

Abstract
Fourteen novel amino-quinoline-5,8-dione derivatives (6a-h and 7a-h) were designed and synthesized by coupling different alkyl-or aryl-amino fragments at the C6-or C7-position of quinoline-5,8-dione. All target compounds showed antiproliferative potency in the low micromolar range in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines. Compounds 6h, 6d, 7a, and 7d exhibited more potent antiproliferative effects than the other compounds. Especially, compounds 6d and 7d displayed NQO1-dependent cytotoxicity and competitive NQO1 inhibitory effects in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines. Furthermore, compounds 6h, 6d, 7a, and 7d induced a dose-dependent lethal mitochondrial dysfunction in both drug sensitive HeLaS3 and multidrug resistant KB-vin cells by increasing intracellular reactive oxygen species (ROS) levels. Notably, compound 7d selectively inhibited cancer cells, but not non-tumor liver cell proliferation in vitro, and significantly triggered HeLaS3 cell apoptosis by regulating apoptotic proteins of Bcl-2, Bax, and cleaved caspase-3 in a dose-dependent manner. Our findings suggest that these novel C6-or C7-substituted amino-quinoline-5,8-dione derivatives, such as 7d, could be further developed in the future as potent and selective antitumor agents to potentially circumvent multi-drug resistance (MDR).
Keywords: Antiproliferative activity; Quinone oxidoreductase 1 (NQO1); Amino-quinoline-5,8-dione; Reactive oxygen species (ROS); Multidrug resistance; Apoptosis
1. Introduction
Due to aberrant metabolic events and abnormal proliferative growth phenotypes, reactive oxygen species (ROS) levels are commonly elevated in cancer cells [1−3]. To cope with an increase in intrinsic oxidative stress, cancer cells augment their antioxidant capacity through upregulation of antioxidant enzymes [4]. In doing so, cancer cells become more dependent on the elevated antioxidant capacity for growth and survival, which makes them susceptible when oxidative stress is further elevated through production of more ROS and/or inhibition of elevated antioxidant enzymes [4,5]. Therefore, a feasible strategy in cancer chemotherapy is to selectively target cancer cells based on the different redox states between normal and tumor cells [1,6–8], particularly when such targeting involves a redox adaptive mechanism that confers drug resistance [2].
In this context, inhibition of NAD(P)H:quinone oxidoreductase 1 (NQO1) has emerged as a promising redox-modulating strategy for anticancer therapy. NQO1 is a ubiquitous flavoenzyme that catalyzes the two-electron reduction of quinones to hydroquinones. It is intimately involved with cancer and upregulated in several human tumor cells, such as pancreas, colon, breast, lung, liver, stomach, kidney, and ovaries, at levels of up to 50-fold greater than in normal tissue [9,10]. Moreover, NQO1 is found in the cytosol, Golgi complex, nucleus, mitochondria, cellular membrane, and endoplasmic reticulum, as well as extracellularly [11–14]. The NQO1 and glutathione systems are the two major cellular thiol antioxidant systems that can be targeted for anticancer therapy [15–18]. Many human cancers can reportedly be killed selectively through rapid ROS generation mediated by NQO1 bioreduction (Fig. 1).
Fig. 1.
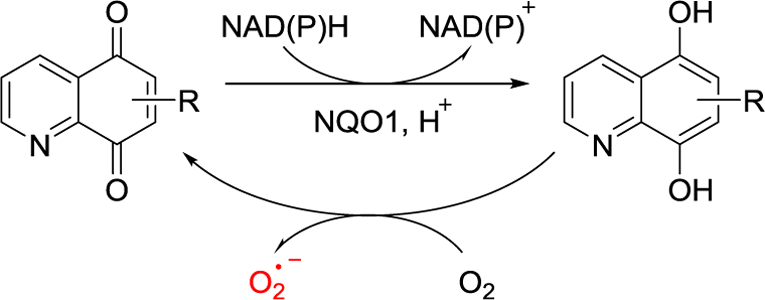
NQO1-mediated redox cycling of quinolinediones.
Recent findings suggest that some quinolinediones are excellent substrates of NQO1 and are selectively cytotoxic to cancer cell lines that overexpress NQO1, but not to normal cell lines [17–23]. These findings form the basis for the development of novel quinoline-5,8-dione derivatives as potential NQO1-directed cytotoxicity agents, which efficiently regulate the intracellular human NQO1 dependent redox cycling and thus the ROS generated in the progress (Fig. 1). Moreover, small substituents such as amine groups at the C-6 or C-7 positions of the quinoline-5,8-dione moiety favorably impacted binding to the active site of NQO1 [16,17,24,25]. In this study, we coupled different alkyl-or aryl-amino fragments to the C6-or C7-position of quinoline-5,8-dione to design novel amino-quinoline-5,8-dione derivatives, and compared the cytotoxic effects of these compounds. In addition, because most quinolinedione derivatives usually have poor water solubility, we introduced hydrophilic groups at the end of arylamino fragments to improve the lipid-aqueous partition coefficient. Herein, we report our synthesis of novel amino-quinoline-5,8-dione derivatives (6a‒6h and 7a‒7h, Table 1), as well as evaluation of their cytotoxicity effects and mechanisms in multiple cancer cell lines.
Table 1.
In vitro antiproliferative data of 6a-h and 7a-h against HeLaS3 and KB-vin cells.
| Compounds | IC50a (µM) |
Compounds | IC50 (µM) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| HeLaS3b | KB-vinc | HeLaS3 | KB-vin | ||||||
| 6a | 6.24 | ± | 1.12 | 5.12 | ± | 0.23 | 7a | 4.53 ± 0.35 | 1.10 ± 0.12 |
| 6b | 5.50 | ± | 0.61 | 6.20 | ± | 0.08 | 7b | 6.12 ± 0.33 | 4.00 ± 0.39 |
| 6c | 6.64 | ± | 0.04 | 4.92 | ± | 0.62 | 7c | 6.21 ± 0.08 | 5.98 ± 0.58 |
| 6d | 0.80 | ± | 0.02 | 1.52 | ± | 0.04 | 7d | 0.59 ± 0.03 | 0.97 ± 0.01 |
| 6e | 5.26 | ± | 0.34 | 4.03 | ± | 0.26 | 7e | 4.37 ± 0.36 | 4.69 ± 0.29 |
| 6f | 5.16 | ± | 0.22 | 5.23 | ± | 0.36 | 7f | 5.07 ± 0.10 | 6.19 ± 0.59 |
| 6g | 5.84 | ± | 0.49 | 3.40 | ± | 0.35 | 7g | 5.67 ± 0.62 | 5.62 ± 0.26 |
| 6h | 5.36 | ± | 0.22 | 0.91 | ± | 0.03 | 7h | 4.99 ± 0.16 | 4.27 ± 0.29 |
| Paclitaxel | 0.013 ± 0.0014 | 1.01 ± 0.11 | |||||||
The inhibitory effects of individual compounds on the proliferation of cancer cell lines were determined by the SRB assay. The data are expressed as the mean ± SD of three independent experiments data.
Human cervical epithelioid carcinoma cell line.
Multi-drug resistant human cervical cancer cell line.
2. Results and discussion
2.1. Chemistry
Scheme 1 summarizes the syntheses of the quinolinedione derivatives 6a‒6h and 7a‒7h. Quinoline-5,8-dione (4) was prepared from 5-hydroxyquinoline (3) by oxidation with iodobenzene diacetate (PIDA) in a mixture of CH3CN:H2O (2:1) according to Barret’s method [26]. Compound 4 was subsequently treated with different substituted 2-(1H-indol-3-yl)ethanamines (5a‒5c) or anilines (5d‒5f) in MeOH to obtain the target compounds 6a‒6c and 7a‒7c or 6d‒6f and 7d‒7f, respectively. This is a non-regioselective Michael addition to give the hydroquinone followed by an autoxidation back to quinone. Often, only catalytic base and air are needed for such oxidations [27]. Fortunately, these two regioisomers were easy separated by column chromatography (CH2Cl2/MeOH = 20/1), and the major isomers were 6a‒6f. In addition, treatment of (E)-methyl 3-(4-(bromomethyl)phenyl) acrylate 8a or (E)-methyl 3-(3-(bromomethyl)phenyl) acrylate 8b in DMF with NaN3 produced intermediate 9a,b. Then intermediate 9a,b was treated with PPh3 in THF to furnish compounds 10a,b, which were further reacted with quinoline-5,8-dione 4 in the presence of MeOH to produce target compound 6g, 7g or 6h, 7h. All target compounds were purified by column chromatography, and their structures were confirmed by 1H NMR, 13C NMR, MS, and HRMS. Each target compound was >95% pure as determined by high-performance liquid chromatography and could be used for subsequent experiments.
Scheme 1.
