

Abstract
As the endpoint for the ubiquitin-proteasome system, the 26S proteasome is the principal proteolytic machine responsible for regulated protein degradation in eukaryotic cells. The proteasome’s cellular functions range from general protein homeostasis and stress response to the control of vital processes such as cell division and signal transduction. To reliably process all the proteins presented to it in the complex cellular environment, the proteasome must combine high promiscuity with exceptional substrate selectivity. Recent structural and biochemical studies have shed new light on the many steps involved in proteasomal substrate processing, including recognition, deubiquitination, and ATP-driven translocation and unfolding. In addition, these studies revealed a complex conformational landscape that ensures proper substrate selection before the proteasome commits to processive degradation. These advances in our understanding of the proteasome’s intricate machinery set the stage for future studies on how the proteasome functions as a major regulator of the eukaryotic proteome.
Keywords: 26S proteasome, AAA+ ATPase, energy-dependent protein degradation, ubiquitin receptor, deubiquitination, ubiquitin code
INTRODUCTION
The 26S proteasome is the major protease in eukaryotic cells, responsible for protein degradation in both the cytosol and the nucleus. Ubiquitin modifications target condemned proteins to the proteasome. These modifications are covalently attached to lysine side chains by a large network of ubiquitin ligases and conjugating enzymes, and they are removed at the proteasome prior to substrate degradation (for more details, see 1). As a compartmental protease of the AAA+ (ATPases associated with various cellular activities) family, the proteasome uses ATP hydrolysis to disrupt higher-order structures of its substrates and translocate the unfolded polypeptides into an internal degradation chamber for proteolytic cleavage. This ability to unravel native structures allows the proteasome to function as a modulator of the eukaryotic proteome and degrade numerous regulatory proteins in addition to damaged or misfolded polypeptides. Therefore, the 26S proteasome not only is essential for general protein and amino acid homeostasis but also controls a myriad of essential cellular processes, including the cell cycle, DNA replication, transcription, signal transduction, and stress responses (2–5).
The high selectivity and tight control required for such promiscuous intracellular proteolysis are accomplished, on the one hand, by the specific ubiquitin labeling of appropriate substrates for degradation and, on the other hand, through the complex architecture of the 26S proteasome holoenzyme (Figure 1a) (6). The holoenzyme’s proteolytic active sites reside within the chamber of the barrel-shaped 20S core particle and are accessible only through narrow axial pores, which exclude folded and even large unfolded polypeptides. Gating of these pores is controlled by the 19S regulatory particle (RP) (7–13), which caps one or both ends of the 20S core peptidase and mechanically translocates appropriate substrates into the degradation chamber. Many years of work have led to an extensive body of knowledge about the 20S core peptidase (4, 14). However, only recently has detailed information about the structure, function, and conformational dynamics of the 19S RP become available, and this review largely focuses on these findings.
Figure 1.

Structure and conformational changes of the proteasome. (a) The 26S proteasome is composed of three subcomplexes: the core (gray); the base (with Rpn2 and the motor subunits Rpt1–Rpt6 in light blue and the ubiquitin-binding subunits Rpn1 and Rpn13 in dark blue); and the lid (with Rpn3, Rpn5, Rpn6, Rpn7, Rpn8, Rpn9, Rpn12, and Sem1 in yellow, and the DUB Rpn11 in orange). The ubiquitin receptor Rpn10 is shown together with the lid (dark blue). (Left) The three subcomplexes are depicted individually; (center and right) the entire 26S proteasome structure is shown (EMDB: 3534) (40). The center orientation allows a view of the entrance to the central pore and the Rpn11 active site, and the right orientation, rotated by 120°, emphasizes the lid subcomplex with its hand-shaped structure of the PCI (proteasome-CSN-initiation factor 3) domain-containing subunits. (b) Conformational switching of the 19S regulatory particle between the s1 state (EMDB: 3534) and the s3 state (EMDB: 3536) (40), with the core particles aligned. Shown are the views from the right and back of the proteasome relative to the center orientation in panel a. In the s1 conformer, the Rpt ring and Rpn2 are depicted in light blue; Rpn1, Rpn10, and Rpn13 in cyan; and the lid in yellow. In the s3 conformer, the Rpt ring and Rpn2 are depicted in medium blue; Rpn1, Rpn10, and Rpn13 in dark blue; and the lid in salmon. For both conformers, the core is shown in gray. During the transition from s1 to s3, the lid and Rpn10 rotate by ~30° relative to the Rpts. (c) Cutaway representations of the proteasome in the conformations s1–s4, emphasizing differences in the location of Rpn11; the width of the central processing channel; and the coaxial alignment of the N-ring, the AAA+ (ATPases associated with various cellular activities) ring, and the 20S core. The central channels through the N-ring and AAA+ ring are highlighted by a solid black line. The coaxial alignment is most pronounced in the s3 and s4 conformers, leading to the formation of a wide continuous channel for substrate translocation, with the Rpn11 active site (red dot) located directly above the entrance. Also shown are top-down views of the 20S core particle, emphasizing the changes in the 20S gate, which has the most density in the s1 state and the least in the s4 state.
