

Abstract
IMPORTANCE
Prenatal exposure to certain medications has been hypothesized to influence the risk of autism spectrum disorders (ASD). However, the underlying effects on the neurotransmitter systems have not been comprehensively assessed.
OBJECTIVE
To investigate the association of early-life interference with different neurotransmitter systems by prenatal medication exposure on the risk of ASD in offspring.
DESIGN, SETTING, AND PARTICIPANTS
This case-control study included children born from January 1, 1997, through December 31, 2007, and followed up for ASD until January 26, 2015, within a single Israeli health maintenance organization. Using publicly available data, 55 groups of medications affecting neurotransmitter systems and prescribed to pregnant women in this sample were identified. Children prenatally exposed to medications were compared with nonexposed children. Data were analyzed from March 1, 2017, through June 20, 2018.
MAIN OUTCOME AND MEASURES
Hazard ratios (HRs) and 95% CIs of ASD risk associated with exposure to medication groups using Cox proportional hazards regression, adjusted for the relevant confounders (eg, birth year, maternal age, maternal history of psychiatric and neurologic disorders, or maternal number of all medical diagnoses 1 year before pregnancy).
RESULTS
The analytic sample consisted of 96 249 individuals (1405 cases; 94 844 controls; mean [SD] age at the end of follow-up, 11.6 [3.1] years; 48.8% female), including 1405 with ASD and 94 844 controls. Of 34 groups of medications, 5 showed nominally statistically significant association with ASD in fully adjusted models. Evidence of confounding effects of the number of maternal diagnoses on the association between offspring exposure to medication and ASD was found. Adjusting for this factor, lower estimates of ASD risk among children exposed to cannabinoid receptor agonists (HR, 0.72; 95% CI, 0.55–0.95; P = .02), muscarinic receptor 2 agonists (HR, 0.49; 95% CI, 0.24–0.98; P = .04), opioid receptor κ and ε agonists (HR, 0.67; 95% CI, 0.45–0.99; P = .045), or α2C-adrenergic receptor agonists (HR, 0.43; 95% CI, 0.19–0.96; P = .04) were observed. Exposure to antagonists of neuronal nicotinic acetylcholine receptor α was associated with higher estimates of ASD risk (HR, 12.94; 95% CI, 1.35–124.25; P = .03).
CONCLUSIONS AND RELEVANCE
Most of the medications affecting neurotransmitter systems in this sample had no association with the estimates of ASD risk. Replication and/or validation using experimental techniques are required.
Lack of rigorous and comprehensive studies of the effects of medications on the developing fetus may have critical public health implications, precluding informed decisions about maternal discontinuation of use of certain drugs during pregnancy. Identifying pharmacologic factors associated with a risk of adverse developmental outcomes may also help expose the biology underpinning the latter and thus focus the efforts toward their prevention and treatment. However, because pregnant women are routinely excluded from the clinical trials, the effects of prenatal exposure to most marketed drugs remain unknown.
Leveraging the scenario of a natural experiment whereby some individuals are exposed to potentially developmentally disruptive agents, epidemiological studies1–4 have been instrumental in uncovering associations between maternal use of antidepressants and anticonvulsants and adverse outcomes in offspring. These findings have supported the notions that early interference with serotonergic, γ-aminobutyric acid GABAergic, or glutaminergic systems could underlie some neurodevelopmental disorders.5–7 However, pregnant women are exposed to a much wider range of drugs than those considered to date, including medications with potentially protective effects on the fetus. Furthermore, although those earlier studies tried to mitigate against the confounding effects of maternal indication, their designs inherently involved a tight link between offspring exposure and maternal disorder (eg, maternal diagnosis of bipolar disorder and/or epilepsy in studies on the prenatal effects of valproic acid8).
To address those issues, our study extended this standard methodology by integrating knowledge from pharmacology to redefine the exposure categories (eFigure 1 in the Supplement). Grouping the drugs prescribed to pregnant women in our cohort and known to affect neurotransmitter systems based on their targets, we could (1) comprehensively assess the effects of medications known to target neurotransmitter systems, considerably widening the range of evaluated exposures; (2)reduce confounding by indication by clustering medication with overlapping functions but prescribed for various conditions; and(3)exploit the functional similarities between different drugs, making their biological targets explicit in the analytical procedures.
The rationale behind our approach is that if certain types of pharmaceuticals affect the risk of the disorder by interfering with some facets of neurodevelopment, they will exert their effects regardless of maternal indication or the internal system on which they were designed to act. Our aim was thus to test neurotransmitter systems to find out disruption of which system—via pharmacologic agents—is linked with higher or lower estimates of the risk of autism spectrum disorders (ASD). To address our aim, we tested the associations between the risk of ASD and prenatal exposure to medications in a large population-based sample from Israel.
Methods
Sample
We performed a population-based case-control cohort study using data from a large health maintenance organization in Israel (Meuhedet). All Israeli citizens are required to purchase a medical insurance plan from one of several health maintenance organizations, which offer equivalent medical provision and fees, limiting potential ascertainment bias in our study. We verified the representativeness of our data by confirming that the age-specific ASD prevalence in our sample is similar to that reported previously in Israel9(eFigure2 in the Supplement) and that the prescription rates approximated those in other national registers(eTable1 in the Supplement). This study was approved by the institutional review board of the University of Haifa and the Helsinki Ethics Committee. Those bodies waived the need for informed consent because the study data were fully deidentified.
