

Abstract
Background
Hydroxyurea (hydroxycarbamide) promotes the production of foetal haemoglobin (HbF) by reactivating gamma‐genes. Evidence has shown clinical benefits of hydroxyurea in people with sickle cell anemia; however, only a few studies have assessed this treatment in people with beta (β)‐thalassaemia.
Objectives
The primary objective is to review the efficacy of hydroxyurea in reducing or ameliorating the requirement of blood transfusions in people with transfusion‐dependent β‐thalassaemia. The second objective is to review the safety of hydroxyurea with regards to severe adverse effects in this population.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register, compiled from electronic database searches and hand searching of journals and conference abstract books. We also searched electronic databases and trial registries, including ClinicalTrials.gov, the WHO ICTRP and PubMed (09 October 2018).
Date of last search of the Group's haemoglobinopathies trials register: 04 March 2019.
Selection criteria
Randomised controlled trials of hydroxyurea in people with transfusion‐dependent β‐thalassaemia, compared with placebo or standard treatment or comparing different doses of hydroxyurea.
Data collection and analysis
Two authors independently assessed trials for inclusion in the review, which was verified by a third author.
Main results
No trials were eligible for inclusion in this review.
Authors' conclusions
Currently, there is no high‐quality evidence to support or challenge the continued use of hydroxyurea for managing people with transfusion‐dependent β‐thalassaemia. Multicentre, randomised controlled trials (compared to placebo or other available treatment, i.e. blood transfusion and iron chelation) are needed in order to assess the efficacy and safety of hydroxyurea for reducing the need for blood transfusion, for maintaining or improving mean haemoglobin levels, as well as for determining its cost‐effectiveness.
Plain language summary
Hydroxyurea (hydroxycarbamide) for treating people with transfusion‐dependent beta‐thalassaemia
Review question
We reviewed the evidence about the effect of hydroxyurea (hydroxycarbamide) in people with transfusion‐dependent beta (β)‐thalassaemia.
Background
β‐thalassaemia is a blood disorder which is transferred by both parents to their children. In this condition less haemoglobin is produced. According to the WHO, almost 60,000 children with thalassaemia are born every year. Transfusion‐dependent β‐thalassaemia is the severe form of β‐thalassaemia, in which symptoms are clearly visible in the first six months of life. It can be fatal if left untreated, i.e. with blood transfusions. However, blood transfusions increase the chance of life‐threatening viral infections and of iron overload in the body, which affects major organs. Medicines for removing iron from the body can reduce the risk of iron overload, but the cost of managing the disease can often become too high for individuals and their families. Hydroxyurea is reported to be effective in treating β‐thalassaemia as it increases haemoglobin (blood) production and its benefits have previously been demonstrated.
Search date
The evidence is current to 04 March 2019.
Study characteristics
We found no trials for inclusion in the review.
Key results
We found no eligible trials for inclusion in the review. Multicentre, randomised controlled trials (compared to placebo or other available treatment, i.e. blood transfusion and iron chelation) are needed to assess the effectiveness and safety of hydroxyurea for reducing the need for blood transfusion, for maintaining or improving average levels of haemoglobin, as well as for assessing whether treatment is cost‐effective.
Background
Description of the condition
Beta (β)‐thalassaemia is the inherited blood disorder that results when an individual is unable to produce sufficient levels of haemoglobin (Hb) (Zamani 2009). It is due to the imbalance of the alpha (α)‐ to β‐globin chain ratio due to decreased production of β‐globin. An excess in the α chain precipitates red blood cell precursors, which leads to their damage in the bone marrow and shortening the survival of the progeny in the peripheral blood (Weatherall 2001). This results in ineffective erythropoiesis and haemolysis. Enhancement of the gamma (γ)‐globin chains can decrease this imbalance (Bradai 2003). One of the major factors in modifying the clinical variability of β‐thalassaemia is a genetically determined ability to produce unusually high levels of foetal haemoglobin (HbF). A second factor that has been clearly identified is that the co‐inheritance of α‐thalassaemia will ameliorate the β‐thalassaemias (Weatherall 2001). In individuals with transfusion‐dependent β‐thalassaemia, the most severe form of β‐thalassaemia, the oxygen depletion in the body becomes apparent within the first six months of life. If left untreated, death usually occurs within a few years since most children with thalassaemia survive on regular blood transfusions. The disorder is prevalent worldwide; the WHO has estimated that approximately 1.5% of the global population might be carriers of β‐thalassaemia and that approximately 60,000 affected children are born every year (Higgs 2012). In the UK, approximately 214,000 to 300,000 people have the β‐thalassaemia trait, whilst 800 to 1000 have the disorder (NHS 2011; RCOG 2014). Approximately 79% of affected births are in the Asian population (NHS 2011; RCOG 2014). In Southeast Asia, 1% to 8% of people have the β‐thalassaemia trait (Rehman 2011), and untreated affected children die before the age of three years (Higgs 2012; Modell 2008). People in low‐ and middle‐income countries have poor access to safe blood and the majority of the people die due to transfusion‐associated complications. It has been reported that there is an increased likelihood of blood transfusion‐transmissible infections in transfusion‐dependent individuals (Vidja 2011). In addition, frequent blood transfusions lead to iron overload which results in various complications such as heart disease (heart failure and arrhythmias), chronic liver hepatitis and fibrosis, endocrine problems (hypogonadism, hypothyroidism), stunted growth, osteoporosis and delayed pubertal growth (Borgna 2011; Rachmilewitz 2011). These complications have been improved by the use of iron chelating agents; however, they still exist in those who are less compliant with iron chelator treatment (Rachmilewitz 2011). Furthermore, the cost of treating these complications, in addition to managing the disease itself, poses a financial burden on both families and healthcare systems.
