

SUMMARY
Despite extensive efforts, oncogenic KRAS remains resistant to targeted therapy. Combined downstream RAL-TBK1 and MEK inhibition induces only transient lung tumor shrinkage in KRAS-driven genetically engineered mouse models (GEMMs). Using the sensitive KRAS;LKB1 (KL) mutant background, we identify YAP1 upregulation and a therapy-induced secretome as mediators of acquired resistance. This program is reversible, associated with H3K27 promoter acetylation, and suppressed by BET inhibition, resensitizing resistant KL cells to TBK1/MEK inhibition. Constitutive YAP1 signaling promotes intrinsic resistance in KRAS;TP53 (KP) mutant lung cancer. Intermittent treatment with the BET inhibitor JQ1 thus overcomes resistance to combined pathway inhibition in KL and KP GEMMs. Using potent and selective TBK1 and BET inhibitors we further develop an effective therapeutic strategy with potential translatability to the clinic.
Graphical Abstract
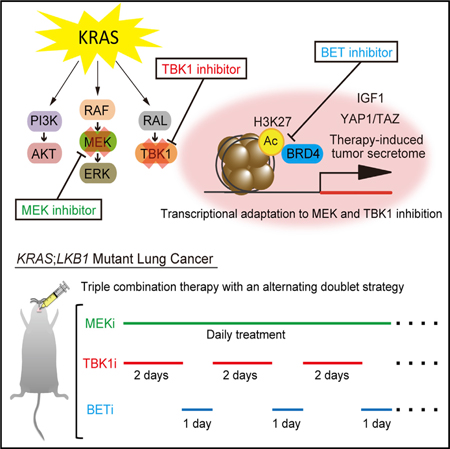
In Brief
Kitajima et al. identify BET-regulated YAP1 upregulation as a mediator of acquired and intrinsic resistance in KRAS;LKB1 and KRAS;TP53 mutant lung cancer cells, respectively, to combined TBK1 and MEK inhibition and show that intermittent BET inhibition overcomes this resistance.
INTRODUCTION
KRAS is one of the most frequently mutated oncogenes in human cancer and is enriched in tumors fueled by inflammatory signaling, such as non-small-cell lung cancer (NSCLC), colorectal cancer (CRC), and pancreatic ductal adenocarcinomas (PDAC) (Kitajima et al., 2016; Pylayeva-Gupta et al., 2011). KRAS mutant cancers have remained refractory to all targeted therapies to date, in part due to the challenges of inhibiting oncogenic KRAS itself (Stephen et al., 2014). While direct targeting of specific KRAS mutants (Hobbs et al., 2016) and immunotherapy (Topalian et al., 2015; Tran et al., 2016) have shown promise, an equally important strategy is to identify optimal combinations of therapy that ablate KRAS signaling downstream of key mediators such as MAPK, PI3K, and RAL-GDS (Stephen et al., 2014).
Although well validated as downstream targets, MAPK and PI3K pathway inhibitors have thus far failed to affect KRAS mutant NSCLC in the clinic, even when used in combination (Hata et al., 2014). Direct inhibitors of RAL-GDS, an equally critical oncogenic KRAS effector (Bodemann and White, 2008), also remain in preclinical development (Yan et al., 2014). Importantly, RAL-GDS activation of RALB engages the more targetable innate immune signaling kinase TBK1, inducing the secretion of IL-6 and CCL5, which promote cancer cell survival via the STAT3 and NF-κB pathways (Barbie et al., 2009; Chien et al., 2006; Zhuetal., 2014a).
MAPK and innate immune signaling pathways are tightly linked by feedback regulation. For example, treatment of KRAS mutant NSCLC cells with the MEK inhibitor selumetinib induces IL-6/STAT3 activation, which contributes to drug resistance (Lee et al., 2014), while TBK1 inhibition rapidly induces MEK/ERK activation (Zhu et al., 2014a). This interdependence of MEK and innate immune signaling downstream of RAS provides a strong rationale for combinatorial therapy (Zhu et al., 2014b). Indeed, we previously reported that combination of selumetinib with the TBK1/JAK inhibitor momelotinib synergistically induces tumor regression in aggressive KRAS-driven lung cancer mouse models (Zhu et al., 2014a). Synergy between MEK and TBK1 inhibition has also been observed downstream of NRAS signaling in melanoma (Vu and Aplin, 2014). Despite these anti-tumor responses, it is likely that higher order drug combinations targeting additional pathways will be required for long-term durable activity.
It is also increasingly clear that KRAS mutant NSCLC is a heterogeneous disease and that co-mutation of the tumor suppressor genes TP53 or STK11/LKB1 (hereafter LKB1) defines different subtypes (Skoulidis et al., 2015). For example, KRAS;TP53 mutant (KP) or KRAS;LKB1 mutant (KL) NSCLC cells exhibit divergent gene expression profiles and sensitivity to targeted or immune directed therapies (Kottakis et al., 2016; Koyama et al., 2016; Skoulidis et al., 2018). LKB1 deficiency specifically has been reported to promote resistance to MEK inhibition (Chen et al., 2012) but sensitivity to IL-6 neutralization (Koyama et al., 2016).
We therefore sought to explore the relative efficacy of combined innate immune and MAPK signaling in these different KRAS genetic backgrounds and to uncover additional pathways that might limit the overall activity of this therapy.
RESULTS
LKB1 Inactivation Engages Innate Immune Cytokines and Momelotinib Sensitivity in KRAS-Driven NSCLC
We noted that elevated IL-6 secretion at baseline in KL or KRAS;LKB1;TP53 mutant (KLP) human NSCLC cells correlated directly with enhanced sensitivity to momelotinib treatment, as compared with KP cells (Figure 1A). Indeed, single-agent momelotinib treatment induced apoptosis in KL and KLP but not in KP cells (Figure 1B). Conversely, KL and KLP cells were resistant to MEK inhibitor treatment relative to KP cells, in consonance with prior work (Chen etal., 2012)(Figure 1A). Given this relative resistance, we explored whether MEK inhibitor-induced innate immune cytokine expression was also higher in KL than in KP cells. Treatment of the human KL NSCLC cell line A549 with the MEK inhibitor trametinib, especially in combination with the PI3K inhibitor buparlisib, resulted in pronounced induction of IL-6, CCL2, and CCL5 expression (Figure S1A). Consistent with prior work (Zhu et al., 2014a), this feedback response was potently suppressed by co-treatment with the dual TBK1/JAK inhibitor momelotinib as compared with the selective JAK1/2 inhibitor ruxolitinib, even though JAK/STAT3 signaling was inactivated to the same degree (Figure S1B). We further confirmed downregulation of IL-6 secretion following momelotinib but not ruxolitinib treatment in multiple KL NSCLC lines, irrespective of whether or not they also had mutated TP53. On the other hand, KP NSCLC cell lines with intact LKB1 (H2009, H441, and H358) secreted much lower IL-6 at baseline and did not show downregulation of IL-6 secretion following momelotinib treatment (Figure S1C). Together these data suggested that LKB1 loss might engender preferential dependence on innate immune cytokine signaling downstream of oncogenic KRAS.
Figure 1. LKB1 Regulates Innate Immune Signaling and Sensitivity to MMB/MEK Inhibitor Therapy in KRAS-Driven NSCLC.
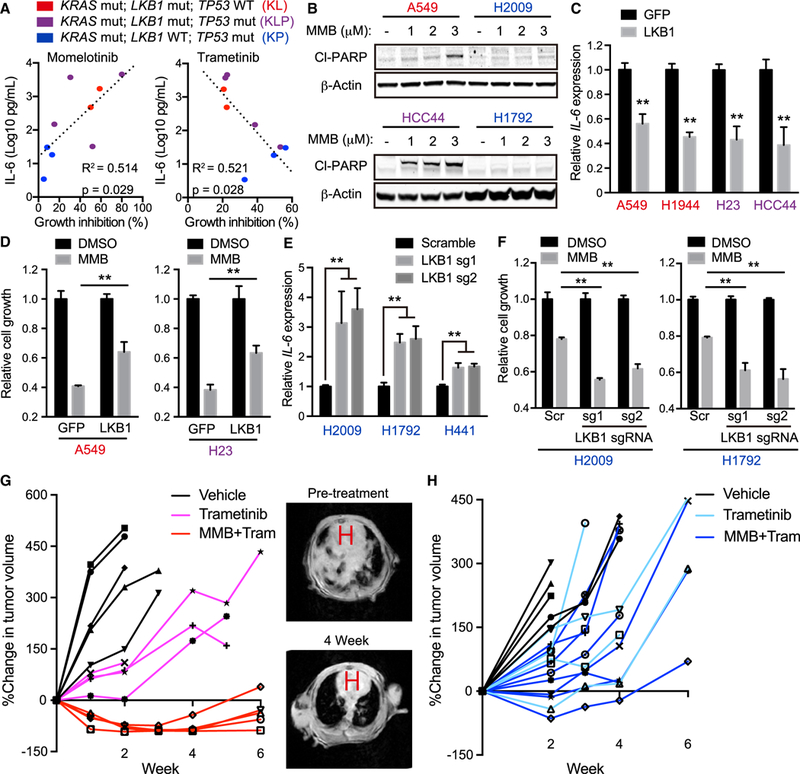
(A) Percentage growth inhibition (x axis) of KL (red, A549 and H1944 cells), KLP (purple, HCC44, H23, H2122, and H2030 cells), and KP (blue, H2009, H441, and H358 cells) human NSCLC lines treated for 72 hr with 2.5 μM momelotinib or 10 nM trametinib plotted versus degree of baseline IL-6 secretion in conditioned medium (y axis). R2 values and p values for the correlation shown.
(B) Immunoblot (IB) ofthe indicated proteins in A549, H2009, HCC44, and H1792 cells treated with indicated concentration of momelotinib (MMB) for24 hr. Red cell lines, KL; blue cell lines, KP.
(C) qRT-PCR of IL-6 in A549, HCC44, H23, and H1355 transduced with the indicated vector (n = 4).
(D) Relative cell growth of A549 and H23 cells transduced with the indicated vector in the presence of 2.5 μM MMB for 96 hr (n = 3).
(E) qRT-PCR of IL-6 in H2009, H1792, and H441 cells transduced with the indicated single guide RNA (sgRNA) (n = 4).
(F) Relative cell growth of H2009 and H1792 cells transduced with the indicated sgRNA in the presence of 2.5 μM MMB for 96 hr (n = 3).
(G) Percentage change in MRI tumor volume of LSL-KrasG12D;Lkb1f/f-induced lung cancer following daily treatment of vehicle, 2 mg/kg trametinib (Tram), or 10 mg/kg MMB + 2 mg/kg Tram (left), with representative MRI images before treatment and 4 weeks following MMB and Tram therapy (right). H, heart.
(H) Percentage change in MRI tumor volume of LSL-KrasG12D;Trp53f/f-induced lung cancer following daily treatment of vehicle, 2 mg/kg Tram, or 10 mg/kg MMB + 2 mg/kg Tram.
All quantitative data are represented as mean ± SD; **p < 0.01; see also Figure S1.