The sample included 35.6% of the Meuhedet cohort of children born in Israel from January 1, 1997, through December 31, 2007 (n = 270799). To create this sample, 19.5% of the birth cohort was first sampled at random without stratification. In addition, we included all individuals from the birth cohort diagnosed with ASD and all siblings of those cases and members of the subcohort who were born within the cohort years (eFigure3 in the Supplement). The subcohort thus consisted of the 19.5% of individuals from the birth cohort, their siblings, and siblings of the cases. Due to random sampling, this group included some ASD cases and thus overlapped with the set of individuals specifically sampled for their positive ASD status.
Medication Exposures
Recording of Dispensed Medications
All medications prescribed through the health provider were recorded in the Meuhedet database and identified by their Anatomical Therapeutic Chemical (ATC) code.10 The exposure interval was defined as the pregnancy period (280 days before the child’s birth). Women were considered exposed regardless of the number of prescriptions or their redemption date. The target annotation of all medications was performed using DrugBank11 and PubChem.12 Our data did not include nonprescribed (over-the-counter) drugs.
Medication Groups
Given the relevance of the early brain development to ASD etiology, 13–15 we focused on the medication targeting neurotransmitter systems (55 categories [eTables 2–4 in the Supplement]). Children were classified as exposed to a given group if their mother received a prescription for any medication from this group during pregnancy. We verified within-individual correlations between the exposures using the Spearman rank correlation and merged together the groups, exposures to which always co-occurred (R = 1.0). All drugs could belong to multiple groups, reflecting their diverse actions on maternal and fetal systems.
Outcome
Cases were ascertained using the criteria for ASD from the International Classification of Diseases, Ninth Revision and International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (see eTable 5 in the Supplement for the codes). All children with suspected ASD underwent evaluation by a panel of social workers, a psychologist, and one of a trained psychiatrist, a developmental behavioral pediatrician, or a child neurologist. The final diagnosis was made by a board-certified developmental behavioral pediatrician. All children were followed up until January 26, 2015, first ASD diagnosis, or death, whichever occurred first.
Covariates
Information about all covariates was obtained from the Meuhedet records except for socioeconomic status (SES), which was obtained from Central Bureau of Statistics Registry.16 For maternal psychiatric and neurological disorders, we considered all diagnoses made from 5 years before pregnancy to the child’s birth (yes/no)(see Table 6 in the Supplement for the diagnostic codes).
Maternal number of diagnoses was defined as the total number of medical diagnoses and/or reported health issues from 1 year before pregnancy to the child’s birth (eg, wheezing, acute bronchitis). Multiple diagnoses of the same condition within the same calendar year were counted as one to avoid bias due to recurrent or chronic issues. Distinct diagnoses received during a single appointment were counted separately. Residential SES was derived from household census data and represented an index of the number of electrical appliances per person and per capita income.16
Statistical Analysis
Data were analyzed from March 1, 2017, through June20, 2018. To describe potential confounding by the indications for each medication group, we calculated the proportion of patients exposed to drugs with different ATC level 3 codes. ATC level 3 often informs about the indication (eg, level 1, subgroup N: drugs prescribed for nervous system-related conditions; level 2, subgroup N06:psychoanaleptics; or level 3, subgroup N06A: antidepressants).
Relative risks of ASD and the associated 2-sided 95% CIs were estimated by the hazard ratios (HRs) from Cox proportional hazards regression models. We examined the proportional hazards assumption using Schoenfeld residuals.17 In all models, we adjusted for correlated data, possibly introduced by including siblings, by calculating robust standard error estimates.18–21
To adjust for the increasing rates of ASD and temporal changes in medication pattern, birth year was adjusted for in all models. Models were then fitted with an increasing degree of control for potential confounding (eFigure 4 in the Supplement). Crude models included variables for birth year and medication group. We then adjusted for maternal age, a well-established confounder, and maternal diagnoses of affective, anxiety, nonaffective psychotic, and neurological disorders (entering separate terms for each of those) because maternal medication is by default associated with the underlying disorder, and a family history of psychiatric and/or neurological conditions may be associated with the risk of ASD. Last, we adjusted for total number of maternal diagnoses (definition in the Covariates subsection of the Methods section).Earlier studies have suggested that the number of maternal psychiatric diagnoses can confound the estimates of ASD risk associated with antidepressant exposure.22 Because the drugs included in the present study were prescribed for diverse conditions, we controlled for the total number of diagnoses to adjust for the underlying broad susceptibility.
We applied inverse probability weighting to account for the differential probability of being included in the study among individuals in the birth cohort,23 depending on ASD status and family size. All cases with ASD and their siblings were included with a probability of 1. Controls were selected for the subcohort as a representative sample (35.6%) with known selection probabilities (see eFigure 5 in the Supplement for a graphic illustration and examples). In the regression, subcohort cases and controls were given different weights.
We conducted several sensitivity analyses. First, we included residential SES (categories 1–20)16 and second, paternal age at birth (continuous variable) in adjusted models. Next, we varied the time windows for maternal diagnoses accounted for (psychiatric and neurologic as well as those contributing to the count of total diagnoses).Pregnancy period was included in all models, but in separate analyses, we also included diagnoses that occurred within 1 and 5 years before pregnancy. Finally, we performed analyses only in the 19.5% of the cohort originally entered into the subcohort (ie, without the sibling matches). All records with missing values on any covariate were excluded from the analyses.
Given the exploratory character of the study and our hypothesis-free design, we did not correct for multiple testing. We reran analyses after randomly reassigning the diagnostic status (ASD or control) to compare our findings with the pattern of results expected when there is no association between ASD and the regression predictors.