Description of the intervention
Various therapeutic agents such as erythropoietin, 5‐azacytidine, butyrate derivatives, and hydroxyurea (hydroxycarbamide), have been investigated for their potential role in augmenting HbF and also their therapeutic role in treating haemoglobinopathies (Ansari 2011; Perrine 2005). Hydroxyurea induces the production of HbF. The clinical benefits of this agent in people with sickle cell disease has been demonstrated in several studies, but only a limited number of studies have assessed its role in people with β‐thalassaemia (Bradai 2003). Such observational studies have shown a therapeutic response in large numbers of people, resulting in an increase in the Hb level and a decrease or elimination of the transfusion requirements in around 30% to 40% of individuals (Ansari 2011; Italia 2009; Yavarian 2004). In β‐thalassaemia, due to a mutation of β‐globin gene, no β‐globin protein is produced. There are four α‐globin proteins which are associated with ineffective erythropoiesis and red blood cell lysis. The beneficial effect of augmenting HbF through hydroxyurea has been associated with the reactivation of the γ‐globin gene, which leads to γ‐globin chain formation. This γ‐globin chain combines with two α chains resulting in the formation of HbF.
How the intervention might work
Hydroxyurea is one of several agents capable of activating the γ‐globin gene associated with an improvement in α to non‐α globin ratios, thereby leading to an increase in the effectiveness of erythropoiesis after several months of therapy (Fucharoen 1996; Zeng 1995).
Mechanisms proposed for the induction of HbF by hydroxyurea include rapid erythroid regeneration, increased erythropoietin (EPO) production, apoptosis, nitric oxide production, increased guanylate cyclase activity and activation of the p38 MAPK pathway. Induction of HbF by hydroxyurea in people with β‐thalassaemia was reported to be of similar magnitude as found in the cells of healthy individuals (1.3‐fold to 3.5‐fold) and people with sickle cell disease (two‐fold to five‐fold). In erythroid progenitor cells treated with hydroxyurea in vitro, HbF induction was comparable to the increase of HbF in the peripheral blood of people with sickle cell disease following hydroxyurea therapy in vivo. The majority of individuals increase HbF production after treatment with hydroxyurea; however, HbF baseline levels and the size of the response varies greatly. The absolute response to hydroxyurea and the HbF baseline level is likely dependent on genetic factors that modulate different regulatory pathways and trans‐acting factors involved in γ‐globin production (Pourfarzad 2013). Hydroxyurea has been widely used, due to the fact it has the lowest toxicity profile among the chemotherapeutic agents (Zeng 1995). It has been found to increase total Hb and HbF levels and reduce the need for blood transfusion in a people with transfusion‐dependent β‐thalassaemia (Arruda 1997).
Hydroxyurea therapy is associated with an improvement in the ineffective erythropoiesis, as reflected by an increase in the Hb level, a reduction in red blood cell transfusion requirement and a decreasing trend in the serum lactate dehydrogenase (LDH) and serum unconjugated bilirubin levels. This favourable effect of hydroxyurea was observed as early as one to three months post treatment. In contrast, a significant decrease in spleen size reflecting a reduction in extramedullary erythropoiesis appeared to take place much later, after six months of hydroxyurea therapy with a maximal benefit observed at 12 months of therapy. These favourable effects of hydroxyurea appeared to be associated with an increase in the γ‐globin biosynthesis, because the percentage of HbF increased as well as the total Hb. This observation is in agreement with a short‐term study that showed a good correlation of Hb levels with the amount of HbF production in people with βº‐thalassaemia and haemoglobin E (HbE) treated with hydroxyurea for five months (Fucharoen 1996; Wahid 2007). In addition to the modest increase in the Hb, most people reported an increase in exercise tolerance and sense of well being in comparison with their symptoms before therapy. This feeling may reflect the significant decrease in ineffective erythropoiesis resulting in the replacement of the poorly haemoglobinised, prematurely dying erythroid progenitor and red blood cell population by another population of cells with higher Hb content and longer survival, the regeneration of which requires less energy and consumption (Loukopoulos 1998).
