED clusters or, based on the molecular etiology, to become the anchor of a new ED cluster.
Figure 1:
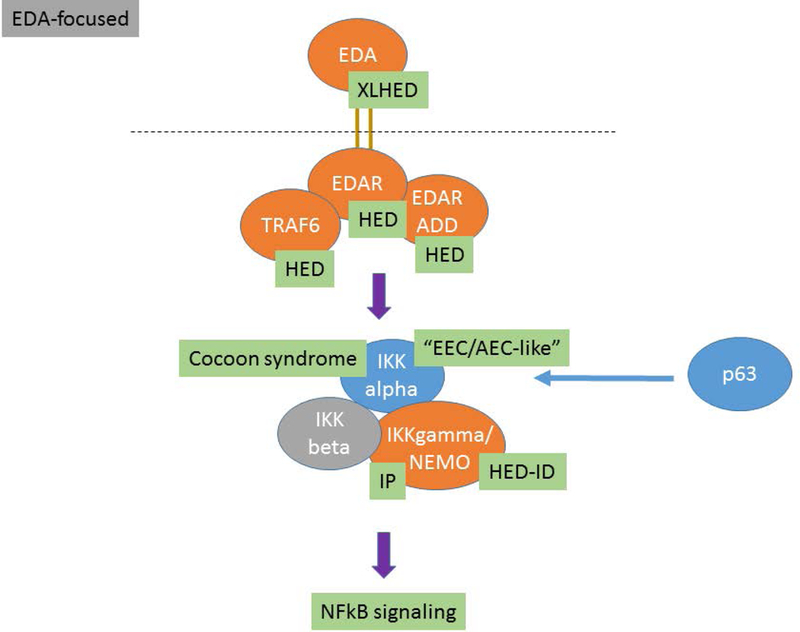
The EDA molecular pathways and the interrelationships between different genes and known associated EDs are presented. Causative genes appear in orange ovals and abbreviations for the ED conditions are shown in green boxes.
Figure 2:
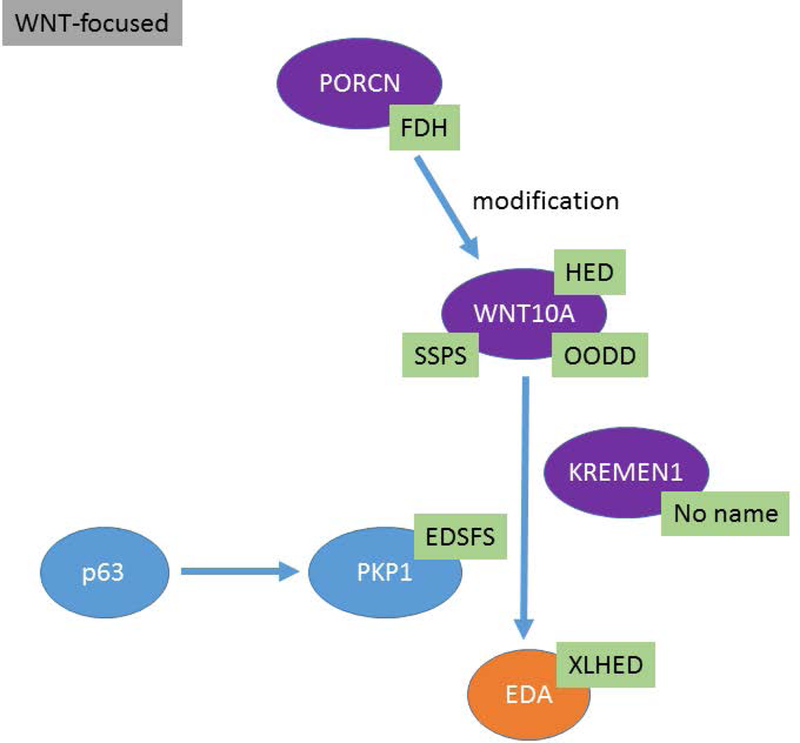
The WNT molecular pathways and the interrelationships between different genes are presented. Causative genes appear in purple ovals and abbreviations for the ED conditions are shown in green boxes.