To isolate the role of LKB1 in this phenotype more specifically, we next reconstituted wild-type LKB1 in KL (A549 and H1944) or KLP (H23 and HCC44) cells (Figure S1D). As compared with GFP-expressing control cells, LKB1 re-expression downregulated IL-6 expression and induced relative resistance to momelotinib treatment in both KL and KLP cells (Figures 1C, 1D, and S1E). Furthermore, apoptosis induced by momelotinib treatment was suppressed by LKB1 re-expression in KL A549 cells (Figure S1F). Conversely, we performed LKB1 knockout by the CRISPR/Cas9 system in KP cell lines (H2009, H1792 and H441) (Figure S1G), and observed increased IL-6 expression and sensitivity to momelotinib treatment (Figures 1E, 1F, and S1H). Together, these results confirm that LKB1 status itself is tightly linked with IL-6 expression and momelotinib sensitivity in KRAS mutant NSCLC cells.
Previously, we found that high-dose momelotinib (100 mg/kg) daily could synergize with MEK inhibition in the aggressive LSL-KrasG12D;Trp53f/f (KP) lung cancer GEMM (Zhu et al., 2014a). To understand further the effect of Lkb1 status on momelotinib/MEK inhibitor sensitivity in vivo, we compared treatment response using a clinically achievable lower dose of momelotinib (10 mg/kg) with trametinib (2 mg/kg) in established lung tumors from the KP and LSL-KrasG12D;Lkb1f/f (KL) GEMMs. Consistent with our in vitro observations, treatment of mice with this dosing schedule induced tumor regressions in the KL GEMM (Figure 1G) in contrast to the KP GEMM, which exhibited a more modest slowing of tumor growth (Figure 1H). Despite a near complete response of lung tumors in KL mice, acquired resistance to this combination developed rapidly by around 4 weeks of treatment (Figure 1G). Thus, while KL lung cancer is particularly sensitive to inhibition of innate immune and MEK signaling, these data suggested that activation of alternative pathways can still mediate acquired resistance.
IGF1/IGF1R Activation as a Bypass Pathway in KL Cells
To identify additional pathways that become activated in response to TBK1/JAK inhibition in KL cells, we used a phospho-receptor tyrosine kinase (RTK) array. In A549 cells, momelotinib treatment for 24 hr increased phosphorylation of IGF1 receptor (IGF1R), in contrast to the JAK1/2-specific inhibitor ruxolitinib (Figure S2A). We confirmed that IGF1R activation following momelotinib treatment was at least as pronounced as that observed following MEK inhibitor treatment with selumetinib (Figure 2A). Feedback IGF1R activation was biphasic: early induction occurred without upregulation of IGF1R ligands such as IGF1 and IGF2 but coincided with suppression of S6K and activation of IRS1, suggesting intracellular feedback activation (Figures S2B and S2C). In contrast, treatment with momelotinib plus or minus MEK inhibition for several days strongly and specifically induced IGF1 (Figures 2B and S2D). Upregulation of IGF1 following combination treatment also increased over time (Figure S2E). We further confirmed this observation in vivo, since murine KL tumors that developed acquired resistance to combination therapy with momelotinib and trametinib upregulated Igf1 expression (Figure 2C).
Figure 2. Inhibition of IGF1/IGF1R Pathway Is Effective in KL Cells but Fails to Achieve Prolonged Therapeutic Effect In vivo.
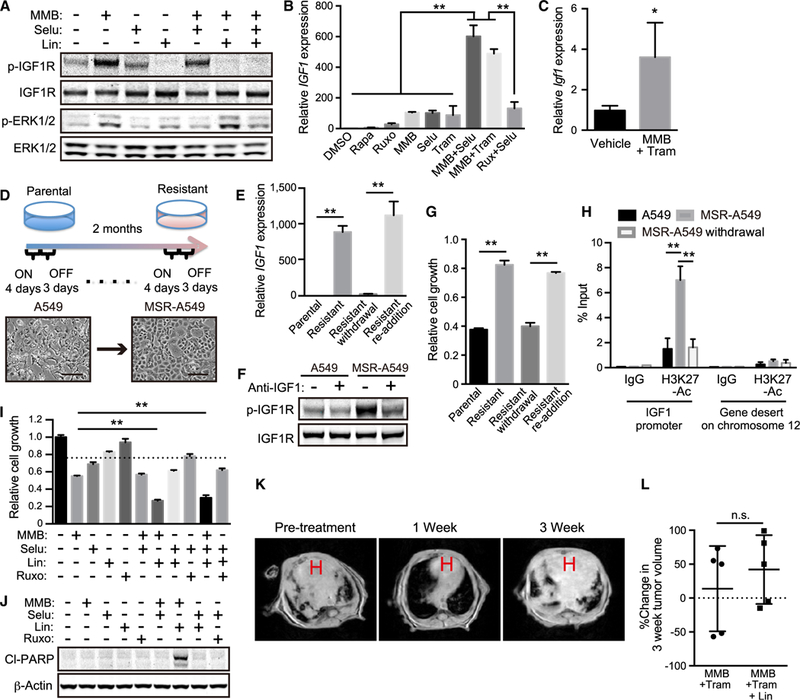
(A) IB of the indicated proteins in A549 cells treated with 5 μM MMB ±1 μM selumetinib (Selu) ± 1 μM linsitinib (Lin) for 48 hr.
(B) qRT-PCR of IGF1 in A549 treated with 1 μM rapamycin (Rapa), 1 μM Ruxo, 5 μM MMB, 1 μM Selu, and/or 10 nM Tram for 12 days (n = 4).
(C) qRT-PCR of IGF1 in tumor nodules (n = 4) from KL mice treated with 10 mg/kg MMB + 2 mg/kg Tram for 6 weeks.
(D) Schematic of resistant cell line generation over 2 months of MMB and Selu treatment. A549 cells were cultured in the presence of 5 μM MMB and 1 μM Selu over 4 days (ON), and then in the absence of these inhibitors over 3 days (OFF). This 7-day cycle was repeated over 2 months (upper). Representative phase-contrast images of A549 and MSR-A549 cells (lower). Scale bars: 100 μm.
(E) qRT-PCR of IGF1 in A549, MSR-A549, MSR-A549 drug withdrawal and drug re-addition cells (n = 4) (See STAR Methods).
(F) IB of the indicated proteins in A549 and MSR-A549 cells in the presence of 1 μg/mL anti-IGF1 antibody treatment for 24 hr.
(G) Relative cell growth of A549, MSR-A549, MSR-A549 withdrawal, and MSR-A549 re-addition cells in the presence of 5 μM MMB for 72 hr (n = 3).
(H) qPCR of IGF1 promoter and gene desert on chromosome 12 (see STAR Methods) on DNA purified from ChIP-H3K27 acetylation in A549, MSR-A549, and MSR-A549 withdrawal cells (n = 3).
(I) Relative cell growth of A549 treated with 2.5 μM MMB, 1 μM Selu, 1 μM Lin, and/or 1 μM Ruxo for 48 hr (n = 3).
(J) IB of the indicated proteins in A549 cells treated with 2.5 μM MMB, 1 μM Selu, 1 μM Lin, and/or 1 μM Ruxo for 24 hr.
(K) Representative MRI images from LSL-KrasG12D;Lkb1f/f mice treated with MMB + Tram + Lin therapy for the indicated time. H, heart.
(L) Percentage change in MRI tumor volume of LSL-KrasG12D;Lkb1f/f-induced lung cancer 4 weeks following MMB + Tram or MMB + Tram + Lin therapy. MMB 10 mg/kg, Tram 2 mg/kg, Lin 7.5 mg/kg. n.s., not significant.
All quantitative data are represented as mean ± SD; *p < 0.05, **p < 0.01; see also Figure S2.
Given these findings, we next assessed the role of IGF1/IGF1R signaling during chronic acquisition of resistance in A549 cells (Figure 2D). IGF1 mRNA and IGF1 secretion was highly upregulated following 2-month culture to develop momelotinib/selumetinib-resistant A549 cells (MSR-A549 cells) (Figures 2E and S2F). The expression of innate immune cytokines such as IL-6, CCL2, and CCL5 remained suppressed (Figure S2G). MSR-A549 cells also exhibited high levels of phospho-IGFIR that were sensitive to treatment with an IGF1 neutralizing antibody (Figure 2F). Drug withdrawal reversed IGF1 mRNA and IGF1 secretion in MSR-A549 cells after only 10 days, while subsequent re-addition of momelotinib and selumetinib rapidly re-induced IGF1 expression (Figures 2E and S2F), which tracked with drug resistance to momelotinib (Figure 2G). Because these findings suggested epigenetic alteration, rather than stable acquisition of a mutation, we examined histone H3K27 acetylation of the IGF1 promoter by chromatin immunoprecipitation (ChIP)-qPCR. Indeed, as compared with a control region, histone H3K27 acetylation of the IGF1 promoter tracked closely with drug resistance of MSR-A549 cells and was highly reversible (Figure 2H). Together, these results suggested an important role for IGF1 activation during momelotinib or selumetinib resistance but also the potential for broader epigenetic reprogramming.
IGF1R Inhibition Is Insufficient to Prevent Acquired Resistance
To determine the specific contribution of IGF1/IGF1R activation to acquired resistance to momelotinib or MEK inhibition, we next treated cells with the IGF1R/insulin receptor inhibitor linsitinib. As compared with parental A549 cells, MSR-A549 cells with established resistance showed enhanced, although partial, sensitivity to linsitinib (Figure S2H). Addition of recombinant human IGF1 to parental A549 and H23 cells reduced cell growth inhibition following momelotinib treatment, which was completely antagonized by linsitinib treatment (Figures S2I and S2J). Conversely, while linsitinib treatment had little effect on parental A549 cells on its own, co-treatment with momelotinib enhanced inhibition of cell growth (Figures 2I and S2K). Indeed, linsitinib treatment potently induced apoptosis together with lower concentrations of momelotinib that were unable to induce apoptosis in A549 cells (Figures 2J and S2L). These synergistic effects of linsitinib treatment were not observed in combination with selumetinib or ruxolitinib (Figures 2I and 2J). Notably, linsitinib synergy with momelotinib was more pronounced in KL and KLP cells compared with KP cells (Figures S2M and S2N). Together, these results suggested that activation of IGF1/ IGF1R signaling could represent a critical survival pathway that facilitates escape from combined innate immune and MEK inhibition in KL cells.
We therefore sought to determine whether IGF1R inhibition could overcome resistance to momelotinib/MEK inhibition in vivo, by pursuing triple combination therapy with momelotinib, trametinib, and linsitinib in both KL and KP GEMMs. To minimize toxicity, we utilized an intermittent combination treatment schedule consisting of 3 days with drug treatment and 4 days off in a week, which fully suppressed cell growth in KL cells in vitro and did not result in significant body weight loss (Figures S2O and S2P). As expected, linsitinib treatment clearly enhanced tumor shrinkage in response to intermittent momelotinib and trametinib dosing in KL GEMMs but not in KP GEMMs (Figures 2K, S2Q, and S2R). However, despite this synergy, KL tumors still recovered after several weeks of therapy, and tumor volume with triple combination therapy was comparable with that with the two-drug combination by 4 weeks (Figures 2K and 2L). These results revealed that engagement of additional pathways beyond IGF1/IGF1R signaling are likely to compensate and promote acquired resistance to this therapy.
