

Summary
This protocol describes a three-dimensional culture method for generating inner ear sensory epithelia, which comprises sensory hair cells and a concurrently arising neuronal population. Mouse embryonic stem cells are initially plated in 96-well plates with differentiation media; following aggregation, Matrigel is added in order to promote epithelialization. A series of small molecule applications is then used over the first 14 days of culture to guide differentiation towards an otic lineage. After 16–20 days, vesicles containing inner ear sensory hair cells and supporting cells arise from the cultured aggregates. Aggregates may be analyzed using immunohistochemistry and electrophysiology techniques. This system serves as a simple and relatively inexpensive in vitro model of inner ear development.
Keywords: Cell differentiation, inner ear, hair cell, vestibular, neurogenesis, cell culture techniques, three-dimensional cell culture, stem cell
1. Introduction
During development, formation of the cranial sensory ganglia occurs as a result of a series of signaling cues that guide definitive ectoderm to the formation of non-neural ectoderm and a subsequent pre-placodal epithelium. This pre-placodal region has the capacity to give rise to six neurogenic placodes, from which many structures of the peripheral nervous system are derived. The placode of particular interest here is the otic placode, from which the entire inner ear develops. The otic placode is derived from a region common with the epibranchial placode known as the otic-epibranchial placode domain (OEPD). After induction, the otic placode invaginates to form the otic pit. This pit eventually pinches off from the overlying ectoderm to form the otic vesicle, or otocyst.
The protocol described here aims to illustrate these aspects of inner ear development in vitro, from the induction of non-neural and pre-placodal ectoderm to the formation of the otic placode and subsequent derivation of otic vesicles containing inner ear sensory epithelia (Figure 1). Mouse pluripotent stem cells are employed in a three-dimensional culture system to recapitulate this differentiation process.
Figure 1: Schematic of inner ear development, highlighting key signaling mechanisms.
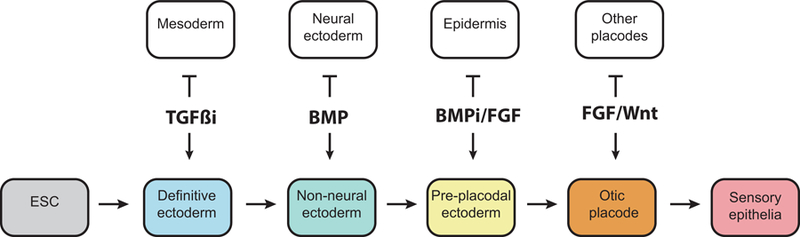
This protocol takes advantage of key signaling mechanisms in order to guide definitive ectoderm to an inner ear-like sensory fate. A TGF-β inhibitor blocks formation of mesoderm/endoderm and promotes induction of definitive ectoderm. BMP signaling is used to induce non-neural ectoderm. BMP inhibition and FGF activation are required for pre-placodal ectoderm induction. Subsequent FGF signaling and endogenous Wnt signaling guides further development of otic placodes, otic vesicles and inner ear sensory epithelia.
Three-dimensional cell culture systems have been previously used to effectively generate complex epithelia, including the generation of the optic cup, mouse and human cortical tissue, and intestinal tissue (1–7). In traditional monolayer (2D) culture, cell growth is restricted to the culture plate, which ultimately affects the shape and development of the cell population. Three-dimensional or “floating” cell culture systems offer the advantage of freedom of growth; this promotes cellular interactions that allow for self-organization that more closely recapitulates an in vivo environment. This is particularly useful in pluripotent stem cell culture, wherein an aggregate of cells may be manipulated with signaling molecules in order to guide differentiation.
In this protocol, mouse embryonic stem cells are initially aggregated in U-bottom 96-well plates (Figure 2). Matrigel, which contains extracellular matrix proteins, is applied on day one of the culture protocol in order to stimulate the development of a basement membrane around the surface of each aggregate. Shortly after Matrigel addition, the surface layer of cells organizes into an epithelium reminiscent of the definitive ectoderm in the embryo.
Figure 2: Schematic overview of the inner ear organoid protocol.
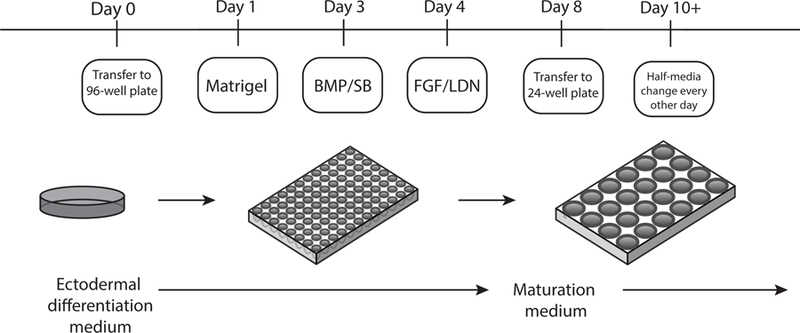
Small molecule application and culture conditions are highlighted here. Cells are initially plated in a 96-well plate in Ectodermal differentiation medium. Matrigel and small molecule application proceeds over days 1–4. Transfer to 24-well plate in Maturation medium occurs on day 8, with subsequent half-media changes every other day starting on day 10.