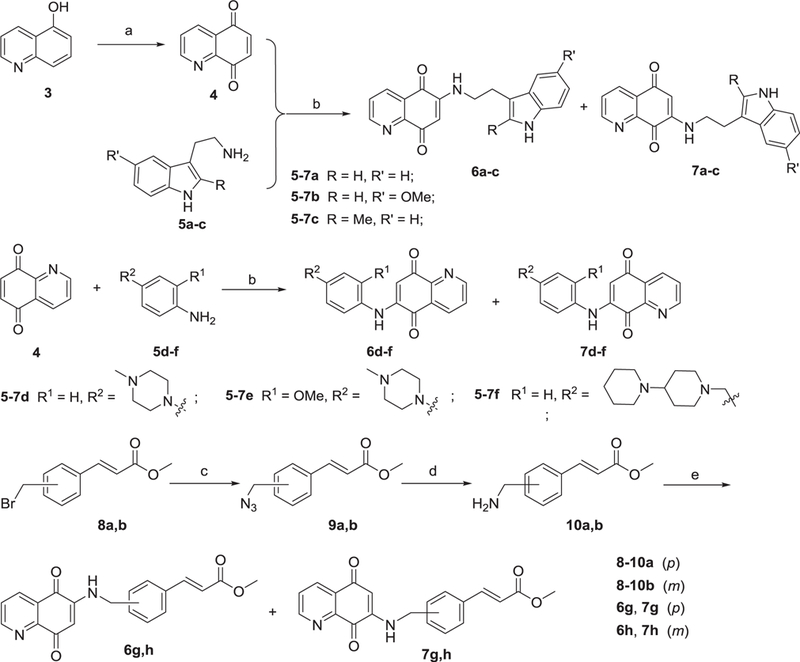
Reagents and conditions: (a) iodobenzene diacetate, CH3CN:H2O = 2:1, 0 °C, 2 h, 86%; (b) MeOH, rt, 6 h, 31−43%; (c) NaN3, DMF, rt, 2 h; (d) PPh3, anhydrous THF, rt, 2 h; (e) quinolinedione 4, MeOH, rt, 6 h, 22−41%.
2.2. Antiproliferative activity against both sensitive and resistant human cervical cancer cells
To evaluate the antiproliferative activity of the target compounds, we initially tested their effects on the proliferation of parental sensitive cervical epithelioid carcinoma cells HeLaS3 and multi-drug resistant human cervical cancer cells KB-vin (both cell lines are used to investigate the cytotoxicity effect and antidrug resistance of potential drugs in vitro) by the SRB assay using paclitaxel as a positive control. The IC50 values of individual compounds against each tumor cell line are presented in Table 1. The results showed that all target compounds displayed antiproliferative potency in the low micromolar range against both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines. Some compounds, such as 6h, 6d, 7a, and 7d, exhibited significant antiproliferative effects with IC50 values between 0.80 and 1.52 µM. These four compounds displayed comparable or greater potency than paclitaxel (IC50 1.01 µM) against multidrug resistant KB-vin cells. Structurally, the addition of a 4-(4-methylpiperazin-1-yl) phenylamino group to position-6 (6d) or ‒ 7 (7d) of quinoline-5,8-dione led to the greatest antiproliferative potency against both HeLaS3 (IC50 0.80 and 0.59 µM, respectively) and KB-vin (IC50 1.52 and 0.97 µM, respectively) cell lines. Compounds 6h (6-amino-(3-methylphenyl)acrylic ester) and 7a (7-aminoethylindole) showed comparable potency to 7d against KB-vin, but not HeLasS3, cells. Indeed, all other compounds were significantly less potent (IC50 3.40−6.24 µM) against the two tested cancer cell lines, even those with only slight structural differences (e.g., 6e and 7e have a 2-methoxy group in addition to the 4-(4-methylpiperazin-1-yl) group found on the phenylamino side chain of 6d and 7d.
2.3. NQO1-dependency assessments
Within the drug concentration range tested, the compounds 6d and 7d showed the most potent cytotoxicities against both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines. To deepen our knowledge about the mechanism of action of compounds 6d and 7d, we examined their potential NQO1-dependent cytotoxicities, with or without dicoumarol (DIC, an NQO1 inhibitor) and N-acetylcysteine (NAc, a free radical scavenger). Based on the survival curves (Fig. 2), the cell viabilities of compound-treated HeLaS3 and KB-vin cells were significantly increased by the addition of DIC and NAc. The cytotoxic IC50 values were greater than 3 µM against both cell lines in the presence of 6 or 7, DIC, and NAc, but much lower in the presence of compound alone (6d only: 0.8 µM, HeLaS3 and 1.52 µM, KB-vin; 7d only: 0.59 µM HeLaS3 and 0.97 µM, KB-vin). These results suggested that the cytotoxic effects of 6d and 7d were NQO1-specific in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines.
Fig. 2.
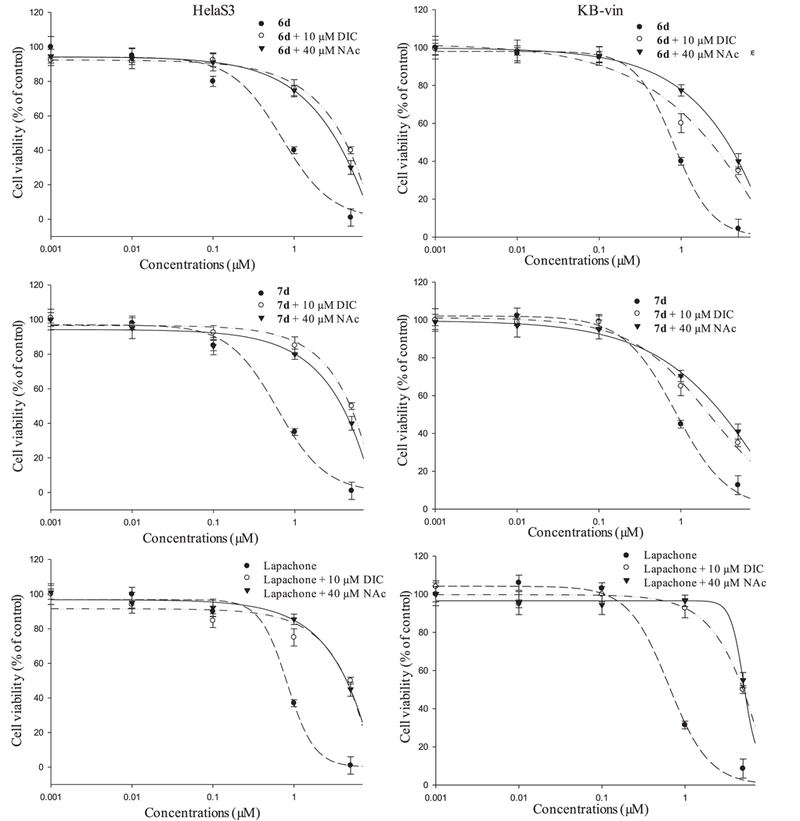
Evaluations of NQO1-dependent cytotoxicity of testing compounds. The cell viability was evaluated by combination treatment of 10 mM DIC or 40 mM NAc with testing compounds, using SRB assay after 72 h treatment.
2.4. NQO1 inhibitory activities and kinetics
Based on the encouraging antiproliferative activity of 6a-h and 7a-h against HeLaS3 and KB-vin cells, we further tested a selected group of compounds (6h, 6d, 7a, and 7d) for in vitro inhibitory activity against NQO1. As shown in Fig. 3, the NQO1 activities in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines were inhibited dose-dependently after 24 h treatment with 6h, 6d, 7a, and 7d. Particularly, compounds 6d and 7d were more potent NQO1 inhibitors than 6h and 7a in drug sensitive HeLaS3 cells. On the other hand, 6h, 6d, 7a, and 7d showed similar NQO1 inhibitory effects in multidrug resistant KB-vin cells (Table 2). Therefore, these four compounds displayed dose-dependent NQO1 inhibitory activity.
Fig. 3.
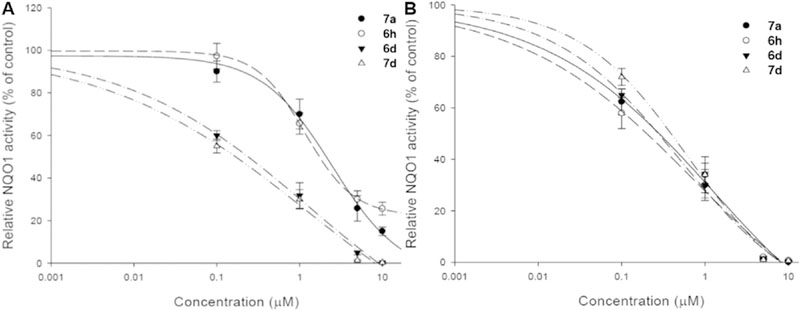
NQO1 activity of (a) HeLa cells (b) KB-vin cells after compound treatment. The HeLa and KB-vin cells were treated with serial concentrations of 6h, 6d, 7a, and 7d for 24 h. NQO1 activity was detected by the NQO1 activity assay kit (catalog number: ab184867), and was significantly altered after treatment with 6h, 6d, 7a, and 7d for 24 h.
Table 2.