Hydroxyurea can be used as an alternative option to blood transfusion for people with transfusion‐dependent β‐thalassaemia in countries having limited blood supplies (Bradai 2007; Karimi 2012), particularly in those people having a favourable molecular background (Yavarian 2004). Bradai reported better response to hydroxyurea with older age groups at the first transfusion and Xmn1 polymorphism (Bradai 2007). Xmn 1 polymorphism is a polymorphism at position ‐158 in γ‐globin gene which is associated with increase HbF formation and hence a better response to a HbF augmenting agent like hydroxyurea. However, few studies have not found any significant association of response to hydroxyurea with Xmn1 polymorphism (Karimi 2012) or any specific β‐thalassaemia mutations or α‐globin deletions (Yavarian 2004).
Why it is important to do this review
Hydroxyurea is a low‐cost medicine which may reduce the need for blood transfusions. According to various studies, significant numbers of people with β‐thalassaemia have responded to this treatment and physicians regard this treatment positively (Ansari 2007; Koren 2008; Zamani 2009). Therefore, this Cochrane Review aims to gather evidence regarding the efficacy of hydroxyurea which can then inform guidelines that in turn can formulate evidence‐based recommendations impacting upon current treatment protocols.
Objectives
The primary objective is to review the efficacy of hydroxyurea in reducing or ameliorating the requirement of blood transfusions in people with transfusion‐dependent β‐thalassaemia. The second objective is to review the safety of hydroxyurea with regards to severe adverse effects in this population.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs).
Types of participants
People with transfusion‐dependent β‐thalassaemia regardless of age and setting (e.g. country, primary or secondary care).
Types of interventions
Hydroxyurea, irrespective of dose, frequency and duration, versus control (no treatment, existing or standard treatment, including blood transfusions and iron chelation therapy). Trials of hydroxyurea comparing different dosage levels were also eligible.
Types of outcome measures
Primary outcomes
Volume of blood required (red blood cell units)
Frequency of blood transfusion
Mean Hb level
Secondary outcomes
Interval between two blood transfusions
Mean HbF level
Quality of life (as reported by trial authors)
Side effects and toxicities (as reported by trial authors)
Cost‐effectiveness of treatment
Search methods for identification of studies
There were no restrictions regarding language or publication status.
Electronic searches
The review authors identified relevant trials from the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register using the following terms: (thalassaemia OR (haemoglobinopathies AND general)) AND hydroxyurea.
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Public Health Agency Annual Scientific Meeting (formerly the Caribbean Health Research Council Meeting); and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of the last search of the Cochrane Cystic Fibrosis and Genetic Disorders Haemoglobinopathies Trials Register: 04 March 2019.
We undertook further electronic searches from:
PubMed (www.ncbi.nlm.nih.gov/pubmed) (searched 1946 to 09 October 2018);
US National Institutes of Health Ongoing Trials Register (Clinicaltrials.gov) (www.clinicaltrials.gov) (searched 09 October 2018);
WHO International Clinical Trials Registry Platform (WHO ICTRP) (apps.who.int/trialsearch) (searched 09 October 2018).
We adopted the search terms according to the terminology used in each database. The terms included are listed in an appendix (Appendix 1).
Searching other resources
Handsearching
We searched the Thalassemia International Federation website (www.thalassaemia.org.cy/) (Appendix 2). We planned to review the bibliographies of any included trials for any further trials and citations; however, there were no eligible trials included in the review.
Personal contacts and grey literature
We contacted researchers known to be experts in the field who may have conducted relevant research in order to identify unpublished research. We searched for other grey literature including theses, dissertations, meetings and conference proceedings particularly those conducted by the Thalassemia International Federation via their website and also a grey literature website (www.greylit.org) (searched 09 October 2018) (Appendix 1).
Data collection and analysis
Selection of studies
Two review authors (SMA, SOA) independently screened the titles and abstracts of the identified trials. Two authors (SMA, SOA) obtained the full text of all potentially relevant articles and independently assessed the eligibility by completing eligibility forms designed in accordance with the specified inclusion criteria. We resolved any disagreements by discussion.
Data extraction and management
We planned to independently assess extracted data from the included trials using standardised data extraction forms and to use the Review Manager software to enter the data for analysis (RevMan 2014). For those trials were a review author is also a trial investigator, that investigator will not be involved in data extraction.
Assessment of risk of bias in included studies
We planned to independently assess the risk of bias for each trial using the criteria listed in the Cochrane Handbook for Systematic Reviews of Interventions. We planned to resolve any disagreement by discussion with the third author (SHA). For those trials were a review author is also a trial investigator, that investigator will not be involved in the assessment of the risk of bias.