Small molecule treatments are then applied on day 3 and day 4.5 in order to activate or inhibit several key signaling pathways. Day 3 treatment modulates BMP and TGF-β signaling. BMP signaling has been shown to play an important role in the specification of ectoderm (8–13). Here, BMP signaling is activated in order to promote induction of non-neural ectoderm. Since TGF-β signaling has been shown to promote the induction of mesoderm and endoderm (14–16), SB-431542, a TGF-β inhibitor, is applied to block mesoderm and endoderm formation.
It is important to note that BMP signaling, while essential for non-neural ectoderm induction, needs to be attenuated in order to guide induction of pre-placodal epithelia (12, 17–20). In this system, LDN-193189 serves as a BMP inhibitor. FGF signaling has been shown to be critical for pre-placodal induction (12, 18–20). The co-application of FGF and LDN-193189 on day 4.5 promotes induction of pre-placodal epithelia. FGF signaling further plays a role in specification of the otic-epibranchial placode domain (21–22).
On day 8, cultured aggregates are transferred from 96-well to 24-well plates. From here, endogenous Wnt signaling guides induction of the otic placode, and subsequent formation of the otic vesicle (23–25). Morphological cues can be used to track the progression of the aggregates (Figure 3). From day 6–8, aggregates typically display a thickening of the outer epithelium, indicative of OEPD formation. From day 8–11, the surface of each aggregate appears to “smooth,” as the cells in the interior of the aggregate migrate out to the surface where they surround the outer epithelium. The first sign of vesicle formation should be apparent on day 12. Typically, vesicles are visible along the outer edges of the aggregates, or towards the interior. These vesicles may stay embedded in the aggregate, or may breach the surface epithelium and protrude from the aggregate (days 14–30). It is within these otic vesicles that the sensory hair cells develop. We designate these hair cell-containing vesicles as inner ear organoids (26).
Figure 3: Cell aggregate morphology during inner ear organoid culture.
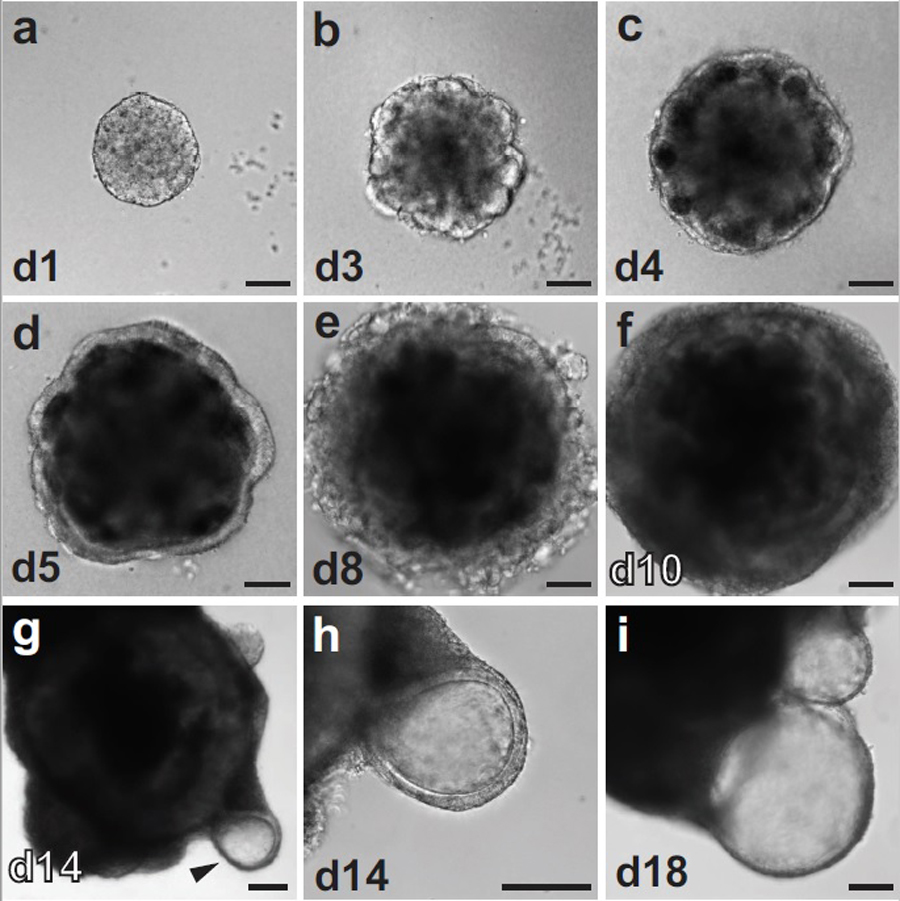
DIC images captured from R1 ESC otic differentiation cultures highlighting the morphological progression. a-f, Early morphology from day 1 to day 10. g-h, Later morphology; day 14 aggregate, arrow head indicating vesicle formation. i, Day 18 vesicle; site of inner ear sensory epithelia formation. Scale bars, 100 μm.