Effects of 6h, 6d, 7a, and 7d on NQO1 activity in HeLaS3 (parental) and KB-vin (MDR) cancer cell lines.
| Compound | IC50 (µM) |
|||||
|---|---|---|---|---|---|---|
| HeLaS3 | KB-vin | |||||
| 7a | 6.25 | ± | 1.25 | 0.55 | ± | 0.01 |
| 6h | 5.25 | ± | 0.90 | 0.53 | ± | 0.05 |
| 6d | 0.47 | ± | 0.07 | 0.56 | ± | 0.01 |
| 7d | 0.44 | ± | 0.08 | 0.57 | ± | 0.02 |
The NQO1 inhibitory kinetics of 6d and 7d indicated competitive inhibition as demonstrated by the Lineweaver-Burk plots (Fig. 4). As the concentration of 6d increased, the maximum rate (Vmax) remained the same (12.5 ± 1.2 nmol/mg protein/20s) and the affinity (Km) increased (no treatment control: 30.22 ± 2.5 µM; addition of 1.0 µM 6d: 45.27 ± 5.33 µM; addition of 2.0 µM 6d: 60.15 ± 3.51 µM). In terms of 7d, as the concentration of 7d increased, the maximum rate (Vmax) remained the same (11.03 ± 1.5 nmol/mg protein/20s) and the affinity (Km) increased (no treatment control: 20.30 ± 0.97 µM; addition of 1.0 µM 7d: 40.17 ± 2.31 µM; addition of 2.0 µM 7d: 55.17 ± 1.49 µM). These results indicated that 6d and 7d are competitive inhibitors of NQO1.
Fig. 4.
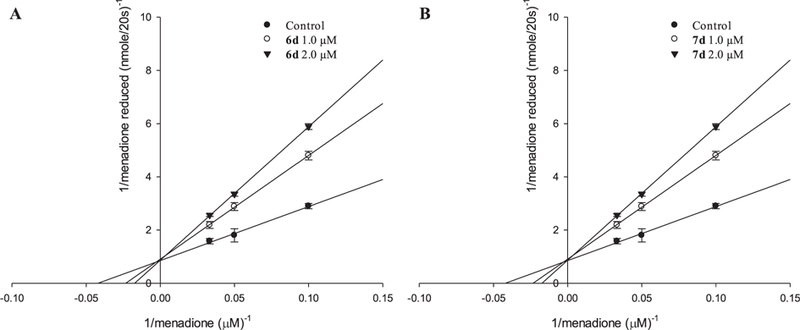
Enzyme kinetics for compound 6d and 7d using recombinant human NQO1. Lineweaver-Burk plot for NQO1 inhibition demonstrated that 6d and 7d competitively inhibited NQO1 activity.
2.5. Treatment with quinolinedione derivatives increases ROS levels
We next examined whether the killing effect of quinolinedione derivatives on cervical cancer cells involved increasing ROS levels. ROS levels in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines were assessed by flow cytometry using the redox-sensitive fluorescent probe 2’,7’-dichlorofluorescein diacetate (DCFH-DA). As shown in Fig. 5, the ROS production increased dose-dependently in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines after 24 h treatment with 6h, 6d, 7a, and 7d. These findings suggested that treatment with 6h, 6d, 7a, and 7d lead to increased oxidative stress in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines, which causes significant cytotoxicity in tumor cells.
Fig. 5.
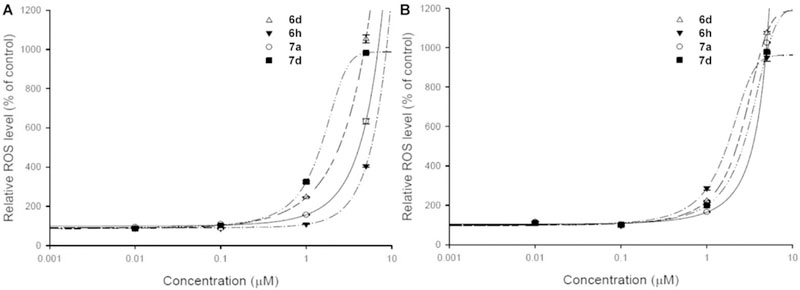
Reactive oxygen species (ROS) generation of (a) HeLa cells (b) KB-vin cells after compound treatment. The HeLa and KB-vin cells were treated with serial concentrations of PL, 6h, 6d, 7a, and 7d for 24 h. The generated ROS were detected by the ROS-Glo™ H2O2 assay kit (catalog number: G8821), and increased significantly after treatment with 6h, 6d, 7a, and 7d for 24 h.
2.6. Quinolinedione derivatives induce mitochondrial dysfunction
It is well known that mitochondria are central to the regulation of apoptosis. Loss of mitochondrial membrane potential (Δψm) is catastrophic for cells and leads to the release of cytochrome C into the cytosol [28]. The mitochondrial membrane potential of both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines were significantly decreased in a dose-dependent manner after 24 h treatment with 6h, 6d, 7a, and 7d (Fig. 6).
Fig. 6.
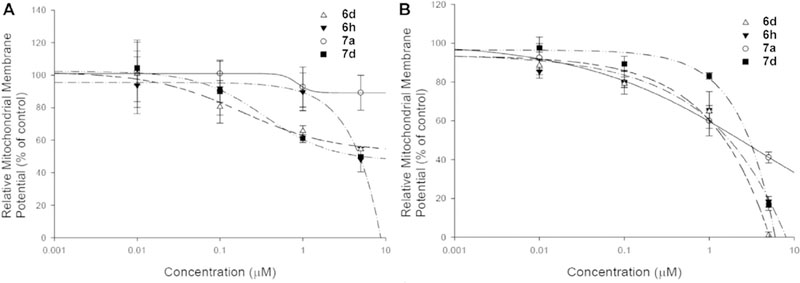
Changes of mitochondrial membrane potential (MMP) in (a) HeLa cells (b) KB-vin cells after compound treatment. The HeLa and KB-vin cells were treated with serial concentrations of PL, 6h, 6d, 7a, and 7d for 24 h. The MMP was detected by the MITO-ID® membrane potential cytotoxicity kit (catalog number: ENZ-51019-KP002), and decreased significantly after treatment with 6h, 6d, 7a, and 7d for 24 h in drug-resistant KB-vin cells.
2.7. The selectivity of quinolinedione derivatives to tumor cells
Given the strong antiproliferative and NQO1 inhibitory effects of 7d in vitro, we investigated the selectivity profile by examining inhibitory effects on the growth of both cancer HeLaS3 cells and normal endometrial epithelial cells (EECs). Treatment with increasing doses of 7d had no significant effect on the survival of non-tumor EECs, while the same treatment regimen induced major HeLaS3 cell death (Fig. 7), suggesting that 7d might possess selective antiproliferative activity against tumor cells.
Fig. 7.
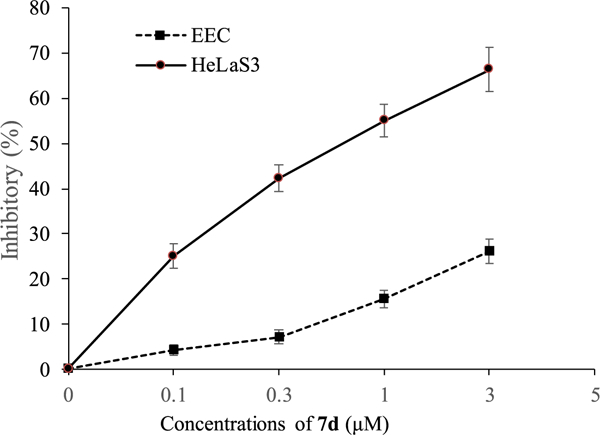
Inhibitory effects of 7d on the proliferation of HeLaS3 and EEC cells. Cells were incubated with the indicated concentrations of 7d for 72 h. Cell proliferation was assessed using the MTT assay. Data are expressed as means ± SD of the inhibition (%) from three independent experiments.
2.8. Quinolinedione derivatives induce apoptosis in HeLaS3 cells
To determine whether the inhibitory effects of 7d on cervical carcinoma cell proliferation are accompanied by enhanced cancer cell apoptosis, HeLaS3 cells were incubated with different concentrtions of 7d for 72 h, and the percentages of apoptotic cells were determined by FITC-Annexin V/PI double staining and flow cytometry. As shown in Fig. 8, the percentage of annexin V + apoptotic HeLaS3 cells gradually increased for those cells exposed to increasing concentrations of 7d (12.8% for 0.2 µM; 27.6% for 0.6 µM; and 66.3% for 1.8 µM), demonstrating that incubation with 7d induced HeLaS3 cell apoptosis in a dose-dependent manner.
Fig. 8.
