

Abstract
Fractionated whole-brain irradiation for the treatment of intracranial neoplasia causes progressive neurodegeneration and neuroinflammation. The long-term consequences of single-fraction high-dose irradiation to the brain are unknown. To assess the late effects of brain irradiation we compared transcriptomic gene expression profiles from non-human primates (NHP; rhesus macaques Macaca mulatta) receiving single-fraction total-body irradiation (TBI; n = 5, 6.75–8.05 Gy, 6–9 years prior to necropsy) to those receiving fractionated whole-brain irradiation (fWBI; n = 5, 40 Gy, 8 × 5 Gy fractions; 12 months prior to necropsy) and control comparators (n = 5). Gene expression profiles from the dorsolateral prefrontal cortex (DLPFC), hippocampus (HC) and deep white matter (WM; centrum semiovale) were compared. Stratified analyses by treatment and region revealed that radiation-induced transcriptomic alterations were most prominent in animals receiving fWBI, and primarily affected white matter in both TBI and fWBI groups. Unsupervised canonical and ontologic analysis revealed that TBI or fWBI animals demonstrated shared patterns of injury, including white matter neuroinflammation, increased expression of complement factors and T-cell activation. Both irradiated groups also showed evidence of impaired glutamatergic neurotransmission and signal transduction within white matter, but not within the dorsolateral prefrontal cortex or hippocampus. Signaling pathways and structural elements involved in extracellular matrix (ECM) deposition and remodeling were noted within the white matter of animals receiving fWBI, but not of those receiving TBI. These findings indicate that those animals receiving TBI are susceptible to neurological injury similar to that observed after fWBI, and these changes persist for years postirradiation. Transcriptomic profiling reaffirmed that macrophage/microglial-mediated neuroinflammation is present in radiation-induced brain injury (RIBI), and our data provide novel evidence that the complement system may contribute to the pathogenesis of RIBI. Finally, these data challenge the assumption that the hippocampus is the predilection site of injury in RIBI, and indicate that impaired glutamatergic neurotransmission may occur in white matter injury.
INTRODUCTION
There have been 57 major nuclear accidents since the Chernobyl disaster (1) despite safety regulations to limit radiation exposure. Occupational, medical and malicious exposures also pose a significant threat to public health and safety. Those who survive the acute radiation syndrome are at risk of developing late-delayed sequelae, which may not appear until several years postirradiation, including pulmonary and cardiac fibrosis, cataracts, muscle wasting and cancer (2–6). The brain is considered relatively radioresistant (ED50 12–14 Gy) (7); however, recently published studies in non-human primate (NHP) long-term radiation survivors indicate that cognitive impairment may be present at lower doses (8). A clear understanding of the molecular mechanisms of radiation injury is necessary to mitigate the health risks associated with radiation exposure; however, little is known regarding the long-term consequences of single-fraction, high-dose radiation exposure to the brain.
In patients receiving fractionated whole-brain irradiation (fWBI) for the treatment of intracranial neoplasia, progressive, neuroinflammatory and neurodegenerative brain disease occurs. Characterized by multifocal cerebrovascular injury and white matter necrosis (9–24), late-delayed radiation-induced brain injury (RIBI) results in cognitive dysfunction and memory impairment, which negatively affects patient quality of life (25–27). A subset of patients will develop profound dementia, incontinence and ataxia (28).
Studies in which the long-term consequences of single-fraction, high-dose irradiation in humans are investigated are confounded by variations in radiation source, dose rate, shielding and the concurrent effects of thermal and mechanical injury (5, 29). While rodent models have been used to investigate the late effects of radiation exposure, the radiation response of rodents differs from that of humans. Dissimilarities in cerebrovascular structure and white matter investiture (e.g., rodents have a lower white:gray matter ratio) (30) may limit the translatability of findings. Rodents do not develop white matter necrosis as a component of RIBI after fWBI (31–34), thus lacking a key histologic feature of the disease. In contrast to rodents, non-human primates possess greater genomic homology to humans; and better recapitulate the histopathologic manifestations of late-delayed RIBI after fWBI (9, 11, 12, 35).
We hypothesized that non-human primates receiving single-fraction, high-dose total-body irradiation (TBI) would develop similar patterns of brain injury as seen in fWBI, but with lower severity and longer latency. Herein, we investigate patterns of change in the cerebral transcriptome from animals receiving single-fraction, high-dose TBI, and compare them to animals with histologically confirmed radiation-induced brain injury, as well as thorax-only irradiated control comparators. In our previously published targeted gene expression analysis (35), we found macrophage/microglial-mediated neuroinflammation, extracellular matrix (ECM) deposition, evidence of hypoxia, impaired glutamatergic neurotransmission within white matter and vascular maturation. We anticipated that RNA sequencing would reveal similar patterns of injury, and a broader view of contributing mechanisms.
MATERIALS AND METHODS
Animals
Three groups of young adult, post-pubertal, male rhesus macaques (n = 5 per group) were compared (Table 1). Five animals (aged 10–13 years; 6.9–19.7 kg) received high-dose TBI (median: 7.2 Gy; range: 6.75–8.05 Gy) a median of 7.8 years prior to necropsy (6.2–8.8 years). Three to four months postirradiation, these animals were enrolled in a cohort of long-term irradiation survivors. Monitoring included daily observations and annual brain magnetic resonance imaging (MRI).
TABLE 1.
Subject Demographics
| Group | Age (years) | Weight (kg) | Dose to brain (Gy) | Survival interval (years) |
|---|---|---|---|---|
| Control | 5.5 ± 0.7 | 6.66 ± 1.24 | 0.1 | 0.7 |
| TBI | 11.5 ± 1 | 11.84 ± 5.47 | 7.19 ± 0.53 | 7.8 ± 1.1 |
| fWBI | 9.7 ± 2.2 | 8.6 ± 0.80 | 40 | 1.2 ± 0.1 |
Note. Mean ± standard deviation, (△) = animal with type 2 diabetes.
Whole-brain irradiated animals (n = 5; aged 7–13 years; 8.3–9.5 kg) received 40 Gy fWBI, 12–15 months prior to necropsy as part of a separate experiment including cognitive assessments. Additional experimental procedures are described in detail Hanbury et al. (12) and Robbins et al. (36).
Control comparators (n = 5; aged 5–7 years; 5.5–8.3 kg) received 10 Gy thoracic-only irradiation as part of an unrelated experiment (37).
All animals were clinically observed daily by trained veterinary personnel. All procedures were performed in accordance with the Guide for Care and Use of Laboratory Animals, and approved by the Wake Forest University School of Medicine Institutional Animal Care and Use Committee (IACUC). The Wake Forest School of Medicine is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and adheres to all state and federal animal welfare laws.
Diet
Animals receiving TBI and control comparators were fed a typical American primate diet [(TAD) diet no. 5L0P, LabDiett®; Land O’Lakes Inc. St. Louis, MO], which is designed to approximate the macronutrients of a Western dietary profile. Animals T2 and T5 were maintained on TAD for 3 and 2 years respectively, and then transitioned back to commercially available monkey chow (diet no. 5038, LabDiet) for the management of type 2 diabetes mellitus (hemoglobin A1C > 6.5%). Additional details are provided elsewhere (4). Animals receiving fWBI were fed diet no. 5038.
Irradiation
Irradiation procedures for control comparators and animals receiving fWBI are described elsewhere (35), and further details about fWBI are provided by Hanbury et al. (12) and Robbins et al. (36). The irradiation procedure for animals receiving TBI is described by Yu et al. (38).
Briefly, animals receiving fWBI were given a total of 40 Gy mid-plane (8 fractions × 5 Gy/fraction, 2 fractions/week × 4 weeks) of 6- MV X rays at a nominal dose rate of 4.9–5.4 Gy/min from a clinical linear accelerator (LINAC) at the WFUSM. The biologically effective dose (BED) of this regimen is 106.7 Gy, and thus comparable to a brain tumor treatment of 30 fractions of 2 Gy in 6 weeks (BED: 100.2 Gy) (39). The opposed lateral fields were designed to match human whole-brain irradiation fields, enclosing the cranial contents (nominal field size of 11 × 7 cm2, with the field edge tangential to the base of the skull). The central axis of each lateral beam was placed at the respective outer canthus, and the eyes and olfactory region were shielded from radiation by cylindrical eye blocks. Non-human primates receiving fWBI were sedated with ketamine HCl (15 mg/kg body weight, intramuscularly) and maintained on isoflurane gas (3% induction, 1.5% maintenance) in 100% oxygen during irradiation.
Total-body irradiated animals received 6.75–8.05 Gy (median: 7.2 Gy; range: 6.75–8.05 Gy) administered via a 6-MV LINAC at a nominal dose rate of 0.8 ± 0.025 Gy/min. Fifty percent of the dose was delivered from the anterior-posterior direction, and the remaining dose was delivered from the posterior-anterior direction (38).
Control comparators received 10 Gy thoracic irradiation to the anterior-posterior thoracic mid-plane. X rays (6 MV) were delivered with parallel opposed anterior-posterior fields from a clinical linear accelerator at a nominal dose rate of 4 Gy/min. The field of irradiation included the heart, mediastinum and lung fields extending up to 4 cm caudal to the xyphoid. The maximum dose to the brain is estimated to be 0.1 Gy.
Tissue Collection and Histopathology
Tissue collection, processing, archival and histopathology procedures were the same for all subjects. At the time of necropsy, the animals were humanely euthanized in accordance with the American Veterinary Medical Association’s Guidelines on Euthanasia (40) by deep anesthesia with pentobarbital, followed by exsanguination and perfusion of the vascular system with 2 liters of cold normal saline. The brain was removed intact and sectioned coronally in 4-mm intervals using a stainless-steel brain matrix with cutting guides. Once removed from the matrix, all slices were photographed. Alternating sections were either immediately frozen on dry ice or immersed in 4% cold paraformaldehyde for 24 h. Fixed tissues were embedded in paraffin and sectioned coronally at 4 µm, then stained with hematoxylin and eosin. All animals were assessed by a board-certified veterinary pathologist (JMC) and received a full histopathologic assessment in accordance with the Society for Toxicologic Pathology’s Recommended Practices for Sampling and Processing the Nervous System (41), with the addition of a section of prefrontal cortex containing Brodmann area 46. Lesions were scored as described elsewhere (12, 35): absent (0), minimal (1 = inflammatory or vascular changes without disruption of the neuropil or clear neuronal loss); mild (2 = focal vascular injury and inflammation with loss of neuropil or neurons and microglial activation); moderate (3 = extensive or multifocal vascular injury, hemorrhage, disruption of the neuropil, neuronal loss and microglial activation); or severe (4 = extensive or multifocal vascular injury, hemorrhage, disruption of the neuropil, neuronal loss and microglial activation, with additionally extensive zones of necrosis within neural tissue). The presence of vascular lesions in each region was recorded as follows: a: endothelial hypertrophy; b: perivascular ECM deposition, c: perivascular edema, d: disorganized vascular morphology. The assessor was blinded to the irradiation status of the animals.