YAP1 Activation in MSR-A549 Cells Contributes to Acquired Resistance and Abrogates the Synergy between Momelotinib and Linsitinib
To uncover additional pathways that could promote acquired resistance to momelotinib/MEK inhibition, we performed RNA sequencing (RNA-seq) of MSR-A549 cells. We found that, in addition to IGF1, multiple YAP1 target genes, including SERTAD4, CTGF, ANKRD1, and CYR61 (Dupont et al., 2011), were upregulated as compared with parental A549 cells (Figure 3A). We confirmed the YAP1-dependent nature of this transcriptional program, since YAP1 depletion antagonized this upregulation (Figures 3B and S3A). Moreover, as compared with parental A549 cells, MSR-A549 cells strongly expressed YAP1 and its related protein TAZ, which primarily localized to the nucleus (Figures 3C and 3D). YAP1 and its targets were also upregulated following momelotinib and MEK inhibitor treatment over time in parental A549 cells (Figures S3B and S3C). We further confirmed that exogenous expression of constitutively active YAP1 (YAP1 5SA) in parental A549 cells induced these YAP1 target genes but not IGF1 (Figures S3D and S3E), suggesting that YAP1 and TAZ activation in this context was independent of IGF1/IGF1R activation. Indeed, YAP1 depletion did not affect IGF1 upregulation in MSR-A549 cells (Figure S3F). Conversely, linsitinib treatment failed to suppress YAP1/TAZ and its downstream genes in MSR-A549 cells (Figures S3G and S3H). Recombinant IGF1 treatment also failed to upregulate YAP1 and TAZ expression, consistent with the independence of these two signaling pathways (Figure S3I).
Figure 3. YAP1 Activation in MSR-A549 Cells Abrogates the Synergy between MMB and Linsitinib.
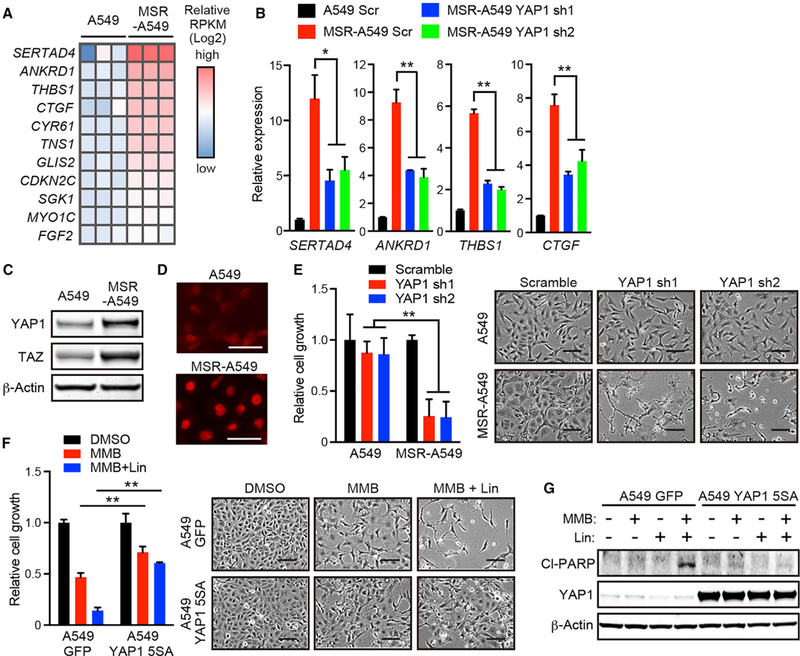
(A) Heatmap of reads per kilobase of exon per million mapped fragments (RPKM) values of YAPI target genes (Dupont et al., 2011) in A549 and MSR-A549 cells.
(B) qRT-PCR of SERTAD4, ANKRD1, THBS1, CTGF, and CYR61 in A549 and MSR-A549 cells transduced with the indicated small hairpin RNA (shRNA) (n = 4).
(C) IB of the indicated proteins in A549 and MSR-A549 cells.
(D) Immunofluorescence of YAPI in A549 and MSR-A549 cells. Scale bars: 100 μm.
(E) Relative cell growth (left) and phase-contrast images (right) of A549 and MSR-A549 cells transduced with the indicated shRNA (n = 3). Scale bars: 100 μm.
(F) Relative cell growth (left) and phase-contrast images (right) of A549 cells transduced with the indicated vector in the presence of 2.5 μM MMB and/or 1 μM Lin for 96 hr (n = 3). Scale bars: 100 μm.
(G) IB of the indicated proteins in A549 cells transduced with the indicated shRNA and treated with 2.5 μM MMB and/or 1 μM Lin for 24 hr.
All quantitative data are represented as mean ± SD; *p < 0.05, **p < 0.01; see also Figure S3.
Importantly, YAP1 depletion dramatically suppressed cell growth of MSR-A549 cells in the presence of momelotinib and selumetinib, even though YAP1-depleted parental A549 cells did not show significant growth inhibition (Figure 3E). On the other hand, A549 and HCC44 cells expressing YAP1 5SA showed resistance to both momelotinib and selumetinib (Figure S3J). Further consistent with the idea that this YAP1 resistance program is distinct from IGF1/IGF1R, YAP1 5SA overexpression in A549 cells strongly antagonized cell growth inhibition and apoptosis induced by treatment with linsitinib and momelotinib (Figures 3F and 3G). Together, these results demonstrate that YAP1 is activated in response to inhibition of innate immune and MEK signaling downstream of KRAS, YAP1-mediated acquired resistance to momelotinib and selumetinib is independent of IGF1/IGF1R activation, and YAP1 activation promotes survival despite IGF1R inhibition in KL cells.
IGF1 and YAP1 Are Components of a Broader BET-Driven Adaptive Transcriptional Program
Given the epigenetic nature of the resistance to this combination therapy, we next performed histone H3K27 acetylation ChIP sequencing (ChIP-seq) in parental A549 and MSR-A549 cells and integrated this with RNA-seq data to correlate gene expression with enhancer acquisition during resistance. This broader analysis revealed that 659 genes were upregulated in MSR-A549 cells and associated with higher histone H3K27 acetylation (Figure 4A and Table S1). As expected, these resistance-related genes contained IGF1 and YAP1 target genes (Figure 4B and Table S1). We also found that MSR-A549 cells upregulated genes encoding multiple other RTKs and secreted factors that have been linked to drug resistance and a therapy-induced secretome (TIS) (Anastas et al., 2014; Obenauf et al., 2015) such as KIT, WNT5A, and PDGFB (Figure 4B and Table S1). These findings suggested that, instead of dual targeting of IGF1/ IGF1R and YAP1, inhibition of this broader transcriptional adaptation might be a more effective strategy to overcome resistance.
Figure 4. BET Inhibitor Abrogates Broader Transcriptional Activation during Acquisition of Resistance to MMB/MEK Inhibitor Treatment.
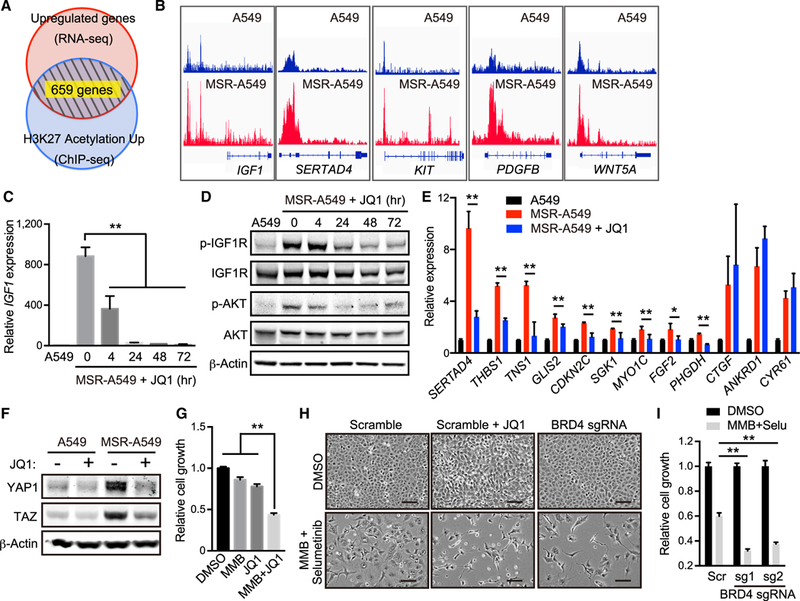
(A) Venn diagram showing overlap of genes upregulated by RNA-seq and enriched by histone H3K27 acétylation ChIP-seq in MSR-A549 cells compared with A549 cells.
(B) ChIP-seq signals visualized by integrative genome viewer (IGV) for histone H3K27 acetylation at IGF1, SERTAD4, KIT, PDGFB, and WNT5A loci in A549 and MSR-A549 cells.
(C) qRT-PCR of IGF1 in A549 and MSR-A549 cells treated with 200 nM JQ1 for the indicated time (n = 4).
(D) IB of the indicated proteins in A549 and MSR-A549 cells treated with 200 nM JQ1 for the indicated times.
(E) qRT-PCR of SERTAD4, THBS1, TNS1, GLIS2, CDKN2C, SGK1, MYO1C, FGF2, PHGDH, CTGF, ANKRD1, and CYR61 in A549 and MSR-A549 cells treated with or without 200 nM JQ1 for 24 hr (n = 4).
(E) IB of the indicated proteins in A549 and MSR-A549 cells treated with or without 200 nM JQ1 for 24 hr.
(G) Relative cell growth of MSR-A549 cells in the presence of 5 μM MMB and/or 500 nM JQ1 for 96 hr (n = 3).
(H) Phase-contrast images of A549 transduced with sgControl (Scramble) or sgBRD4 and treated with 5 μM MMB, 1 μM Selu, and/or 200 μM JQ1 for 96 hr. Scale bars: 100 μm.
(I) Relative cell growth of A549 transduced with the indicated sgRNA in the presence of 5 μM MMB and 1 μM Selu for 96 hr (n = 3).
All quantitative data are represented as mean ± SD; *p < 0.05, **p < 0.01; see also Figure S4 and Table S1.
BET family proteins recognize H3K27 acetylated histones and activate the RNA pol II machinery at promoters and enhancers (Shi and Vakoc, 2014). Indeed, genes suppressed by treatment with the BET inhibitor JQ1 were significantly enriched in 659 resistance-related genes (Figure S4A). JQ1 was also recently found to inhibit IGF1/IGF1R resistance mechanisms in breast cancer and Ewing sarcoma (Loganathan et al., 2016; Stratikopoulos et al., 2015). Consistent with these results and reports, IGF1 expression and downstream IGF1R and AKT activation were immediately suppressed following JQ1 treatment of MSR-A549 cells (Figures 4C and 4D). In addition, most but not all YAP1 target genes, RTKs, and secreted factors upregulated in MSR-A549 cells were strongly downregulated by JQ1 treatment (Figures 4E and S4B). Unexpectedly, YAP1 and TAZ upregulation in MSR-A549 cells were also suppressed by JQ1 treatment, suggesting reinforcement of YAP1/TAZ activation by this transcriptional program (Figure 4F and S4C). Consistent with these findings, JQ1 treatment also re-sensitized MSR-A549 cells to momelotinib treatment (Figure 4G). More-over, JQ1 treatment or genetic BRD4 depletion enhanced growth inhibition following momelotinib and MEK inhibitor combination treatment in parental A549 cells (Figures 4H, 4I, and S4D). Together, these results suggested that BET inhibition might represent a particularly potent approach to overcome transcriptional adaptation to inhibition of MEK and innate immune signaling downstream of oncogenic KRAS.