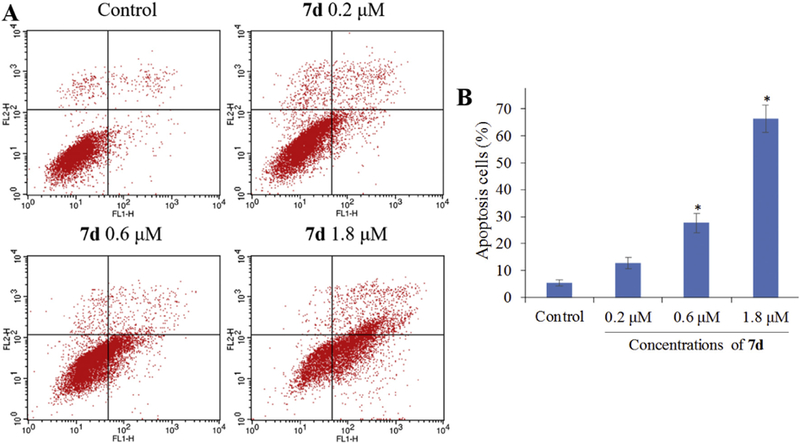
Compound 7d induces HeLaS3 cell apoptosis in vitro. HeLaS3 cells were incubated with the indicated concentrations of 7d for 72 h, and the cells were stained with FITC-Annexin V/PI, followed by flow cytometry analysis. (A) Flow cytometry analysis. (B) Quantitative analysis of apoptotic cells. Data are expressed as means ± SD of the percentages of apoptotic cells from three independent experiments. *P < 0.01 vs control.
To further investigate the induction of apoptosis by 7d, we examined the expression of apoptotic proteins Bax, Bcl-2, and the cleavage states of caspase-3 [29], in response to 7d treatment (Fig. 9). Sub-confluent HeLaS3 cervical carcinoma cells were treated with or without 7d for 72 h, and then lysed and analyzed by western blot. β-Actin expression was used as an internal control. The treatment with 7d dramatically increased the relative levels of pro-apoptotic Bax expression but reduced the levels of anti-apoptotic Bcl-2 expression in a dose-dependent manner. Furthermore, compound 7d treatment also induced more cleavage of caspase-3 than the control group (Fig. 9A and B). Taken together, these results confirm that 7d treatment induced apoptosis in HeLaS3 cells.
Fig. 9.
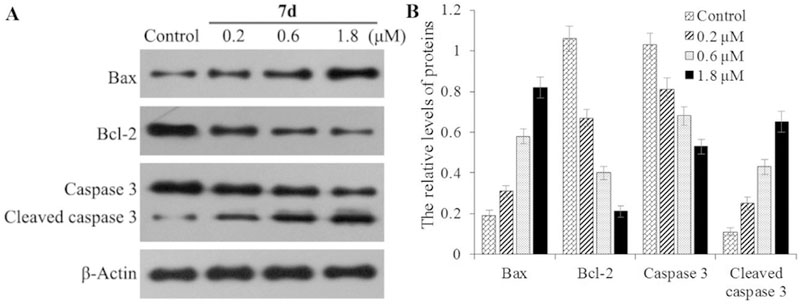
(A) The expression of Bax, Bcl-2, Caspase 3, and b-actin was examined by western blot analysis. HeLaS3 cells were incubated with or without 7d at the indicated concentrations for 72 h and the levels of protein expression were detected using specific antibodies. Data shown are representative images of each protein for three separate experiments. (B) Quantitative analysis of Bax, Bcl-2, and Caspase 3. The relative levels of each protein compared to control b-actin were determined by densimetric scanning. Data are expressed as means ± SD from three separate experiments. *P < 0.01 vs respective control.
3. Conclusion
In summary, we have designed and synthesized novel amino-quinoline-5,8-dione derivatives 6a-h and 7a-h by coupling different alkyl-or aryl-amino fragments at the C6-or C7-position of quinoline-5,8-dione, and then evaluated their biological activities in various in vitro assays. All target compounds showed antiproliferative potency in the low micromolar range in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines. Among them, compounds 6d and 7d exhibited NQO1-dependent antiproliferative effects with IC50 values between 0.59 and 1.52 µM, as well as competitive NQO1 inhibitory effects in both drug sensitive HeLaS3 and multidrug resistant KB-vin cell lines. Furthermore, compounds 6h, 6d, 7a, and 7d induced a dose-dependent lethal mitochondrial dysfunction in both drug sensitive HeLaS3 and multidrug resistant KB-vin cells by increasing intracellular ROS levels. Notably, compound 7d selectively inhibited cancer cells but not non-tumor liver cell proliferation in vitro, and significantly induced HeLaS3 cell apoptosis by reducing the anti-apoptotic protein levels of Bcl-2 and up-regulating the pro-apoptotic proteins of Bax and cleaved caspase-3 in a concentration dependent manner. Taken together, these results suggest that these novel C6-or C7-substituted amino-quinoline-5,8-dione derivatives, such as 7d, could be further developed as potent and selective antitumor agents to potentially circumvent MDR.
4. Experimental section
The synthesized compounds were purified by column chromatography using silica gel (300−400 mesh) except for recrystallization and thin-layer chromatography (TLC) using silica gel 60 F254 plates (250 mm; Qingdao Ocean Chemical Company, China). 1H NMR and 13C NMR spectra were recorded with a Bruker Avance 400 MHz spectrometer at 300 K, using TMS as an internal standard. MS spectra were recorded on a Mariner Mass Spectrum (ESI). High resolution mass spectra were recorded with a hybrid LTQ FT (ICR 7 T) (ThermoFisher, Bremen, Germany) mass spectrometer coupled with a Waters Acquity H-class liquid chromatograph system. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Solutions after reactions and extractions were concentrated using a rotary evaporator operating at a reduced pressure of ca. 20 Torr. Organic solutions were dried over anhydrous sodium sulfate.
4.1. General procedure for synthesis of quinoline-5,8-dione (4)
A solution of iodobenzene diacetate (10.35 mmol, 3.3 g) in 12 mL CH3CN:H2O (2:1) was added to quinolin-5-ol (3) (3.45 mmol, 0.50 g) at 0 °C under N2 and stirred for 1 h. After the reaction was completed, water (40 mL) was added, and the solution was extracted with EtOAc (3 × 20 mL). The organic phase was washed by brine and concentrated in vacuo to give 4 as a yellow solid, yield 82%. MS (ESI) m/z = 160 [M+H]+.
4.2. General procedure for synthesis of 10a, b
4.2.1. (E)-Methyl 3-(4-(aminomethyl)phenyl)acrylate (10a)
To a solution of (E)-methyl 3-(4-(bromomethyl)phenyl)acrylate 8a (0.1 g, 0.39 mmol) in 4 mL DMF, NaN3 (38.2 mg, 0.59 mmol) was added and the mixture was stirred at rt for 2 h. After the reaction was completed, 20 mL water were added and the solution was extracted with EtOAc (3 × 20 mL). The organic phase was washed by brine, dried over anhydrous sodium sulfate, and evaporated in vacuo to give 9a, which was then dissolved in 3 mL THF. Triphenylphosphine was added and the mixture was stirred at rt for 2 h. Then, 1 mL water was added and the mixture was stirred for 1 h. The solution was extracted with EtOAc (3 × 5 mL). The organic phase was combined and washed with brine, dried over anhydrous sodium sulfate, and evaporated in vacuo. The residue was purified by column chromatography (EA/PE = 2/1) on silica gel to afford 10a as a clear translucent oil in 52% yield. MS (ESI) m/z = 192 [M+H]+.
4.2.2. (E)-Methyl 3-(3-(aminomethyl)phenyl)acrylate (10b)
Compound 10b was synthesized from (E)-methyl 3-(3-(bromomethyl)phenyl)acrylate 8b, according to the synthetic procedure for 10a in a yield of 52%. MS (ESI) m/z = 192 [M+H]+.
4.3. General procedure for synthesis of 6a, 7a
Quinoline-5,8-dione (4) (149.1 mg, 0.94 mmol) was added to a solution of 2-(1H-indol-3-yl) ethanamine (5a) (100.0 mg, 0.63 mmol) in MeOH (4 mL) at rt. The mixture was stirred at rt for 16 h and evaporated in vacuo. The residue was purified by column chromatography (CH2Cl2/MeOH 20/1) to give 6a and 7a in yields of 40% and 26%, respectively.
4.3.1. 6-((2-(1H-Indol-3-yl)ethyl)amino)quinoline-5,8-dione (6a)
Analytical data for 6a: 1H NMR (DMSO-d6, 400 MHz): δ 10.87 (s, 1H, NH), 8.91 (m, 1H, Ar-H), 8.31 (m, 1H, Ar-H), 7.82 (m, 2H, NH, Ar-H), 7.78 (d, 1H, J = 8.0 Hz, Ar-H), 7.36 (m, 2H, Ar-H), 7.28 (m, 2H, Ar-H), 5.80 (s, 1H, CH), 3.53 (m, 2H, CH2), 3.06 (m, 2H, CH2). 13C NMR (DMSO-d6, 100 MHz): δ 180.8, 180.2, 153.0, 149.4, 147.1, 136.7, 133.9, 130.7, 128.9, 127.5, 123.5, 123.5, 121.5, 118.6, 111.9, 111.6, 99.1, 43.2, 23.7. MS (ESI) m/z = 318 [M+H]+; MS (ESI) m/z = 318 [M+H]+; HRMS (ESI): m/z calcd for C19H16N3O2: 318.1243; found: 318.1231 [M+H]+.