Interestingly, we have observed the development of a neuronal population in the culture coinciding with inner ear organoid development (26). Our previous evidence suggests that these neurons form synapse-like structures with sensory hair cells in the derived inner ear epithelia. Further analysis, however, is necessary to confirm the identity of these sensory-like neurons and whether the synapses are functional.
Aggregates may be fixed and analyzed at any point during the culture period. It may be helpful to fix and analyze aggregates at early time-points to ensure that differentiation is taking place along the desired lineage. We suggest performing immunohistochemical analysis using antibodies for markers of the OEPD and inner ear sensory epithelia to characterize the derived tissue (Figure 4).
Figure 4: Immunohistochemistry of OEPD-like epithelium on day 6/8 and inner ear organoids on day 20.
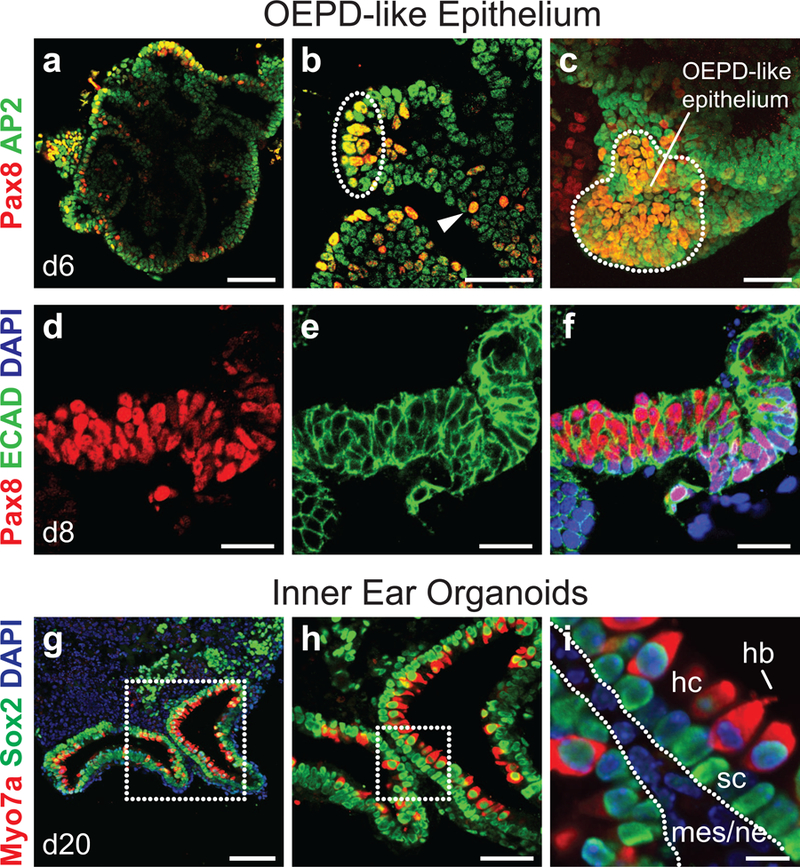
a-c, Wholemount staining for Pax8 and AP2 on day 6 reveals patches of Pax8+ AP2+ cells (dotted outline) reminiscent of placodes. Individual Pax8+ AP2+ (arrowhead) cells were observed throughout the epithelium. Panel c contains a z-projection through a representative Pax8+ AP2+ patch. d-f, Representative Pax8+ E-cadherin+ epithelium on day 8. g-i, Protruding inner ear organoids with Sox2+ supporting cells (sc) and Sox2+ Myo7a+ hair cells (hc) on day 20. A hair bundle (hb) extends from the apical end of the hair cell in panel i. Mesenchymal and neural cells surround the inner ear organoids (mes/ne). Scale bars, 100 µm (a, g), 50 µm (b, c, h), 25 µm (d-f), 10 µm (i).
In summary, this protocol presents an in vitro model for the derivation of inner ear sensory epithelia. Protein and small molecule treatments are used to guide mouse embryonic stem cell differentiation towards an otic lineage. The resulting tissue contains vestibular hair cells as well as a neuronal population that has been shown to innervate the derived epithelia (26).
2. Materials
2.1. Reagents
Mouse ES cells: Prior to use, cells should be acclimated to growth in LIF-2i medium and 80% confluent. Any well-established mouse pluripotent cell line is suitable for use with this protocol. Although we primarily use a feeder-free ES cell line derived from R1 mice, this protocol has been performed with a mouse iPS cell line and several transgenic ES cell lines, yielding comparable results.