Figure 3:
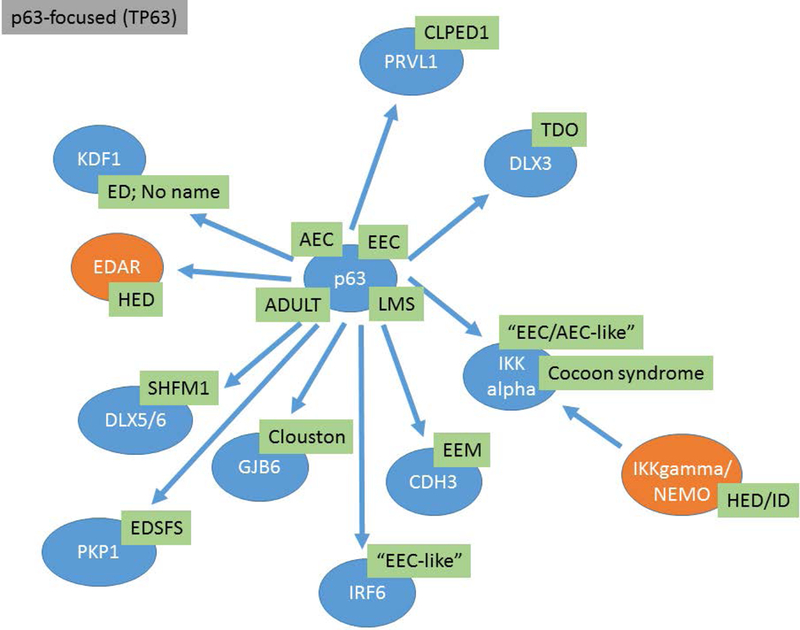
TP63 molecular pathways and the interrelationships between different genes are presented. Causative genes appear in blue ovals and abbreviations for the ED conditions are shown in green boxes.
Table 1.
Organization of ED conditions based on molecular pathways
| EDA/NFKappaB Pathway | |||
| OMIM Number | Syndrome Name(s) | Gene | Distinguishing Features |
| 305100 | Hypohidrotic Ectodermal Dysplasia; ED1; Christ-Siemens-Touraine Syndrome | Ectodysplasin A; EDA (300451) | Hypohidrosis, hypotrichosis, hypodontia, smooth dry skin, craniofacial dysmorpholgy, periorbital pigmentation |
| 129490 | Hypohidrotic Ectodermal Dysplasia 10A | Ectodysplasin A Receptor EDAR (604095) or EDARADD (606603) | Hypohidrosis, hypotrichosis, hypodontia, smooth dry skin, craniofacial dysmorphology, periorbital pigmentation |
| 224900 | Hypohidrotic Ectodermal Dysplasia 10B | Ectodysplasin A Receptor EDAR (604095) or EDARADD (606603) | Hypohidrosis, hypotrichosis, hypodontia, smooth dry skin, craniofacial dysmorpholgy, periorbital pigmentation |
| 308300 | Incontinentia Pigmenti; IP |
IKBKG
(300248) |
Short stature, cataract, micropthalmia,, hypodontia, extra ribs, breast aplasia, staged skin involvement, nail dystrophy, atrophic hair |
| 300291 | Ectodermal Dysplasia and Immunodeficiency 1: EDAID1 |
IKBKG
(300248) |
Hypohidrosis, hypotrichosis, morbidity/mortality secondary to immunodeficiency |
| WNT Pathway | |||
| OMIM Number | Syndrome Name(s) | Gene | Distinguishing Features |
| 305600 | Focal Dermal Hypoplasia, Goltz Syndrome |
PORCN (300651) |
Short stature, facial asymmetry, narrow auditory canals, hearing loss, oral papillomas, hypodontia, syndactyly, sparse hair, skin atrophy |
| 257980 | Odontoonychodermal Dysplasia; OODD | WNT10A (606268) | sparse eyebrows, severe hypodontia, smooth tongue, hyperhidrosis, hyperkeratosis, dystrophic nails, sparse eyebrows, thin hair |
| 224750 | Schopf-Schulz-Passarge Syndrome | WNT10A (606268) | hypodontia, eyelid cysts, keratoderma, hypoplastic nails, hypotrichosis |
| TP63 Pathway | |||
| OMIM Number | Syndrome Name(s) | Gene | Distinguishing Features |
| 103285 | Acro-Dermato-Ungual-Lacrimal-Tooth Syndrome (ADULT Syndrome) |
TP63
(603273) |
Lacrimal obstruction, hypodontia, dysplastic teeth, breast hypoplasia, ectrodactyly, thin skin, dysplastic nails |
| 106260 | Ankyloblepharon-Ectodermal Defects-Cleft Lip/Palate (AEC; Hay-Wells Syndrome) |
TP63
(603273)) |
scalp erosions, conductive hearing loss, maxillary hypoplasia, lacrimal duct atresia, hypotrichosis, ankyloblepharon, cleft l/p, hypodontia |
| 129400 | Rapp-Hodgkin Syndrome |
TP63
(603273) |
short stature, maxillary hypoplasia, hearing loss, cleft l/p, hypodontia, syndactyly, thin skin, hypohidrosis |
| 604292 | Ectrodactyly, Ectodermal Dysplasia, and Cleft Lip/Palate Syndrome 3; EEC3 |
TP63
(603273) |
blepharophimosis, cleft l/p, microdontia, hypodontia, syndactyly, hypokeratosis, dystrophic nails hypotrichosis |
| 603543 | Limb-Mammary Syndrome; LMS |
TP63 (603273) |