Random sequence generation (checking for possible selection bias)
For each included trial, we planned to describe the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups. We planned to assess the method as:
low risk of bias (any truly random process, e.g. random number table, computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth, hospital or clinic record number);
unclear risk of bias.
Allocation concealment (checking for possible selection bias)
For each included trial, we planned to describe the method used to conceal allocation to interventions prior to assignment and to assess whether this could have been foreseen in advance of, or during recruitment, or changed after assignment. We planned to assess the methods as:
low risk of bias (e.g. telephone or central randomisation, consecutively numbered sealed opaque envelopes);
high risk of bias (e.g. open random allocation, unsealed or non‐opaque envelopes, alternation, date of birth);
unclear risk of bias.
Blinding of participants and personnel (checking for possible performance bias)
For each included trial, we planned to describe any methods used to blind participants and personnel from knowing which intervention a participant received. We would have considered that trials are at a low risk of bias if they were blinded, or if we judged that the lack of blinding would be unlikely to affect results. We would have assessed blinding separately for different outcomes or classes of outcomes. We planned to assess the methods as:
low, high or unclear risk of bias for participants;
low, high or unclear risk of bias for personnel.
Blinding of outcome assessment (checking for possible detection bias)
We planned to describe any methods used to blind outcome assessors from knowing which intervention a participant received. We would have assessed blinding separately for different outcomes or classes of outcomes. We planned to assess methods used to blind outcome assessment as:
low, high or unclear risk of bias.
Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
For each included trial and for each outcome or class of outcomes, we planned to describe the completeness of data including any attrition or exclusions from the analysis. We planned to state whether these were reported and the numbers included in the analysis at each stage (compared with the total randomised participants), the reasons for attrition or exclusion, and whether missing data were balanced across groups or were related to outcomes. Where reports presented sufficient information, or if this was available from trial authors, we planned to include these missing data in our analyses. We planned to assess methods as:
low risk of bias (e.g. no missing outcome data, missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups, 'as treated' analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
Selective reporting (checking for reporting bias)
For each included trial we planned to describe how we investigated the possibility of selective outcome reporting bias and what we found. We planned to assess the methods as:
low risk of bias (where it is clear that all of the pre‐specified outcomes and all expected outcomes of interest to the review have been reported);
high risk of bias (where not all the pre‐specified outcomes have been reported, one or more reported primary outcomes were not pre‐specified, outcomes of interest are reported incompletely and so cannot be used, trial fails to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
Other bias
We planned to describe any important concerns we had about other possible sources of bias. We planned to assess each trial as having:
a low risk of other bias;
a high risk of other bias;
an unclear risk of other bias.
Overall risk of bias
We planned to make explicit judgements about whether trials were at a high risk of bias, according to Cochrane criteria (Higgins 2011a). We planned to assess the likely magnitude and direction of the bias and whether we considered it is likely to impact on the findings. We also planned to explore the impact of the level of bias through undertaking sensitivity analyses.
Measures of treatment effect
Dichotomous data
For dichotomous data (for the outcomes such as side effects and toxicities), we planned to present results as risk ratios (RR) with corresponding 95% confidence intervals (CIs).
Continuous data
For continuous data (for the outcomes such as volume of blood required, mean haemoglobin level, interval between two blood transfusions, mean HbF level, quality of life), we planned to use the mean difference (MD) and corresponding CIs if outcomes were measured in the same way by the included trials. We planned to use the standardized mean difference (SMD) to combine data from trials that have measured the same outcome using different units.
Unit of analysis issues
We planned to analyse cluster‐randomised trials or trials with more than two intervention groups in the analyses using the methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b).
Dealing with missing data
We planned to contact trial authors to retrieve missing data. We planned to perform a sensitivity analysis to evaluate the impact of our assumption on the results.
Assessment of heterogeneity
We planned to assess statistical heterogeneity in each meta‐analysis using the I² statistic and the Chi² test. We planned to regard heterogeneity as substantial if the I² statistic was greater than 50%, or if there was a low P value (< 0.10) in the Chi² test for heterogeneity. If we detected substantial statistical heterogeneity, we planned to explore this by subgroup analysis and use a random‐effects meta‐analysis to produce an overall summary.
Assessment of reporting biases
We planned to investigate reporting biases such as publication biases using a funnel plot. We planned to assess funnel plot asymmetry visually. If asymmetry was suggested by a visual assessment we would have performed an exploratory analysis to investigate this.
Data synthesis
We planned to carry out statistical analysis using the Review Manager software (RevMan 2014). We would have used a fixed‐effect meta‐analysis for combining data in the absence of significant heterogeneity where trials were sufficiently similar.
Subgroup analysis and investigation of heterogeneity
We planned to investigate any substantial heterogeneity using the following subgroup analyses:
genetic mutations;
Xmn1 polymorphism, i.e. heterozygous or homozygous or absent;
age at first transfusion;
gender;
duration for which the treatment was given;
number of transfusions per year in responders (those who become transfusion independent after treatment) and non‐responders before starting the treatment.