GMEM (Gibco, cat. no. 11710–035)
Advanced DMEM/F12 (Gibco, cat. no. 12634–010)
Sodium Pyruvate, 100 mM (Stemcell Technologies, cat. no. 07000)
Non Essential Amino Acids, 10 mM (Stemcell Technologies, cat. no. 07600)
Penicillin-Streptomycin (Stemcell Technologies, cat. no. 07500)
Knockout Serum Replacement (KSR; Gibco, cat. no. 10828–010). NOTE: KSR is light sensitive. Store in the dark at −20°C per the manufacturer’s recommendation)
2-Mercaptoethanol (Gibco, cat. no. 21985–023)
Normocin (Invivogen, cat. no. ant-nr-1)
N2 Supplement (Gibco, cat. no. 17502–048)
GlutaMax (Gibco, cat. no. 35050–079)
Matrigel (BD Biosciences cat. no. 354230)
Recombinant BMP4 (Stemgent, cat. no. 03–0007)
FGF-2 (Peprotech, cat. no. 100–18B)
SB-431542 in solution (Stemgent, cat. no. 04–0010-05)
LDN-193189 in solution (Stemgent, cat. no. 04–0074-02). Note that LDN-193189 is light sensitive, and should be stored in the dark at −20°C per the manufacturer’s recommendation.
Neurobasal Medium (Gibco, cat. no. 21103–049)
B27 Supplement minus Vitamin A (Gibco, cat. no. 12587–010)
PD-0325901 in solution (Stemgent, cat. no. 04–0006-02)
CHIR99021 in solution (Stemgent, cat. no. 04–0004-02)
0.1% Gelatin (Stemcell Technologies, cat. no. 07903)
0.25% Trypsin-EDTA (Gibco, cat. no. 25200–072)
Phosphate Buffered Saline (PBS; Gibco, cat. no. 10010–023)
ESGRO® Leukemia Inhibitory Factor (LIF; Millipore, cat. no. ESG1106)
Dimethyl Sulfoxide (DMSO; Sigma, cat. no. D8418)
Paraformaldehyde (PFA, Electron Microscopy Sciences, cat. no. 15710)
Vacuum grease (Fisher Scientific, cat. no. S41718)
Normal goat serum (Vector laboratories, cat. no. S-1000)
Urea (Sigma, cat. no. U5378)
Glycerol (Sigma, cat. no. G5516)
Triton X-100 (Sigma, cat. no. T8787)
Antibodies
2.2. Equipment
Cell culture dish (60 mm; BD Falcon, cat. no. 353002)
Bacterial dish (100 mm; Fisher Scientific, cat. no. 0875712)
U-bottom 96-well plate (Lipidure-coat; Gel Company, cat. no. LCU96)
24-well plate (Lipidure-coat; Gel Company, cat. no. LCMD24)
Safe-Lock centrifuge tubes (1.5 and 2 mL; Eppendorf, cat. no. 022363204, 022363352)
Transfer pipets (Fisherbrand, cat. no. 13–711-7M)
Polystyrene round bottom centrifuge test tubes with cell strainer tops (5 mL; BD Falcon, cat. no. 352235)
Reagent Reservoirs (VistaLab, cat. no. 21–381-27F)
Tissue-Tek Cryomolds (Electron Microscopy Sciences, cat. no. 62534–10)
Olympus FV1000 multiphoton microscope or equivalent inverted confocal microscope equipped with a long-working distance objective (20–25X)
Cover glasses (24 × 50–1 and 25 × 25 or equivalent; Fisher Scientific, cat. no. 12–544-14 and 12–548-C)
15 mm diameter round cover glasses (thickness no. 0; Assistant, cat. no. 01105509)
Biosafety cabinet
CO2 incubator
Hotplate
Water bath
Desiccator
Cryostat
2.3. Preparation of small molecules
Human recombinant BMP4 stock solution (100 ng/µL): 10 µg of BMP in 100 µL of sterile 4 mM HCl. Vortex and spin down the solution in a tabletop centrifuge. Aliquot BMP4 solution at 5 uL per tube. Aliquots can be stored at −20°C for 6 months or at −80°C for 1 year.
Human recombinant FGF-2 stock solution (200 ng/µL): 50 µg of FGF-2 in 250 µL of sterile PBS or 5 mM Tris (pH 7.6). Vortex and spin down the solution in a tabletop centrifuge. Aliquot at 6 µL per tube. Aliquots can be store at −20°C for 6 months or at −80°C for 1 year.