All hypotheses were tested with the 2-sided 5% level of significance, and P < .05 indicated significance. Analyses were performed using R software (version 3.3.3)24 and R packages survival (version 2.41–3),25 corrplot (version 0.77),26 and ggplot2 (version 2.2.1).27
Results
Our data set consisted of 96270 children (mean [SD] age at the end of follow-up, 11.6 [3.1] years; 48.8% female and 51.2% male). After removing records with missing values on maternal age (n = 21), the primary analysis subsample consisted of 96 249 individuals (35.54% of the birth cohort), including 1405 cases with ASD and 94844 members of the subcohort without an ASD diagnosis (controls). Sample characteristics are presented in Table 1 (see eTable 7 in the Supplement for the sample before exclusions).
Table 1.
Demographic Characteristics of the Subsample Used for the Main Analysesa
| Characteristic | Cases With ASD (n = 1405) | Controls (n = 94844) | P Value |
|---|---|---|---|
| Female, No. (%) | 269 (19.15) | 45819 (49.25) | 2.20 × 10−15 |
| SES, mean (SD)b | 10.28 (4.17) | 7.63 (4.27) | 2.20 × 10−15 |
| Maternal age, mean (SD), y | 29.92 (5.42) | 29.74 (5.41) | .23 |
| Paternal age, mean (SD), y | 32.82 (6.17) | 32.35 (6.08) | .02 |
| Maternal anxiety disorders, No. (%) | 78 (5.55) | 4012 (4.20) | .03 |
| Maternal mood disorders, No. (%) | 55 (3.91) | 1815 (1.90) | 1.27 × 10−4 |
| Maternal nonaffective psychotic disorders, No. (%) | 1 (0.07) | 203 (0.20) | .05 |
| Maternal neurologic disorders, No. (%) | 7 (0.50) | 312 (0.30) | .36 |
| No. of maternal diagnoses, mean (SD) | 16.21 (15.18) | 12.30 (8.66) | 2.20 × 10−15 |
Abbreviations: ASD, autism spectrum disorders; SES, socioeconomic status.
No data were missing for any of the covariates.
Categories range from 1 to 20.16
The proportions of individuals exposed to drugs with different ATC codes within each medication group is presented in eFigures 6 through 12 in the Supplement. Of the initial 55 medication groups, we excluded 16 for which we did not observe any ASD cases(eFigure 13 in the Supplement). Among the remaining 39 exposures, we identified 4 clusters where medication groups correlated at R = 1.0 (Figure), and thus tested 34 prenatal exposures for an association with subsequent ASD. Inspection of the Schoenfeld residuals did not provide support for nonproportional hazards(eTable8 in the Supplement).
Figure.
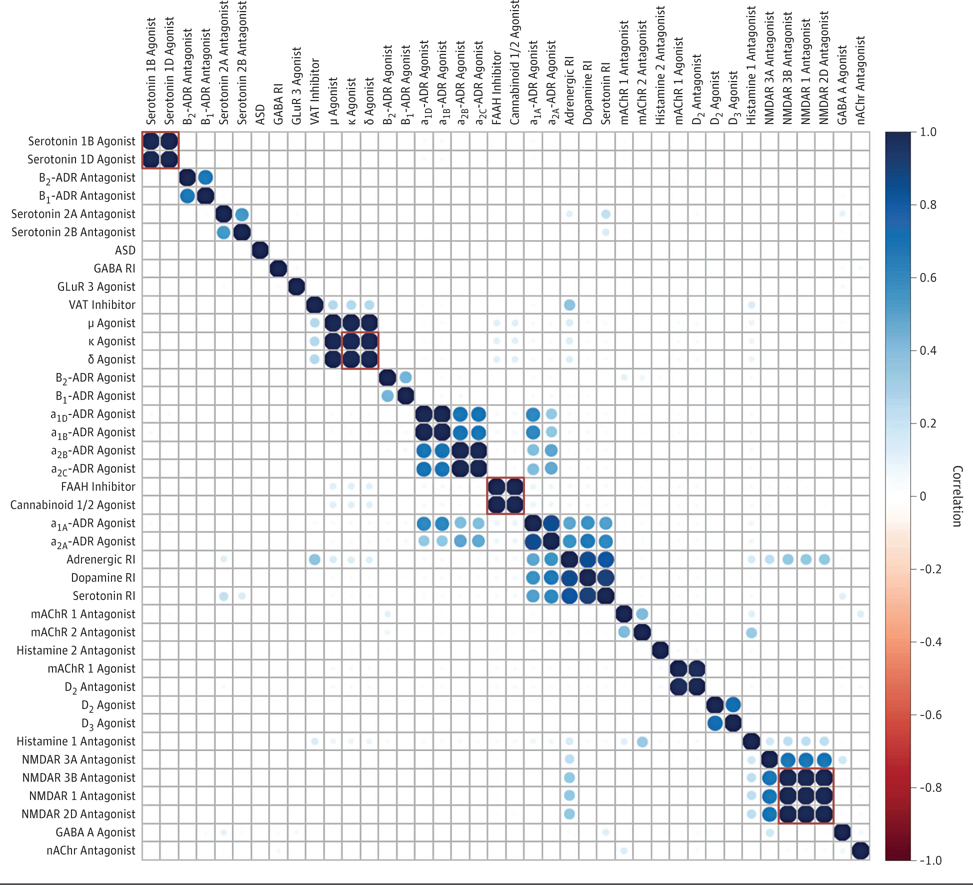
Within-Individual Correlations Between Exposures
Correlations were computed using Spearman rank correlation test (all input values were 0s and 1s) with the subset of exposures for which we recorded cases of autism spectrum disorders (ASD) among exposed individuals. Exposures enclosed within red squares were fully correlated with each other and therefore analyzed as a single factor. ADR indicates adrenergic receptor; FAAH, fatty acid amide hydrolase; GABA, ϒ-aminobutyric acid; GluR, glutamate receptor; mAChR, muscarinic acetylcholine receptor; nAChR, neuronal nicotinic acetylcholine receptor; NMDAR, N-methyl-D-aspartate receptor; RI, reuptake inhibitor; and VAT, vesicular amine transporter.