We planned to assess subgroup differences by the interaction test available within Review Manager (RevMan 2014). We planned to report the results of each subgroup analysis, quoting the Chi² test and P value, and the I² statistic.
Sensitivity analysis
We planned to carry out sensitivity analyses by examining the effect on the results of excluding trials at a high overall risk of bias (but most importantly in relation to sequence generation, allocation concealment, blinding and incomplete outcome data).
Summary of findings table
If we include trials in future versions of this review, we plan to present the main findings in summary of findings tables prepared using the GRADE approach (Guyatt 2008). We plan to list the three primary outcomes and side effects and toxicities for each comparison, with estimates of the relative effects along with the number of participants and trials contributing data for those outcomes. For each individual outcome, we plan to assess the quality of the evidence using the GRADE approach, which involves consideration of within‐trial risk of bias (methodological quality), directness of evidence, heterogeneity, precision of effect estimates and risk of publication bias. We plan to rate the quality of the body of evidence for each key outcome as 'high', 'moderate', 'low' or 'very low'.
Results
Description of studies
Results of the search
Please refer to the study flow diagram (Figure 1). We identified 376 records from our searches. After removing duplicates, we were left with 373 results. Out of these, we discarded 321 records which were not relevant and screened the abstracts of the remaining 52 records. At this stage we discarded 37 records (not relevant) and we further assessed 15 trials for inclusion in this review; however, 13 of these were subsequently excluded (reasons given below) and two refer to ongoing trials (see below).
1.
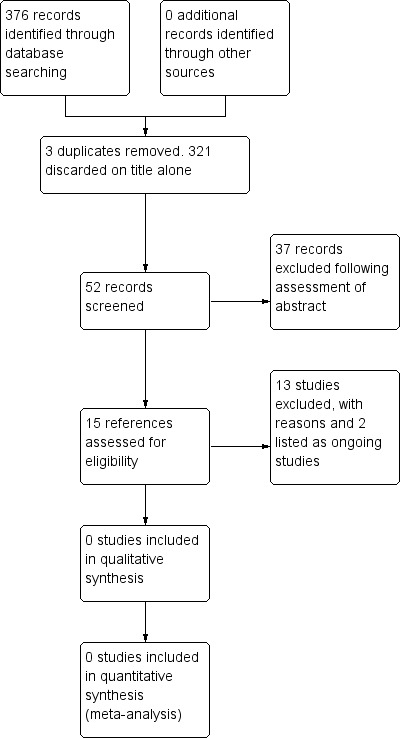
Study flow diagram.
Included studies
No trial fulfilled our inclusion criteria.
Excluded studies
After initial trial selection, 13 trials were excluded from the review, since on closer inspection, trials were found not to be RCTs, or the population did not include people with transfusion‐dependent β‐thalassaemia, or both. Please refer to the relevant table for further details (Characteristics of excluded studies).
Ongoing studies
We identified two ongoing trials, which we will assess for eligibility in a future update (NCT03183375; SLCTR/2018/024). The lead author is currently the principal investigator on one of these recently registered trials (NCT03183375). Please refer to a table for further details (Characteristics of ongoing studies).
Risk of bias in included studies
No trials were included in the review.
Effects of interventions
No trials were included in the review.
Discussion
Summary of main results
No trials were included in the review.
Potential biases in the review process
We were aware of the potential of introducing bias in the searching and selection of studies, therefore, two review authors independently searched and assessed the eligibility of trials for inclusion.
Agreements and disagreements with other studies or reviews
There were no trials that fulfilled the inclusion criteria. However, we found low‐quality evidence including case reports, case series, observational studies and before‐and‐after design or non‐RCTs and systematic reviews of observational studies that report on significant benefits of hydroxyurea in this population (particularly in the countries where substandard management of blood transfusion and blood banking practices and substandard quality exists) (Ansari 2007; Ansari 2011; Bradai 2003; Bradai 2007; Italia 2009; Karimi 2012; Koren 2008; Saxon 1998; Yavarian 2004; Zamani 2009). These have shown that a significant number of individuals have become transfusion independent or have decreased transfusion needs after taking hydroxyurea.
Authors' conclusions
Implications for practice.
Currently, we do not have any high‐quality evidence to support or challenge the continued use of this low‐cost treatment for managing people with transfusion‐dependent beta (β)‐thalassaemia.
Implications for research.
Multicentre, parallel‐group design, randomised controlled trials (comparing hydroxyurea (hydroxycarbamide) to placebo or another available treatment, i.e. blood transfusion and iron chelation) should be carried out to assess the efficacy and safety of hydroxyurea for reducing blood transfusion needs or maintaining or improving mean haemoglobin levels, as well as determining its cost‐effectiveness. Also, it is important to assess its impact on the quality of life of people with this disease. Such trials must look for the specific determinants that affect the efficacy of this drug in this population, such as age at first transfusion and the presence of specific genetic mutations (e.g. IVSI‐I, cap+1, cd30) or secondary genetic modifiers (e.g. XmnI polymorphism).