2.4. Preparation of media
LIF-2i ES cell maintenance medium: For 100 mL basal LIF-2i medium, combine 48 mL Advanced DMEM/F12, 48 mL Neurobasal medium, 2 mL B27 supplement, 1 mL N2 supplement, and 1 mL GlutaMax in a sterile 250 mL bottle. Basal LIF-2i medium can be used for 3–4 weeks. Immediately before use in culture, prepare complete LIF-2i medium by adding 100 µL Penicillin-Streptomycin, 10 µL LIF, 1 µL PD-0325901, and 3 µL CHIR99021 to 10 mL of basal LIF-2i medium. Complete LIF-2i medium can be used for up to 1 week. It is important to note that B27 should not contain Vitamin A when used in LIF-2i preparation for ES cell culture. (See Note 1)
Ectodermal differentiation medium: For 100 mL ectodermal differentiation medium, combine 95.5 mL GMEM, 1.5 mL KSR, 1 mL sodium pyruvate, 1 mL non-essential amino acids, 1 mL Penicillin-Streptomycin, and 180 µL 2-Mercaptoethanol in a sterile 250 mL bottle. Complete ectodermal differentiation medium can be used for the duration of one experiment or up to 1 week. Adjust the volume of the medium as needed for the number of 96-well plates being prepared.
Maturation medium: For 100 mL maturation medium, combine 97.9 mL Advanced DMEM/F12, 1 mL N2 supplement, 1 mL GlutaMax, 100 µL Normocin in a sterile 250 mL bottle. Complete maturation medium can be used for up to 2 weeks. (See Note 2)
ScaleA2 Solution: For 1 L of ScaleA2 solution, dissolve 240.24g (4M final concentration) of urea powder in 750 mL of Milli-Q water. Add 1 mL of Triton X-100 (0.1% vol/vol final concentration) and 100 g of glycerol (10% wt/vol final concentration). Add Milli-Q water to a final of volume of 1 L. Solution can be stored at room temperature for at least one year.
3. Methods
3.1. ESC dissociation and plating
Warm 0.25% trypsin and LIF-2i medium to 37°C in water bath.
In a Biosafety cabinet, add gelatin to completely cover bottom of 60mm plate for new passage (See Note 3). Let the plate sit approximately 20 minutes; in the meantime, proceed to step 3.
Aspirate media from ESC plate (cells at ~80% confluency) and wash one time with 5 mL PBS.
Add 500 µL trypsin; incubate at RT or in 37°C incubator for 1–2 minutes. Agitate the plate to break up cell clumps and confirm via microscope that cells have rounded and detached from the surface of the plate.
Add 1 mL LIF-2i medium to plate and transfer to 2 mL microcentrifuge tube.
Pipet in microcentrifuge tube to dissociate cell clumps into single-cells, taking care not to introduce air bubbles into the medium.
Centrifuge at RT for 2.5 minutes at 1400 rpm.
While cells are in centrifuge, aspirate gelatin from the new 60mm plate; let plate sit at least 5 minutes to dry.
Remove supernatant from cell suspension by aspirating or pouring; avoid disturbing the pellet.
Re-suspend in 1 mL LIF-2i medium.
Split the cells 1:10 for re-plating in approximately 2 days, or 1: 50 for re-plating in approximately 4 days. (See Note 4)
3.2. ES cell differentiation
Warm Ectodermal differentiation medium in 37°C water bath.
Aspirate LIF-2i media from plate of ~80% confluent ESCs; wash three times with 5 mL PBS (See Note 5).
Add 500 µL of 0.25% Trypsin-EDTA. Incubate at RT or at 37°C for 1–2 minutes. Agitate plate to break up cell clumps and confirm via microscopy that cells have rounded and detached from the surface of the plate.
Add 1 mL warmed Ectodermal differentiation medium to plate; transfer cells to a 2 mL microcentrifuge tube.
Dissociate cell clumps into single cells by pipetting up and down with a P1000 tip. Centrifuge for 2.5 minutes at 1400 rpm to pellet the cells.
Remove supernatant by pipetting or aspirating; avoid disturbing the pellet. Re-suspend in 1 mL Ectodermal differentiation medium.
To a cell-strainer-top test tube, forcefully pipet 1 mL fresh Ectodermal differentiation medium. This will prime the strainer. Then pipet 1 mL ES cell suspension into the cell strainer, followed by 1 mL fresh Ectodermal differentiation medium, for a total of 3 mL in the tube. (See Note 6)
With a P1000 tip, pipet the cell suspension to mix. Use a hemacytometer to determine cell concentration.
For two 96-well plates, dilute the appropriate volume of cell suspension in 22 mL fresh ectodermal differentiation medium. The desired final concentration is 30,000 cells per mL. Invert to mix. (See Note 7)
Transfer the cell suspension to a reservoir. Using a multichannel pipette, dispense 100 uL cell suspension per well into two 96-well plates.
Centrifuge the plates at RT for 5 minutes at 800 rpm.
Store the plates in a 37°C incubator with 5.0% CO2.
3.3. Day 1: Addition of Matrigel
Add 440 µL Matrigel to 10.56 mL Ectodermal differentiation medium for a total of 11 mL complete medium. It is important to keep the Matrigel on ice or at 4°C immediately prior to mixing, and to add Matrigel to ice-cold medium, as Matrigel will become gelatinous at temperatures above 15°C. After mixing, hold the medium up to a light source to confirm that the Matrigel has been fully incorporated.