Associations Between Prenatal Exposure to Medication and ASD
Table 2 presents associations between prenatal exposures and ASD. The HRs of the disorder associated with the use of antagonists of neuronal nicotinic acetylcholine α receptor increased after adjustments (HR, 12.94; 95% CI, 1.35–124.25; P = .03) and remained statistically significant in all sensitivity analyses. The estimates of the risk of ASD associated with the use of GABA transaminase inhibitors (solely valproate sodium) diminished after successive adjustments; although the HRs remained high in both main and sensitivity analyses, they were not statistically significant after adjustments (HR, 3.15; 95% CI, 0.82–12.06; P = .09)
Table 2.
Top Associations Between Prenatal Exposure to Medications and Subsequent Diagnosis of ASD
| Medication Group | Model 1a | Model 2b | Model 3c | Model 4d | ||||
|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | |
| Serotonin transporter inhibitor | 1.35 (0.96–1.89) | .08 | 1.37 (0.97–1.91) | .07 | 1.26 (0.90–1.76) | .17 | 1.06 (0.76–1.48) | .73 |
| Serotonin receptor 1B and 1D agonist | 0.72 (0.10–5.20) | .75 | 0.76 (0.11–5.43) | .78 | 0.72 (0.10–5.21) | .75 | 0.57 (0.08–4.14) | .58 |
| Serotonin receptor 2A antagonist | 1.07 (0.15–7.69) | .95 | 1.10 (0.15–7.93) | .92 | 0.87 (0.12–6.30) | .89 | 0.79 (0.11–5.76) | .82 |
| Serotonin receptor 2B antagonist | 3.56 (0.48–26.27) | .22 | 3.70 (0.50–27.48) | .20 | 2.57 (0.34–19.42) | .36 | 2.43 (0.32–18.77) | .39 |
| GABA transaminase inhibitor | 4.09 (1.01–16.60)e | .05 | 4.06 (1.00–16.46)e | .05 | 3.36 (0.86–13.22) | .08 | 3.15 (0.82–12.06) | .09 |
| GABA A receptor agonist | 1.47 (0.91–2.38) | .11 | 1.53 (0.95–2.48) | .08 | 1.32 (0.82–2.11) | .25 | 1.17 (0.73–1.86) | .52 |
| GluR 3 agonist | 3.32 (0.47–23.52) | .23 | 3.27 (0.46–23.51) | .24 | 2.42 (0.33–17.60) | .39 | 2.67 (0.36–19.68) | .34 |
| NMDAR 1, 2D, and 3B antagonists | 0.80 (0.25–2.49) | .70 | 0.80 (0.26–2.51) | .71 | 0.79 (0.25–2.48) | .69 | 0.57 (0.18–1.81) | .34 |
| NMDAR 3A antagonist | 0.63 (0.26–1.51) | .30 | 0.63 (0.26–1.52) | .30 | 0.62 (0.26–1.49) | .28 | 0.48 (0.20–1.15) | .10 |
| nAChR α antagonist | 5.83 (0.77–44.27) | .09 | 6.15 (0.82–46.11) | .08 | 10.01 (1.08–92.87)e | .04 | 12.94 (1.35–124.25)e | .03 |
| mAChR 1 agonist | 1.01 (0.78–1.30) | .96 | 1.00 (0.77–1.29) | >.99 | 0.99 (0.76–1.28) | .92 | 0.80 (0.62–1.03) | .09 |
| mAChR 1 antagonist | 0.42 (0.14–1.31) | .14 | 0.43 (0.14–1.34) | .14 | 0.42 (0.14–1.32) | .14 | 0.33 (0.11–1.03) | .06 |
| mAChR 2 antagonist | 0.61 (0.30–1.23) | .17 | 0.62 (0.31–1.24) | .18 | 0.62 (0.31–1.24) | .18 | 0.49 (0.24–0.98)e | .04 |
| CBR 1 and 2 agonists/FAAH inhibitor | 0.89 (0.68–1.17) | .40 | 0.81 (0.68–1.17) | .40 | 0.89 (0.68–1.16) | .38 | 0.72 (0.55–0.95)e | .02 |
| OR κ and ε agonists | 0.84 (0.57–1.25) | .40 | 0.85 (0.57–1.27) | .42 | 0.85 (0.57–1.26) | .41 | 0.67 (0.45–0.99)e | .045 |
| OR μ agonist | 0.86 (0.58–1.28) | .46 | 0.87 (0.59–1.29) | .49 | 0.87 (0.59–1.28) | .48 | 0.69 (0.46–1.01) | .06 |
| Dopamine transporter inhibitor | 1.15 (0.78–1.70) | .49 | 1.16 (0.78–1.71) | .47 | 1.14 (0.77–1.69) | .51 | 0.93 (0.63–1.38) | .73 |
| Dopamine receptor D2 agonist | 1.22 (0.58–2.58) | .60 | 1.24 (0.59–2.61) | .58 | 1.22 (0.58–2.58) | .61 | 1.08 (0.51–2.29) | .83 |
| Dopamine receptor D2 antagonist | 0.97 (0.76–1.26) | .84 | 0.97 (0.75–1.25) | .80 | 0.96 (0.74–1.23) | .73 | 0.78 (0.60–1.00) | .052 |