RNA Extraction Methods
Total mRNA was extracted from 100 mg of brain tissue from three brain regions [dorsolateral prefrontal cortex (DLPFC; Brodmann area 46), hippocampus (HC) and deep white matter (centrum semiovale)] for all animals. Tissue was placed into a 1.4-mm ceramic bead tube with 1 ml QIAzolt® lysis reagent (QIAGENt®, Valencia, CA), and homogenized using a Bead Ruptor 24 (Omni International, Kennesaw, GA). The tissue sample tube was processed on the Bead Ruptor for 1 cycle at a speed of 4.7 m/s for 20 s, and repeated up to three times until the sample was completely homogenized. Aliquots of homogenized lysates equivalent to 40 mg tissue were extracted for total RNA using the RNeasy Microarray Tissue Mini kit (QIAGEN). Extracted RNA was DNase-treated and purified using the RNA Clean and Concentrator-5 kit (Zymo Research Corp., Irvine, CA), then assessed for RNA quality using an Agilent 2100 Bioanalyzer and the RNA 6000 Nano Kit (Agilent Technologies Inc., Santa Clara, CA).
cDNA Library Preparation and Sequencing Methods
Total RNA was used to prepare cDNA libraries using the Illuminat® TruSeq Stranded Total RNA with Ribo-Zero Gold Preparation kit (San Diego, CA) and the SciClone NGS Work Station (PerkinElmert® Inc., Waltham, MA). RIN values for the RNA samples ranged from 7.1 to 9.3. Briefly, 750 ng of total RNA was rRNA depleted followed by enzymatic fragmentation, reverse-transcription and double-stranded cDNA purification using AMPure XP magnetic beads (Beckman Coulter® Inc., Fullerton, CA). The cDNA was end repaired, 30 adenylated, with Illumina sequencing adaptors ligated onto the fragment ends, and the stranded libraries were pre-amplified with PCR. The library size distribution was validated and quality inspected on a Bioanalyzer DNA 1000 chip (Agilent Technologies). The quantity of each cDNA library was measured using the Qubit® 3.0 (Thermo Fisher Scientific™ Inc., Rockford, IL). Three library pools were formed, each containing 15 libraries. Library pools were sequenced to a target read depth of 28M reads per library using single-end 76 cycle sequencing with the High Output 75-cycle kit on the Illumina NextSeq® 500.
Data Analysis
Raw read quality was assessed using FASTQC analysis (Babraham Bioinformatics, Cambridge, UK). Uniquely mapped reads ranged from 21M–36M reads per sample. Reads with >Q20 quality score were aligned to the Ensembl Macaca mulatta genome build Mmul_1 using the STAR aligner (42) and gene counts were determined using featureCounts software (43). Differentially expressed genes (DEGs) were identified by DESeq2 (44). Significant DEGs were conservatively defined as log2 fold change ratios ≥ ±1 and P < 0.05 after adjustment for false discovery (Benjamini-Hochberg). Gene expression analysis data are deposited in the Gene Expression Omnibus database (accession no. GSE120901). Gene identities were preliminarily mapped in Ingenuity® Pathway Analysis (IPA®) (45) using Ensembl identification numbers for the rhesus macaque and assigned the corresponding HGNC identifier. As the rhesus gene identifiers were transcribed to human identifiers for analysis, the MHC class II molecules referred to herein as HLA-transcripts reference the MAMU sequence. Genes that did not map in IPA were identified by secondary screening in PANTHER (46, 47) and UniProt (48), then backmapped to the encoding gene by the corresponding HGNC identifier. Ensembl IDs that did not correspond to an identified gene were recorded (Supplementary Table S1; http://dx.doi.org/10.1667/RR15263.1.S1) and excluded from pathway analysis.
Enriched biological pathways and signaling networks were identified in IPA via unsupervised analysis of significant DEGs. Genes that contributed to the top 10 canonical pathways (Supplementary Table S2; http://dx.doi.org/10.1667/RR15263.1.S1)) were evaluated for contributing molecular functions in Gene Ontology using the GO Enrichment Analysis tool powered by PANTHER (46, 47) with Fisher’s exact test with false discovery rate (FDR) correction for multiple comparisons. As there were no enriched canonical pathways detected within the dorsolateral prefrontal cortex or hippocampus of TBI animals, these regions were omitted from ontologic analysis. Biological process and molecular function analyses were hierarchically clustered and the most specific subclass is reported. Hierarchies were sorted by fold-enrichment value, and the 10 greatest fold-enriched hierarchies are reported. FDR-corrected P values ≤ 0.05 were considered significant.
RESULTS
Demographics
Control comparators were younger than groups receiving fWBI (mean difference: 4.18 years; P ≤ 0.002) and TBI (mean difference: 6.06 years; P ≤ 0.0001) (Fig. 1A). The ages of animals receiving fWBI and TBI were comparable (P = 0.18). There was no difference in animal weight between groups (Fig. 1B); the TBI animals weighing >15 kg were those with type 2 diabetes mellitus (Fig. 1B, open triangles). Differences in radiation dose (Fig. 1C) and survival interval between groups (Fig. 1D) were due to differences in experimental design.
FIG. 1.

Subject demographics. Mean ± standard deviation, (△) = Animal with type 2 diabetes
Clinical Findings
Causes of death and comorbid conditions are summarized in Table 2. Neurologically, all animals (n = 15) were normal. Four out of five (4/5) animals receiving TBI developed neoplasia during the study period. Three out of four (3/4) tumors were of neuroendocrine origin, and the remaining neoplasm was a hemangiosarcoma (n = 1); there was no evidence of intracranial metastasis for any animal. Pulmonary bullous emphysema, with bulla rupture and resultant pneumothorax necessitated the euthanasia of two out of five (2/5) animal receiving TBI. Two out of five (2/5) animals receiving TBI developed type 2 diabetes mellitus four years postirradiation.
TABLE 2.
Subject Causes of Death and Comorbid Illness
| Group | Animal ID | Cause of death/comorbid illness |
|---|---|---|
| Control | C1 | Experimental euthanasia |
| C2 | Experimental euthanasia | |
| C3 | Experimental euthanasia | |
| C4 | Experimental euthanasia | |
| C5 | Experimental euthanasia | |
| fWBI | F1 | Experimental euthanasia |
| F2 | Experimental euthanasia | |
| F3 | Experimental euthanasia | |
| F4 | Experimental euthanasia | |
| F5 | Experimental euthanasia | |
| TBI | T1 | Clinical euthanasia |
| Pulmonary bullous emphysema; with rupture and pneumothorax | ||
| Neoplasia, hemangiosarcoma, pulmonary | ||
| T2 | Clinical euthanasia | |
| Fatal fasting syndrome of obese macaques | ||
| Type 2 diabetes mellitus | ||
| T3 | Clinical euthanasia | |
| Pulmonary bullous emphysema; with rupture and pneumothorax | ||
| Neoplasia, seminal vesicle, poorly-differentiated neuroendocrine tumor | ||
| T4 | Clinical euthanasia | |
| Neoplasia, renal tubular carcinoma with polycythemia | ||
| Neoplasia, subcutis, chondrolipoma | ||
| Neoplasia, subcutis, poorly-differentiated neuroendocrine tumor | ||
| T5 | Clinical euthanasia | |
| Type 2 diabetes mellitus | ||
| Bacterial pneumonia | ||
| Chronic renal disease Hypertension | ||
| Hypertension | ||
| Neoplasia, neuroendocrine tumor, heart base |
Since control comparators had received high-dose thoracic irradiation as part of a previous study, clinical data regarding respiratory and cardiac function were reviewed prior to selection as control comparators. Respiratory rate, heart rate and pulse oximetry were within normal limits until euthanasia.
Histopathology
All animals receiving fWBI (5/5) developed multifocal white matter necrosis and cerebrovascular injury (Table 3).
TABLE 3.
Histopathologic Grading of RIBI in Animals Receiving fWBI
| F1 | F2 | F3 | F4 | F5 | |
|---|---|---|---|---|---|
| White matter necrosis | |||||
| Forebrain: white matter | + | ++++ | ++ | +++ | +++ |
| Forebrain: cortical gray matter | − | +++ | + | + | ++ |
| Prefrontal cortex | − | − | − | + | ++ |
| Basal ganglia/striatum | − | +++ | − | − | − |
| Hippocampus | − | − | − | − | − |
| Thalamus | − | ++ | − | ++ | ++ |
| Midbrain | − | ++ | − | + | +++ |
| Cerebellum | − | + | − | ++ | ++ |
| Brainstem | − | ++ | − | + | − |
| Spinal cord | − | ++ | na | + | − |
| Vascular | |||||
| Forebrain: white matter | a, c | a, b, d | − | a, b, d | b, d |
| Forebrain: cortical gray matter | − | a, d | d | a, d | b |
| Prefrontal cortex | − | − | − | − | − |
| Basal ganglia/striatum | − | − | a | − | − |
| Hippocampus | c | − | − | − | − |
| Thalamus | − | − | − | − | − |
| Midbrain | − | a, d | − | − | b |
| Cerebellum | − | − | − | − | b |
| Brainstem | − | a, d | − | − | − |
| Spinal cord | − | − | − | − | − |
Notes.
= Endothelial hypertrophy.
= Perivascular extracellular matrix deposition.
= Perivascular edema.
= Disorganized vascular morphology.
There were no significant lesions in control comparators. There was no evidence of white matter necrosis in animals receiving TBI. In animal no. T002, perivascular spaces surrounding penetrating arterioles in the rostral frontal cortex contained multifocal aggregates of hemosiderin. A few arterioles within the caudal caudate nucleus were surrounded by homogeneous eosinophilic ECM. In animal no. T004 histopathologic evaluation revealed a focal vascular malformation consistent with a cavernous hemangioma within the subcortical white matter of the right inferior occipital lobe.