4.3.2. 7-((2-(1H-Indol-3-yl)ethyl)amino)quinoline-5,8-dione (7a)
Analytical data for 7a: 1H NMR (d6-CD3OD, 400 MHz): δ 8.83 (m, 1H, NH), 8.42 (m, 1H, Ar-H), 7.79 (dd, 1H, J = 4.0 Hz, J = 4.0 Hz, Ar-H), 7.60 (s, 1H, Ar-H), 7.31 (m, 2H, NH, Ar-H), 6.99–7.15 (m, 4H, Ar-H), 5.78 (s, 1H, CH), 3.51 (m, 2H, CH2), 3.04 (m, 2H, CH2). 13C NMR (DMSO-d6, 100 MHz): δ 180.8, 180.2, 153.1, 149.4, 147.1, 136.7, 133.9, 130.8, 129.1, 127.6, 123.6, 123.5, 121.4, 118.6, 111.9, 111.6, 99.2, 43.1, 24.0. MS (ESI) m/z = 318 [M+H]+; MS (ESI) m/z = 318 [M+H]+; HRMS (ESI): m/z calcd for C19H16N3O2: 318.1243; found: 318.1241 [M+H]+.
4.4. General procedure for synthesis of 6b, 7b
Compounds 6b and 7b were synthesized from quinoline-5,8-dione (4), 2-(5-methoxy-1H-indol-3-yl)ethanamine (5b), according to the synthetic procedure for compounds 6a and 7a in yields of 41% and 23%, respectively.
4.4.1. 6-((2-(5-Methoxy-1H-indol-3-yl)ethyl)amino)quinoline-5,8-dione (6b) [30]
Analytical data for 6b: 1H NMR (DMSO-d6, 400 MHz): δ 10.71 (s, 1H, NH), 8.96 (m, 1H, Ar-H), 8.32 (d, 1H, J = 4.0 Hz, Ar-H), 7.72 (m, 2H, Ar-H, NH), 7.22 (m, 2H, Ar-H), 7.06 (d, 1H, J = 4.0 Hz, Ar-H), 6.71 (m, 1H, Ar-H), 5.87 (s, 1H, CH), 3.76 (s, 3H, OCH3), 3.48 (m, 2H, CH2), 3.00 (m, 2H, CH2). 13C NMR (DMSO-d6, 100 MHz): δ 182.0, 180.3, 155.0, 153.5, 149.2, 148.4, 134.3, 131.8, 128.0, 127.9, 127.9, 126.9, 124.2, 112.5, 111.6, 111.4, 101.1, 100.4, 55.8, 43.1, 23.8. MS (ESI) m/z = 348 [M+H]+; HRMS (ESI): m/z calcd for C20H18N3O3: 348.1348; found: 348.1344 [M+H]+.
4.4.2. 7-((2-(5-Methoxy-1H-indol-3-yl)ethyl)amino)quinoline-5,8-dione (7b)
Analytical data for 7b: 1H NMR (d6-CD3OD, 400 MHz): δ 8.79 (m, 1H, Ar-H), 8.36 (d, 1H, J = 4.0 Hz, Ar-H), 7.76 (m, 1H, Ar-H), 7.18 (m, 3H, J = 6.0 Hz, NH, Ar-H), 6.70 (m, 1H, Ar-H), 5.75 (s, 1H, CH), 3.75 (s, 3H, OCH3), 3.45 (m, 2H, CH2), 3.08 (m, 2H, CH2). 13C NMR (DMSO-d6, 100 MHz): δ 182.0, 180.3, 155.0, 153.5, 149.3, 148.4, 134.3, 131.8, 128.0, 127.9, 126.9, 124.2, 112.5, 111.6, 111.4, 101.1, 100.4, 55.7, 41.1, 23.8. MS (ESI) m/z = 348 [M+H]+; HRMS (ESI): m/z calcd for C20H18N3O3: 348.1348; found: 348.1343 [M+H]+.
4.5. General procedure for synthesis of 6c, 7c
Compounds 6c and 7c were synthesized from quinoline-5,8-dione (4), 2-(2-methyl-1H-indol-3-yl) ethanamine (5c), according to the synthetic procedure for compounds 6a and 7a in yields of 38% and 25%, respectively.
4.5.1. 6-((2-(2-Methyl-1H-indol-3-yl)ethyl)amino)quinoline-5,8-dione (6c)
Analytical data for 6c: 1H NMR (DMSO-d6, 400 MHz): δ 10.78 (s, 1H, NH), 8.96 (d, 1H, J = 4.0 Hz, Ar-H), 8.33 (d, 1H, J = 8.0 Hz, Ar-H), 7.73 (m, 3H, NH, Ar-H), 7.24 (d, 1H, J = 8.0 Hz, Ar-H), 7.00 (m, 2H, Ar-H), 5.80 (s, 1H, CH), 3.39 (m, 2H, CH2), 2.98 (m, 2H, CH2), 2.34 (s, 3H, CH3). 13C NMR (DMSO-d6, 100 MHz): δ 182.0, 180.3, 155.0, 149.2, 135.6, 134.4, 134.2, 132.8, 128.6, 127.8, 126.9, 117.6, 117.5, 110.9, 107.2, 101.0, 43.1, 22.8, 11.7. MS (ESI) m/z = 332 [M+H]+; HRMS (ESI): m/z calcd for C20H18N3O2: 332.1399; found: 332.1390 [M+H]+.
4.5.2. 7-((2-(2-Methyl-1H-indol-3-yl)ethyl)amino)quinoline-5,8-dione (7c)
Analytical data for 7c: 1H NMR (DMSO-d6, 400 MHz): δ 10.78 (s, 1H, NH), 8.90 (d, 1H, J = 4.0 Hz, Ar-H), 8.31 (d, 1H, J = 8.0 Hz, Ar-H), 7.81 (m, 1H, Ar-H), 7.78 (m, 1H, Ar-H), 7.48 (d, 1H, J = 8.0 Hz, Ar-H), 7.24 (d, 1H, J = 8.0 Hz, Ar-H), 6.98 (m, 2H, Ar-H), 5.74 (s, 1H, CH), 3.41 (m, 2H, CH2), 2.98 (m, 2H, CH2), 2.34 (s, 3H, CH3). 13C NMR (DMSO-d6, 100 MHz): δ 180.7, 180.2, 153.0, 149.3, 147.0, 135.6, 134.0, 132.8, 130.7, 129.0, 128.9, 128.6, 117.7, 117.6, 110.9, 107.3, 99.0, 43.2, 22.8, 11.6. MS (ESI) m/z = 332 [M+H]+; HRMS (ESI): m/z calcd for C20H18N3O2: 332.1399; found: 332.1389 [M+H]+.
4.6. General procedure for synthesis of 6d, 7d
Compounds 6d and 7d were synthesized from quinoline-5,8-dione (4), 4-(4-methyl piperazin-1-yl)aniline (5d), according to the synthetic procedure for compounds 6a and 7a in yields of 39% and 24%, respectively.
4.6.1. 6-((4-(4-Methylpiperazin-1-yl)phenyl)amino)quinoline-5,8-dione (6d)
Analytical data for 6d: 1H NMR (DMSO-d6, 400 MHz): δ 9.14 (s, 1H, NH), 8.94 (d, 1H, J = 4.0 Hz, Ar-H), 8.36 (d, 1H, J = 8.0 Hz, Ar-H), 7.72 (m, 1H, Ar-H), 7.19 (d, 2H, J = 8.0 Hz, Ar-H), 6.97 (d, 2H, J = 8.0 Hz, Ar-H), 6.09 (s, 1H, CH), 3.13 (m, 4H, CH2), 2.52 (m, 4H, CH2), 2.18 (s, 3H, CH3). 13C NMR (DMSO-d6, 100 MHz): δ 182.1, 181.1, 154.9, 149.1, 148.8, 146.6, 134.4, 129.0, 128.0, 127.0, 125.2, 116.0, 102.4, 54.9, 48.3, 46.1. MS (ESI) m/z = 349 [M+H]+; HRMS (ESI): m/z calcd for C20H21N4O2: 349.1665; found: 349.1660 [M+H]+.
4.6.2. 7-((4-(4-Methylpiperazin-1-yl)phenyl)amino)quinoline-5,8-dione (7d)
Analytical data for 7d: 1H NMR (DMSO-d6, 400 MHz): δ 9.30 (s, 1H, NH), 8.94 (s, 1H, Ar-H), 8.31 (s, 1H, J = 8.0 Hz, Ar-H), 7.82 (s, 1H, Ar-H), 7.23 (d, 2H, J = 8.0 Hz, Ar-H), 7.02 (d, 2H, J = 8.0 Hz, Ar-H), 5.99 (s, 1H, CH), 3.16 (m, 4H, CH2), 2.40 (m, 4H, CH2), 2.26 (m, 3H, CH3). 13C NMR (DMSO-d6, 100 MHz): δ 181.6, 180.3, 153.1, 149.2, 147.7, 147.1, 133.7, 130.3, 129.1, 128.9, 125.3, 116.0, 100.5, 54.9, 48.3, 46.1. MS (ESI) m/z = 349 [M+H]+; HRMS (ESI): m/z calcd for C20H21N4O2: 349.1665; found: 349.1658 [M+H]+.