YAP1 Activation Mediates Intrinsic Resistance to Momelotinib and MEK Inhibitor Treatment in KP Cells
Since KP cells were relatively insensitive to momelotinib treatment, we also considered the possibility that activation of similar pathways at baseline could contribute to intrinsic resistance to JAK/TBK1 inhibition. We therefore compared baseline levels of YAP1 and markers of downstream pathway activation in KP versus KL NSCLC cell lines. As compared with KL cells, KP cells exhibited increased YAP1 protein levels, which were sensitive to JQ1 treatment, as in MSR-A549 cells (Figure 5A). YAP1 expression was also downregulated in BRD4-depleted KP cells (Figure S5A). To assess more systematically whether YAP1 signaling is activated in KP versus KL cells, we analyzed gene expression profiling data from NSCLC cell lines in the Cancer Cell Line Encyclopedia (CCLE) database (Figures 5B and S5B and Table S2). KP-induced genes were significantly enriched for having binding motifs for the DNA-binding partner of YAP1 and TAZ, TEAD1, in their promoters (Figure 5B). Moreover, a survey of The Cancer Genome Atlas (TCGA) database revealed that gene expression in KP lung adenocarcinomas was also highly enriched in genes involved in YAP1 signature activation (Dupont et al., 2011) and downstream targets of TEAD1 (Figure 5C and Table S3).
Figure 5. YAP1 Downregulation Following BET Inhibitor Treatment Sensitizes KP Cells to MMB/MEK Inhibitor Treatment.
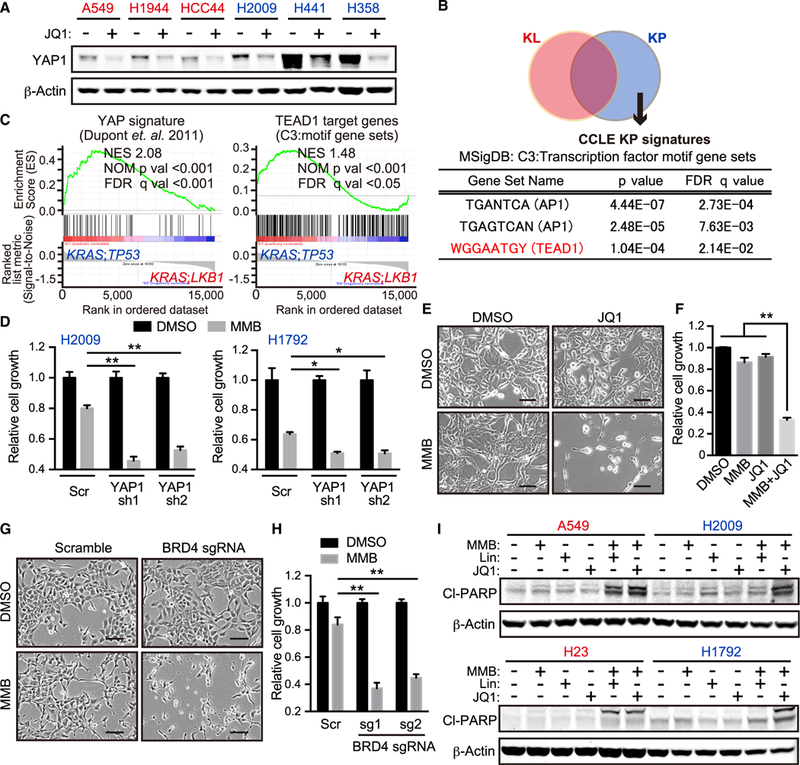
(A) IB of the indicated proteins in A549, H1944, HCC44, H2009, H441, and H358 cells treated with or without 500 nM JQ1 for 24 hr. Red cell lines, KL; blue cell lines, KP.
(B) Venn diagram showing differentially expressed genes between KL and KP in Cancer Cell Line Encyclopedia (CCLE). Analysis of transcription factor motif gene sets from Molecular Signatures Database (MSigDB) and significantly enriched in KP lung adenocarcinoma cell lines (See STAR Methods).
(C) Gene set enrichment analyses of published YAP1 signature (Dupont et al., 2011) (left) and TEAD1 target genes (WGGAATGY_V$TEF1_Q6) (right) in KRAS;TP53 versus KRAS;LKB1 lung adenocarcinomas (see STAR Methods). NES, normalized enrichment score; NOM, nominal; FDR, false discovery rate.
(D) Relative cell growth of H2009 and H1792 cells transduced with the indicated shRNA in the presence of 2.5 μM MMB for 96 hr (n = 3).
(E) Phase-contrast images of H2009 cells treated with 2.5 μM MMB and/or 200 nM JQ1 for 96 hr. Scale bars: 100 μm.
(F) Relative cell growth of MSR-A549 cells in the presence of 2.5 μM MMB and/or 200 nM JQ1 for 96 hr (n = 3).
(G) Phase-contrast images of H2009 transduced with the indicated sgRNA and treated with 5 μM MMB for 96 hr. Scale bars: 100 μm.
(H) Relative cell growth of H2009 transduced with the indicated sgRNA in the presence of 5 μM MMB for 96 hr (n = 3).
(I) IB of the indicated proteins in A549, H2009, H23, and H1792 cells treated with 2.5 μM MMB, 1 μM Lin, and/or 500 nM JQ1 for 24 hr. Red cell lines, KLand KLP; blue cell lines, KP.
All quantitative data are represented as mean ± SD; *p < 0.05, **p < 0.01; see also Figure S5 and Tables S2 and S3.
To elucidate whether higher YAP1 expression in KP cells contributes to intrinsic resistance to momelotinib, we established YAP1-depleted KP cells (Figure S5C). YAP1-depleted KP cells showed higher sensitivity to momelotinib or MEK inhibitor treatment compared with control cells (Figures 5D and S5D). Further-more, JQ1 treatment or BRD4 depletion strongly sensitized KP cells to momelotinib treatment in vitro (Figures 5E-5H and S5E). Combination treatment with momelotinib and JQ1 potently induced apoptosis both in KL and KP cells at the same level even though combination treatment with momelotinib and linsitinib induced apoptosis only in KL cells and not in KP cells (Figure 5I). These combination therapies did not significantly induce apoptosis in normal cells, such as mouse embryonic fibroblasts (Figure S5F). Taken together, these results suggested that higher YAP1 expression in KP cells at least partially contributes to intrinsic resistance to momelotinib and MEK inhibition, and that combination therapy with JQ1 might be broadly effective in both KL and KP backgrounds.
Intermittent BET Inhibitor Treatment Overcomes Resistance to Momelotinib/MEK Inhibition in KL and KP GEMMs
Since BET inhibitor treatment broadly antagonized acquired resistance to momelotinib and MEK inhibitor therapy in KL cells and intrinsic resistance in KP cells, we explored the potential of higher order combination therapy as a strategy to achieve durable therapeutic responses in vivo. While sustained JQ1 treatment in KL, KLP, and KP cells strongly enhanced cell death following momelotinib plus or minus trametinib in vitro (Figure S6A), we anticipated potential toxicity of this triple combination in vivo. We therefore tested the possibility that interruption of this transcriptional program by intermittent pulses of JQ1 might be equally effective. Indeed, we tested several different schedules of JQ1 in MSR-A549 cells and found that treatment every 2 days was sufficient to maintain IGF1 suppression (Figure S6B). We thus examined combination treatment by alternating daily JQ1 treatment with daily momelotinib and trametinib treatment in vitro, which fully suppressed cell growth in both KL and KP cells as compared with normal cells (Figure 6A).
Figure 6. Triple Combination Therapy Shows Durable Therapeutic Effect both in KL and KP GEMMs.
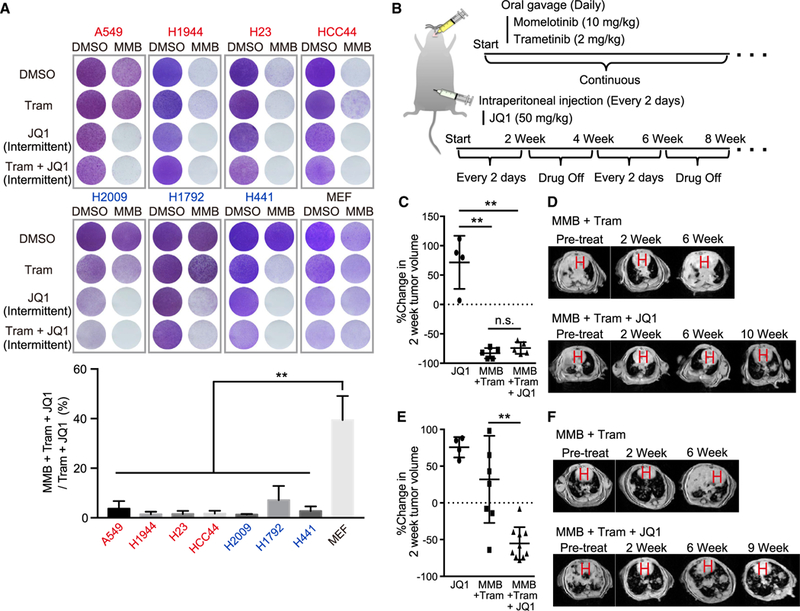
(A) Crystal violet staining of A549, H1944, HCC44, H23, H2009, H1792, H441, and mouse embryonic fibroblast (MEF) cells In the presence of 2.5 μM MMB and 10 nM Tram each day and/or 500 nM JQ1 every other day (intermittent) for 12 days (See STAR Methods). Red cell lines, KL and KLP; blue cell lines, KP (upper). Quantification of extracted crystal violet in MMB + Tram + JQ1 treated cells was normalized to Tram + JQ1 treated cells (n = 3) (lower).
(B) Schematic of in vivo MMB + trametinib + JQ1 combination therapy schedule in GEMMs.
(C) Percentage change in MRI tumor volume of LSL-KrasG12D;Lkb1f/f-induced lung cancer 2 weeks following JQ1, MMB + Tram, or MMB + Tram + JQ1 therapy. n.s., not significant.
(D) Representative MRI images from LSL-KrasG12D;Lkb1f/f mice treated with MMB + Tram therapy (upper) and MMB + Tram + JQ1 therapy (lower) for the indicated time. H, heart.
(E) Percentage change in MRI tumor volume of LSL-KrasG12D;Trp53f/f-induced lung cancer 2 weeks following JQ1, MMB + Tram, or MMB + Tram + JQ1 therapy.
(F) Representative MRI images from LSL-KrasG12D;Trp53f/f mice treated with MMB + trametinib therapy (upper) and MMB + trametinib + JQ1 therapy (lower) for the indicated time. H, heart.
All quantitative data are represented as mean ± SD; **p < 0.01; see also Figure S6.
Based on these in vitro findings, we treated both KL and KP GEMMs with momelotinib and trametinib every day together with an intermittent pulse schedule of JQ1 (Figure 6B), which did not result in significant body weight loss (Figure S6C). As expected, triple combination therapy induced marked tumor regressions in KLGEMM by 2 weeks without severe toxicity (Figures 6C and 6D). Because momelotinib and trametinib combination therapy already induced strong tumor regressions in the KL GEMM, there was no significant difference between two- and three-drug combinations at that point (Figure 6C). However, in contrast to two-drug therapy, triple combination therapy inhibited tumor volume below baseline even after 10 weeks in the KL GEMM (Figure 6D). In addition, triple combination therapy achieved deep tumor regressions by 2 weeks of treatment in the KP GEMM, with prolonged tumor shrinkage up to 9 weeks of therapy, even though KP GEMMs showed intrinsic resistance to momelotinib and trametinib dual treatment (Figures 6E and 6F). These findings reveal that inhibiting transcriptional adaptation to MEK and innate immune pathway inhibition can result in durable responses in difficult-to-treat mouse KRAS lung cancer models.