Acknowledgements
We thank the editorial team of the Cochrane Cystic Fibrosis and Genetic Disorders Group for their guidance provided during the protocol and review development.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Database searching
| Database/ Resource | Strategy |
| PubMed (1946 onwards) | (beta thalassemia OR beta thalassaemia) AND (hydroxyurea OR hydroxycarbamide OR hydrea OR droxia OR mylocel OR oncocarbide) |
| ClinicalTrials.gov | thalassaemia AND (hydroxyurea OR hydroxycarbamide) |
| WHO ICTRP | [Advanced search option] CONDITION: Thalassaemia OR thalassemia AND INTERVENTION: hydroxyurea OR hydroxycarbamide OR hydrea OR droxia OR mylocel OR oncocarbide RECRUITMENT STAUS: All |
| Greylit.org | SEARCH 1: hydrox SEARCH 2: droxia SEARCH 3: hydrea SEARCH 4: mylocel SEARCH 5: oncocarbide |
Appendix 2. Handsesarching
| Journal/ Website | Strategy |
| Thalassemia International Federation website (www.thalassaemia.org.cy/) | SEARCH 1: hydroxyurea SEARCH 2: hydroxycarbamide SEARCH 3: droxia SEARCH 4: hydrea SEARCH 5: mylocel SEARCH 6: oncocarbide |
Characteristics of studies
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alebouyeh 2004 | Not a RCT, not all the study population had transfusion‐dependent beta‐thalassaemia |
| Arruda 1997 | Clinical report, no comparison group |
| Bordbar 2014 | Not a RCT and has no control group |
| Bradai 2003 | Not a RCT and has no control group |
| Bradai 2007 | Not a RCT, a before and after study, so there is no comparison group |
| Italia 2009 | Not a RCT and has no control group |
| Loukopoulos 1998 | The study population does not have transfusion‐dependent beta‐thalassaemia, also the comparison involves combination of hydroxyurea (hydroxycarbamide) with other drug |
| NCT00809042 | The study population does not have transfusion‐dependent beta‐thalassaemia |
| Olivieri 1998 | Not a RCT, the study population does not have transfusion‐dependent beta‐thalassaemia |
| Saxon 1998 | Not a RCT but a case report |
| Voskaridou 1995 | Not a RCT and so no comparison group and the study population does not have transfusion‐dependent beta‐thalassaemia |
| Yavarian 2004 | Not a RCT, no control group |
| Zamani 2009 | Not a RCT, no control group |
RCT: randomised controlled trial
Characteristics of ongoing studies [ordered by study ID]
NCT03183375.
| Trial name or title | The Efficacy and Safety of HYDROXYUREA in Management of Beta Thalassemia Patients in Karachi Pakistan |
| Methods | Open‐label RCT |
| Participants | People with transfusion‐ and non‐transfusion‐dependent beta‐thalassaemia |
| Interventions | Hydroxyurea (hydroxycarbamide) |
| Outcomes | Number of participants who become responder (become transfusion independent or maintain a mean haemoglobin level or result in increase in haemoglobin by 1 ‐ 2 g/dL) or partial responder (transfusion need decreases of at least 50% as compared to baseline) |
| Starting date | 20 June 2017 |
| Contact information | muddasirsaqib@yahoo.com |
| Notes |
SLCTR/2018/024.
| Trial name or title | Efficacy and safety of oral hydroxyurea in transfusion‐dependent beta‐thalassaemia: a randomised double‐blind placebo‐controlled clinical trial |
| Methods | RCT |
| Participants | Beta‐thalassaemia major or HbE beta‐thalassaemia patients who required more than 10 transfusions during the preceding year |
| Interventions | Hydroxyurea |
| Outcomes | Complete response to treatment is defined as complete cessation of blood transfusions with a baseline haemoglobin > 9.0g/dL during the treatment period Partial response is defined as over 50% reduction in transfusion requirement during treatment period (≦ 3 transfusions during treatment 6 months) with baseline haemoglobin > 7.0g/dL |
| Starting date | 01 September 2018 |
| Contact information | sachith.mettananda@kln.ac.lk |
| Notes |
RCT: randomised controlled trial
Contributions of authors
Saqib Ansari: inception and design of the review and commenting on it critically for intellectual content.
Zohra Lassi: design of the review and commenting on it critically for intellectual content.
Salima Muhammad Khowaja: drafting the protocol, systematic search of literature and draft the review.
Syed Omair Adil: drafting the protocol, systematic search of literature and draft the review.
Tahir Shamsi: final approval of the protocol and later the review, to be published.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Saqib Ansari: none known.
Zohra Lassi: none known.
Salima Muhammad Khowaja: none known.