| Dopamine receptor D3 agonist | 1.48 (0.55–4.00) | .44 | 1.46 (0.54–3.95) | .46 | 1.42 (0.52–3.86) | .49 | 1.22 (0.45–3.31) | .69 |
| Adrenergic transporter inhibitor | 1.08 (0.76–1.51) | .68 | 1.08 (0.77–1.53) | .65 | 1.07 (0.76–1.50) | .71 | 0.86 (0.61–1.21) | .38 |
| α1A-ADR agonist | 1.18 (0.94–1.47) | .15 | 1.18 (0.95–1.48) | .14 | 1.17 (0.94–1.46) | .16 | 0.96 (0.77–1.20) | .71 |
| α1B-ADR agonist | 1.27 (0.90–1.80) | .18 | 1.27 (0.90–1.80) | .18 | 1.26 (0.89–1.79) | .20 | 1.05 (0.74–1.48) | .80 |
| α1D-ADR agonist | 1.24 (0.87–1.76) | .24 | 1.24 (0.87–1.76) | .24 | 1.23 (0.86–1.75) | .26 | 1.02 (0.72–1.45) | .91 |
| α2A-ADR agonist | 1.06 (0.81–1.39) | .69 | 1.06 (0.81–1.39) | .66 | 1.06 (0.81–1.38) | .69 | 0.86 (0.66–1.13) | .28 |
| α2B-ADR agonist | 0.59 (0.28–1.25) | .17 | 0.59 (0.28–1.24) | .16 | 0.59 (0.28–1.24) | .16 | 0.50 (0.24–1.04) | .06 |
| α2C-ADR agonist | 0.51 (0.23–1.15) | .10 | 0.51 (0.23–1.14) | .10 | 0.51 (0.23–1.14) | .10 | 0.43 (0.19–0.96) | .04 |
| β1-ADR agonist | 1.28 (0.41–4.00) | .68 | 1.31 (0.42–4.10) | .65 | 1.32 (0.42–4.13) | .64 | 1.05 (0.34–3.29) | .93 |
| β2-ADR agonist | 0.75 (0.38–1.49) | .42 | 0.77 (0.39–1.53) | .46 | 0.76 (0.39–1.51) | .44 | 0.59 (0.30–1.18) | .14 |
| β1-ADR antagonist | 1.12 (0.53–2.37) | .76 | 1.21 (0.57–2.56) | .62 | 1.18 (0.56–2.49) | .67 | 0.87 (0.41–1.84) | .71 |
| β2-ADR antagonist | 1.57 (0.58–4.24) | .37 | 1.65 (0.61–4.45) | .32 | 1.56 (0.58–4.24) | .38 | 1.14 (0.42–3.08) | .80 |
| Histamine receptor 1 antagonist | 1.09 (0.85–1.40) | .50 | 1.10 (0.86–1.41) | .46 | 1.09 (0.85–1.40) | .51 | 0.89 (0.69–1.14) | .35 |
| Histamine receptor 2 antagonist | 1.17 (0.72–1.90) | .52 | 1.19 (0.73–1.93) | .49 | 1.16 (0.72–1.89) | .54 | 0.86 (0.53–1.39) | .53 |
| VAT inhibitor | 1.19 (0.49–2.89) | .70 | 1.20 (0.50–2.91) | .68 | 1.20 (0.50–2.90) | .69 | 1.00 (0.42–2.41) | >.99 |
Abbreviations: ADR, adrenergic receptor; ASD, autism spectrum disorders; CBR, cannabinoid receptor; FAAH, fatty acid amide hydrolase; GABA, γ-aminobutyric acid; GluR, glutamate receptor; HR, hazard ratio; mAChR, muscarinic acetylcholine receptor; nAChR, neuronal nicotinic acetylcholine receptor; NMDAR, N-methyl-D-aspartate receptor; OR, opioid receptor; VAT, vesicular amine transporter.
Adjusted for exposure and year of birth
Adjustedforcovariatesinmodel1, maternal age at birth, and paternal age at birth.
Adjusted for covariates in model 2, maternal affective disorders, maternal anxiety disorders, maternal psychotic disorders, and maternal neurologic disorders.
Adjusted for covariates in model 3 and number of maternal diagnoses.
Indicates statistically significant.
When we included the number of maternal diagnoses in the model, we found cannabinoid receptors agonists/fatty acid amide hydrolase inhibitors (solely paracetamol; HR, 0.72; 95% CI,0.55–0.95;P = .02), muscarinic receptor 2 agonists (HR, 0.49; 95% CI, 0.24–0.98; P =. 04), opiod receptor κ and ε agonists (HR, 0.67; 95% CI, 0.45–0.99; P = .045), and α2C-adrenergic receptor agonists (HR, 0.43;95%CI,0.19–0.96;P = .04)were all associated with lower estimates of ASD risk. However, in at least 1 of the sensitivity analyses, all models became statistically nonsignificant (eTables 9–11 in the Supplement).We observed similar pattern of results in the sample without the sibling matches (eTable 12 in the Supplement). Results from the permuted data sets were all statistically nonsignificant, with no effects of successive adjustments on the effect sizes or their P values (eTable 13 in the Supplement).