4.7. General procedure for synthesis of 6e, 7e
Compounds 6e and 7e were synthesized from quinoline-5,8-dione (4), 2-methoxy-4-(4-methylpiperazin-1-yl)aniline (5e), according to the synthetic procedure for compounds 6a and 7a in yields of 36% and 22%, respectively.
4.7.1. 6-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino) quinoline-5,8-dione (6e)
Analytical data for 6e: 1H NMR (d6-CD3OD, 400 MHz): δ 8.89 (d, 1H, J = 4.0 Hz, Ar-H), 8.48 (d, 1H, J = 8.0 Hz, Ar-H), 7.73 (m, 1H, Ar-H), 7.23 (d, 1H, J = 8.0 Hz, Ar-H), 6.68 (s, 1H, Ar-H), 6.62 (d, 1H, J = 8.0 Hz, Ar-H), 6.04 (s, 1H, CH), 4.53 (m, 4H, CH2), 3.88 (s, 3H, OCH3), 3.28 (m, 4H, CH2), 2.35 (s, 3H, NCH3). 13C NMR (DMSO-d6, 100 MHz): δ 182.0, 181.6, 155.0, 148.6, 147.9, 146.3, 141.9, 135.9, 130.3, 130.1, 128.1, 125.2, 124.0, 113.9, 103.4, 56.5, 55.1, 48.3, 46.1. MS (ESI) m/z = 379 [M+H]+; HRMS (ESI): m/z calcd for C21H23N4O3: 379.1770; found: 379.1759 [M+H]+.
4.7.2. 7-((2-Methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino) quinoline-5,8-dione (7e)
Analytical data for 7e: 1H NMR (d6-CD3OD, 400 MHz): δ 8.85 (d, 1H, J = 4.0 Hz, Ar-H), 8.42 (m, 1H, Ar-H), 7.80 (m, 1H, Ar-H), 7.22 (d, 1H, J = 8.0 Hz, Ar-H), 6.68 (m, 2H, Ar-H), 5.94 (s, 1H, CH), 4.62 (m, 4H, CH2), 3.85 (s, 3H, OCH3), 2.61 (m, 4H, CH3), 2.34 (s, 3H, NCH3). 13C NMR (DMSO-d6, 100 MHz): δ 182.0, 181.7, 155.1, 155.0, 148.6, 146.3, 142.3, 134.7, 130.5, 130.0, 128.1, 127.4, 124.0, 114.9, 103.5, 56.8, 55.3, 49.6, 46.2. MS (ESI) m/z = 379 [M+H]+; HRMS (ESI): m/z calcd for C21H23N4O3: 379.1770; found: 379.1767 [M+H]+.
4.8. General procedure for synthesis of 6f, 7f
Compounds 6f and 7f were synthesized from quinoline-5,8-dione (4), 4-([1,4’-bipiperidin]-1’-ylmethyl)aniline (5f), according to the synthetic procedure for compounds 6a and 7a in yields of 38% and 23%, respectively.
4.8.1. 6-((4-([1,4’ -Bipiperidin]-1’ -yl)phenyl)amino)quinoline-5,8-dione (6f)
Analytical data for 6f: 1H NMR (DMSO-d6, 400 MHz): δ 9.30 (s, 1H, NH), 9.00 (m, 1H, Ar-H), 8.43 (m, 1H, Ar-H), 7.80 (m, 1H, Ar-H), 7.37 (m, 2H, Ar-H), 6.72 (d, 2H, J = 8.0 Hz, Ar-H), 6.20 (s, 1H, CH), 3.44 (s, 2H, CH2), 2.51 (m, 8H, CH2), 2.19 (s, 1H, CH), 1.94 (m, 2H, CH2), 1.68 (m, 2H, CH2), 1.47 (m, 6H, CH2). 13C NMR (DMSO-d6, 100 MHz): δ 182.1, 181.5, 155.0, 154.8, 148.7, 146.5, 135.2, 134.5, 132.8, 130.2, 128.1, 127.2, 124.2, 103.2, 62.5, 57.1, 53.0, 51.3, 29.4, 24.9, 22.5. MS (ESI) m/z = 431 [M+H]+; HRMS (ESI): m/z calcd for C26H31N4O2: 431.2447; found: 431.2433 [M+H]+.
4.8.2. 7-((4-([1,4’ -Bipiperidin]-1’ -yl)phenyl)amino)quinoline-5,8-dione (7f)
Analytical data for 6f: 1H NMR (DMSO-d6, 400 MHz): δ 9.37 (s, 1H, NH), 8.96 (m, 1H, Ar-H), 8.32 (m, 1H, Ar-H), 7.85 (d, 1H, J = 4.0 Hz, Ar-H), 7.42 (m, 2H, Ar-H), 6.46 (d, 2H, J = 8.0 Hz, Ar-H), 6.07 (s, 1H, CH), 3.46 (s, 2H, CH2), 2.50 (s, 8H, CH2), 2.19 (m, 1H, CH), 1.89 (m, 2H, CH2), 1.67 (m, 2H, CH2), 1.43 (m, 6H, CH2). 13C NMR (DMSO-d6, 300 MHz): δ 182.1, 174.9, 153.3, 147.6, 147.2, 135.7, 135.3, 133.8, 130.2, 128.9, 124.2, 101.4, 61.1, 55.9, 53.3, 49.9, 29.4, 24.9, 22.5. MS (ESI) m/z = 431 [M+H]+; HRMS (ESI): m/z calcd for C26H31N4O2: 431.2447; found: 431.2433 [M+H]+.
4.9. General procedure for synthesis of 6g, 7g
Compounds 6g and 7g were synthesized from quinoline-5,8-dione (4), (E)-methyl 3-(4-(aminomethyl)phenyl)acrylate (10a), according to the synthetic procedure for compounds 6a and 7a in yields of 40% and 23%, respectively.
4.9.1. (E)-Methyl 3-(4-(((5,8-dioxo-5,8-dihydroquinolin-6-yl) amino)methyl)phenyl)acrylate (6g)
Analytical data for 6g: 1H NMR (d6-CD3OD, 400 MHz): δ 8.87 (m, 1H, Ar-H), 8.43 (m, 1H, Ar-H), 7.59e7.71 (m, 4H, Ar-H), 7.40 (m, 2H, Ar-H), 6.51 (d, 1H, J 16.0 Hz, CH), 5.77 (s, 1H, CH), 4.52 (s, 2H, CH2), 3.74 (s, 3H, OCH3). 13C NMR (DMSO-d6, 100 MHz): δ 181.9, 180.6, 167.0, 154.9, 149.0, 148.6, 144.8, 138.6, 134.6, 129.6, 128.1, 127.6, 127.5, 126.9, 118.6, 102.1, 52.0, 45.2. MS (ESI) m/z = 349 [M+H]+; HRMS (ESI): m/z calcd for C20H17N2O4: 349.1188; found: 349.1177 [M+H]+.
4.9.2. (E)-Methyl 3-(4-(((5,8-dioxo-5,8-dihydroquinolin-7-yl) amino)methyl)phenyl) acrylate (7g)
Analytical data for 7g: 1H NMR (d6-CD3OD, 400 MHz): δ 8.82 (d, 1H, J 4.0 Hz, Ar-H), 8.38 (d, 1H, J 4.0 Hz, Ar-H), 7.67 (m, 1H, Ar-H), 7.66 (s, 1H, Ar-H), 7.58 (s, 1H, CH), 7.52 (d, 1H, J 6.0 Hz, Ar-H), 7.38 (m, 2H, Ar-H), 6.51 (d, 1H, J 16.0 Hz, CH), 5.68 (s, 1H, CH), 4.50 (s, 2H, CH2), 3.73 (s, 3H, OCH3). 13C NMR (DMSO-d6, 100 MHz): δ 181.1, 180.1, 167.0, 153.0, 149.6, 147.2, 144.8, 138.5, 134.6, 133.9, 130.5, 129.6, 128.8, 127.5, 118.3, 100.2, 52.0, 45.3. MS (ESI) m/z = 349 [M+H]+; HRMS (ESI): m/z calcd for C20H17N2O4: 349.1188; found: 349.1177 [M+H]+.
4.10. General procedure for synthesis of 6h, 7h
Compounds 6h and 7h were synthesized from quinoline-5,8-dione (4), (E)-methyl 3-(3-(aminomethyl)phenyl)acrylate (10b), according to the synthetic procedure for compounds 6a and 7a in yields of 35% and 22%, respectively.