Development of Combination Therapy with Optimized Inhibitors
Recently, more potent and specific TBK1 (compound 1) and BET (GS-626510) inhibitors have been described (Jenkins et al., 2018; Shi et al., 2016). We therefore utilized an aggressive KL patient-derived xenograft (PDX) model (DFCI-366) to design a combination therapy schedule for potential future translation to the clinic. First, we confirmed that GS-626510 treatment suppressed IGF1 and YAP1 activation in MSR-A549 and also decreased high levels of endogenous YAP1 expression in KP cells in vitro (Figures S7A-S7C). We next compared the relative efficacy of each single-agent, double combination, ortriplecombination therapy in the KL PDX model, at doses guided by prior pharmacokinetic (PK)/pharmacodynamic (PD) and tolerability studies (Figures 7A and S7D). Although we observed a statistically significant benefit of compound 1 and trametinib treatment over trametinib alone, concurrent triple combination therapy failed to provide benefit, despite being well tolerated (Figures 7B and 7C). We therefore considered the possibility of drug-drug interaction (DDI) and/or subtherapeutic dosing of GS-626510 in NSG mice. Indeed, a single-dose PK study confirmed suboptimal dosing of 10 mg/kg (data not shown), prompting a PD study that identified 40 mg/kg as the minimum daily dose that completely inhibited Ccr2 expression in blood, which returned to baseline after 24 hr (Figures 7D and 7E). To minimize any effect of DDI, we tested an alternating doublet strategy, including 2 days of compound 1 and trametinib alternating with 1 day GS-626510 and trametinib guided by effective suppression of feedback IGF1 induction with this schedule in vitro (Figures 7F and S7E). This combination approach in particular was especially effective, causing over 90% tumor growth inhibition with sustained benefit even after drug withdrawal at 3 weeks (Figures 7G, 7H, and S7F). Moreover, treatment was well tolerated with only minor transient weight loss (Figure S7G). Together, these data confirm the clinical potential of using intermittent pulse BET inhibitor therapy to sustain response to dual innate immune and MAPK pathway inhibition.
Figure 7. Development of Combination Therapy Using Optimized TBK1 and BET Inhibitors in KL PDX Model.
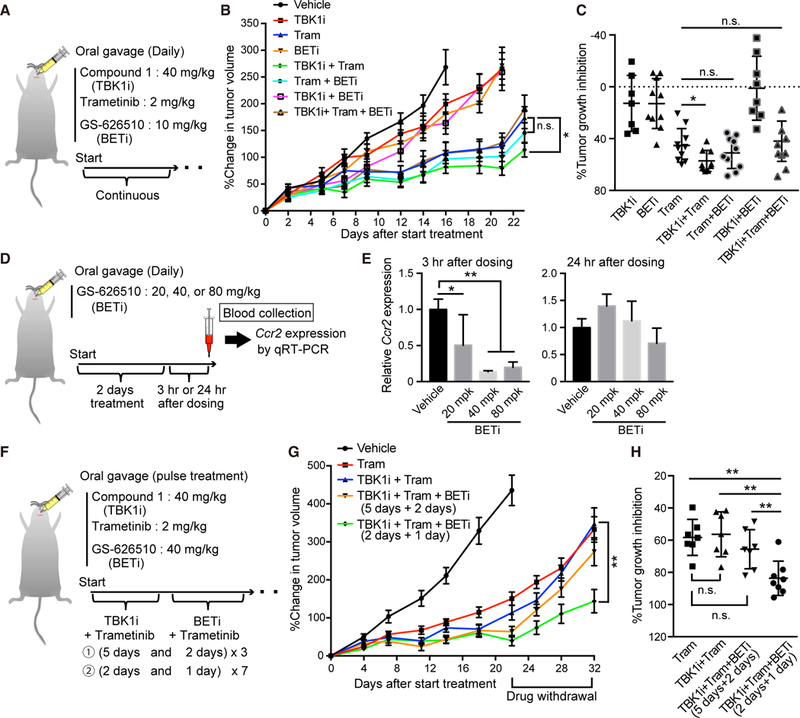
(A) Schematic of in vivo compound 1 (TBK1 inhibitor) + trametinib + GS-626510 (BET inhibitor) combination therapy schedule in KL PDX model. BETi, BET inhibitor.
(B) Percentage change in tumor volume over time of DFCI366 PDX following daily treatment of vehicle, 40 mg/kg compound 1 ± 2 mg/kg Tram ±10 mg/kg GS-626510. n.s., not significant (each group, n = 10).
(C) Percentage tumor growth inhibition at the end of treatment of DFCI366 PDX following each therapy for 3 weeks. n.s., not significant.
(D) Schematic of pharmacodynamics (PD) study of GS-62510 in NSG mice. (E) qRT-PCR of Ccr2 in blood samples derived from mice treated with the indicated amount of GS-626510 (n = 6). mpk, mg/kg.
(F) Schematic of alternating doublet compound 1, trametinib, and GS-626510 combination therapy schedules in KL PDX model.
(G) Percentage change in tumor volume over time of DFCI366 PDX following pulse treatment of 40 mg/kg compound 1, 2 mg/kg Tram, and/or 40 mg/kg GS-626510 in accordance with the indicated schedule (See Figure 7F) (each group, n = 8).
(H) Percentage tumor growth inhibition after drug withdrawal of DFCI366 PDX following each therapy for 3 weeks and then 10 days off treatment. n.s., not significant.
All quantitative data are represented as mean ± SD; *p < 0.05, **p < 0.01; see also Figure S7.
DISCUSSION
Despite decades of effort, effective targeted therapies for oncogenic KRAS-driven malignancies remain to be identified. While downstream pathway-targeted therapy represents a rational approach, single agents or dual combinations have been limited by escape pathways or dose-limiting toxicities (Stephen et al.,2014). Here we follow up on an approach that targets downstream RAL and MEK signaling but leaves PI3K and other pathways unchecked. Thus, despite initial efficacy in aggressive murine Kras mutant lung cancer GEMMs, momelotinib and rametinib therapy fails to achieve durable responsiveness. By focusing on LKB1 -deficient KRAS-driven models that are particularly sensitive to this combination, we uncovered YAP1 pathway activation as a key component of resistance programs in both KL and KP models. Moreover, interruption of YAP1 signaling and additional transcriptional adaptation through pulsed BET inhibitor treatment limits the ability of cells to develop the plasticity required to escape dual therapy, andresults in prolonged preclinical activity in challenging therapeutic models.
YAP1 is a key effector of Hippo signaling, and hyperactivation of the YAP1 pathway has been implicated in a variety of human cancers (Moroishi et al., 2015). Of note, recent studies have established that YAP1 activation bypasses oncogenic Kras addiction in both NSCLC and PDAC models (Garcia-Rendueles et al., 2015; Kapoor et al., 2014; Lin et al., 2015; Shao et al., 2014). In one study, a systematic rescue screen of cellular survival following KRAS suppression in a KRAS-dependent colorectal cancer cell line identified YAP1 as a dominant mediator of resistance (Shao et al., 2014). These results were confirmed in the KP lung cancer GEMM, and converged upon activation of the epithelial to mesenchymal transition. In the other study, tumors that arose after suppression of mutant KRAS expression in a PDAC GEMM were found to have YAP1 amplification (Kapoor et al., 2014). Activation of a YAP-TEAD transcriptional program together with E2F mechanistically fueled growth in place of KRAS signaling. Together, these studies anticipate the findings reported here, whereby robust inhibition of multiple downstream KRAS signaling pathways might be predicted to uncover a similar resistance program. Indeed, this study provides evidence that multitargeted KRAS pathway therapy enforces YAP1 activation as a means of escape from inhibition.
We further determined that LKB1 or TP53 status dictates important differences in KRAS signaling and resistance to pathway-targeted therapy. While it has been known for some time that loss of these tumor suppressors alters the phenotypic outcome of KRAS-driven tumorigenesis in GEMMs (Chen et al., 2012), it has recently become clear that LKB1 loss has a unique impact on the tumor immune microenvironment and comprises a distinct subclass of human NSCLC tumors (Koyama et al., 2016; Skoulidiset al., 2015). The observation that proliferation and survival of KL cells are dependent on innate immune cytokine inhibition is consistent with recent work that highlighted a critical role for IL-6 in GEMMs (Koyama et al., 2016). LKB1 loss impairs autophagy, which negatively regulates pTBK1, likely contributing to this effect and potentially explaining the preferential dependence of KL cells on momelotinib (Yang et al., 2016). We further discovered that feedback activation of innate immune signaling following the inhibition of the MEK and/or PI3K pathway is particularly pronounced in KL cells. Previous reports have indicated that feedback activation of innate immune cytokines promotes drug resistance to RTK, MEK, and RAF inhibitors (Korkaya et al., 2012; Lee et al., 2014; Sos et al., 2014), consistent with the relative resistance to MEK inhibitor treatment in KL cells compared with KP cells.
On the other hand, the acquired resistance program we observed in KL cells was also uniquely driven by a TIS response (Obenauf et al., 2015), in addition to YAP1. Although innate immune cytokines remained suppressed in MSR-A549 cells, several other secreted factors related to the TIS such as IGF1 and WNT5A were highly upregulated. In addition, several RTKs themselves, such as c-KIT, were upregulated in MSR-A549 cells, suggesting the potential for ligand-independent activation of RTK signaling (Stratikopoulos et al., 2015; Stuhlmiller et al.,2015). Activation of many of these pathways has been implicated in resistance to kinase-targeted cancer therapy in KRAS-driven NSCLC and other types of cancer cells (Anastas et al., 2014; Ebi et al., 2011; Molina-Arcas et al., 2013). These data suggest the need to shut off a broad array of ligand-dependent and -independent kinases simultaneously to overcome resistance. For example, while IGFI-dependent IGF1R/AKT activation directly contributed to acquired resistance to combination therapy in KL cells, isolated inhibition of the IGF1R/AKT pathway was not sufficient to prevent acquisition of resistance over time.
Although more selective IGF1R/AKT and YAP1 inhibitors are being developed, this TIS response poses a challenge to pharmacologic targeting of the multifaceted output. However, the observation that this coordinated response is driven by transcriptional reprogramming highlights the potential of targeting YAP1 and the TIS at the root via BET inhibition. Indeed, several recent papers have demonstrated that transcriptional reprogramming during adaptive bypass of kinase-targeted therapies, including MEK and PI3K inhibition, is susceptible to pharmacological targeting, preventing cancer cells from acquiring resistance (Stratikopoulos et al., 2015; Zawistowski et al., 2017). Consistent with these and other reports, elevated histone H3K27 acetylation, a marker of active transcription, was enriched in many of resistance-related genes, including IGF1 and YAP1 target genes during acquisition of resistance to combination therapy. Thus, inhibition of BET family proteins, which recognize H3K27 acetylated histones and activate transcription, by JQ1 treatment clearly suppressed acquisition of resistance to combination therapy in A549 cells. Furthermore, JQ1 treatment rapidly decreased expression of resistance-related genes in MSR-A549, re-sensitized MSR-A549 cells to momelotinib treatment, and significantly attenuated acquisition of resistance in the highly aggressive KL GEMM.