Discussion
Our study suggests that prenatal exposure to most of the medications targeting neurotransmitter systems, including typical targets of antidepressants and antipsychotics, is not associated with increased estimates of ASD risk. Two medication groups in our sample that were associated with higher estimates of ASD risk included valproate (inhibiting GABA transaminase, involved in the neurotransmitter’s breakdown)and antagonists of neuronal nicotinic acetylcholine receptor α, both used in the treatment of epilepsy. The extent to which the observed effects arose due to shared mechanisms pertinent to epilepsy is unclear; however, a diagnosis of neurologic disorders itself was not statistically significantly associated with ASD in our analyses, and previous studies3 did not find evidence for the effects of many other anticonvulsants on the risk of ASD. Owing to the low number of exposed individuals (eTable 4 in the Supplement) and statistically nonsignificant results for valproate after adjustments, those associations warrant further examination.
After adjusting for the number of maternal diagnoses, several medication groups were associated with reduced estimates of ASD risk, suggesting that this factor was likely confounding the results in unadjusted models. All groups of medications associated with reduced risk of ASD are used as antianalgesic and/or anti-inflammatory agents, consistent with the previous reports of associations between maternal fever,28 inflammation,29 and immune activation30 during pregnancy and ASD in her offspring. Given that those associations appeared to be independent of other measured maternal factors, the causal nature of those observations will be amenable to experimental validation. Increased and decreased estimates of ASD risk have been previously observed in association with maternal medication use during pregnancy.3,31
Our study sheds new light on the role of maternal general health and its potentially confounding effects in pharmacoepidemiologic studies on ASD. We found no support in the data for the role of serotonin transporter in ASD risk, in line with the reported mixed pattern of statistical significance among different selected serotonin reuptake inhibitors.1 Rather, the already weak evidence for their association with ASD (HR, 1.26; 95%CI, 0.90–1.76;P = .17)was further substantially diminished after adjusting for the maternal number of diagnoses (HR, 1.06; 95%CI, 0.76–1.48;P = .73). Using the set of covariates typically applied in pharmacoepidemiologic studies (model 3) would have led us to conclude that maternal use of this group of medications during pregnancy may increase the odds of ASD in offspring by approximately 25%. Although the absolute risk remains extremely low (assumingabaselineof1.1%, exposed mothers would have on average a 1.38%chance of having a child with ASD), the perception of risk can still have implication son the clinical practice, is discouraging physicians from prescribing those medications to pregnant women,32 and misguide formulation of research hypotheses.
Strengths and Limitations
The strengths of the study include its large sample size, population-based records, and application of a new approach integrating knowledge from pharmacology with epidemiologic analysis. Such a biology-first method, whereby the shared biological properties of medications are explicitly acknowledged in the analytical procedures, aims to understand the causal mechanisms that underlie the effects of prenatal exposure to medications in public health research, and as such could be pertinent to studies on other conditions that originate in utero. We confirmed the validity of our approach by showing that, with adjustment for similar covariates, our risk estimates are in line with those reported previously for selective serotonin reuptake inhibitors.1 Furthermore, we highlighted the importance of consideration of maternal factors accompanying medication use and identified groups of medications warranting further interest in the context of prenatal ASD risk factors.
Nevertheless, the study remains exploratory in nature, and we acknowledge the limitations associated with our approach and the need to validate our findings through other epidemiologic (because the rates of ASD in Israel are low, the generalizability of our findings remains to be verified) and experimental studies. We confirmed that our findings are robust under different model assumptions and showed that statistically significant results are no longer present when assuming random associations between ASD diagnosis and the covariates, indicating our findings were different from those expected under the null hypothesis. Nevertheless, an increase in standard errors by 2% to 3% for some associations would be sufficient to preclude rejection of the null hypothesis (eTable 14 in the Supplement), strongly warranting additional validation of those results. We also acknowledge that our findings should be interpreted in the context of the developmental framework of ASD and that the factors uncovered by our study, if validated in future research, represent only triggers in disruption of brain maturation, the remaining causes and trajectory of which remain to be explained.
Owing to incompleteness of data on medication redemption date, our analyses were based on the date when the medication was prescribed, and we did not consider medication prescribed before pregnancy, potentially resulting in some misattribution of prenatal exposures. Given the differential time scales at which the drugs we have considered operate and the likely varying sensitivity periods for the processes they affect, we did not consider the effects of timing or length of maternal exposures. Similarly, the pharmacologic actions of many prescription drugs and their interactions have not been fully characterized, and therefore our method will be progressively refined as we will continue to learn more about them. Furthermore, medication groups underlaid by a single type of medication (eg, muscarinic receptor 1 agonist, GABA transaminase inhibitor [eFigures 6–12 in the Supplement]) remained in the analysis, in which the potential for confounding by indication may be more severe.
The data set available for the study was created for different purposes and thus used a sibling sampling approach. Although sibling comparisons could have helped disentangle family effects given the rarity of some exposures and the outcome, our study was underpowered to perform those comparisons.
Finally, we verified that the number of maternal diagnoses had a strong association with the risk of ASD in the subcohort (eFigure 14 in the Supplement), confirming that our results were not owing to higher completeness of information on maternal diagnoses in cases vs subcohort families. Nevertheless, elucidating the active ingredient of this measure will be important for future research. Those effects might have been driven by a particular group of diagnoses, capture factors associated with maternal health anxiety, or converge on a factor that itself acts as an autism risk factor (eg, obstetric complications), and we acknowledge that the nature of the associations among maternal disorder, maternal medication use, and offspring outcomes still requires elucidation. Pursuing those further research questions will be instrumental to our understanding of the consequences—or lack thereof—of maternal medication use during pregnancy.