Transcriptomic Profiling
In all brain regions examined, the number of DEGs was greatest in animals that received fWBI (Fig. 2). White matter contained the greatest number of DEGs within treatment groups, followed by the dorsolateral prefrontal cortex and then the hippocampus. A total of 133 DEGs were expressed within the white matter of both fWBI and TBI groups. Two DEGs (MEIS3, SCEL) were expressed within the dorsolateral prefrontal cortex of both fWBI and TBI groups (Fig. 3).
FIG. 2.
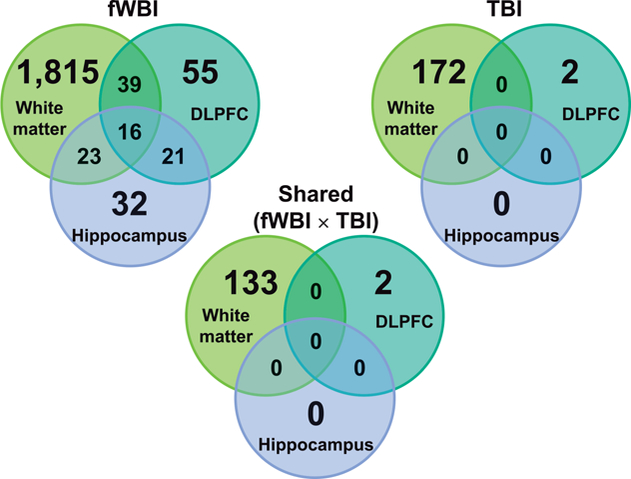
Venn diagrams of regional differences in differential gene expression by region, across irradiation groups. Both fWBI and TBI groups are normalized to the expression values of the control group. The number of genes differentially regulated in all regions in animals receiving fWBI, was greater than comparable regions in animals receiving TBI. White matter contained the highest number of DEGs within both irradiation groups.
FIG. 3.
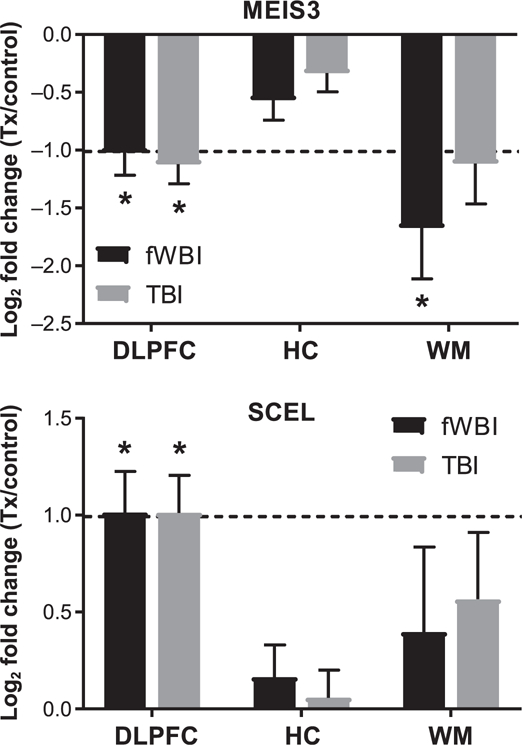
MEIS3 and SCEL are differentially expressed within the dorsolateral prefrontal cortex of both fWBI and TBI animals. MEIS3 was downregulated within the dorsolateral prefrontal cortex (log2 fold change: –1.02) and white matter (log2 fold change: –1.67) of animals receiving fWBI, and the dorsolateral prefrontal cortex (log2 fold change: –1.12) of animals receiving TBI. SCEL was upregulated within the dorsolateral prefrontal cortex in animals receiving fWBI (log2 fold change: 1.01) and animals receiving TBI (log2 fold change: 1.01). Bars denote mean log2 fold change and error bars are set to log2 fold standard error. Dashed line indicates log2 fold change of ±1.0. DLPFC = dorsolateral prefrontal cortex; HC = hippocampus; WM = temporal white matter. *FDR adjusted P value ≤ 0.05
Canonical Pathway Analysis
The top 10 enriched canonical pathways are reported in order of decreasing statistical significance, with all pathways reaching at least P ≤ 0.05 (Fig. 4). Shared canonical pathways were detected between regions and treatments (Fig. 5A and B). Since overall patterns suggested shared processes involving inflammation, ECM remodeling and neurotransmission/signal transduction, for further analysis the contributing genes were grouped by broader function (Table 4).
FIG. 4.
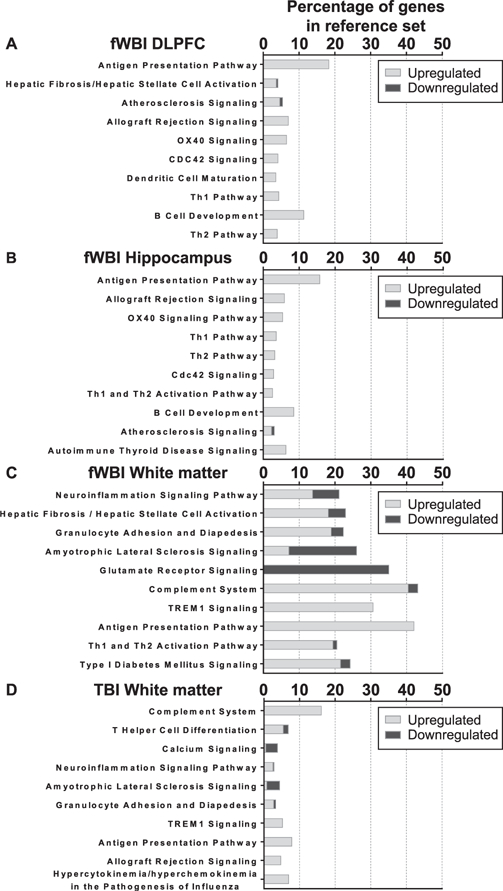
Top 10 enriched canonical pathways in ingenuity pathway analysis. All comparisons are normalized to the expression values of the matching region from the control group. Pathways are listed in descending order of decreasing statistical significance, all FDR adjusted P values ≤ 0.05. The stacked bars indicate the percentage of up- or downregulated DEGs out of a reference gene set curated by IPA. Data are not shown for dorsolateral prefrontal cortex or hippocampus comparisons, as zero (0) enriched canonical pathways were detected.
FIG. 5.
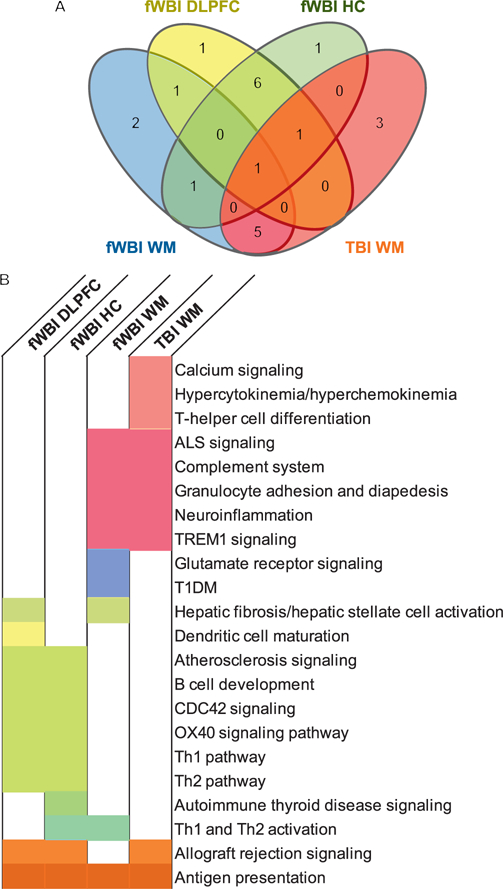
Overlapping canonical pathways across brain regions and irradiation groups. Patterns in canonical pathway expression were shared across regions, and across treatments. Panel A: Shared and exclusive pathways are quantitated, and displayed by region and treatment. Panel B: Boxes are color coded to the corresponding Venn-diagram sector. White indicates that the following canonical pathway was not within the top 10 enriched pathways for that region and treatment.
TABLE 4.
Functional Grouping of Canonical Pathways
| Inflammation | ECM | Neurotransmission and signal transduction |
|---|---|---|
| Neuroinflammation | CDC42 signaling | Calcium signaling |
| Granulocyte adhesion and diapedesis | Atherosclerosis signaling | ALS signaling |
| Hepatic fibrosis/hepatic stellate cell activation | Hepatic fibrosis/hepatic stellate cell activation | Glutamate receptor signaling |
| Complement system | ||
| TREM1 signaling | ||
| Antigen presentation pathway | ||
| Th1 and Th2 activation pathway | ||
| Type 1 diabetes mellitus signaling | ||
| Allograft rejection signaling | ||
| OX40 signaling | ||
| Th1 pathway | ||
| Th2 pathway | ||
| B-cell development | ||
| Dendritic cell maturation | ||
| Autoimmune thyroid disease signaling | ||
| T-helper cell differentiation | ||
| Role of hypercytokinemia/hyperchemokinemia in the pathogenesis of influenza |
Canonical pathway analysis indicated enrichment of inflammation and immunologic signaling patterns within all regions in animals receiving fWBI and within the white matter of animals receiving TBI. ECM-associated pathways were enriched within fWBI animals only. Neurotransmission and signal transduction-associated pathways were enriched within the white matter of fWBI and TBI animals. Comparisons with the greatest numbers of shared pathways were between the dorsolateral prefrontal cortex and hippocampus of fWBI animals (significantly enriched pathway n = 6), and the white matter of fWBI and TBI animals (significantly enriched pathways n = 5), suggesting that the radiation response in RIBI is differentially regulated by region.
Nineteen genes were implicated in the top 10 canonical pathways within fWBI dorsolateral prefrontal cortex, 10 in fWBI hippocampus, and 192 in fWBI white matter. Thirty-one contributing genes were identified in the TBI white matter, and there were no enriched canonical pathways detected within the TBI dorsolateral prefrontal cortex or hippocampus.
Gene Ontology
Inflammation-associated canonical pathways were identified in all brain regions from animals receiving fWBI and the white matter of animals receiving TBI. The contributing DEGs (fWBI DLPFC: 15 DEGs; fWBI HC: 6 DEGs; fWBI WM: 172 DEGs; and TBI WM: 22 DEGs) were evaluated for patterns in molecular function (Table 5). Complement system transcripts were increased in the white matter of both irradiated groups. Transcripts common to major histocompatibility complex (MHC) class II-mediated peptide antigen presentation were noted in the fWBI dorsolateral prefrontal cortex, fWBI hippocampus and TBI white matter. Ontologic patterns suggesting pro-inflammatory chemokine signaling were present in the white matter of both irradiated groups. Patterns consistent with modulation of neurotransmission noted in the white matter of animals receiving fWBI (glutamate-gated calcium ion channel activity, NMDA glutamate receptor activity and calcium-dependent protein kinase C activity) suggest cross-talk between neuroinflammatory signaling pathways and neurotransmission.