Although KP cells failed to induce IL-6 and lacked this secreted response, we instead found that they expressed elevated YAP1 activity at baseline. These findings suggest that inactivation of LKB1 or TP53 promotes distinct paths for KRAS adaptability to downstream pathway inhibition. In the former scenario, YAP1 activity is low, and cells are particularly reliant on innate immune cytokines as an adaptive response, but, when these are suppressed together with MEK inhibition, YAP1 and the TIS resistance program emerges. In the setting of TP53 inactivation, this innate immune program and secreted factors such as IGF1 remain low, but YAP1 signaling is engaged and promotes intrinsic resistance to downstream pathway therapy on its own. Thus, while existing YAP1 inhibitors such as verteporfin (Liu-Chittenden et al., 2012) are limited by non-specific cytotoxic effects, it is possible that newer highly selective and potent YAP1 inhibitors may reveal preferential synergy with momelotinib in KP cells. On the other hand, JQ1 treatment sensitized KP cells to momelotinib treatment to a greater degree than YAP1 depletion by small hairpin RNA, despite equivalent YAP1 suppression, suggesting that intrinsic resistance to momelotinib treatment in KP cells may also depend on additional transcriptional factors beyond YAP1 alone.
Finally, we also used recently described potent and selective TBK1 and BET inhibitors (Jenkins et al., 2018; Shi et al., 2016) as tool compounds to develop a therapeutic strategy with translatable potential for KRAS-driven NSCLC patients. The alternating doublet strategy minimizes the toxicity of inhibiting each target, limits DDI, and also provides a faster path to the clinic compared with triple combination therapies that require complex dose escalation schedules. Thus, KRAS pathway-targeted therapy that inhibits innate immune cytokines, suppresses MEK signaling, and accounts for this adaptive transcriptional response could potentially translate into effective combination therapy for these refractory tumors.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
For further information and requests for reagents generated in this study, please contact David A. Barbie (dbarbie@partners.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient-Derived Xenograft Model
DFCI 366 (KRASG12A; LKB1A205fs) is a patient derived xenograft model of lung cancer established at the Belfer Center at Dana Farber Cancer Institute. The human PDX tumor sample used to generate this PDX was collected and analyzed according to Dana-Farber/ Harvard Cancer Center IRB-approved protocol 02–180. Informed consent was obtained from the patient, and studies were conducted according to the Declaration of Helsinki and approved by the DFCI IRB. Tumor cells from a lung cancer patient were implanted subcutaneously in Matrigel® (BD BioSciences, Bedford, MA) in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (Jackson Labs Stock No: 00557; NSG). The resulting tumor was expanded and frozen tumor fragments were stored under liquid nitrogen.
Animal Studies
All mouse experiments were conducted in accord with a Dana-Farber Cancer Institute and NYU Langone Medical Center Institutional Animal Care and Use Committee (IACUC) approved protocol. Lung tumors were induced in LSL-KrasG12D/WT;Lkb1Ftox/Ftox or LSL-KrasG12D/WT;Trp53Ftox/Ftox mice by intra-nasal adenoviral Cre and measured by serial MRI as described (Chen et al., 2012). Momelotinib was dissolved in 0.5% methylcellulose, 0.4% polysorbate-80 at 1 mg/mL for daily oral gavage dose of 10 mg/kg. Trametinib was dissolved in 0.5% hydroxypropyl methyl cellulose (HPMC) and dose was 2 mg/kg daily by oral gavage. Linsitinib was dissolved in HPMC and dose was 7.5 mg/kg daily by oral gavage. JQ1 was dissolved in 10% DMSO and 10% cyclodextrin with water and dose was 50 mg/kg every two days by intraperitoneal injection. Passage 3 DFCI 366 tumors were used for all experiments. Briefly, tumor fragments (2 mm3) in Matrigel® were implanted in subcutaneously NSG mice using an 11 gauge trocar needle. Tumors were measured by caliper, allowed to grow to ~100 mm3 and were then grouped for each arm of the study. Trametinib was formulated 0.5% HPMC + 0.2% Tween 80; the TBK1i, Compound 1, was formulated in 0.5% HPMC, 0.4% Tween 80 and 99.1% 0.05 N HCl and the BETi, GS-626510, was formulated in 10% ethanol, 40% PEG300, 50% water. All compounds were dosed by oral gavage as indicated.
Cell Lines
A549, H2009 and HEK293T cells were cultured in DMEM (Thermo Fisher Scientific, Cat.# 11965–118) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-products, Cat# 100–106) and 1x penicillin-streptomycin (Gemini Bio-products, Cat# 400–109), and H1944, HCC44, H23, H1355, H2030, H2122, H1792, H441 and H358 cells were cultured in RPMI 1640 (Thermo Fisher Scientific, Cat.# 11875–119) supplemented with 10% FBS and 1x penicillin-streptomycin. A549, H1944, HCC44, H23, H2030, H1355, H2122, H1792 and H2009 cells were authenticated by short tandem repeats genotyping. HEK293T, H441 and H358 were purchased from ATCC and used for all experiments before reaching 10 passages. Primary MEFs were prepared as described previously (Kitajima et al., 2015) from day E14 embryo, maintained in RPM11640 supplemented with 10% FBS, and were used for all experiments before reaching 10 passages. Mycoplasma infection was regularly checked by PCR using the conditioned media derived from each cell line. The sequences of the primers used for checking mycoplasma infection are listed in Table S4.
Establishment of Momelotinib/Selumetinib Resistant (MSR)-A549 Cells
To establish MSR-A549 cells, A549 cells were cultured in the presence of 5 μM MMBand 1 μM selumetinib or in growth media (DMEM supplemented with 10% FBS) alternately for 2 months. (See Figure 2D regarding accurate schedule). After 2 months culture, MSR-A549 cells were maintained with 5 μM momelotinib and 1 μM selumetinib continuously. MSR-A549 withdrawal cells were established from MSR-A549 cells cultured without MMB and selumetinib treatment for 10 days. MSR-A549 re-addition cells were established from MSR-A549 withdrawal cells cultured with 5 μM MMB and 1 μM selumetinib treatment for 12 days.
METHOD DETAILS
ELISA
Human IL-6and IGF1 ELISAs (R&D systems) were performed according to manufacture’s instructions. Conditioned media from each cell lines was collected after 24 hr culture in the presence of the indicated concentration of drugs.
Immunoblotting
Cells were lysed in RIPA buffer containing 1x protease inhibitors (Roche, Cat# 11–836-145–001) and phosphatase inhibitors (50 mM NaF and 100 mM Na3VO4). Immunoblotting was performed as described (Zhu et al., 2014a). See the Key Resources Table for primary antibodies used. Secondary antibodies were from LICOR Biosciences: IRDye 680LT Goat anti-Mouse IgG (#926–68020), IRDye 800CW Goat anti-Rabbit IgG (#926–32211). HIKARI Signal Enhancer Solutions 1 (Nacalai USA, Inc. # NU00101) and Solution 2 (Nacalai USA, Inc. # NU00102) was used to dilute primary and secondary antibodies. Imaging of blots and quantitation of bands was performed using the LICOR Odyssey system.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-cleaved PARP | Cell signaling technology | Cat# 5625S; RRID: AB_10699459 |
| Mouse monoclonal anti-β-actin | Cell signaling technology | Cat# 3700S; RRID: AB_2242334 |
| Rabbit monoclonal anti-LKB1 | Cell signaling technology | Cat# 3047S; RRID: AB_2198327 |
| Rabbit monoclonal anti-phospho IGF1R β (Tyr1135) | Cell signaling technology | Cat# 3918S; RRID: AB_10548764 |
| Rabbit monoclonal anti-IGF1R β | Cell signaling technology | Cat# 9750; RRID: AB_10950969 |
| Rabbit monoclonal anti-IGF1 | Abcam | Cat# ab9572; RRID: AB_308724 |
| Rabbit monoclonal anti-phospho IRS1 (Ser307) | Cell signaling technology | Cat# 2381S; RRID: AB_330342 |
| Rabbit monoclonal anti-IRS1 | Cell signaling technology | Cat# 3407S; RRID: AB_2127860 |
| Rabbit monoclonal anti-phospho S6K (Thr389) | Cell signaling technology | Cat# 9205S; RRID: AB_330944 |
| Rabbit monoclonal anti-S6K | Cell signaling technology | Cat# 2708S; RRID: AB_390722 |
| Rabbit monoclonal anti-YAP1/TAZ | Cell signaling technology | Cat# 8418S; RRID: AB_10950494 |
| Rabbit monoclonal anti-YAP1 | Cell signaling technology | Cat# 14074S; RRID: AB_2650491 |
| Rabbit monoclonal anti-phospho AKT (Ser473) | Cell signaling technology | Cat# 4060S; RRID: AB_2315049 |
| Rabbit polyclonal anti-AKT | Cell signaling technology | Cat# 9272S; RRID: AB_329827 |
| Rabbit monoclonal anti-phospho STAT3 (Tyr705) | Cell signaling technology | Cat# 9145S; RRID: AB_2491009 |
| Rabbit monoclonal anti-STAT3 | Cell signaling technology | Cat# 4904S; RRID: AB_331269 |
| Rabbit polyclonal anti-phospho Erk1/2 (Thr202/Tyr204) | Cell signaling technology | Cat# 9101S; RRID: AB_331646 |
| Rabbit polyclonal anti-Erk1/2 | Cell signaling technology | Cat# 9102S; RRID: AB_330744 |
| Rabbit polyclonal anti-histone H3 (acetyl K27) ChIP grade | Abcam | Cat# ab4729; RRID: AB_2118291 |
| ChromPure Rabbit IgG, whole moleculre (control IgG for ChIP assay) | Jackson ImmunoResearch | Cat# 011–000-003; RRID: AB_2337118 |
| Biological Samples | ||
| Patient-derived xenografts (PDX) | Belfer Center at Dana Farber Cancer Institute | http://belfercenter.dfci.harvard.edu/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Momelotinib | Shanghai Haoyuan Chemexpress Co. Ltd. | Cat# HY-10961, CAS#: 1056634–68-04 |
| Trametinib | Selleck chemicals | Cat# S2673, CAS#: 871700–17-3 |
| Selumetinib | Selleck chemicals | Cat# S1008, CAS#: 606143–52-6 |
| Buparlisib | Chemietek | Cat# CT-BKM120, CAS#: 944396–07-0 |
| Ruxolitinib | Selleck chemicals | Cat# S1378, CAS#: 941678–49-5 |
| Linsitinib | Selleck chemicals | Cat# S1091, CAS#: 867160–71-2 |
| JQ1 | Gifted from Dr. James E. Bradner | CAS#: 1268524–70-4 |
| Recombinant human IGF1 | R&D systems | Cat# 291-G1–200 |
| Compound 1 | Gilead Sciences | N/A |
| GS-626510 | Gilead Sciences | N/A |
| Critical Commercial Assays | ||
| Human IL-6 Quantikine ELISA kit | R&D systems | Cat# D6050 |
| Human IGF1 Quantikine ELISA kit | R&D systems | Cat# DG100 |
| Human phosphor-RTK array kit | R&D systems | Cat# ARY001B |