4.10.1. (E)-Methyl 3-(3-(((5,8-dioxo-5,8-dihydroquinolin-6-yl) amino)methyl)phenyl)acrylate (6h)
Analytical data for 6h: 1H NMR (d6-CD3OD, 400 MHz): δ 8.80 (m, 1H, Ar-H), 8.37 (d, 1H, J 8.0 Hz, Ar-H), 7.62 (m, 3H, Ar-H, CH), 7.53 (s, 1H, Ar-H), 7.33 (m, 2H, Ar-H), 6.48 (d, 1H, J 16.0 Hz, CH), 5.74 (s, 1H, CH), 4.45 (s, 2H, CH2), 3.68 (s, 3H, OCH3). 13C NMR (DMSO-d6, 100 MHz): δ 181.9, 180.5, 167.0, 154.8, 148.9, 148.5, 144.8, 138.5, 134.5, 134.3, 129.6, 129.5, 128.0, 127.5, 127.4, 126.9, 118.4, 102.0, 51.9, 45.2. MS (ESI) m/z = 349 [M+H]+; HRMS (ESI): m/z calcd for C20H17N2O4: 349.1188; found: 349.1179 [M+H]+.
4.10.2. (E)-Methyl 3-(3-(((5,8-dioxo-5,8-dihydroquinolin-7-yl) amino)methyl)phenyl)acrylate (7h)
Analytical data for 7h: 1H NMR (d6-CD3OD, 400 MHz): δ 8.81 (m, 1H, Ar-H), 8.35 (d, 1H, J 4.0 Hz, Ar-H), 7.62 (m, 3H, Ar-H, CH), 7.58 (s, 1H, Ar-H), 7.38 (m, 2H, Ar-H), 6.53 (d, 1H, J 16.0 Hz, CH), 5.70 (s, 1H, CH), 4.53 (m, 2H, CH2), 3.74 (s, 3H, OCH3). 13C NMR (DMSO-d6, 100 MHz): δ 181.6, 169.5, 152.1, 149.4, 144.3, 134.8, 134.1, 131.6, 131.5, 129.0, 128.9, 128.5, 128.4, 126.9, 126.6, 117.7, 109.9, 99.8, 50.7, 45.3. MS (ESI) m/z = 349 [M+H]+; HRMS (ESI): m/z calcd for C20H17N2O4: 349.1188; found: 349.1182 [M+H]+.
4.11. Cell culture
The multidrug resistant cancer cell line, KB-vin, was a generous gift from Dr. Kuo-Hsiung Lee (University of North Carolina, Chapel Hill, U.S.A) and maintained with vincristine. The drug sensitive cell line, HeLaS3, was purchased from Bioresource Collection and Research Center (Hsinchu, Taiwan). Human endometrial epithelial cells (EEC) were purchased from Shanghai Institute of Cell Biology (Shanghai, China). All cells were cultured in RPMI-1640 containing 10% FBS at 37 °C in a humidified atmosphere of 5% CO2.
4.12. Cell viability assay
The effects of test compounds on cell viability were evaluated by SRB and MTT assays. For the SRB assay, after 72 h treatment with serial concentrations of test compounds, 50% trichloroacetic acid (TCA) was added to fix cells for 30 min, and then the cells were washed with water and air-dried. Subsequently, cells were stained with 0.04% sulforhodamine B (SRB) for 30 min and washed with 1% acetic acid. The bound stain was solubilized in 10 mM Tris Base and the absorbance was measured by BioTek Synergy HT Multi-Mode Microplate Reader at 515 nm. For the MTT assay, 100 mL of different cancer cells were plated in a 96-well flat bottom tissue culture plate at a density of 104 cells/mL in DMEM medium containing 10% fetal bovine serum and allowed to adhere overnight at 37 °C in 5% CO2. The cells were treated by adding 100 mL of different compounds at various concentrations into the respective well. The reagent DMSO (0.1%) was used as a negative control. The cell viability assay (MTT assay) was carried out at 72 h after drug treatment. The concentration that inhibited 50% of cellular growth (IC50 value) was calculated by the following formula: Cell inhibition rate (%) (1 OD of treatment group/OD of control group) 100%. To evaluate the NQO1 dependency, cell viabilities were evaluated by combination treatment of 10 mM dicoumarol or 40 mM N-acetylcysteine with test compounds. The cell viability was evaluated by SRB assay after 72 h treatment. The cytotoxic potency of test compounds on cancer cells was expressed as IC50 values or by a histogram (The bars are the mean ± SD). All data were derived from three independent measurements.
4.13. NQO1 activity assay
The effects of test compounds (0.1 mMe10 µM) on endogenous NQO1 activity were evaluated by the NQO1 activity assay kit (Abcam, Cambridge, MA, USA). Briefly, cell pellets were solubilized in 1× extraction buffer on ice for 20 min and then centrifuged at 18,000 × g for 20 min at 4 °C. Supernatants were transferred to new tubes and protein concentrations were determined using the BioRad protein assay method. Samples were diluted with supplemented buffer to 2 × the working concentration. Two wells were prepared for each sample (with and without inhibitor) in triplicate. The reaction buffer and the reaction buffer plus inhibitor were prepared according to the instruction of manufacturer. Absorbance was measured at 440 nm every 20 s for 5 min using the BioTek Synergy HT Multi-Mode Microplate Reader with shaking before reading.
4.14. NQO1 inhibitory kinetic assay
The inhibitory activity of recombinant human NQO1 was measured by the NQO1 activity assay kit (Abcam, Cambridge, MA, USA). The inhibitory kinetics of NQO1 were analyzed by Lineweaver-Burk plots and compared to no treatment control. To calculate the kinetic parameters of inhibition mechanism of NQO1, steady-state rates were obtained at various concentrations of test compounds and substrate.
4.15. Detection of ROS
The ROS-Glo H2O2 Assay (Promega®, Southampton, UK) was used to measure changes of ROS by directly detecting H2O2 in cells. The cells were plated in 96-well white tissue culture plates and then treated with test compounds (0.01 µM−5 µM) for 24 h. The H2O2 substrate solution was added to each well and incubated for 6h at 37 °C in a CO2 incubator. After the incubation with H2O2 substrate, ROS-Glo Detection Solution was added to each well and incubated at room temperature for 20 min. The luminescence was detected by BioTek Synergy HT Multi-Mode Microplate Reader.
4.16. Mitochondrial membrane potential assay
The effects of test compounds on changes in the mitochondrial membrane potential (MMP) were detected by using the Mito-ID membrane potential cytotoxicity and detection kit (Enzo Life Sciences, Lausen, Switzerland) according to the manufacturer’s instructions. Briefly, 2.5 × 105/mL cells were plated in 96-well black wall and clear bottom plates. The cells were treated with test compounds (0.01 µM−5 µM), CCCP (positive control) and DMSO (vehicle control), respectively. After incubation for 24 h at 37 °C, each well received 100 µL of the MITO-ID®MP Dye Loading Solution and was protected from light. Then, the plate was read with a BioTek Synergy HT Multi-Mode Microplate Reader using excitation at 490 nm and emission at 590 nm after incubation at 37 °C for 30 min.
4.17. Flow cytometry assay of cell apoptosis
HeLaS3 cells were cultured overnight and incubated in triplicate with different concentrations of test compounds (0.2, 0.6, and 1.8 µM) or vehicle for 72 h. The cells were harvested and stained with FITC-Annexin V and PI at room temperature for 15 min. The percentage of apoptotic cells was determined by flow cytometry (Epics XL-MCL, Beckman Coulter, Indianapolis, USA) analysis. The FITC signal detector (FL1) and PI staining signal detector (FL3) were used to detect the cells with the flow cytometer (Ex = 488 nm; Em 530 nm). Ten thousand cells were counted for three independent experiments. The data were analyzed using WinList 3D (version7.1) and the histogram was plotted using Excel 2010.
4.18. Western blot analysis
HeLaS3 cells at 1.5 × 105/mL were treated with different dosage of test compounds or vehicle control (0.1% of DMSO) in 2 mL DMEM medium supplemented with 10% FBS for 72 h. After being harvested and lyzed, the cell lysates (50 µg/lane) were separated by SDS-PAGE (12% gel) and transferred onto nitrocellulose membranes. After being blocked with 5% fat-free milk, the target proteins were probed with anti-Bax, anti-Bcl-2, anti-caspase 3, anticleaved-caspase 3, and anti-β-actin antibodies (Cell Signaling Technology, MA, USA), respectively. The bound antibodies were detected by HRP-conjugated second antibodies and visualized using the enhanced chemiluminescent reagent. The relative levels of each signaling event to control b-actin were determined by densimetric scanning.
Supplementary Material
Acknowledgement
We gratefully acknowledge the financial support from the Natural Science Foundation of China (Grant Nos. 81302628), the Project of “Jiangsu Six Peaks of Talent” (2014-SWYY-044), China Pharmaceutical University for the Open Project Program of State Key Laboratory of Natural Medicines (SKLNMKF201415), China and Jiangsu Province Postdoctoral Science Foundation (2016M590488 and 1601136B), Jiangsu Government Scholarship for Overseas Studies (JS-2014–212), and partial support by NIH grant CA177584 from the National Cancer Institute awarded to K.H. Lee, and also thank a project funded by the Priority Academic Programs Development of Jiangsu Higher Education Institutions (PAPD) and the testing service provided by Analysis and Testing Center in Nantong University.
Footnotes
A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ejmech.2018.05.025.