TABLE 5.
Molecular Function Analysis of Neuroinflammatory Genes in Irradiated Brain
| Fold enrichment | P value | |
|---|---|---|
| fWBI, dorsolateral prefrontal cortex | ||
| aMHC class II receptor activity | >100.00 | 1.50E–04 |
| Platelet-derived growth factor binding | >100.00 | 1.50E–04 |
| aMHC class II protein complex binding | >100.00 | 2.66E–04 |
| aPeptide antigen binding | >100.00 | 5.29E–05 |
| Integrin binding | 34.91 | 3.63E–02 |
| fWBI, hippocampus | ||
| aMHC class II receptor activity | >100.00 | 1.26E–05 |
| aPeptide antigen binding | >100.00 | 1.03E–06 |
| aMHC class II protein complex binding | >100.00 | 1.11E–02 |
| fWBI, white matter | ||
| bOpsonin receptor activity | 89.16 | 1.20E–03 |
| Tumor necrosis factor binding | 79.25 | 2.78E–02 |
| ICAM-3 receptor activity | 79.25 | 2.75E–02 |
| Glutamate-gated calcium ion channel activity | 71.33 | 1.76E–03 |
| NMDA glutamate receptor activity | 67.93 | 1.42E–04 |
| Calcium-dependent protein kinase C activity | 59.44 | 3.81E–02 |
| bComplement component C3b binding | 59.44 | 3.78E–02 |
| Benzodiazepine receptor activity | 59.44 | 2.39E–03 |
| Lipoteichoic acid binding | 59.44 | 3.75E–02 |
| CCR2 chemokine receptor binding | 59.44 | 3.72E–02 |
| TBI, white matter | ||
| aMHC class II receptor activity | >100.00 | 2.78E–02 |
| Platelet-derived growth factor receptor binding | >100.00 | 4.06E–02 |
| CXCR chemokine receptor binding | >100.00 | 4.13E–02 |
| Chemokine activity | 53.68 | 1.06E–02 |
| Cytokine binding | 34.38 | 5.49E–03 |
| Cytokine receptor activity | 28.28 | 3.99E–02 |
| bSerine-type endopeptidase activity | 16.99 | 7.86E–03 |
| aPeptide binding | 13.13 | 4.87E–02 |
Notes. Analyses completed in Gene Ontology enrichment analysis tool. Maximum fold-enrichment values are truncated at 100. The top 10 enriched ontologies are reported in order of decreasing fold-enrichment and statistical significance, multiplicity adjusted P values ≤ 0.05 were considered significant. Overarching molecular function:
MHC class II peptide antigen presentation
complement activation.
ECM-associated canonical pathways were detected in all regions of animals receiving fWBI and none from the animals receiving TBI. The contributing genes (fWBI DLPFC: 12 DEGs; fWBI HC: 4 DEGs; fWBI WM: 43) were evaluated for patterns in molecular function (Table 6). Enriched ECM-pertinent molecular functions were not detected in the hippocampus of animals receiving fWBI. Transcripts related to the platelet derived growth factor (PDGF) signaling pathway were increased in the dorsolateral prefrontal cortex and white matter. Transcripts corresponding to ECM deposition and remodeling were detected in the fWBI white matter. Enrichment of the PDGF signaling pathway is partially due to decreased PDGF receptor alpha (PDGFRA) mRNA expression (Fig. 8), presumed reflective of oligodendrocyte progenitor depletion. However, the differential transcript expression of collagen subunits in the dorsolateral prefrontal cortex and white matter, and epidermal growth factor (EGF), epidermal growth factor receptor (EGFR), fibroblast growth factor 1 (FGF1), platelet derived growth factor beta (PDGFB) and hepatocyte growth factor (HGF) in white matter suggest that PDGF signaling may also mediate ECM deposition in RIBI.
TABLE 6.
Molecular Function Analysis of Extracellular Matrix and Remodeling-Associated Genes in fWBI Brain
| Fold enrichment | P value | |
|---|---|---|
| fWBI, dorsolateral prefrontal cortex | ||
| aPlatelet-derived growth factor binding | >100.00 | 2.96E–04 |
| fWBI, white matter | ||
| Tumor necrosis factor binding | >100.00 | 5.93E–03 |
| aPlatelet-derived growth factor binding | >100.00 | 6.18E–06 |
| Interleukin-1 receptor activity | >100.00 | 1.80E–02 |
| Tumor necrosis factor-activated receptor activity | 99.63 | 1.50E–06 |
| aPlatelet-derived growth factor receptor binding | 95.65 | 1.16E–03 |
| Phosphatidylinositol-4,5-bisphosphate 3-kinase activity | 43.48 | 2.51E–06 |
| bExtracellular matrix structural constituent | 42.92 | 5.27E–07 |
| bIntegrin binding | 29.62 | 1.85E–06 |
| bProtease binding | 24.98 | 3.89E–06 |
| bCollagen binding | 22.77 | 3.80E–02 |
Notes. Analyses completed in Gene Ontology enrichment analysis tool. Maximum fold-enrichment values are truncated at 100. The top 10 enriched ontologies are reported in order of decreasing fold-enrichment and statistical significance, multiplicity adjusted P values ≤ 0.05 were considered significant. Overarching molecular function:
Platelet derived growth factor (PDGF) signaling
ECM remodeling.
FIG. 8.
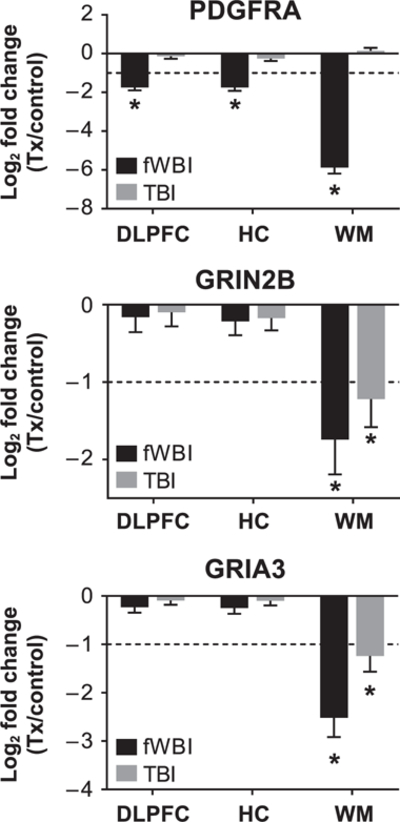
Decreases in glutamatergic neurotransmitter receptor subunits in RIBI occur independently of PDGFRA expression. Supervised analysis of oligodendrocyte precursor marker, platelet derived growth factor alpha (PDGFRA) and glutamatergic neurotransmitter receptor subunits GRIN2B and GRIA3. PDGFRA was decreased in dorsolateral prefrontal cortex, hippocampus and white matter 12 months after fWBI but not 6–9 years after TBI. Glutamatergic NMDA receptor subunit GRIN2B and AMPA receptor subunit GRIA3 expression were decreased in white matter in both fWBI and TBI groups. Bars denote mean log2 fold change and error bars are set to log fold change standard error. *Log2 fold changes < ±1 and multiplicity-adjusted P values (Benjamini-Hochberg) < 0.05.
Alteration of canonical pathways involved in neurotransmission and signal transduction were detected within the white matter of both irradiated groups. Analysis of the contributing genes to neurotransmission and signal transduction-associated canonical pathways (32 fWBI DEGs, 10 TBI DEGs) demonstrated alteration in mechanisms modulating glutamatergic neurotransmission and calcium channel activity (Table 7).
TABLE 7.
Molecular Function Analysis of Neurotransmission and Signal Transduction Associated Genes in White Matter
| Fold enrichment | P value | |
|---|---|---|
| fWBI, white matter | ||
| aNeurotransmitter receptor activity involved in regulation of postsynaptic cytosolic calcium ion concentration | >100.00 | 2.64E–03 |
| bGlutamate-gated calcium ion channel activity | >100.00 | 4.11E–05 |
| bNMDA glutamate receptor activity | >100.00 | 2.13E–06 |
| bAdenylate cyclase inhibiting G-protein coupled glutamate receptor activity | >100.00 | 4.97E–03 |
| bGlutamate binding | >100.00 | 2.05E–04 |
| bExtracellularly glutamate-gated ion channel activity | >100.00 | 7.71E–07 |
| bHigh voltage-gated calcium channel activity | >100.00 | 2.34E–04 |
| Glycine binding | >100.00 | 3.94E–04 |
| bL-glutamate transmembrane transporter activity | 88.41 | 2.30E–02 |
| aCalcium channel regulator activity | 53.05 | 3.19E–03 |
| TBI, white matter | ||
| bIonotropic glutamate receptor activity | >100.00 | 6.99E–03 |
| bExtracellularly glutamate-gated ion channel activity | >100.00 | 6.75E–03 |
| aLigand-gated calcium channel activity | >100.00 | 1.01E–02 |
| Actinin binding | 97.87 | 2.87E–02 |
| Neurotransmitter binding | 80.93 | 4.01E–02 |
Notes. Analyses completed in Gene Ontology enrichment analysis tool. Maximum fold-enrichment values are truncated at 100. The top 10 enriched ontologies are reported in order of decreasing fold-enrichment and statistical significance, multiplicity adjusted P values ≤ 0.05 were considered significant. Overarching molecular function:
Calcium ion regulation
glutamatergic neurotransmission.
Supervised Analyses
Neuroinflammatory pathway analyses indicated that MHC class II peptide antigen presentation and the complement system play a role in RIBI. As antigen presentation may lead to T-cell activation, and the adaptive immune system may regulate complement activation, transcripts indicating T-cell activation and differentiation were also assessed.
Gene expression associated with MHC class II antigen presenting molecules (HLA-DPB1, HLA-DQA1, HLA-DMA) was increased within all brain regions in fWBI and white matter in TBI (Fig. 6A). HLA-DPA1 and HLA-DRB5 were also expressed within all brain regions in fWBI animals, and HLA-DMB within the dorsolateral prefrontal cortex and white matter of fWBI animals.
FIG. 6.