Warm Ectodermal differentiation medium with Matrigel in 37°C water bath.
Remove 50 µL of medium from each well using a multichannel pipette, making sure to hold the pipette at an angle so as not to disturb the cell aggregate at the bottom of each well.
To each well, add 50 µL of Ectodermal differentiation medium plus Matrigel and pipette 4–6 times to mix. (See Note 8)
3.4. Day 3: Addition of BMP4 and SB-431542
For each experimental condition, prepare an appropriate amount of Ectodermal differentiation medium. For two 96-well plates, 6 mL of medium is typically adequate.
For 6 mL medium, add 3 µL BMP4 and 3 µL SB-431542 (0.5 µL per 1 mL). This results in a 5X concentration of BMP4 and SB.
To each well, add 25 µL of Ectodermal differentiation medium plus BMP/SB. Note there is a final volume of 125 uL and a final concentration of 10 ng/mL BMP4 and 1 uM SB-431542 per well. (See Note 9)
3.5. Day 4.5: Addition of FGF and LDN-193189
For each experimental conditional, prepare an appropriate amount of Ectodermal differentiation medium. For two 96-well plates, 6 mL of medium is typically adequate.
For 6 mL, add 4.5 µL of FGF-2 (0.75 µL per 1 mL) and 3.6 µL of LDN-193189 (0.6 µL per 1 mL). This results in FGF-2 and LDN-193189 at a 6X concentration.
To each well, add 25 µL of Ectodermal differentiation medium plus FGF/LDN. Note there is a final volume of 150 uL and a final concentration of 25 ng/mL FGF-2 and 1 µM LDN-193189 per well. (See Note 10)
3.6. Day 8: Transfer to long-term culture
Cut the end of a P1000 pipette tip and transfer the aggregates from each well into a 15 mL conical tube (See Note 11). Separate different experimental conditions into different 15 mL conical tubes if necessary.
Let the aggregates settle to the bottom of the tube; then aspirate the media, taking care not to disturb the aggregates.
Wash the aggregates in 5 mL DMEM/F12 (pre-warmed to 37° C).
Repeat steps 2 and 3 at least 3 times.
Remove any excess DMEM/F12 and then re-suspend the aggregates in Maturation medium containing 1% Matrigel.
Using a wide-mouth P1000, transfer the aggregates to a 100 mm bacterial dish. Suspend in a sufficient amount of Maturation medium to account for medium spreading out across the surface of the place.
Pipet 1–3 aggregates in 1 mL medium into the wells of a 24-well plate. Alternatively, single aggregates may be transferred to a new 96-well plate (suspend in 150–200 µL Maturation medium containing Matrigel). By this time point, aggregates in the bacterial plate should be visible without use of a microscope.
Beginning on day 10 and continuing every other day (day 12, 14, 16, etc.), replace half of the medium in each well with Maturation medium that does not contain Matrigel. For aggregates plated in 96-well plates, replace half of the medium every day beginning on day 9. Aggregates can be incubated for up to 22 days in 24-well plates, or 12 days in 96-well plates. This results in a total optimal culture period of 30 days (in 24-well plates) or 20 days (in 96-well plates). Aggregates can be fixed at any point for imaging as a cryosection or as a wholemount. After the prescribed durations, further culture will likely require changes in culture medium or format.
3.7. Preparation of aggregates for immunohistochemistry
Using a P1000 tip (for collection days 1–6) or a wide-mouth P1000 tip (for collection after day 6), pipet aggregates into a 2 mL tube.
Aspirate the medium and add 4% PFA (vol/vol) in PBS to the tube (See Note 12)
Incubate the aggregates in 4% PFA to fix the tissue. For aggregates collected day 1–8 samples, incubate for 20 minutes at RT. For day 9–30 aggregates, incubate overnight at 4°C.
Remove PFA and wash aggregates at least three times with PBS. Fixed aggregates can be stored in PBS at 4° C for several months. (See Note 13)
Incubate the aggregates in a series of sucrose-PBS solutions of increasing sucrose concentration (i.e. 10%, 20%, 30% (wt/vol) sucrose). Incubate in each concentration for 30 minutes while rocking. Sucrose treated aggregates can be stored at 4° C for up to one week.
Using a wide-mouth pipette, transfer the aggregates into a cryomold and carefully remove the sucrose solution using a P20 pipette. Gently position the aggregates in the center of the cryomold.
Slowly add tissue-freezing medium along the outer walls of the cryomold, taking care to make sure the aggregates do not float away from the bottom of the cryomold.
Place the cryomolds in a vacuum desiccator for 30 minutes. This will remove bubbles from the tissue-freezing medium.
Embedded aggregates should be placed on dry ice for 30 minutes, or until the tissue-freezing medium becomes opaque. Place frozen tissue blocks in a sealed container or zip-lock bag and store at −80° C. Frozen tissue blocks can be stored for several years.