Syed Omair Adil: none known.
Tahir Shamsi: none known.
New
References
References to studies excluded from this review
Alebouyeh 2004 {published data only}
- Alebouyeh M, Moussavi F. Hydroxyurea in the treatment of major beta‐thalassemia and importance of genetic screening. Annals of Hematology 2004;83(7):430‐3. [DOI] [PubMed] [Google Scholar]
Arruda 1997 {published data only}
- Arruda VR, Lima CS, Saad ST, Costa FF. Letter to the Editor. Successful use of hydroxyurea in β‐thalassemia major. New England Journal of Medicine 1997;336(13):964‐5. [DOI] [PubMed] [Google Scholar]
Bordbar 2014 {published data only}
- Bordbar MR, Silavizadeh S, Haghpanah S, Kamfiroozi R, Bardestani M, Karimi M. Hydroxyurea treatment in transfusion‐dependent β‐thalassemia patients. Iranian Red Crescent Medical Journal 2014;16(6):1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bradai 2003 {published data only}
- Bradai M, Abad MT, Pissard S, Lamraoui F, Skopinski L, Montalembert M. Hydroxyurea can eliminate transfusion requirements in children with severe beta‐thalassemia. Blood 2003;102(4):1529–30. [DOI] [PubMed] [Google Scholar]
Bradai 2007 {published data only}
- Bradai M, Pissard S, Abad MT, Dechartres A, Ribeil JA, Landais P, et al. Decreases transfusion need associated with hydroxyurea therapy in Algerian patients with thalassemia major or intermedia. Transfusion 2007;47(10):1830‐6. [DOI] [PubMed] [Google Scholar]
Italia 2009 {published data only}
- Italia KY, Jijina FJ, Merchant R, Panjwani S, Nadkarni A, Sawant PM, et al. Response to hydroxyurea in beta thalassemia major and intermedia: experience in western India. Clinica Chimica Acta 2009;407(1‐2):10‐5. [DOI] [PubMed] [Google Scholar]
Loukopoulos 1998 {published data only}
- Loukopoulos D, Voskaridou E, Stomoulakatou A, Papassotiriou Y, Kalotychou V, Loutradi A, et al. Hydroxurea therapy in thalassemia. Annals of the New York Academy of Sciences 1998;850:120–8. [DOI] [PubMed] [Google Scholar]
NCT00809042 {published data only}
- NCT00809042. Combination Therapy of Hydroxyurea With L‐Carnitine and Magnesium Chloride in Thalassemia Intermedia. www.clinicaltrials.gov/show/NCT00809042 Date first received: 16 December 2008.
Olivieri 1998 {published data only}
- Olivieri NF, Rees DC, Ginder GD, Thein SL, Waye JS, Chang L, et al. Elimination of transfusions through induction of fetal hemoglobin synthesis in Cooley’s anemia. Annals New York Academy of Sciences 1998;850:100‐9. [DOI] [PubMed] [Google Scholar]
Saxon 1998 {published data only}
- Saxon BR, Waye JS, Olivieri NF. Increase in hemoglobin concentration during therapy with hydroxyurea in Cooley’s anemia. Annals New York Academy of Sciences 1998;850:459‐60. [DOI] [PubMed] [Google Scholar]
Voskaridou 1995 {published data only}
- Voskaridou E, Kalotychou V, Loukopoulos D. Clinical and laboratory effects of long‐term administration of hydroxyurea to patients with sickle‐cell/b‐thalassaemia. British Journal of Hematology 1995;89(3):479‐84. [DOI] [PubMed] [Google Scholar]
Yavarian 2004 {published data only}
- Yavarian M, Karimi M, Bakker E, Harteveld CL, Giordano PC. Response to hydroxyurea treatment in Iranian transfusion‐dependent b‐thalassemia patients. Haematologica 2004;89(10):1172‐78. [PubMed] [Google Scholar]
Zamani 2009 {published data only}
- Zamani F, Shakeri R, Eslami SM, Razavi SM, Basi A. Hydroxyurea therapy in 49 patients with major beta‐thalassemia. Archives of Iranian Medicine 2009;12(3):295‐7. [PubMed] [Google Scholar]
References to ongoing studies
NCT03183375 {published data only}
- NCT03183375. The Efficacy and Safety of HYDROXYUREA in Management of Beta Thalassemia Patients in Karachi Pakistan. www.clinicaltrials.gov/show/NCT03183375 Date first received: 05 June 2017.
SLCTR/2018/024 {published data only}
- SLCTR/2018/024. Effectiveness of hydroxyurea treatment compared to placebo in reducing transfusions in beta‐thalassaemia patients. www.slctr.lk/trials/1286 Date first received: 01 August 2018.