Conclusions
Our data indicate that, in the current sample, most of the medications known to affect the neurotransmitters and used by women during pregnancy may not themselves influence the estimates of offspring risk of ASD via effects on their known pharmacologic targets. However, maternal number of diagnoses can confound associations between prenatal exposures and ASD and as such should be accounted for in the future studies. Those results are pending replication in the independent data sets as well by using alternative designs.
Supplementary Material
Key Points.
Question
Do prenatal exposures to medication affecting neurotransmitter systems increase the risk of autism spectrum disorders?
Findings
This population-based case-control cohort study of 95 978 individuals from the population of a single health maintenance organization assessed the effects of prenatal exposure to medications that affect major neurotransmitter systems. Most of the associations were substantially modified when accounting for maternal characteristics.
Meaning
Higher estimates of autism spectrum disorder risk among the offspring of mothers taking certain medications during pregnancy are most likely not owing to pharmacologic effects of those drugs.
Acknowledgments
Funding/Support: This study was supported in part by a Seaver Postdoctoral Fellowship (Dr Janecka) and Seaver Faculty Scholar funding (Drs Sandin and Reichenberg) from the Seaver Foundation; grants HD073978 (Dr Reichenberg), MH097849 (Dr Buxbaum), GM108911 (Dr Schlessinger), and T32 GM062754 (Mr Rahman) from the National Institutes of Health; and from the Fredrik and Ingrid Thuring Foundation, the Swedish Society of Medicine, and the Swedish Brain Foundation (Dr Viktorin).
Role of the Funder/Sponsor: The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Conflict of Interest Disclosures: Dr Levine reported receiving research support from Shire Pharmaceuticals outside the submitted work. Dr Lusskin reported being a director of psychopharmacologic agents for the Reproductive Toxicology Foundation, a nonprofit foundation; receiving royalties as a peer reviewer for UpToDate; and consulting for Forest Laboratories regarding escitalopram litigation and Pfizer, Inc, regarding sertraline and venlafaxine litigation outside the submitted work. No other disclosures were reported.
Contributor Information
Magdalena Janecka, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York, Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, New York.
Arad Kodesh, Department of Community Mental Health, University of Haifa, Haifa, Israel, Meuhedet Health Services, Tel Aviv, Israel.
Stephen Z. Levine, Department of Community Mental Health, University of Haifa, Haifa, Israel.
Shari I. Lusskin, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York, Department of Obstetrics, Gynecology, and Reproductive Science, Icahn School of Medicine at Mount Sinai, New York, New York.
Alexander Viktorin, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York, Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, New York, Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden.
Rayees Rahman, Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, New York.
Joseph D. Buxbaum, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York, Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, New York, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, New York, Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, New York.
Avner Schlessinger, Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, New York.
Sven Sandin, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York, Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, New York, Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden.
Abraham Reichenberg, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York, Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York, New York, Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, New York, Department of Preventive Medicine, Icahn School of Medicine at Mount Sinai, New York, New York.
REFERENCES
- 1.Viktorin A, Uher R, Reichenberg A, Levine SZ, Sandin S. Autism risk following antidepressant medication during pregnancy. Psychol Med 2017; 47 (16):2787–2796. doi: 10.1017/S0033291717001301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rai D, Lee BK, Dalman C, Golding J, Lewis G, Magnusson C. Parental depression, maternal antidepressant use during pregnancy, and risk of autism spectrum disorders: population based case-control study. BMJ 2013;346(April):f2059. doi: 10.1136/bmj.f2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christensen J, Grønborg TK, Sørensen MJ, et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013; 309(16):1696–1703. doi: 10.1001/jama.2013.2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasalam AD, Hailey H, Williams JHG, et al. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol 2005; 47(8):551–555. doi: 10.1017/S0012162205001076 [DOI] [PubMed] [Google Scholar]
- 5.Croen LA, Najjar DV, Fireman B, Grether JK. Maternal and paternal age and risk of autism spectrum disorders. Arch Pediatr Adolesc Med 2007; 161(4):334–340. doi: 10.1001/archpedi.161.4.334 [DOI] [PubMed] [Google Scholar]
- 6.Kim KC, Lee DK, Go HS, et al. Pax6-dependentcortical glutamatergic neuronal differentiation regulates autism-like behavior in prenatally valproic acid-exposed rat offspring. Mol Neurobiol 2014; 49 (1):512–528. doi: 10.1007/s12035-013-8535-2 [DOI] [PubMed] [Google Scholar]
- 7.Banerjee A, García-Oscos F, Roychowdhury S, et al. Impairment of cortical GABAergic synaptic transmission in an environmental rat model of autism. Int J Neuropsychopharmacol 2013; 16(6): 1309–1318. doi: 10.1017/S1461145712001216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wyszynski DF, Nambisan M, Surve T, Alsdorf RM, Smith CR, Holmes LB; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology 2005; 64(6):961–965. doi: 10.1212/01.WNL.0000154516.43630.C5 [DOI] [PubMed] [Google Scholar]
- 9.Davidovitch M, Hemo B, Manning-Courtney P, Fombonne E. Prevalence and incidence of autism spectrum disorder in an Israeli population. J Autism Dev Disord 2013; 43(4):785–793. doi: 10.1007/s10803-012-1611-z [DOI] [PubMed] [Google Scholar]
- 10.WHO Collaborating Centre for Drug Statistics Methodology. ATC structure and principles https://www.whocc.no/atc/structure_and_principles/ Updated February 15, 2018. Accessed March 1, 2017.