lacrimal duct atresia, hypodontia cleft p, hypoplastic breasts, syndactyly, ectrodactyly, nail dysplasia |
| Structure Group | |||
| OMIM Number | Syndrome Name(s) | Gene | Distinguishing Features |
| 225280 | Ectodermal dysplasia, ectrodactyly, and macular dystrophy syndrome; EEMS |
CDH3 Cadherin 3 (114021) |
Sparse scalp hair, eyebrows and eyelashes, hypodontia, small teeth, ectrodactyly, syndactyly, camptodactyly, normal sweating |
| 602032 | Ectodermal dysplasia 4, hair/nail typez ; ECTD4 |
KRT85
Keratin 85 (602767) |
Nail dystrophy, onycholysis, absent eybrows/eyelashes, alopecia, normal skin/teeth |
| 604536 | Ectodermal dysplasia/skin fragility syndrome |
PKP1 Plakophilin 1 (601975) |
Nail dystrophy and thickening, hypotrichosis, sweat glands, skin fragility |
| 158000 | Monilethrix MNLIX |
Keratins 81, 86, 83 KRT81, KRT86, KRT83 (602153, 601928 602765) |
Follicular keratosis, nail dystrophy, hypotrichosis, brittle hear |
| 225060 | Cleft lip/Palate-Ectodermal Dysplasia (CLPED1) | Nectin 1, NECTIN1(600644) | Malar hypoplasia, hypotrichosis, cleft lip/palate, hypodontia, syndactyly, onychodysplasia |
| Other/Unknown | |||
| OMIM Number | Syndrome Name(s) | Gene | Distinguishing Features |
| 601701 | Arthorgryposis and Ectodermal Dysplasia | Unknown | Short statue, microcephaly, cataract, cleft lip/palate, oligodontia, enamel defects, arthrogryposis, hypohidrosis, onychodysplasia |
| 125640 | Dermoonontodysplasia | Unknown | trichodysplasia, onychodysplasia, dental anomalies |
Discussion
The omics era ushered in entirely new and rapidly developing areas of knowledge that continue to change our view of health and disease. Conditions previously thought to be unrelated can result from allelic mutations that lead to markedly different clinical phenotypes (e.g. TP63-associated syndromes).[Rostagno and others 2010; Wisniewski and Trzeciak 2012] Prior to the explosion of knowledge in the molecular era, the foundation for classifying conditions was phenotype and mode of inheritance. More recent classificatory approaches include a variety of molecular data that aids in understanding the relationship of the underlying molecular defect, protein alteration and resulting phenotype. For example, the inherited epidermolysis bullosa conditions are clustered based on the histological level of tissue separation, the clinical phenotype, the gene involved and, when possible, the specific genetic alteration.[Fine and others 2014] Other classification systems, such as the nosology proposed for inherited ichthyosis, remain clinically based.[Oji and others 2010] The international ichthyosis workgroup recognized, however, that a pathophysiologic classification of inherited ichthyoses should be developed as additional information becomes available. The Nosology Group of the International Skeletal Dysplasia Society clusters conditions based on their molecular basis as well as using phenotype clustering.[Bonafe and others 2015]