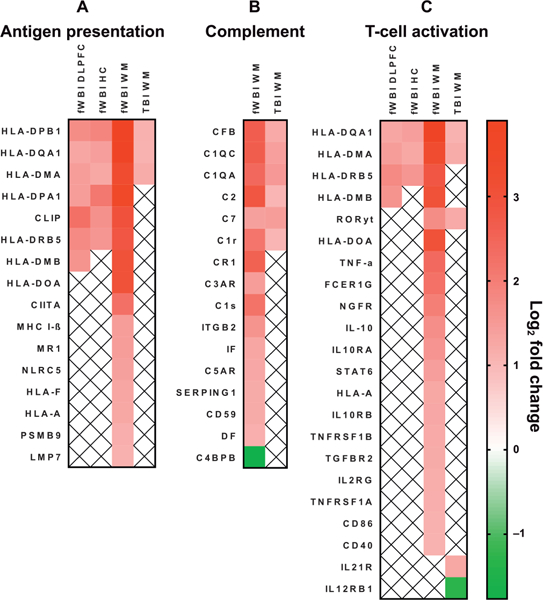
Differentially regulated gene contributors to canonically-enriched pathways of neuroinflammation are shown. Panel A: Antigen presentation. Panel B: The complement system. Panel C: T-cell activation. Gene lists were curated by Ingenuity Pathway Analysis. Gene IDs are HGCN identifiers. Red indicates upregulation and green indicates downregulation. White boxes containing ‘‘x’’ indicate the gene was not differentially regulated within that region. All log2 fold changes < ±1 and multiplicity-adjusted P values (Benjamini-Hochberg) < 0.05.
Complement factor associated gene expression was increased within the white matter of fWBI and TBI animals (Fig. 6B). CFB and C7 transcripts were also expressed within the fWBI dorsolateral prefrontal cortex, and C3AR within the fWBI hippocampus. Complement regulatory transcripts CD59, C4BP, SERPING1, and CR1 were differentially regulated in fWBI, but not in TBI.
Gene expression related to T-cell activation and differentiation was most prominent within the white matter of fWBI animals. CD3D and CD3E gene expression was increased within the white matter of fWBI animals (Fig. 6C). RAR-related orphan gamma receptor (RORyt), a molecule involved in chronic inflammatory signaling through Th17 cells, was transcriptionally upregulated within the white matter of fWBI and TBI animals. STAT6 mRNA, involved in Th2 differentiation, was upregulated in the white matter of animals receiving fWBI. Co-stimulatory molecules essential for the activation of T cells (CD2, CD86) and B cells (CD40) were transcriptionally upregulated within fWBI white matter. These data provide preliminary evidence that MHC class II antigen presentation may induce T-cell activation in RIBI.
Ontologic evaluation suggests that ECM deposition and remodeling in the white matter may contribute to RIBI in fWBI animals. A comprehensive list of ECM contributors was compiled by literature review (48–57). Of several classes of ECM molecules, differential gene regulation was noted in basement membrane contributors, proteoglycans, fibrous proteins, glycoproteins and ECM molecules generally enriched in CNS, bone, cartilage and teeth (Fig. 7). No changes were noted in any of the keratan sulfate proteoglycans, vascular-associated fibrinogen chains, vitronectin or von Willebrand factor, small interstitial proteoglycans (decorin, biglycan, asporin), laminins, agrin, aggrecan or nidogen.
FIG. 7.
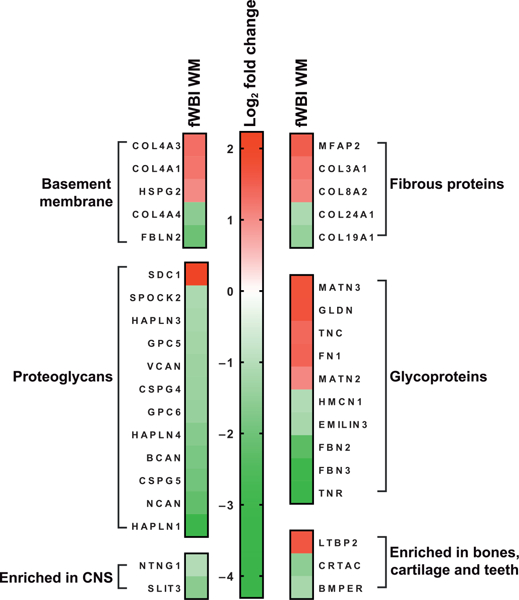
Differentially regulated ECM transcripts in white matter after fWBI. Supervised analysis of differentially expressed extracellular matrix genes in white matter in RIBI after fWBI. Gene IDs are HGCN identifiers. Red indicates upregulation and green indicates downregulation. All log2 fold changes < ±1 and multiplicity adjusted P values (Benjamini-Hochberg) < 0.05.
Since previous analyses suggested that radiation exposure alters neurotransmission and signal transduction pathways in white matter, we completed a supervised review of DEGs in the white matter animals receiving TBI or fWBI for contributing molecular functions. The oligodendrocyte precursor marker platelet derived growth factor alpha (PDGFRA) was decreased in all regions after fWBI but not TBI (Fig. 8). Glutamatergic NMDA receptor subunit GRIN2B and AMPA receptor subunit GRIA3 were decreased in white matter but not dorsolateral prefrontal cortex or hippocampus in both irradiated groups (Fig. 8).
Additional downregulated neurotransmission-associated genes detected in white matter of both irradiated groups were bassoon presynaptic cytomatrix protein (BSN), calcium voltage-gated channel subunit alpha 1 G (CACNA1G), CACNA1I, calcium voltage-gated channel auxillary subunit beta 1 (CACNB1), calcium/calmodulin-dependent protein kinase 2 beta (CAMK2B), potassium voltage-gated channel subfamily B member 1 (KCNB1), potassium calcium-activated channel subfamily M alpha 1 (KCNMA1), potassium sodium-activated channel subfamily T member 1 (KCNT1), synaptotagamin 2 (SYT2), SYT5 and SYT6 (Supplementary Fig. S1; http://dx.doi.org/10.1667/RR15263.1.S1). CACNA1D, KCNK12, synaptic Ras-GTPase activating protein 1 (SYNGAP1), sodium voltage-gated channel beta subunit 2 (SCN2B) were differentially regulated in the white matter of animals receiving TBI. The gene encoding glutamate transporter excitatory amino acid transporter 2 (EAAT2; gene: SLC1A2) was downregulated in the white matter of animals receiving fWBI. Potassium two pore domain channel subfamily K member (KCNK5) was upregulated within the white matter of both irradiated groups.
DISCUSSION
Herein we have characterized the transcriptomic signature of radiation-induced brain injury in the rhesus macaque and demonstrate that fWBI and TBI manifest shared patterns of injury months and years postirradiation, respectively. Our analyses are in agreement with previously published studies indicating involvement of neuroinflammatory processes in RIBI (58–64) and we present novel evidence indicating that the complement system may contribute to injury. Our findings also suggest that ECM deposition and remodeling are affected after fWBI but not TBI. Lastly, regional stratification indicates that radiation-induced impairment of neurotransmission and signal-transduction transcripts is most prominent in white matter. Coupled with the greater proportion of DEGs within white matter compared to hippocampus, these data challenge the hypothesis that radiation-induced brain injury is primarily hippocampal.
Our data reaffirm that neuroinflammatory processes play a role in RIBI and agree with previously published studies in rodents, which demonstrate microglial activation present 4 h to six months after single-fraction dose (58, 59, 63, 65, 66 and fractionated doses (67) brain irradiation. Such activation includes MHC class II antigen presentation (68–72). In the absence of exogenous antigens, we presume that MHC class II molecules must present endogenously derived antigens, e.g., exposed intracellular antigens or proteins altered by radiation (73). We hypothesize that sustained microglial activation after irradiation may perpetuate RIBI via antigen presentation and direction of the neuroimmune response against endogenous targets.
RIBI-induced ECM deposition and remodeling was noted in the white matter of animals receiving fWBI but not in the white matter of those receiving TBI. It is unclear as to whether this is an effect of dose or fractionation. In an effort to understand radiation-induced alterations in the matrix-ome, we have provided an assessment of all differentially regulated ECM contributors to fWBI white matter (Fig. 7). Notably, fWBI was associated with cognitive and neurological impairment while animals receiving TBI were neurologically normal.
Regional stratification of DEGs reveals fewer gene expression changes within the hippocampus compared to white matter within both treatment groups, consistent with our previously reported gene expression analyses (35). Numerous published studies in cell culture and rodent models have demonstrated deleterious, radiation-induced effects on hippocampal structure and function, including: reduced neuronal stem cell number, survival and maturation (58, 65, 74), changes in dendritic spine morphology and density (75–78), alterations in glutamatergic ionotropic receptor subunits (32, 79–81) and neuronal cytoskeletal alterations (78, 82). Thus, hippocampal gene expression data were carefully reviewed for the involvement of neurotransmission, signal transduction and stem cell signaling pathways. Differentially regulated signaling pathways within the hippocampus were consistent with inflammatory processes, and we did not detect alteration in neurogenic or synapse markers. These data suggest that hippocampal-mediated dysfunction may contribute less to RIBI in primates than in rodents, or that hippocampal injury may be dependent on factors not examined here (e.g., neurogenesis in juvenile subjects).
An unexpected white matter-specific reduction in synaptic neurotransmission and signal transduction-associated mRNAs was observed in both groups of irradiated animals (fWBI and TBI). As mentioned previously, studies in rodent models of RIBI have also demonstrated radiation-induced synaptic pathology, including changes in: expression levels of synaptic proteins, neurotransmitter receptors and structure (32, 77, 78, 80, 81, 83–85); however, these analyses were restricted to examination of the hippocampus and/or cortex.
Although synaptic neurotransmission is generally considered restricted to gray matter, myelination may be regulated by action-potential mediated vesicular release of glutamate between unmyelinated axons (presynaptic initiator) and adjacent NG2+ oligodendroglial progenitors (post-synaptic recipient) (86–89). We hypothesize that the reduction in neurotransmission-associated transcripts within white matter are related to radiation-induced loss or dysfunction of these deep white matter synapses. Although radiation (90–93) and ischemia-induced (86–89) PDGFRA+ oligodendrocyte progenitor loss may correspond to reduced numbers of deep white matter synapses, the reduction in glutamatergic neurotransmitter receptors (GRIN2B and GRIA3) in the face of normal levels of PDGFRA in animals receiving TBI suggests that deep white matter synaptic impairment may occur independently of oligodendrocyte progenitor cell loss. Reduction in glutamatergic neurotransmitter receptors and failure of scavenging mechanisms [decreased expression of the encoding gene for glutamate scavenger EAAT2 (SLC1A2) in animals receiving fWBI; Supplementary Fig. S1; http://dx.doi.org/10.1667/RR15263.1.S1] may also contribute to RIBI via excitotoxic injury due to extracellular glutamate accumulation. A similar mechanism has been shown to result in oligodendroglial loss, demyelination and severe axonal damage in optic nerves of rats (94).