Using a cryostat, cryosection tissue blocks into sections of 10–15 uM in thickness.
Cryosections may now be analyzed using a standard immunohistochemistry protocol. (See Note 14)
3.8. Preparation of aggregates for whole-mount staining
Collect and fix the tissue, as described in steps 1–4 of previous section (3.7, Preparation of aggregates for immunohistochemistry).
Add PBS containing 10% (vol/vol) normal goat serum and 0.1% (vol/vol) Triton X-100 to block the fixed aggregates. Shake overnight at RT. Note that BSA or normal horse serum can be used in place of goat serum, depending on the type of antibody being used.
Remove the blocking solution and apply primary antibody solution: PBS containing 3% (vol/vol) normal goat serum, 0.1% (vol/vol) Triton X-100, plus primary antibodies. Shake for 2–3 d at RT.
Remove primary antibody solution and suspend aggregates in PBS containing 0.1 PBS containing 0.1% (vol/vol) Triton X-100 to wash. Shake for 1 hour at RT. Repeat three times.
Apply secondary antibody solution: PBS containing 3% (vol/vol) normal goat serum, 0.1 % (vol/vol) Triton X-100, plus secondary antibodies. Shake for 2–3 d at RT. (See Note 15)
Repeat step 3.
Apply scaleA2 solution to aggregates and for 4–5 days prior to imaging. (See Note 16)
To image, transfer ~250–500 uL of scaleA2 solution containing an aggregate to an imaging chamber. The volume of solution needed will be determined by the size of the imaging chamber used.
Affix the coverslip slowly; note that a small amount of liquid may escape. If an air bubble should form, it will likely not interfere with imaging.
Flip the chamber while imaging in order to view the specimen from multiple orientations.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R21DC012617, R01DC013294 and an Action of Hearing Loss Research Grant (to E.H.). The authors would like to thank Andrew Mikosz and Sreeparna Majumdar for their technical assistance and comments on the manuscript.
4. Notes
It is important that the LIF-2i maintenance medium is free of Vitamin A. Vitamin A is capable of converting into retinoic acid, which can cause spontaneous differentiation in culture. When adapting ES cells to culture in LIF-2i medium, serial passaging can eliminate any undesirable differentiation that may arise.
Note that during the maturation phase of culture, penicillin-streptomycin (a component of Ectodermal differentiation medium) is replaced with normocin. Streptomycin is an aminoglycoside, which can be toxic to inner ear hair cells (27). Normocin does not contain aminoglycosides. Alternative choice is Ampicillin, which is also devoid of aminoglycosides.
Coating the plate with gelatin prior to plating promotes adherence of cells to the surface of the plate. If ES cells do not attach to the plate, it may be due to insufficient gelatin coating. Re-plate the cells on a new plate with fresh gelatin coating.
If there is an issue with cell attachment or growth, the cells may not be adapted to growth in LIF-2i medium. In this case, we recommend passaging the cells several times before use.
Take care to wash thoroughly with PBS, as residual Lif-2i medium can affect differentiation.
This step is important for ensuring that the cells are completely dissociated into single cells. Take care to pipet forcefully through the cell-top strainer.
For example, if the cell suspension contains 1×106 cells, dilute 0.66 mL of cell suspension in 21.34 mL Ectodermal differentiation medium.
Matrigel is critical for the formation of an epithelium on the surface of the aggregates. If an epithelium does not form or thicken over time, Matrigel may not have been properly incorporated into the ectodermal differentiation medium. It is essential to add Matrigel to cold medium and mix immediately for complete incorporation.
Non-neural ectoderm is induced via the application of BMP4. Simultaneous application of SB-431542, a TGF-β inhibitor, serves to block the formation of mesoderm.
FGF-2 and LDN application are necessary for induction of pre-placodal ectoderm. LDN serves to attenuate BMP signaling.
Wide-mouth P1000 tips are most effective in the transfer of day 8 aggregates. To prepare, use scissors to cut approximately 2 mm off the end of a tip. A whole box of tips may be prepared in this manner, and autoclaved prior to use.
It is important to note that PFA is toxic. Apply and remove PFA in a fume hood.
For long-term storage, suspend aggregates in PBS containing 0.02% (wt/vol) sodium azide. This will suppress bacterial growth.
A table of antibodies for markers of placodal and otic development may be found in Koehler et al 2014 (28).
Exposure to light may cause photo-bleaching of the fluorophores. To protect samples, wrap samples in foil during this and subsequent steps.
Note that Scale A2 medium may cause slight enlargement of the tissue.