Additional references
Ansari 2007
- Ansari SH, Shamsi TS, Siddiqui FJ, Irfan M, Parveen K, Farzana T, et al. Efficacy of hydroxyurea (HU) in reduction of pack red cell (PRC) transfusion requirement among children having beta‐thalassemia major: Karachi HU trial (KHUT). Journal of Pediatric Hematology Oncology 2007;29(11):743‐6. [DOI] [PubMed] [Google Scholar]
Ansari 2011
- Ansari SH, Shamsi TS, Ashraf M, Parveen K, Farzana T, Bohray M, et al. Efficacy of hydroxyurea in providing transfusion independence in β‐thalassemia. Journal of Pediatric Hematology Oncology 2011;33(5):339‐43. [DOI] [PubMed] [Google Scholar]
Borgna 2011
- Borgna PC, Gamberini MR. Complications of thalassemia major and their treatment. Expert Review Hematology 2011;4(3):353‐66. [DOI] [PubMed] [Google Scholar]
Fucharoen 1996
- Fucharoen S, Siritanaratkul N, Winichagoon P, Chowthaworn J, Siriboon W, Muangsup W, et al. Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in β‐thalassemia/Hemoglobin E disease. Blood 1996;87(3):887–92. [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Wyer P, Ioannidis J. When to believe a subgroup analysis. In: Guyatt GH, Rennie D, Meade MO, Cook DJ editor(s). The Users’ Guides to the Medical Literature: A Manual for Evidence‐Based Clinical Practice. 2nd Edition. New York, NY: McGraw‐Hill, 2008:571‐96. [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC, editor(s) on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG, editor(s) on behalf of the Cochrane Statistical Methods Group. Chapter 16: Special topics in statistics. In: Higgins JPT, Green S, editor(s). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgs 2012
- Higgs DR, Engel JD, Statmatoyannopoulos G. Thalassemia. Lancet 2012;379:373‐83. [DOI] [PubMed] [Google Scholar]
Karimi 2012
- Karimi M, Haghpanah S, Farhadi A, Yavarian M. Genotype‐phenotype relationship of patients with β‐thalassemia taking hydroxyurea: a 13‐year experience in Iran. International Journal of Hematology 2012;95(1):51‐6. [DOI] [PubMed] [Google Scholar]
Koren 2008
- Koren A, Levin C, Dgany O, Kransnov T, Elhasid R, Zalman L, et al. Response to hydroxyurea therapy in beta‐thalassemia. American Journal of Hemataology 2008;83(5):366‐70. [DOI] [PubMed] [Google Scholar]
Modell 2008
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization 2008;86:480‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
NHS 2011
- UK National Steering Committee. Sickle cell and thalassaemia screening programme: standards for the linked antenatal and newborn screening program. http://sct.screening.nhs.uk/standardsandguidelines (accessed 26 September 2014).
Perrine 2005
- Perrine SP. Fetal globin induction can it cure beta thalassemia?. Hematology. American Society of Hematology. Education Program 2005;2005 no.1:38‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pourfarzad 2013
- Pourfarzad F, Lindern MV, Azarkeivan A, Hou J, Kia SK, Esteghamat F, et al. Hydroxyurea responsiveness in β‐thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity. Haematologica 2013;98(5):696‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rachmilewitz 2011
- Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood 2011;118(13):3479‐88. [DOI] [PubMed] [Google Scholar]
RCOG 2014
- Royal College of Obstetricians and Gynaecologists. New guideline for managing thalassaemia in pregnancy announced at the RCOG World Congress in India. www.rcog.org.uk/en/news/new‐guideline‐for‐managing‐thalassaemia‐in‐pregnancy‐announced‐at‐the‐rcog‐world‐congress‐in‐india/ (accessed 26 September 2014).
Rehman 2011
- Rehman A. Beta thalassemia prevention and Pakistan: review article. Pakistan Paediatric Journal 2011;35(2):55‐62. [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Vidja 2011
- Vidja PJ, Vachhani JH, Sheikh SS, Santwani PM. Blood transfusion transmissible infections in multiple blood transfused patients of beta thalassemia. Indian Journal of Hematology and Blood Transfusion 2011;27(2):65‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wahid 2007
- Wahid SF, Rahmat R, Azhar S, Hussin NH, Keng CS. Hydroxyurea appears beneficial in patients with beta‐thalassaemia major and intermedia. Medical Journal of Indonesia 2007;16(2):78‐83. [Google Scholar]
Weatherall 2001
- Weatherall D. Thalassemias. Encylopedia of Life Sciences. Chichester: John Wiley & Sons Ltd, 2001:1‐3. [Google Scholar]
Zeng 1995
- Zeng YT, Huang SZ, Ren ZR, Lu ZH, Zeng FY, Schechter AN, et al. Hydroxyurea therapy in beta‐thalassemia intermedia: improvement in hematological parameters due to enhanced beta‐globin synthesis. British Journal of Haematology 1995;90(3):557‐63. [DOI] [PubMed] [Google Scholar]