Synaptic loss and dysfunction may also explain the downregulation of ion channels in the current study (Supplementary Fig. S1; http://dx.doi.org/10.1667/RR15263.1.S1). Although KCNK channels (KCNK3 and KCNK2) are neuroprotective against excitotoxic injury (95–97), upregulation of KCNK5 exacerbates ischemic brain damage by inducing apoptosis in a rodent model of transient middle cerebral artery occlusion (98). Upregulation of KCNK5 in the white matter of animals receiving fWBI and TBI (Supplementary Fig. S1) may reflect similar physiologic processes.
Complement is a component of the innate immune system responsible for the destruction and clearance of pathogens and damaged cells from the body, and complement-mediated synaptic destruction has been implicated as a mechanism of neurodegeneration and cognitive dysfunction in aging (99) and Alzheimer’s disease (100–102). While complement is known to mediate radiation-induced cell killing (103–105), to our knowledge we are the first to demonstrate that complement activation may play a role in the pathogenesis of RIBI. Additional studies are needed to verify proteolytic activation of the complement system and to identify the cellular and molecular targets of complement fixation in RIBI.
The significance of differential regulation of MEIS3 and SCEL in the dorsolateral prefrontal cortex of animals receiving fWBI or TBI is unclear. MEIS3 encodes a homeobox protein presumed to play a role in transcriptional regulation (106, 107). SCEL is a component of the cornified envelope of keratinocytes (108–112).
The current study indicates that the brain is vulnerable to injury from single-fraction TBI (6.05–8.5 Gy), much lower than the previously estimated threshold for CNS injury (7). This suggests that even patients that do not develop the acute radiation CNS syndrome (113) after exposure may be at risk of developing more subtle long-term neurological injury. Cognitive testing in our laboratory indicates animals receiving similar TBI doses may demonstrate less cognitive flexibility (8). Thus, ARS survivors may benefit from more aggressive surveillance and monitoring of cognitive function.
We acknowledge that differing diet, age and comorbidities between groups may have affected cerebral gene expression. Nevertheless, to our knowledge, this is the first published work reporting on cerebral transcriptomic data from NHP years after single-fraction high-dose TBI. In the event of a large-scale nuclear accident or malicious exposure, anatomic sites of radiation exposure and extent of shielding within the exposed population will be heterogeneous, often with multiple organ systems affected, as has been demonstrated by published studies of the Hiroshima and Chernobyl survivors, and the NHP RSC (4, 114). Therefore, radiation-associated comorbidities are anticipated in survivor populations, and related alterations in cerebral gene expression are relevant to considerations for long-term survivors of radiation exposure.
Despite statistical differences in age, all groups of animals were within similar life stages (young adult, post-pubertal) and thus biologically comparable. Since previously published studies have demonstrated age and development-associated changes in hippocampal gene expression in rodents (115–117) and rhesus macaques (118), the agreement in hippocampal gene expression between animals receiving TBI and control animals (0 DEGs, mean age difference 6.01 years) supports that these animals were in similar life stages.
In summary, to our knowledge, we are the first to report whole transcriptomic profiling from a NHP model of RIBI after fWBI and to assess transcriptomic alterations in NHP survivors of single-fraction high-dose TBI (6.75–8.05 Gy). We present novel evidence of CNS injury years after TBI that shares gene expression patterns with fWBI. These analyses reaffirm the involvement of macrophage/microglial-mediated neuroinflammation in RIBI and indicate novel evidence that complement system activation and impaired synaptic neurotransmission within white matter may contribute to RIBI after fWBI and TBI. Our regional analyses challenge the belief that alterations in hippocampal structure and function are solely responsible for cognitive impairment in RIBI, and provide evidence that alterations in neurotransmission may be most prominent within white matter.
Supplementary Material
Table S1. Unmappable Ensembl ID.
Table S2. Contributory genes to the top 10 IPA pathways, by region.
Fig. S1. Additional differentially regulated neurotransmission and signal transduction-associated transcripts in white matter after brain irradiation. Several neurotransmission and signal transduction transcripts were downregulated in white matter after brain irradiation. Potassium two pore domain channel subfamily K member (KCNK5) was upregulated within both irradiated groups. Differential expressed genes are listed in order of direction and then alphabetically within treatment groups. Bar: Mean log2 fold change. Error bars: log fold change standard error. *Log2 fold changes < ± 1 and multiplicity adjusted P values (Benjamini-Hochberg) < 0.05.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health [NIH grant nos. U19-AI67798 awarded to Nelson Chao, Duke University (JMC, primate core leader, Wake Forest School of Medicine), and R01 CA155293 (M Robbins and SA Deadwyler). Analyses were performed with assistance from the Cancer Genomics Shared Resource supported by the Wake Forest Baptist Comprehensive Cancer Center’s NCI Cancer Center Support Grant P30 CA012197, P30 CA012197–39 (Cancer Center grant) and 1S10OD023409–01 (instrumentation grant). Support in part was provided by NIH Training Grant T32 OD010957 (RMA and JMC).
Footnotes
Editor’s note. The online version of this article (DOI: 10.1667/RR15263.1) contains supplementary information that is available to all authorized users.
REFERENCES
- 1.Sovacool BK. A critical evaluation of nuclear power and renewable electricity in Asia. J Contemp Asia 2010; 40:369–400. [Google Scholar]
- 2.Cui W, Bennett AW, Zhang P, Barrow KR, Kearney SR, Hankey KG, et al. A non-human primate model of radiation-induced cachexia. Sci Rep 2016; 6:23612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unthank JL, Miller SJ, Quickery AK, Ferguson EL, Wang M, Sampson CH, et al. Delayed effects of acute radiation exposure in a murine model of the H-ARS: Multiple-organ injury consequent to <10 gy total body irradiation. Health Phys 2015; 109:511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBo RJ, Lees CJ, Dugan GO, Caudell DL, Michalson KT, Hanbury DB, et al. Late effects of total-body gamma irradiation on cardiac structure and function in male rhesus macaques. Radiat Res 2016; 186:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnett DJ, Parker CL, Blodgett DW, Wierzba RK, Links JM. Understanding radiologic and nuclear terrorism as public health threats: preparedness and response perspectives. J Nucl Med 2006; 47:1653–61. [PubMed] [Google Scholar]
- 6.Prasanna PG, Stone HB, Wong RS, Capala J, Bernhard EJ, Vikram B, et al. Normal tissue protection for improving radiotherapy: Where are the gaps? Transl Cancer Res 2012; 1:35–48. [PMC free article] [PubMed] [Google Scholar]
- 7.Fike JR, Cann CE, Turowski K, Higgins RJ, Chan AS, Phillips TL, et al. Radiation dose response of normal brain. Int J Radiat Oncol Biol Phys 1988; 14:63–70. [DOI] [PubMed] [Google Scholar]
- 8.Hanbury DB, Peiffer AM, Dugan G, Andrews RN, Cline JM. Long-term cognitive functioning in single-dose total-body gamma-irradiated rhesus monkeys (Macaca mulatta). Radiat Res 2016; 186:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caveness WF. Pathology of radiation damage to the normal brain of the monkey. Natl Cancer Inst Monogr 1977; 46:57–76. [PubMed] [Google Scholar]
- 10.Nakagaki H, Brunhart G, Kemper TL, Caveness WF. Monkey brain damage from radiation in the therapeutic range. J Neurosurg 1976; 44:3–11. [DOI] [PubMed] [Google Scholar]
- 11.Wakisaka S, O’Neill RR, Kemper TL, Verrelli DM, Caveness WF. Delayed brain damage in adult monkeys from radiation in the therapeutic range. Radiat Res 1979; 80:277–91. [PubMed] [Google Scholar]
- 12.Hanbury DB, Robbins ME, Bourland JD, Wheeler KT, Peiffer AM, Mitchell EL, et al. Pathology of fractionated whole-brain irradiation in rhesus monkeys (Macaca mulatta). Radiat Res 2015; 183:367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furuse M, Nonoguchi N, Kawabata S, Miyatake S, Kuroiwa T. Delayed brain radiation necrosis: pathological review and new molecular targets for treatment. Med Mol Morphol 2015; 48:183–90. [DOI] [PubMed] [Google Scholar]
- 14.Yoshii Y Pathological review of late cerebral radionecrosis. Brain Tumor Pathol 2008; 25:51–8. [DOI] [PubMed] [Google Scholar]
- 15.Schindler MK, Forbes ME, Robbins ME, Riddle DR. Aging-dependent changes in the radiation response of the adult rat brain. Int J Radiat Oncol Biol Phys 2008; 70:826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan H, Gaber MW, Boyd K, Wilson CM, Kiani MF, Merchant TE. Effects of fractionated radiation on the brain vasculature in a murine model: blood-brain barrier permeability, astrocyte proliferation, and ultrastructural changes. Int J Radiat Oncol Biol Phys 2006; 66:860–6. [DOI] [PubMed] [Google Scholar]
- 17.Price RE, Langford LA, Jackson EF, Stephens LC, Tinkey PT, Ang KK. Radiation-induced morphologic changes in the rhesus monkey (Macaca mulatta) brain. J Med Primatol 2001; 30:81–7. [DOI] [PubMed] [Google Scholar]
- 18.Vigliani MC, Duyckaerts C, Hauw JJ, Poisson M, Magdelenat H, Delattre JY. Dementia following treatment of brain tumors with radiotherapy administered alone or in combination with nitrosourea-based chemotherapy: a clinical and pathological study. J Neurooncol 1999; 41:137–49. [DOI] [PubMed] [Google Scholar]
- 19.Schultheiss TE, Kun LE, Ang KK, Stephens LC. Radiation response of the central nervous system. Int J Radiat Oncol Biol Phys 1995; 31:1093–112. [DOI] [PubMed] [Google Scholar]
- 20.Reinhold HS, Calvo W, Hopewell JW, van der Berg AP. Development of blood vessel-related radiation damage in the fimbria of the central nervous system. Int J Radiat Oncol Biol Phys 1990; 18:37–42. [DOI] [PubMed] [Google Scholar]