Classification systems for ED have been proposed that categorize the conditions within groups based on the underlying functional defect such as abnormal developmental regulation or structural protein.[Priolo and Lagana 2001] We are aware that the proposed ED nosology will require additions and modifications due to the identification of new genes and genetic alterations. Nonetheless, we believe that this current construct has utility for the clinician struggling with a differential diagnosis and the researcher hoping to elucidate underlying causality. With the therapeutic management of XLHED now a reality, through the intra-amniotic delivery of a fusion protein that substitutes for the function of the abnormal EDA protein, the need to establish the correct diagnosis of the EDs has never been greater.[Schneider and others 2018]
Supplementary Material
Acknowledgments:
We acknowledge the support from the NFED and NIDCR for helping move this agenda forward over the past decade and their ongoing assistance to improve our understanding of the ectodermal dysplasias.
Footnotes
Dedication: We dedicate this manuscript to the late Dr. Carlos Salinas, long-time leader who sought continuous improvement in our understanding and classification of the ectodermal dysplasias.
Contributor Information
Wright Timothy, Bawden Distinguished Professor, Department of Pediatric Dentistry, Bauer Hall CB#7450 School of Dentistry, University of North Carolina Chapel Hill, NC 27599-7450.
Mary Fete, National Foundation for Ectodermal Dysplasias, Fairview Heights IL. USA.
Holm Schneider, Department Head of Molecular Pediatrics at University Hospital Erlangen, Erlangen Germany.
Madeline Zinser, Research Intern, NFED, Fairview Heights IL. USA.
Maranke I. Koster, NFED Scientific Advisory Council / Dermatology and Ophthalmology at University of Colorado Anschutz Medical Campus in Aurora, CO.
Angus J. Clarke, Clinical Genetics, School of Medicine, Cardiff University, Wales, UK..
Smail Hadj Rabia, Université Paris Descartes, Sorbonne Paris Cité, Paris, France..
Gianluca Tadini, Dept of Pathophysiology and Transplantation, Center for Inherited Cutaneous Diseases, Università degli Studi di Milano, Foundation IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Milan Italy..
Nina Pagnan, Department of Genetics, Federal University of Parana, Curitiba, Brazil.
Atila Fernando Visinoni, Positivo University, Curitiba, Brazil.
Birgitta Bergendal, National Oral Disability Center for Rare Disorders, The Institute for Postgraduate Dental Education, Jönköping, Sweden.
Becky Abbott, mother of affected son / contracted by NFED for treatment & research, Fairview Heights IL. USA.
Timothy Fete, Department of Child Health, NFED Scientific Advisory Council University of Missouri, Columbia, MO.
Clark Stanford, NFED Scientific Advisory Council / University of Illinois at Chicago College of Dentistry in Chicago, IL.
Clayton Butcher, Clinical Medicine and Child Health, University of Missouri School of Medicine, Columbia, MO.
Rena D’ Souza, for Academic Affairs and Education, Health Sciences, Professor of Dentistry at The University of Utah in Salt Lake City, UT.