| CellTiter-Glo luminescent cell viability assay kit | Promega | Cat# G7572 |
| High sensitivity DNA kit | Agilent Technologies | Cat# 5067–4626 |
| Qubit dsDNA HS assay kit | Thermo Fisher Scientific | Cat# Q32851 |
| Deposited Data | ||
| RNA-seq in parental A549 and MSR-A549 cells | This paper | GSE96779 |
| Histone H3K27 acetylation ChIP-seq in parental A549 and MSR-A549 cells |
This paper | GSE96780 |
| Experimental Models: Cell Lines | ||
| Human: A549 | ATCC | CCL-185 |
| Human: NCI-H1944 | ATCC | CRL-5907 |
| Human: HCC44 | Broad Institute | N/A |
| Human: NCI-H23 | ATCC | CRL-5800 |
| Human: NCI-H1355 | ATCC | CRL-5865 |
| Human: NCI-H2122 | ATCC | CRL-5985 |
| Human: NCI-H2030 | ATCC | CRL-5914 |
| Human: NCI-H2009 | ATCC | CRL-5911 |
| Human: NCI-H1792 | ATCC | CRL-5895 |
| Human: NCI-H441 | ATCC | HTB-174 |
| Human: NCI-H358 | ATCC | CRL-5807 |
| Human: HEK293T | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: LSL-KrasG12D/WT; Lkb1flox/flox | Ji et al., 2007 | N/A |
| Mouse: LSL-KrasG12D/WT; Trp53flox/flox | Chen et al., 2012 | N/A |
| Mouse: NOD.Cg-Prkdcscid II2rgtm1WJl/SzJ | The Jackson Laboratory | Stock No. 00557 |
| Recombinant DNA | ||
| pLX304-GFP | Rosenbluh et al., 2012 | N/A |
| pLX304-human LKB1 | This paper | N/A |
| pLX304-human YAP1 5SA | Rosenbluh et al., 2012 | N/A |
| pXPR_BRD111 (Cas9) | Broad Institute | N/A |
| plentiGuide-puro-scramble sgRNA control | Broad Institute | N/A |
| plentiGuide-puro-human LKB1 sgRNA1, see Table S4 | This paper | N/A |
| plentiGuide-puro-human LKB1 sgRNA2, see Table S4 | This paper | N/A |
| pCRISPRv2-puro-scramble sgRNA control | Broad Institute | N/A |
| pCRISPRv2-puro-human BRD4 sgRNA1, see Table S4 | This paper | N/A |
| pCRISPRv2-puro-human BRD4 sgRNA2, see Table S4 | This paper | N/A |
| pLKO.1-Non-Target shRNA control | Sigma-Aldrich | SHC002 |
| pLKO.1-human YAP1 shRNA1, see Table S4 | Rosenbluh et al., 2012 | N/A |
| pLKO.1-human YAP1 shRNA2, see Table S4 | Rosenbluh et al., 2012 | N/A |
| pCMV-dR8.91 (lentivirus packaging) | Broad Institute | N/A |
| pCMV-VSV-G | Broad Institute | N/A |
| Oligonucleotides | ||
| Primers for qRT-PCR, see Table S4 | This paper | N/A |
| Taqman probe for human IGF1 (Hs01547656_m1) | Thermo Fisher Scientific | Cat# 4331182 |
| Taqman probe for human YAP1 (Hs00902712_g1) | Thermo Fisher Scientific | Cat# 4331182 |
| Taqman probe for mouse Ccr2 (Mm99999051_gH) | Thermo Fisher Scientific | Cat# 4331182 |
| Primers for ChIP-qPCR, see Table S4 | This paper | N/A |
| Human negative control primer set 1 (for ChIP) | Active Motif | Cat# 71001 |
| Software and Algorithms | ||
| GraphPad Prism7 | GraphPad Software, Inc. | http://www.graphpad.com/ |
| R version 3.2.2 | The Comprehensive R Archive Network | https://cran.r-project.org/ |
| TCC, an R package | Sun et al., 2013 | http://www.iu.a.u-tokyo.ac.jp/~kadota/TCC/ |
| sgRNA designer | Broad Institute | http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| IGV, Integrative genomics viewer | Broad Institute | http://software.broadinstitute.org/software/igv/ |
| GSEA, Gene set enrichment analysis | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| Tophat | Johns Hopkins University | https://ccb.jhu.edu/software/tophat/index.shtml |
| Bowtie2 | Johns Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Cufflinks | University of Washington | http://cole-trapnell-lab.github.io/cufflinks/ |
| MAnorm | Dana-Farber Cancer Institute | http://bcb.dfci.harvard.edu/~gcyuan/MAnorm/MAnorm.htm |
| MACS, Model-based Analysis for ChIP-Seq | Dana-Farber Cancer Institute | http://liulab.dfci.harvard.edu/MACS/ |
| BETA, Binding and Expression Target Analysis | Dana-Farber Cancer Institute | http://cistrome.org/BETA/ |
Generation of Lentivirus
2 × 106 HEK293T cells were plated onto 60 mm dish and transfected using X-tremeGENE HP DNA Transfection Reagent (Roche, Cat.# 06366236001) with 1.5 μg of lentivirus-based expression vectors (See Key Resources Table) together with 1.5 μg of pCMV-dR8.91 and 1.5 μg of pCMV-VSV-G. After 48 hr incubation, the media containing lentivirus particles were collected, passed through a 0.45 μm filter, and concentrated using Lenti-X Concentrator (Clontech, Cat# 631231). Lentiviral infection of ORFs, sgRNAs and shRNAs was performed as described (Zhu et al., 2014a). For selection of virally infected cells, 0.5–2 μg/ml of Puromycin (pLKO.1 and plentiGuide-puro) or 3–8 μg/ml of Blasticidin (pLX304 and pXPR_BRD111) was used 24 hr post infection.
CRISPR/Cas9 System
Target sequences for CRISPR interference were designed using the sgRNA designer (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). A non-targeting sgRNA from the Gecko library v2 was used as a scramble sgRNA. sgRNA target sequences are listed in Table S4 plentiGuide-puro or pCRISPRv2-puro vectors were cloned as previously described (Sanjana et al., 2014; Shalemetal., 2014).
Quantitative RT-PCR
RNA extraction from cells was performed using RNeasy Mini Kit (Qiagen, Cat.# 74106). RNA extraction from mouse blood samples was performed using RNA protect animal blood tubes (Qiagen, Cat.# 76544) and RNeasy protect animal blood kit (Qiagen, Cat.# 73224). RNA samples (1 μg) were reverse-transcribed using SuperScript® III First-Strand Synthesis SuperMix (Thermo Fisher Scientific, Cat.# 1683483). Quantitative real-time PCR was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Cat.# 4367659) and TaqMan Universal PCR Master Mix (Thermo Fisher Scientific, Cat.# 4304437). The sequences of the primers used for qRT-PCR are listed in Table S4. The values represent the average of four replicates from at least two independent experiments.
Cell Viability Assay
1500–4000 cells were plated onto 96-well plates, and incubated with growth media containing drugs as indicated for 72 or 96 hr. Values of CellTiter-Glo Luminescent Cell Viability assay (Promega) after 72 or 96 hr were normalized to vehicle treated cells. Plates were read on a Tecan Infinite M200 Pro plate reader and analysis was performed using Prism7 (GraphPad Software). All conditions were tested in triplicate. The values represent the average of three replicates and a representative experiment from at least two independent experiments.
Phospho-Receptor Tyrosine Kinase (RTK) Array
Human phospho-RTK arrays were performed according to manufacture’s instructions. Cells were lysed in RIPA buffer containing 1× protease inhibitors (Roche, Cat# 11–836-145–001) and phosphatase inhibitors (20 mM NaF and 100 mM Na3VO4) after 24 hr culture in the presence of the indicated drug.
Chromatin Immunoprecipitation (ChIP) and ChIP-seq
1 × 107 cells were crosslinked by 1% paraformaldehyde in fixing buffer (50 mM HEPES pH 7.5,100 mM NaCl, 1 mM EDTA pH 8.0) for 5 min at room temperature, and then quenched the crosslinking reaction by 1.25 M glycine. Cells were washed once with cold phosphate-buffered saline (PBS), collected into tube by centrifugation, washed once with cold PBS again, and then resuspened in lysis buffer (140 mM NaCl, 50 mM HEPES pH 8.0,1 mM EDTA pH 8.0,10% Glycerol, 0.5% NP-40, 0.25% Triton X-100,1 x protease inhibitor, 1 x phosphatase inhibitor). The subsequent cell pellet was washed with wash buffer (200 mM NaCl, 10mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0,1 x protease inhibitor, 1 x phosphatase inhibitor), resuspended 1 ml shearing buffer (0.1% SDS, 10 mM Tris-HCl pH 8.0,1 mM EDTA pH 8.0,1 x protease inhibitor, 1 x phosphatase inhibitor) and sonicated with Covaris LE220. After DNA shearing, 110 μl 10% Triton X-100 and 33 μl 5 M NaCl were added to 1 ml sonicated lysates. Then lysates were incubated with 2 μg rabbit antihistone H3K27 acetylation antibody (Abcam) overnight at 4°C, and then incubated with 20 μl PureProteome Protein G Magnetic Beads (Millipore, Cat.# LSKMAGG02) for 2 hours at 4°C. Beads were washed once with low salt immune complex buffer (150 mM NaCl, 0.1% SDS, 20 mM Tris-HCl pH 8.0, 1% Triton-X100, 2 mM EDTA pH 8.0), high salt immune complex buffer (500 mM NaCl, 0.1% SDS, 20 mM Tris-HCl pH 8.0, 1% Triton-X100, 2 mM EDTA pH 8.0), LiCl immune complex buffer (250 mM LiCl, 10 mM Tris-HCl pH 8.0, 1% NP-40, 1% Sodium Deoxycholate, 1 mM EDTA pH 8.0) and then TE buffer. Cross-links were reversed overnight at 65°C. RNA and protein were digested using 200 μg/ml RNase A (Thermo Fisher Scientific, Cat.# EN0531) and 200 mg/ml Proteinase K (Thermo Fisher Scientific, Cat.# E00491) respectively, and DNA was purified with phenol chloroform extraction and ethanol precipitation. Quality of DNA sharing was analyzed by High Sensitivity DNA Chips (Agilent Technologies, Cat.# 5067–4626) according to manufacture’s instruction. Total amount of ChIPed DNA was analyzed by Qubit dsDNA HS assay kit (Thermo Fisher Scientific, Cat.# Q32851) according to manufacture’s instruction. The sequences of the primers used for ChIP-qPCR are listed in Table S4. The values represent the average of two technical replicates and a representative experiment from at three independent experiments (biological replicates). Libraries were prepared using Rubicon Genomics Thruplex-DNAseq sample preparation kits from 2 ng immunoprecipitated DNA according to manufacturer’s protocol. The finished double strand DNA (dsDNA) libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200, and RT-qPCR using the Kapa Biosystems library quantification kit according to manufacturer’s protocols. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq500 with single-end 75bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities.
RNA Preparation and RNA-seq
RNA extraction was performed using RNeasy Mini Kit (Qiagen, Cat.# 74106) and PerfectPure RNA cultured cell Enzyme Set DNase (5 PRIME, Cat.# 2900315). Libraries were prepared using Illumina TruSeq Stranded mRNA sample preparation kits from 500 ng of purified total RNA according to the manufacturer’s protocol. The finished dsDNA libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200, and qRT-PCR using the Kapa Biosystems library quantification kit according to manufacturer’s protocols. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq500 with single-end 75bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities.