- 11.Law V, Knox C, Djoumbou Y, et al. Drug Bank 4.0: shedding new light on drug metabolism. Nucleic Acids Res 2014; 42(Database issue):D1091–D1097. doi: 10.1093/nar/gkt1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S, Thiessen PA, Bolton EE, et al. PubChem substance and compound databases. Nucleic Acids Res 2016; 44(D1):D1202–D1213. doi: 10.1093/nar/gkv951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Courchesne E Brain development in autism: early overgrowth followed by premature arrest of growth. Ment Retard Dev Disabil Res Rev 2004; 10 (2):106–111. doi: 10.1002/mrdd.20020 [DOI] [PubMed] [Google Scholar]
- 14.Courchesne E, Pierce K. Brain overgrowth inautism during a critical time in development: implications for frontal pyramidal neuron and interneuron development and connectivity. Int J Dev Neurosci 2005; 23(2–3):153–170. doi: 10.1016/j.ijdevneu.2005.01.003 [DOI] [PubMed] [Google Scholar]
- 15.Cheung C, McAlonan GM, Fung YY, et al. MRI study of minor physical anomaly in childhood autism implicates aberrant neurodevelopment in infancy. PLoS One 2011; 6(6):e20246. doi: 10.1371/journal.pone.0020246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Central Bureau of Statistics. Demographic Characteristics of the Population in Localities and Statistical Areas Jerusalem, Israel: Central Bureau of Statistics; 1995. [Google Scholar]
- 17.Grambsch P, Therneau T. Proportional hazard tests and diagnostics based on weight residuals. Biometrika 1994; 81(3):515–526. doi: 10.1093/biomet/81.3.515 [DOI] [Google Scholar]
- 18.Barlow WE. Robust variance estimation for the case-cohort design. Biometrics 1994; 50(4): 1064–1072. doi: 10.2307/2533444 [DOI] [PubMed] [Google Scholar]
- 19.Kang S, Cai J, Chambless L. Marginal additive hazards model for case-cohort studies with multiple disease outcomes: an application to the Atherosclerosis Risk in Communities (ARIC) study. Biostatistics 2013; 14(1):28–41. doi: 10.1093/biostatistics/kxs025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samuelsen SO, Ånestad H, Skrondal A. Stratified case-cohort analysis of general cohort sampling designs. Scand J Stat 2007; 34(1):103–119. doi: 10.1111/j.1467-9469.2006.00552.x [DOI] [Google Scholar]
- 21.Lu SE, Shih JH. Case-cohort designs and analysis for clustered failure time data. Biometrics 2006; 62(4):1138–1148. doi: 10.1111/j.1541-0420.2006.00584.x [DOI] [PubMed] [Google Scholar]
- 22.Viktorin A, Uher R, Kolevzon A, Reichenberg A, Levine SZ, Sandin S. Association of antidepressant medication use during pregnancy with intellectual disability in offspring. JAMA Psychiatry 2017; 74 (10):1031–1038. doi: 10.1001/jamapsychiatry.2017.1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Therneau TM, Li H. Computing the Cox model for case cohort designs. Lifetime Data Anal 1999;5. [DOI] [PubMed]
- 24.The R Core Team. R: a language and environment for statistical computing (version 3.3.3) https://www.r-project.org. Accessed March 1, 2017.
- 25.Therneau T, Lumley T. Package “survival”(version 2.41–3) https://cran.r-project.org/web/packages/survival/survival.pdf Published 2014. Accessed September 6, 2018.
- 26.Wei T, Simko V. Package “corrplot” (version0.77) https://cran.r-project.org/web/packages/corrplot/corrplot.pdf. Accessed March 1, 2017.
- 27.Package “ggplot2” (version 2.2.1) https://cran.r-project.org/web/packages/ggplot2/ggplot2.pdf 2016. Accessed September 6, 2018.
- 28.Zerbo O, Iosif AM, Walker C, Ozonoff S, Hansen RL, Hertz-Picciotto I. Is maternal influenza or fever during pregnancy associated with autism or developmental delays? results from the CHARGE (Childhood Autism Risks From Genetics and Environment) study. J Autism Dev Disord 2013; 43 (1):25–33. doi: 10.1007/s10803-012-1540-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown AS, Sourander A, Hinkka-Yli-Salomäki S, McKeague IW, Sundvall J, Surcel H-M. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry 2014; 19(2):259–264. doi: 10.1038/mp.2012.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atladóttir HÓ, Thorsen P, Østergaard L, et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 2010; 40(12):1423–1430. doi: 10.1007/s10803-010-1006-y [DOI] [PubMed] [Google Scholar]
- 31.Levine SZ, Kodesh A, Viktorin A, et al. Association of maternal use of folic acid and multivitamin supplements in the periods before and during pregnancy with the risk of autism spectrum disorder in offspring. JAMA Psychiatry 2018; 75 (2):176–184. doi: 10.1001/jamapsychiatry.2017.4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Margulis AV, Kang EM, Hammad TA. Patternsof prescription of antidepressants and antipsychotics across and within pregnancies in a population-based UK cohort. Matern Child Health J 2014; 18(7):1742–1752. doi: 10.1007/s10995-013-1419-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.