Virginia Sybert, Department of Medicine, Division of Medical Genetics, University of Washington School of Medicine, Seattle, WA.
References
- Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. 2015. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 167A(12):2869–2892. [DOI] [PubMed] [Google Scholar]
- Chassaing N, Bourthoumieu S, Cossee M, Calvas P, Vincent MC. 2006. Mutations in EDAR account for one-quarter of non-ED1-related hypohidrotic ectodermal dysplasia. Hum Mutat 27(3):255–259. [DOI] [PubMed] [Google Scholar]
- Cluzeau C, Hadj-Rabia S, Jambou M, Mansour S, Guigue P, Masmoudi S, Bal E, Chassaing N, Vincent MC, Viot G, Clauss F, Maniere MC, Toupenay S, Le Merrer M, Lyonnet S, Cormier-Daire V, Amiel J, Faivre L, de Prost Y, Munnich A, Bonnefont JP, Bodemer C, Smahi A. 2011. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mutat 32(1):70–72. [DOI] [PubMed] [Google Scholar]
- Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, Marinkovich MP, Martinez AE, McGrath JA, Mellerio JE, Moss C, Murrell DF, Shimizu H, Uitto J, Woodley D, Zambruno G. 2014. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 70(6):1103–1126. [DOI] [PubMed] [Google Scholar]
- Freire-Maia N 1971. Ectodermal dysplasias. Hum Hered 21(4):309–312. [DOI] [PubMed] [Google Scholar]
- Freire-Maia N 1977. Nosologic groups. An overview. Hum Hered 27(4):251–256. [DOI] [PubMed] [Google Scholar]
- Freire-Maia N, Pinheiro M. 1984. Ectodermal Dysplasias: A Clinical and Genetic Study. New York: Alan R Liss, Inc; 251 p. [Google Scholar]
- Freire-Maia N, Pinheiro M. 1988. Ectodermal dysplasia-some recollections and a classification In: Salinas CF, Opitz JM, Paul NW, editors. Recent Advances in Ectodermal Dysplasias. New York: Alan R. Liss, Inc; p 3–14. [PubMed] [Google Scholar]
- Headon DJ, Emmal SA, Ferguson BM, Tucker AS, Justice MJ, Sharpe PT, Zonana J, Overbeek PA. 2001. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 414(6866):913–916. [DOI] [PubMed] [Google Scholar]
- Itin PH. 2013. Ectodermal dysplasia: thoughts and practical concepts concerning disease classification - the role of functional pathways in the molecular genetic diagnosis. Dermatology 226(2):111–114. [DOI] [PubMed] [Google Scholar]
- Itin PH. 2014. Etiology and pathogenesis of ectodermal dysplasias. Am J Med Genet A 164A(10):2472–2477. [DOI] [PubMed] [Google Scholar]
- Kantaputra PN, Carlson BM. 2018. Genetic regulatory pathways of split-hand/foot malformation. Clin Genet. [DOI] [PubMed] [Google Scholar]
- Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, Ferguson B, Munoz F, Morgan D, Clarke A, Baybayan P, Chen EY, Ezer S, Saarialho-Kere U, de la Chapelle A, Schlessinger D. 1996. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nature Genet 13:409–416. [DOI] [PubMed] [Google Scholar]
- Koster MI. 2010. p63 in skin development and ectodermal dysplasias. J Invest Dermatol 130(10):2352–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucker GP, Van de Kerkhof PC, Steijlen PM. 1994. The hereditary palmoplantar keratoses: an updated review and classification. Br J Dermatol 131(1):1–14. [DOI] [PubMed] [Google Scholar]
- Naveau A, Seidel K, Klein OD. 2014. Tooth, hair and claw: comparing epithelial stem cell niches of ectodermal appendages. Exp Cell Res 325(2):96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Nielsen M, Skovbo S, Svaneby D, Pedersen L, Fryzek J. 2013. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur J Med Genet 56(5):236–242. [DOI] [PubMed] [Google Scholar]
- Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, Coudiere P, DiGiovanna JJ, Elias P, Fischer J, Fleckman P, Gina M, Harper J, Hashimoto T, Hausser I, Hennies HC, Hohl D, Hovnanian A, Ishida-Yamamoto A, Jacyk WK, Leachman S, Leigh I, Mazereeuw-Hautier J, Milstone L, Morice-Picard F, Paller AS, Richard G, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taieb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. 2010. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol 63(4):607–641. [DOI] [PubMed] [Google Scholar]
- Pagnan NA, Visinoni AF. 2014. Update on ectodermal dysplasias clinical classification. Am J Med Genet A 164A(10):2415–2423. [DOI] [PubMed] [Google Scholar]
- Patthey C, Gunhaga L. 2014. Signaling pathways regulating ectodermal cell fate choices. Exp Cell Res 321(1):11–16. [DOI] [PubMed] [Google Scholar]
- Priolo M, Lagana C. 2001. Ectodermal dysplasias: a new clinical-genetic classification. J Med Genet 38(9):579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostagno P, Wolchinsky Z, Vigano AM, Shivtiel S, Zhou H, Van Bokhoven H, Ferone G, Missero C, Mantovani R, Aberdam D, Virolle T. 2010. Embryonic stem cells as an ectodermal cellular model of human p63-related dysplasia syndromes. Biochem Biophys Res Commun 395(1):131–135. [DOI] [PubMed] [Google Scholar]
- Salinas CF, Irvine AD, Itin PH, Di Giovanna JJ, Schneider H, Clarke AJ, McGovern LS, Fete M. 2014. Second International Conference on a classification of ectodermal dysplasias: development of a multiaxis model. Am J Med Genet A 164A(10):2482–2489. [DOI] [PubMed] [Google Scholar]
- Salinas CF, Jorgenson RJ, Wright JT, DiGiovanna JJ, Fete MD. 2009. 2008 International Conference on Ectodermal Dysplasias Classification: conference report. Am J Med Genet A 149A(9):1958–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H, Faschingbauer F, Schuepbach-Mallepell S, Korber I, Wohlfart S, Dick A, Wahlbuhl M, Kowalczyk-Quintas C, Vigolo M, Kirby N, Tannert C, Rompel O, Rascher W, Beckmann MW, Schneider P. 2018. Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia. N Engl J Med 378(17):1604–1610. [DOI] [PubMed] [Google Scholar]
- van den Boogaard MJ, Creton M, Bronkhorst Y, van der Hout A, Hennekam E, Lindhout D, Cune M, Ploos van Amstel HK. 2012. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet 49(5):327–331. [DOI] [PubMed] [Google Scholar]
- Vera-Carbonell A, Moya-Quiles MR, Ballesta-Martinez M, Lopez-Gonzalez V, Bafalliu JA, Guillen-Navarro E, Lopez-Exposito I. 2012. Rapp-Hodgkin syndrome and SHFM1 patients: delineating the p63-Dlx5/Dlx6 pathway. Gene 497(2):292–297. [DOI] [PubMed] [Google Scholar]
- Wisniewski SA, Trzeciak WH. 2012. A new mutation resulting in the truncation of the TRAF6-interacting domain of XEDAR: a possible novel cause of hypohidrotic ectodermal dysplasia. J Med Genet 49(8):499–501. [DOI] [PubMed] [Google Scholar]
- Wright JT, Morris C, Clements SE, D’Souza R, Gaide O, Mikkola M, Zonana J. 2009. Classifying ectodermal dysplasias: Incorporating the molecular basis and pathways (Workshop II). Am J Med Genet A 149A(9):2062–2067. [DOI] [PubMed] [Google Scholar]
- Yang Y, Luo L, Xu J, Zhu P, Xue W, Wang J, Li W, Wang M, Cheng K, Liu S, Tang Z, Ring BZ, Su L. 2013. Novel EDA p.Ile260Ser mutation linked to non-syndromic hypodontia. J Dent Res 92(6):500–506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.