- 21.Oi S, Kokunai T, Ijichi A, Matsumoto S, Raimondi AJ. Radiation-induced brain damage in children–histological analysis of sequential tissue changes in 34 autopsy cases. Neurol Med Chir (Tokyo) 1990; 30:36–42. [DOI] [PubMed] [Google Scholar]
- 22.Di Chiro G, Oldfield E, Wright DC, De Michele D, Katz DA, Patronas NJ, et al. Cerebral necrosis after radiotherapy and/or intraarterial chemotherapy for brain tumors: PET and neuropathologic studies. AJR Am J Roentgenol 1988; 150:189–97. [DOI] [PubMed] [Google Scholar]
- 23.Delattre JY, Fuks Z, Krol G, Rottenberg DA, Posner JB. Cerebral necrosis following neutron radiation of an extracranial tumor. J Neurooncol 1988; 6:113–7. [DOI] [PubMed] [Google Scholar]
- 24.Calvo W, Hopewell JW, Reinhold HS, Yeung TK. Time- and dose-related changes in the white matter of the rat brain after single doses of X rays. Br J Radiol 1988; 61:1043–52. [DOI] [PubMed] [Google Scholar]
- 25.Imperato JP, Paleologos NA, Vick NA. Effects of treatment on long-term survivors with malignant astrocytomas. Ann Neurol 1990; 28:818–22. [DOI] [PubMed] [Google Scholar]
- 26.Crossen JR, Garwood D, Glatstein E, Neuwelt EA. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol 1994;12:627–42. [DOI] [PubMed] [Google Scholar]
- 27.Johannesen TB, Lien HH, Hole KH, Lote K. Radiological and clinical assessment of long-term brain tumour survivors after radiotherapy. Radiother Oncol 2003; 69:169–76. [DOI] [PubMed] [Google Scholar]
- 28.DeAngelis LM, Delattre JY, Posner JB. Radiation-induced dementia in patients cured of brain metastases. Neurology 1989; 39:789–96. [DOI] [PubMed] [Google Scholar]
- 29.Douple EB, Mabuchi K, Cullings HM, Preston DL, Kodama K, Shimizu Y, et al. Long-term radiation-related health effects in a unique human population: lessons learned from the atomic bomb survivors of Hiroshima and Nagasaki. Disaster Med Public Health Prep 2011; 5:S122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang K, Sejnowski TJ. A universal scaling law between gray matter and white matter of cerebral cortex. Proc Natl Acad Sci U S A 2000; 97:5621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown WR, Thore CR, Moody DM, Robbins ME, Wheeler KT. Vascular damage after fractionated whole-brain irradiation in rats. Radiat Res 2005; 164:662–8. [DOI] [PubMed] [Google Scholar]
- 32.Shi L, Adams MM, Long A, Carter CC, Bennett C, Sonntag WE, et al. Spatial learning and memory deficits after whole-brain irradiation are associated with changes in NMDA receptor subunits in the hippocampus. Radiat Res 2006; 166:892–9. [DOI] [PubMed] [Google Scholar]
- 33.Shi L, Linville MC, Iversen E, Molina DP, Yester J, Wheeler KT, et al. Maintenance of white matter integrity in a rat model of radiation-induced cognitive impairment. J Neurol Sci 2009; 285:178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robbins ME, Payne V, Tommasi E, Diz DI, Hsu FC, Brown WR, et al. The AT1 receptor antagonist, L-158,809, prevents or ameliorates fractionated whole-brain irradiation-induced cognitive impairment. Int J Radiat Oncol Biol Phys 2009; 73:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews RN, Metheny-Barlow LJ, Peiffer AM, Hanbury DB, Tooze JA, Bourland JD, et al. Cerebrovascular remodeling and neuroinflammation is a late effect of radiation-induced brain injury in non-human primates. Radiat Res 2017; 187:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robbins ME, Bourland JD, Cline JM, Wheeler KT, Deadwyler SA. A model for assessing cognitive impairment after fractionated whole-brain irradiation in nonhuman primates. Radiat Res 2011; 175:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghandhi SA, Turner HC, Shuryak I, Dugan GO, Bourland JD, Olson JD, et al. Whole thorax irradiation of non-human primates induces persistent nuclear damage and gene expression changes in peripheral blood cells. PloS One 2018; 13:e0191402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu JZ, Lindeblad M, Lyubimov A, Neri F, Smith B, Szilagyi E, et al. Subject-based versus population-based care after radiation exposure. Radiat Res 2015; 184:46–55. [DOI] [PubMed] [Google Scholar]
- 39.Brown WR, Blair RM, Moody DM, Thore CR, Ahmed S, Robbins ME, et al. Capillary loss precedes the cognitive impairment induced by fractionated whole-brain irradiation: a potential rat model of vascular dementia. J Neurol Sci 2007; 257:67–71. [DOI] [PubMed] [Google Scholar]
- 40.Euthanasia AP. AVMA guidelines for the euthanasia of animals 2013 ed. Schaumburg, IL: American Veterinary Medical Association; 2013. [Google Scholar]
- 41.Bolon B, Garman RH, Pardo ID, Jensen K, Sills RC, Roulois A, et al. STP position paper: Recommended practices for sampling and processing the nervous system (brain, spinal cord, nerve, and eye) during nonclinical general toxicity studies. Toxicol Pathol 2013; 41:1028–48. [DOI] [PubMed] [Google Scholar]
- 42.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014; 30:923–30. [DOI] [PubMed] [Google Scholar]
- 44.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kramer A, Green J, Pollard J Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014;30(4):523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 2017; 45:D331–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res 2018; 46:2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sasaki T, Fassler R, Hohenester E. Laminin: the crux of basement membrane assembly. J Cell Biol 2004; 164:959–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khoshnoodi J, Cartailler JP, Alvares K, Veis A, Hudson BG. Molecular recognition in the assembly of collagens: terminal noncollagenous domains are key recognition modules in the formation of triple helical protomers. J Biol Chem 2006; 281:38117–21. [DOI] [PubMed] [Google Scholar]
- 51.Rhodes JM, Simons M. The extracellular matrix and blood vessel formation: not just a scaffold. J Cell Mol Med 2007; 11:176–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hynes RO, Naba A. Overview of the matrisome – an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol 2012; 4:a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin CQ, Bissell MJ. Multi-faceted regulation of cell differentiation by extracellular matrix. FASEB J 1993; 7:737–43. [DOI] [PubMed] [Google Scholar]
- 54.Ingber DE. Mechanical control of tissue morphogenesis during embryological development. Int J Dev Biol 2006; 50:255–66. [DOI] [PubMed] [Google Scholar]
- 55.Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J 2006; 20:9–22. [DOI] [PubMed] [Google Scholar]
- 56.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al. Essentials of glycobiology 2nd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 57.Alberts B, Bray D, Johnson A, Lewis J, Raff M, Roberts K, et al. Essential cell biology: An introduction to the molecular biology of the cell New York: Garland Publishing; 1998. [Google Scholar]
- 58.Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Res 2003; 63:4021–7. [PubMed] [Google Scholar]
- 59.Chiang CS, McBride WH, Withers HR. Radiation-induced astrocytic and microglial responses in mouse brain. Radiother Oncol 1993; 29:60–8. [DOI] [PubMed] [Google Scholar]
- 60.Schnegg CI, Kooshki M, Hsu FC, Sui G, Robbins ME. PPARdelta prevents radiation-induced proinflammatory responses in microglia via transrepression of NF-kappaB and inhibition of the PKCalpha/MEK1/2/ERK1/2/AP-1 pathway. Free Radic Biol Med 2012; 52:1734–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang SY, Jung JS, Kim TH, Lim SJ, Oh ES, Kim JY, et al. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol Dis 2006; 21:457–67. [DOI] [PubMed] [Google Scholar]
- 62.Kyrkanides S, Moore AH, Olschowka JA, Daeschner JC, Williams JP, Hansen JT, et al. Cyclooxygenase-2 modulates brain inflammation-related gene expression in central nervous system radiation injury. Brain Res Mol Brain Res 2002; 104:159–69. [DOI] [PubMed] [Google Scholar]
- 63.Zhou K, Bostrom M, Ek CJ, Li T, Xie C, Xu Y, et al. Radiation induces progenitor cell death, microglia activation, and blood-brain barrier damage in the juvenile rat cerebellum. Sci Rep 2017; 7:46181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramanan S, Kooshki M, Zhao W, Hsu F-C, Robbins ME. PPARalpha ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-kappaB and AP-1 pathways. Free Radic Biol Med 2008; 45:1695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med 2002; 8:955–62. [DOI] [PubMed] [Google Scholar]
- 66.Greene-Schloesser DM, Kooshki M, Payne V, D’Agostino RB Jr., Wheeler KT, Metheny-Barlow LJ, et al. Cellular response of the rat brain to single doses of (137)Cs gamma rays does not predict its response to prolonged ‘biologically equivalent’ fractionated doses. Int J Radiat Biol 2014; 90:790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cho HJ, Lee WH, Hwang OMH, Sonntag WE, Lee YW. Role of NADPH oxidase in radiation-induced pro-oxidative and pro-inflammatory pathways in mouse brain. Int J Radiat Biol 2017; 93:1257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmitt AB, Brook GA, Buss A, Nacimiento W, Noth J, Kreutzberg GW. Dynamics of microglial activation in the spinal cord after cerebral infarction are revealed by expression of MHC class II antigen. Neuropathol Appl Neurobiol 1998; 24:167–76. [DOI] [PubMed] [Google Scholar]
- 69.Nikodemova M, Watters JJ, Jackson SJ, Yang SK, Duncan ID. Minocycline down-regulates MHC II expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC)alpha/betaII. J Biol Chem 2007; 282:15208–16. [DOI] [PubMed] [Google Scholar]
- 70.Hayes GM, Woodroofe MN, Cuzner ML. Microglia are the major cell type expressing MHC class II in human white matter. J Neurol Sci 1987; 80:25–37. [DOI] [PubMed] [Google Scholar]
- 71.Perlmutter LS, Scott SA, Barron E, Chui HC. MHC class II-positive microglia in human brain: association with Alzheimer lesions. J Neurosci Res 1992; 33:549–58. [DOI] [PubMed] [Google Scholar]
- 72.Ebner F, Brandt C, Thiele P, Richter D, Schliesser U, Siffrin V, et al. Microglial activation milieu controls regulatory T cell responses. J Immunol 2013; 191:5594–602. [DOI] [PubMed] [Google Scholar]