References
- 1.Eiraku M et al. (2011) Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature, 472:51–56 [DOI] [PubMed] [Google Scholar]
- 2.Nakano T et al. (2012) Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10:771–785 [DOI] [PubMed] [Google Scholar]
- 3.Eiraku M et al. (2008) Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3:519–532 [DOI] [PubMed] [Google Scholar]
- 4.Eiraku M, Sasai Y (2012) Mouse embryonic stem cell culture for generation of three-dimensional retinal and cortical tissues. Nature Protocols 7:69–79 [DOI] [PubMed] [Google Scholar]
- 5.Nasu M et al. (2012) Robust formation and maintenance of continuous stratified cortical neuroepithelium by laminin-containing matrix in mouse ES cell culture. PloS ONE 7, e53024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spence JR et al. (2010) Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature, 470:105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCracken KW, Howell JC, Wells JM, Spence JR (2011) Generating human intestinal tissue from pluripotent stem cells in vitro. Nature Protocols 6:1920–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson PA, Hemmati-Brivanlou A (1995) Induction of epidermis and inhibition of neural fate by BMP-4, Nature 376:331–333 [DOI] [PubMed] [Google Scholar]
- 9.Wilson PA, Lagna G, Suzuki A, Hemmati-Brivanlou A (1997) Concentration-dependent patterning of the Xenopus ectoderm by BMP4 and its signal transducer Smad1, Development 124:3177–3184 [DOI] [PubMed] [Google Scholar]
- 10.Neave B, Holder N, Patient R (1997) A graded response to BMP-4 spatially coordinates patterning of the mesoderm and ectoderm in zebrafish, Mech. Dev 62:183–195 [DOI] [PubMed] [Google Scholar]
- 11.Bartha KA et al. (1999) BMP activity establishes a gradient of positional information throughout the entire neural plate, Development 126:4977–4987 [DOI] [PubMed] [Google Scholar]
- 12.Kwon HJ, Bhat N, Sweet EM, Cornell RA, Riley BB (2010) Identification of early requirements for preplacodal ectoderm and sensory organ development, PLoS Genet 6: e1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey NT et al. (2010) Response to BMP4 signaling during ES cell differentiation defines intermediates of the ectoderm lineage, J. Cell Sci 123:1796–1804 [DOI] [PubMed] [Google Scholar]
- 14.James D, Levine AJ, Besser D, Hemmati-Brivanlou A (2005) TGF-B/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132:1273–1282 [DOI] [PubMed] [Google Scholar]
- 15.Camus A, Perea-Gomez A, Moreau A, Collignon J (2005) Absence of Nodal signaling promotes precocious differentiation in the mouse embryo, Dev. Biol 295:743–755 [DOI] [PubMed] [Google Scholar]
- 16.Chambers SM et al. (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27:275–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reichert S, Randall RA, Hill CS (2013) A BMP regulatory network controls ectodermal cell fate decisions at the neural plate border. Development 140:4435–4444 [DOI] [PubMed] [Google Scholar]
- 18.Litsiou A, Hanson S, Streit A (2005) A balance of FGF, BMP, WNT signaling positions the future placode territory in the head, Development 131:4015–4062 [DOI] [PubMed] [Google Scholar]
- 19.Grocott T, Tambalo M, Streit A (2012) The peripheral sensory nervous system in the vertebrate head: a gene regulatory perspective. Dev. Biol 370:3–23 [DOI] [PubMed] [Google Scholar]
- 20.Pieper M, Ahrens K, Rink E, Peter A, Schlosser G (2012) Differential distribution of competence for panplacodal and neural crest induction to non-neural and neural ectoderm, Development 139:1175–1187 [DOI] [PubMed] [Google Scholar]
- 21.Wright TJ, Mansour SL (2010) Fgf3 and Fgf10 are required for mouse otic placode induction, Development 130:1777–1785 [DOI] [PubMed] [Google Scholar]
- 22.Urness LD, Paxton CN, Wang X, Schoenwolf GC, Mansour SL (2010) FGF signaling regulates otic placode induction and refinement by controlling both ectodermal target genes and hindbrain Wnt8a. Dev. Biol 340:595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohyama T, Mohamed OA, Taketo MM, Dufort D, Groves AK (2006) Wnt signals mediate a fate decision between otic placode and epidermis, Development 133:865–875 [DOI] [PubMed] [Google Scholar]
- 24.Riccomango MM, Takada S, Epstein DJ (2005) Wnt-dependent regulation of inner ear morphogenesis is balanced by the opposing and supporting roles of Shh, Genes Dev 19:1612–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freter S, Muta Y, Mak SS, Rinkwitz S, Ladher RK (2008) Progressive restriction of otic fate: the role of FGF and Wnt in resolving inner ear potential, Development 135: 3415–3424 [DOI] [PubMed] [Google Scholar]
- 26.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E (2013) Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature, 500:217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lerner SA, Matz GJ, Hawkins JE (1981) Aminoglycoside Ototoxicity Little Brown and Company [Google Scholar]
- 28.Koehler KR, Hashino E, 3D mouse embryonic stem cell culture for generating inner ear organoids. Nat Protocols vol. 9 (6), 1229–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]