Data Analysis
Sequenced reads from histone H3K27 acetylation ChIPed DNA were aligned to hg19 using Bowtie2 with -k mode 1 and enriched ChIP peaks were called by MACS algorithm at the threshold of p < 1×10-5. Peak signals from two sets of the ChIP peaks (MSR-A549 cells vs. A549 cells) were normalized using MAnorm algorithm and 4,251 peaks were identified to be significantly increased on MSR-A549 cells at the arbitrary threshold at p < 1×10-25. Sequenced reads from RNA-seq were aligned with Tophat aligner to UCSC Refgene hg19. Transcripts were quantified using Cufflinks and differentially expressed transcripts were identified by Cuffdiff. Those 4,251 peaks with increased signal upon resistance were assigned to genes potentially contributed for its expression using Citrome BETA tool. Of those genes, 986 genes were identified to have their expression also significantly increased in MSR-A549 cells to momelotinib/selumetinib treatment at FDR q < 0.25. From the resulting genes, 659 genes were identified by selecting genes with the average of RPKM in MSR-A549 cells > 0.1 and fold change of average RPKM (MSR-A549 cells/A549 cells) > 1.5, and shown in Table S1 as resistance-related genes. Lung adenocarcinoma cell lines in CCLE repository are subdivided into 2 classes, KP (n = 9, H2009, H358, H1792, CALU6, H441, RERFLCAD2, HCC2108, HCC1171 and H2291) harboring KRAS and TP53 mutation/deletion with intact LKB1, and KL (n = 10, H1734, HCC44, H647, H2122, H1573, H1355, A549, H2030, H23 and H1944) harboring KRAS and LKB1 mutation, respectively. The RPKM values for each cell line was obtained from CCLE repository, and then 386 differentially expressed genes between KP and KL cell lines (p < 0.05 and FDR q < 0.25) were identified using R platform and TCC package (Sun et al., 2013). Of those genes, 168 genes were upregulated specifically in KP cell lines (M > 0, A > 0), and shown in Figure 7B (Top 30 genes) and Table S2 (168 genes). The level3 RNA-seq V2 datasets for lung adenocarcinoma samples were downloaded from TCGA data portal and classified into KP samples or KL samples according to their mutation status (See Table S3). Samples having mutations on both LKB1 and TP53 were excluded. TCGA ID: TCGA-78–7160,TCGA-78–7166 andTCGA-78–7540 were further eliminated because they were previously identified as a different subtype from KP or KL (Skoulidis et al., 2015). Then differentially expressed genes between KP samples (n = 21) and KL samples (n = 17) were analyzed by Gene Set Enrichment Analysis (GSEA) with YAP1 signature (Dupont et al., 2011) and MSigDB C3 signature TEAD1 target.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 10 minutes and washed by PBS 3 times. Cells were then blocked and permeabilized with 1% BSA and 0.3% Triton X-100 in PBS for 1 hour at room temperature, and incubated with rabbit polyclonal anti-YAP1 (Cell signaling technologies). Cells were then stained with Goat anti-rabbit IgG conjugated Alexa Fluor 555 (Thermo Fisher Scientific, Cat.# A-21429) and mounted with Prolong Gold antifade reagent with DAPI (Thermo Fisher Scientific, Cat.# P-36935).
Crystal Violet Staining
1×105 cells were plated onto 6-well type plates, and then cultured in the presence of MMB, Tram, and/or JQ1 for 12 days. Media was changed every day with the indicated drugs. For JQ1 treatment, this was only included in the media on alternating days, to mimic intermittent JQ1 exposure in vivo. After 12 days of culture, cells were fixed with 4% paraformaldehyde, and then stained with 0.1% crystal violet as previously described (Zhu et al., 2014a). After washing excessive dye, crystal violet was extracted by using 10% acetic acid for 20 min incubation with shaking, diluted in water as necessary, and then measured at 590 nm in a spectrometer.
Statistical Analysis
Statistical significance was assessed using unpaired two-tailed Student’s t-test, one-way ANOVA followed by Tukey’s post-hoc test, or two-way ANOVA followed by Tukey’s or Sidak’s post-hoc test. p values less than 0.05 were considered significant. Asterisks used to indicate significance correspond with: *p<0.05, **p<0.01. Columns represent means ± standard deviation (SD). In one-way or two-way ANOVA followed by post-hoc tests, we showed * only in pairs of interest. GraphPad Prism7 was used for all statistical analysis, data processing and presentation.
Supplementary Material
Highlights.
LKB1 regulates innate immune signaling in KRAS-driven NSCLC cells
IGF1 and YAP1 promote resistance to TBK1/JAK/MEKi therapy in KL cells
BETi suppresses IGF1, YAP1 signaling and a therapy-induced secretome
Combination therapy with intermittent BETi achieves durable efficacy in KL models
Significance.
Targeted therapy for oncogenic KRAS-driven cancers remains a major unmet clinical need. Here we explore KRAS directed combination therapy targeting TBK1 and MEK signaling, two of the major outputs downstream of RAL and MAPK. We uncover KRAS;LKB1 mutant NSCLC cells as particularly sensitive to this combination, but with YAP activation/transcriptional plasticity as a major resistance mechanism. Blocking this feedback at the root via BET inhibition overcomes acquired and intrinsic resistance, and can achieve long-term tumor control in aggressive Kras lung cancer GEMMs regardless of Lkb1 or Trp53 status. Furthermore, the set of targets identified (TBK1, MEK, and BRD4) are all being optimized with more potent and selective inhibitors, thus highlighting the clinical potential of this combination strategy.
ACKNOWLEDGMENTS
We are grateful to Drs. P. Gao, M. Kim, H. Seav, H. Zhang, Y. Mitsuishi, K. Hinohara, and S. Masuda for providing materials, technical instruction, and useful discussion; and Molecular Biology Core Facilities at Dana-Farber Cancer Institute for the preparation and sequencing of RNA-seq and ChIP-seq. This work was supported by NCI-R01 CA190394–01 (D.A.B.); NCI P01 CA154303 (D.A.B.); the Gloria T. Maheu, Candice Bagby, and Heerwagen Family Funds for Lung Cancer Research (D.A.B.); NIH/NCI P01CA120964 (K.-K.W.), 5R01CA163896–04 (K.-K.W.), 5R01CA140594–07 (K.-K.W.), 5R01CA122794–10 (K.-K.W.), 5R01CA166480–04 (K.-K.W.); the Gross-Loh Family Fund for Lung Cancer Research (K.-K.W., D.A.B.); The Uehara Memorial Foundation Post-Doctoral Fellowship (S.K.); Strategic Young Researcher Overseas Visit Program for Accelerating Brain Circulation (S.K.); and JSPS Postdoctoral Fellowship For Research Abroad (S.K.). Additional support from a Stand Up To Cancer-American Cancer Society Lung Cancer Dream Team Translational Research grant (SU2CAACR-DT1715). Stand Up to Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C.
Footnotes
DECLARATION OF INTERESTS
D.A.B is a consultant for N of One.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and four tables and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.08.009.
DATA AND SOFTWARE AVAILABILITY
Sequence data were deposited at Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/). The accession number for the RNA-seq data reported in this paper is GEO: GSE96779. The accession number for the ChIP-seq data reported in this paper is GEO: GSE96780.
REFERENCES
- Anastas JN, Kulikauskas RM, Tamir T, Rizos H, Long GV, von Euw EM, Yang PT, Chen HW, Haydu L, et al. (2014). WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J. Clin. Invest. 124, 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodemann BO, and White MA (2008). Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat. Rev. Cancer 8, 133–140. [DOI] [PubMed] [Google Scholar]
- Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, Liu Y, Tupper T, Ouyang J, Li J, et al. (2012). A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483, 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M Jr., et al. (2006). RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127, 157–170. [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. [DOI] [PubMed] [Google Scholar]
- Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, Ryan DP, Meyerhardt JA, Benes C, Settleman J, et al. (2011). Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J. Clin. Invest. 121, 4311–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rendueles ME, Ricarte-Filho JC, Untch BR, Landa I, Knauf JA, Voza F, Smith VE, Ganly I, Taylor BS, Persaud Y, et al. (2015). NF2 loss promotes oncogenic RAS-induced thyroid cancers via YAP-dependent transactivation of RAS proteins and sensitizes them to MEK inhibition. Cancer Discov. 5, 1178–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Yeo A, Faber AC, Lifshits E, Chen Z, Cheng KA, Walton Z, Sarosiek KA, Letai A, Heist RS, et al. (2014). Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Res. 74, 3146–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs GA, Wittinghofer A, and Der CJ (2016). Selective targeting of the KRAS G12C mutant: kicking KRAS when it’s down. Cancer Cell 29, 251–253. [DOI] [PubMed] [Google Scholar]
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, et al. (2018). Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al. (2007). LKB1 modulates lung cancer differentiation and metastasis. Nature 448, 807–810. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. (2014). Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima S, Kohno S, Kondoh A, Sasaki N, Nishimoto Y, Li F, Abdallah Mohammed MS, Muranaka H, Nagatani N, et al. (2015). Undifferentiated state induced by Rb-p53 double inactivation in mouse thyroid neuroendocrine cells and embryonic fibroblasts. Stem Cells 33, 1657–1669. [DOI] [PubMed] [Google Scholar]
- Kitajima S, Thummalapalli R, and Barbie DA (2016). Inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin. Cell Dev. Biol. 58, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, Tawakkol N, D’Angelo R, Paulson AK, et al. (2012). Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 47, 570–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T, et al. (2016). LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, et al. (2016). STK11/ LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 76, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, and Settleman J (2014). Drug resistance via feedback activation of Stat3in oncogene-addicted cancer cells. Cancer Cell 26, 207–221. [DOI] [PubMed] [Google Scholar]
- Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, et al. (2015). The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 47, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, and Pan D (2012). Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loganathan SN, Tang N, Fleming JT, Ma Y, Guo Y, Borinstein SC, Chiang C, and Wang J (2016). BET bromodomain inhibitors suppress EWS-FLI1-dependent transcription and the IGF1 autocrine mechanism in Ewing sarcoma. Oncotarget 7, 43504–43517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, and Downward J (2013). Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 3, 548–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, and Guan KL (2015). The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 15, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. (2015). Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 520, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, and Bar-Sagi D (2011). RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, et al. (2012). beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et al. (2014). KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, and Vakoc CR (2014). The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54, 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Mihaylova VT, Kuruvilla L, Chen F, Viviano S, Baldassarre M, Sperandio D, Martinez R, Yue P, Bates JG, et al. (2016). Loss of TRIM33 causes resistance to BET bromodomain inhibitors through MYC- and TGF-beta-dependent mechanisms. Proc. Natl. Acad. Sci. USA 113, E4558–E4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR, et al. (2015). Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 5, 860–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. (2018). STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos ML, Levin RS, Gordan JD, Oses-Prieto JA, Webber JT, Salt M, Hann B, Burlingame AL, McCormick F, Bandyopadhyay S, and Shokat KM (2014). Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 8, 1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, and McCormick F (2014). Dragging ras back in the ring. Cancer Cell 25, 272–281. [DOI] [PubMed] [Google Scholar]
- Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, and Parsons R (2015). Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell 27, 837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KA, Granger DA, Reuther RA, et al. (2015). Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 11, 390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Nishiyama T, Shimizu K, and Kadota K (2013). TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinformatics 14, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, and Pardoll DM (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A,Zheng Z, Ray S, Groh EM, et al. (2016). T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu HL, and Aplin AE (2014). Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol. Cancer Res. 12, 1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Liu D, Li L, Wempe MF, Guin S, Khanna M, Meier J, Hoffman B,Owens C, Wysoczynski CL, et al. (2014). Discovery and characterization of small molecules that target the GTPase Ral. Nature 515, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Imamura Y, Jenkins RW, Canadas I, Kitajima S, Aref A, Brannon A, Oki E, Castoreno A, Zhu Z, et al. (2016). Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol. Res. 4, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU, et al. (2017). Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex. Cancer Discov. 7, 302–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, Moody SE, Shen RR, Schinzel AC, Thai TC, et al. (2014a). Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 4, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Golay HG, and Barbie DA (2014b). Targeting pathways down-stream of KRAS in lung adenocarcinoma. Pharmacogenomics 15,1507–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.