- 73.Moghaddam AE, Gartlan KH, Kong L, Sattentau QJ. Reactive carbonyls are a major Th2-inducing damage-associated molecular pattern generated by oxidative stress. J Immunol 2011; 187:1626–33. [DOI] [PubMed] [Google Scholar]
- 74.Bellinzona M, Gobbel GT, Shinohara C, Fike JR. Apoptosis is induced in the subependyma of young adult rats by ionizing irradiation. Neurosci Lett 1996; 208:163–6. [DOI] [PubMed] [Google Scholar]
- 75.Ji S Cranial irradiation altered dendritic spine complexity in the rat hippocampus and induced memory decline. Int J Radiat Oncol Biol Phys 2017; 99:E599. [Google Scholar]
- 76.Chakraborti A, Allen A, Allen B, Rosi S, Fike JR. Cranial irradiation alters dendritic spine density and morphology in the hippocampus. PloS One 2012; 7:e40844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parihar VK, Limoli CL. Cranial irradiation compromises neuronal architecture in the hippocampus. Proc Natl Acad Sci U S A 2013; 110:12822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shirai K, Mizui T, Suzuki Y, Okamoto M, Hanamura K, Yoshida Y, et al. X irradiation changes dendritic spine morphology and density through reduction of cytoskeletal proteins in mature neurons. Radiat Res 2013; 179:630–6. [DOI] [PubMed] [Google Scholar]
- 79.Machida M, Lonart G, Britten RA. Low (60 cGy) doses of (56)Fe HZE-particle radiation lead to a persistent reduction in the glutamatergic readily releasable pool in rat hippocampal synaptosomes. Radiat Res 2010; 174:618–23. [DOI] [PubMed] [Google Scholar]
- 80.Wu PH, Coultrap S, Pinnix C, Davies KD, Tailor R, Ang KK, et al. Radiation induces acute alterations in neuronal function. PLoS One 2012; 7:e37677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moore ED, Kooshki M, Wheeler KT, Metheny-Barlow LJ, Robbins ME. Differential expression of Homer1a in the hippocampus and cortex likely plays a role in radiation-induced brain injury. Radiat Res 2014; 181:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Puspitasari A, Koganezawa N, Ishizuka Y, Kojima N, Tanaka N, Nakano T, et al. X irradiation induces acute cognitive decline via transient synaptic dysfunction. Radiat Res 2016; 185:423–30. [DOI] [PubMed] [Google Scholar]
- 83.D’Amelio FE, Kraft LM, D’Amelio ED, Zeman W, Doody L, Pfeffer J, et al. Synaptic plasticity in the cerebral cortex of mice: effects of radiation and anesthesia. J Hirnforsch 1983; 24:479–83. [PubMed] [Google Scholar]
- 84.Kempf SJ, Buratovic S, von Toerne C, Moertl S, Stenerlöw B, Hauck SM, et al. Ionising radiation immediately impairs synaptic plasticity-associated cytoskeletal signalling pathways in HT22 cells and in mouse brain: An in vitro/in vivo comparison study. PLoS One 2014; 9:e110464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miao S, Koganezawa N, Hanamura K, Puspitasari A, Shirao T. N-methyl-D-aspartate receptor mediates x-irradiation-induced drebrin decrease in hippocampus. Kita Kanto Igaku 2018; 68:111–5. [Google Scholar]
- 86.Alix JJ, Domingues AM. White matter synapses: form, function, and dysfunction. Neurology 2011; 76:397–404. [DOI] [PubMed] [Google Scholar]
- 87.Gallo V Surprising synapses deep in the brain. Nat Neurosci 2007; 10:267–9. [DOI] [PubMed] [Google Scholar]
- 88.Kukley M, Capetillo-Zarate E, Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci 2007; 10:311–20. [DOI] [PubMed] [Google Scholar]
- 89.Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci 2007; 10:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Panagiotakos G, Alshamy G, Chan B, Abrams R, Greenberg E, Saxena A, et al. Long-term impact of radiation on the stem cell and oligodendrocyte precursors in the brain. PLoS One 2007; 2:e588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chari DM, Gilson JM, Franklin RJM, Blakemore WF. Oligodendrocyte progenitor cell (OPC) transplantation is unlikely to offer a means of preventing X-irradiation induced damage in the CNS. Exp Neurol 2006; 198:145–53. [DOI] [PubMed] [Google Scholar]
- 92.Irvine KA, Blakemore WF. A different regional response by mouse oligodendrocyte progenitor cells (OPCs) to high-dose X-irradiation has consequences for repopulating OPC-depleted normal tissue. Eur J Neurosci 2007; 25:417–24. [DOI] [PubMed] [Google Scholar]
- 93.Begolly S, Shrager PG, Olschowka JA, Williams JP, O’Banion MK. Fractionation spares mice from radiation-induced reductions in weight gain but does not prevent late oligodendrocyte lineage side effects. Int J Radiat Oncol Biol Phys 2016; 96:449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matute C, Alberdi E, Domercq M, Sanchez-Gomez M-V, Perez-Samartíın A, Rodríguez-Antiguedad A, et al. Excitotoxic damage to white matter. J Anat 2007; 210:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meuth SG, Kleinschnitz C, Broicher T, Austinat M, Braeuninger S, Bittner S, et al. The neuroprotective impact of the leak potassium channel TASK1 on stroke development in mice. Neurobiol Dis 2009; 33:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Muhammad S, Aller MI, Maser-Gluth C, Schwaninger M, Wisden W. Expression of the kcnk3 potassium channel gene lessens the injury from cerebral ischemia, most likely by a general influence on blood pressure. Neuroscience 2010; 167:758–64. [DOI] [PubMed] [Google Scholar]
- 97.Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Polyunsaturated fatty acids induce ischemic and epileptic tolerance. Neuroscience 2002; 109:231–41. [DOI] [PubMed] [Google Scholar]
- 98.Gob E, Bittner S, Bobak N, Kraft P, Gobel K, Langhauser F, et al. The two-pore domain potassium channel KCNK5 deteriorates outcome in ischemic neurodegeneration. Pflugers Arch 2015; 467:973–87. [DOI] [PubMed] [Google Scholar]
- 99.Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, et al. Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci 2015; 35:13029–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016; 352:712–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci 2004; 24:6457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hakobyan S, Harding K, Aiyaz M, Hye A, Dobson R, Baird A, et al. Complement biomarkers as predictors of disease progression in Alzheimer’s disease. J Alzheimers Dis 2016; 54:707–16. [DOI] [PubMed] [Google Scholar]
- 103.Regal JF, Dornfeld KJ, Fleming SD. Radiotherapy: killing with complement. Ann Transl Med 2016; 4:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Surace L, Lysenko V, Fontana AO, Cecconi V, Janssen H, Bicvic A, et al. Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response. Immunity 2015; 42:767–77. [DOI] [PubMed] [Google Scholar]
- 105.Elvington M, Scheiber M, Yang X, Lyons K, Jacqmin D, Wadsworth C, et al. Complement-dependent modulation of antitumor immunity following radiation therapy. Cell Rep 2014; 8:818–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dibner C, Elias S, Ofir R, Souopgui J, Kolm PJ, Sive H, et al. The Meis3 protein and retinoid signaling interact to pattern the Xenopus hindbrain. Dev Biol 2004; 271:75–86. [DOI] [PubMed] [Google Scholar]
- 107.Elkouby YM, Polevoy H, Gutkovich YE, Michaelov A, Frank D. A hindbrain-repressive Wnt3a/Meis3/Tsh1 circuit promotes neuronal differentiation and coordinates tissue maturation. Development 2012; 139:1487–97. [DOI] [PubMed] [Google Scholar]
- 108.Champliaud MF, Burgeson RE, Jin W, Baden HP, Olson PF. cDNA cloning and characterization of sciellin, a LIM domain protein of the keratinocyte cornified envelope. J Biol Chem 1998; 273:31547–54. [DOI] [PubMed] [Google Scholar]
- 109.Alibardi L, Toni M. Skin structure and cornification proteins in the soft-shelled turtle Trionyx spiniferus. Zoology (Jena) 2006; 109:182–95. [DOI] [PubMed] [Google Scholar]
- 110.Alibardi L, Toni M. Localization and characterization of specific cornification proteins in avian epidermis. Cells Tissues Organs 2004; 178:204–15. [DOI] [PubMed] [Google Scholar]
- 111.Alibardi L, Toni M. Distribution and characterization of proteins associated with cornification in the epidermis of gecko lizard. Tissue Cell 2005; 37:423–33. [DOI] [PubMed] [Google Scholar]
- 112.Alibardi L, Toni M. Immunolocalization and characterization of cornification proteins in snake epidermis. Anat Rec A Discov Mol Cell Evol Biol 2005; 282:138–46. [DOI] [PubMed] [Google Scholar]
- 113.A brochure for physicians. Acute radiation syndrome Atlanta: Centers for Disease Control and Prevention; 2017. (https://bit.ly/2QsotMM) [Google Scholar]
- 114.Kavanagh K, Dendinger MD, Davis AT, Register TC, DeBo R, Dugan G, et al. Type 2 diabetes is a delayed late effect of whole-body irradiation in nonhuman primates. Radiat Res 2015; 183:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Burger C, Lopez MC, Baker HV, Mandel RJ, Muzyczka N. Genome-wide analysis of aging and learning-related genes in the hippocampal dentate gyrus. Neurobiol Learn Mem 2008; 89:379–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kadish I, Thibault O, Blalock EM, Chen KC, Gant JC, Porter NM, et al. Hippocampal and cognitive aging across the lifespan: a bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci 2009; 29:1805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, et al. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci 2003; 23:3807–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Blalock EM, Grondin R, Chen KC, Thibault O, Thibault V, Pandya JD, et al. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J Neurosci 2010; 30:6058–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Unmappable Ensembl ID.
Table S2. Contributory genes to the top 10 IPA pathways, by region.
Fig. S1. Additional differentially regulated neurotransmission and signal transduction-associated transcripts in white matter after brain irradiation. Several neurotransmission and signal transduction transcripts were downregulated in white matter after brain irradiation. Potassium two pore domain channel subfamily K member (KCNK5) was upregulated within both irradiated groups. Differential expressed genes are listed in order of direction and then alphabetically within treatment groups. Bar: Mean log2 fold change. Error bars: log fold change standard error. *Log2 fold changes < ± 1 and multiplicity adjusted P values (Benjamini-Hochberg) < 0.05.