References
- [1].Waris G, Ahsan H, Reactive oxygen species: role in the development of cancer and various chronic conditions, J. Carcinog 5 (2006) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wu WS, The signaling mechanism of ROS in tumor progression, Canc. Metastasis Rev 25 (2006) 695–705. [DOI] [PubMed] [Google Scholar]
- [3].Lu Y, Zhang R, Liu S, Zhao Y, Gao J, Zhu L, ZT-25, a new vacuolar H -ATPase inhibitor, induces apoptosis and protective autophagy through ROS generation in HepG2 cells, Eur. J. Pharmacol 771 (2016) 130–138. [DOI] [PubMed] [Google Scholar]
- [4].Amoedo ND, Valencia JP, Rodrigues MF, Galina A, Rumjanek FD, How does the metabolism of tumour cells differ from that of normal cells, Biosci. Rep 33 (2013) 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang J, Yang A, Wu Y, Guan W, Xiong B, Peng X, Wei X, Chen C, Liu Z, Stachydrine ameliorates carbon tetrachloride-induced hepatic fibrosis by inhibiting inflammation, oxidative stress and regulating MMPs/TIMPs system in rats, Biomed. Pharmacother 97 (2018) 1586–1594. [DOI] [PubMed] [Google Scholar]
- [6].Gorrini C, Harris IS, Mak TW, Modulation of oxidative stress as an anticancer strategy, Nat. Rev. Drug Discov 12 (2013) 931–947. [DOI] [PubMed] [Google Scholar]
- [7].Ni H, Xia C, Zhao Y, Synthesis, cytotoxicity and pro-apoptosis activity of isoquinoline quinones, Med. Chem. Res 87 (2017) 88–94. [Google Scholar]
- [8].Qin J, Wu M, Yu S, Gao X, Zhang J, Dong X, Ji J, Zhang Y, Zhou L, Zhang Q, Ding F, Pyrroloquinoline quinone-conferred neuroprotection in rotenone models of Parkinson’s disease, Toxicol. Lett 238 (2015) 70–82. [DOI] [PubMed] [Google Scholar]
- [9].Danson S, Ward TH, Butler J, Ranson M, Exploiting tumor hypoxia in cancer treatment, Canc. Treat Rev 30 (2004) 437–449. [DOI] [PubMed] [Google Scholar]
- [10].Malkinson AM, Siegel D, Forrest GL, Gazdar AF, Oie HK, Chan DC, Bunn PA, Mabry M, Dykes DJ, Harrison SD, Ross D, Elevated DT-diaphorase activity and messenger RNA content in human non-small cell lung carcinoma: relationship to the response of lung tumor xenografts to mitomycin C, Canc. Res 52 (1992) 4752–4757. [PubMed] [Google Scholar]
- [11].Nakamura M, Hayashi T, Biochem J, One-and two-electron reduction of quinones by rat liver subcellular fractions, J Biochens 115 (1994) 1141–1147. [DOI] [PubMed] [Google Scholar]
- [12].Winski SL, Koutalos Y, Bentley DL, Ross D, Subcellular localization of NAD(P)H: quinone oxidoreductase 1 in human cancer, Canc. Res 62 (2002) 1420–1424. [PubMed] [Google Scholar]
- [13].Prochaska HJ, Fernandes CL, Elevation of serum Phase II enzymes by anticarcinogenic enzyme inducers: markers for a cemoprotected state? Carcinogenesis 14 (1993) 2441–2445. [DOI] [PubMed] [Google Scholar]
- [14].Schlager JJ, Hoerl BJ, Riebow J, Scott DP, Gasdaska P, Scott RE, Powis G, Increased NAD(P)H:(quinone-acceptor)oxidoreductase activity is associated with density-dependent growth inhibition of normal but not transformed cells, Canc. Res 53 (1993) 1338–1342. [PubMed] [Google Scholar]
- [15].da Cruz EHG, Silvers MA, Jardim GAM, Resende JM, Cavalcanti BC, Bomfim IS, Pessoa C, de Simone CA, Botteselle GV, Braga AL, Nair DK, Namboothiri INN, Boothman DA, da Silva Júnior EN, Synthesis and antitumor activity of selenium-containing quinone-based triazoles possessing two redox centres, and their mechanistic insights, Eur. J. Med. Chem 122 (2016) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Keyari C, Kearns A, Duncan N, Eickholt E, Abbott G, Beall H, Diaz P, Synthesis of new quinolinequinone derivatives and preliminary exploration of their cytotoxic properties, J. Med. Chem 56 (2013) 3806–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hassani M, Cai W, Holley D, Lineswala J, Maharjan B, Ebrahimian G, Seradj H, Stocksdale M, Mohammadi F, Marvin C, Gerdes J, Beall H, Behforouz M, Novel lavendamycin analogues as antitumor agents: synthesis, in vitro cytotoxicity, structure-metabolism, and computational molecular modeling studies with NAD(P)H: quinone Oxidoreductase 1, J. Med. Chem 48 (2005) 7733–7749. [DOI] [PubMed] [Google Scholar]
- [18].Ryu CK, Jeong HJ, Lee SK, You HJ, Choi KU, Shim JY, Heo YH, Lee CO, Effects of 6-arylamino-5,8-quinolinediones and 6-chloro-7-arylamino-5,8-isoquinolinediones on NAD(P)H: quinone oxidoreductase (NQO1) activity and their cytotoxic potential, Arch Pharm. Res. (Seoul) 24 (2001) 390–896. [DOI] [PubMed] [Google Scholar]
- [19].Fryatt T, Pettersson HI, Gardipee WT, Bray KC, Green SJ, Slawin AMZ, Beall HD, Moody CJ, Novel quinolinequinone antitumor agents: structure-metabolism studies with NAD(P)-H:quinone oxidoreductase (NQO1), Bioorg. Med. Chem 12 (2004) 1667–1687. [DOI] [PubMed] [Google Scholar]
- [20].Swann E, Barraja P, Oberlander AM, Gardipee WT, Hudnott AR, Beall HD, Moody CJ, Indolequinone antitumor agents: correlation between quinone structure and rate of metabolism by recombinant human NAD(P)H:quinone oxidoreductase. Part 2, J. Med. Chem 44 (2001) 3311–3319. [DOI] [PubMed] [Google Scholar]
- [21].Hassani M, Cai W, Koelsch KH, Holley DC, Rose AS, Olang F, Lineswala JP, Holloway WG, Gerdes JM, Behforouz M, Beall HD, Lavendamycin antitumor agents: structure-based design, synthesis, and NAD(P)H:quinone oxidoreductase 1 (NQO1) model validation with molecular docking and biological studies, J. Med. Chem 51 (2008) 3104–3115. [DOI] [PubMed] [Google Scholar]
- [22].Cai W, Hassani M, Karki R, Walter ED, Koelsch KH, Seradj H, Lineswala JP, Mirzaei H, York JS, Olang F, Sedighi M, Lucas JS, Eads TJ, Rose AS, Charkhzarrin S, Hermann NG, Beall HD, Behforouz M, Synthesis, metabolism and in vitro cytotoxicity studies on novel lavendamycin antitumor agents, Bioorg. Med. Chem 18 (2010) 1899–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bian J, Li X, Wang N, Wu X, You Q, Zhang X, Discovery of quinone-directed antitumor agents selectively bioactivated by NQO1 over CPR with improved safety profile, Eur. J. Med. Chem 129 (2017) 27–40. [DOI] [PubMed] [Google Scholar]
- [24].Kadela-Tomanek M, Jastrze˛bska M, Pawełczak B, Be˛ benek E, Chrobak E, Latocha M, Ksia˛ z_ ek M, Kusz J, Boryczka S, Alkynyloxy derivatives of 5,8-quinolinedione: synthesis, in vitro cytotoxicity studies and computational molecular modeling with NAD(P)H: quinone oxidoreductase 1, Eur. J. Med. Chem 126 (2017) 969–982. [DOI] [PubMed] [Google Scholar]
- [25].Soedjak H, Hajdu J, Raffetto J, Cano R, Bales B, Prasad L, Kispert L, The structure of streptonigrin semiquinone in solution, Biochim. Biophys. Acta 1335 (1997) 73–90. [DOI] [PubMed] [Google Scholar]
- [26].Barret R, Daudon M, Oxidation of phenols to quinones by bis(tri-fluoroacetoxy)iodobenzene, Tetrahedron Lett 31 (1990) 4871–4872. [Google Scholar]
- [27].Aubart KM, Heathcock CH, A biomimetic approach to the discorhabdin alkaloids: total syntheses of discorhabdins C and E and dethiadiscorhabdin D, J. Org. Chem 64 (1999) 16–22. [DOI] [PubMed] [Google Scholar]
- [28].Weinberg SE, Chandel NS, Targeting mitochondria metabolism for cancer therapy, Nat. Chem. Biol 11 (2015) 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yu Z, Jie H, Li Z, Synthesis and bioevaluation of novel arylnaphthalene lignans as anticancer agents, Med. Chem. Res 22 (2013) 2505–2510. [Google Scholar]
- [30].Brown ML, Paige M, Toretsky JA, Üren A, Kosturko G, Bulut G, Novel ezrin inhibitors and methods of making and using, PCT Int. Appl (2012). WO2012064396 A2, 20120518.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.