The RP can be separated biochemically into the base and lid subcomplexes (Figure 1a) (15,16). The base subcomplex includes three non-ATPase subunits, in Saccharomyces cerevisiae called Rpn1, Rpn2, and Rpn13, with Rpn1 and Rpn2 containing large alpha solenoids that provide multiple binding sites for ubiquitin and ubiquitin-like proteins (UBLs) (on Rpn1) and a binding site for the ubiquitin receptor Rpn13 (on Rpn2) (Table 1) (17–19). An additional ubiquitin-receptor subunit, Rpn10, is not considered part of the base or lid per se, but instead bridges both subcomplexes in the assembled RP (20, 21). At the center of the base are six distinct ATPase subunits (Rpt1–Rpt6 in yeast), whose AAA+ domains form the ring-shaped heterohexameric motor of the proteasome. Each Rpt also contains an N-terminal alpha helix and an OB (oligonucleotide/oligosaccharide binding)-fold domain that in the hexamer assembles into a distinct N-ring above the AAA+ domain ring. The Rpts use conserved loops projecting from their AAA+ domains into the central pore of the motor to engage protein substrates, apply mechanical pulling force for unfolding, and then translocate the unfolded polypeptide into the associated 20S core (22–26).
Table 1.
Proteasome subunit names and function
| Subcomplex | Saccharomyces cerevisiae | Homo sapiens | Function |
| Base | Rpn1 | PSMD2/S2 | Ubp6 and ubiquitin/UBL binding |
| Rpn2 | PSMD1/S1 | Structural | |
| Rpn13 | ADRM1 | Ubiquitin/UBL binding | |
| Rpt1 | PSMC2/S7 | ATPase | |
| Rpt2 | PSMC1/S1 | ATPase | |
| Rpt3 | PSMC4/S6 | ATPase | |
| Rpt4 | PSMC6/S10 | ATPase | |
| Rpt5 | PSMC3/S6a | ATPase | |
| Rpt6 | PSMC5/S8 | ATPase | |
| Lid | Rpn3 | PSMD3/S3 | Structural |
| Rpn5 | PSMD12 | Structural | |
| Rpn6 | PSMD11/S9 | Structural | |
| Rpn7 | PSMD6/S10 | Structural | |
| Rpn8 | PSMD7/S12 | Structural | |
| Rpn9 | PSMD13/S11 | Structural | |
| Rpn11 | PSMD14/Poh1/Pad1 | Deubiquitinase | |
| Rpn12 | PSMD8/S14 | Structural | |
| Sem1 | PSMD9/Dss1/Rpn15 | Structural | |
| Additional cofactor | Rpn10 | PSMD4/S5a | Ubiquitin/UBL binding |
| Associated deubiquitinases | Ubp6 | Usp14 | Deubiquitinase |
| NA | Uch37 | Deubiquitinase |
Abbreviations: NA, not available; UBL, ubiquitin-like.
The lid subcomplex acts as a scaffold that braces one side of the base (27,28) and includes six PCI (proteasome-CSN-initiation factor 3) domain–containing subunits (Rpn3, Rpn5, Rpn6, Rpn7, Rpn9, Rpn12) as well as two subunits (Rpn8 and Rpn11) with an MPN (Mpr1-Pad1 N-terminal) domain (Table 1). Rpn11 is a Zn2+-dependent deubiquitinase (DUB) of the JAMM/MPN family and responsible for the removal of substrate-attached ubiquitin chains before they enter the AAA+ ATPase (29–31). The proteasome also contains one or two additional stably associated DUBs, Ubp6 and Uch37 (Table 1). Ubp6 (Usp14 in mammals) is a ubiquitin-specific protease (USP) that interacts with Rpn1 of the base and uses an active site cysteine to cleave supernumerary ubiquitin chains from substrates (32). The ubiquitin C-terminal hydrolase Uch37 (also called UCHL5) associates with the ubiquitin receptor Rpn13 and likely functions in cleaving or editing distal ubiquitin chains on proteasome substrates (33–36).
The past few years have bestowed a major leap in our structural and functional understanding of the RP, especially through cryo-electron microscopy (EM) studies that have identified several new conformations (37–45). The combination of newly available structural information with a large body of biochemical studies has provided us with an intriguing molecular model for substrate processing by the 19S RP (Figure 2). Substrates are targeted to the proteasome through interactions between attached ubiquitin modifications and several ubiquitin receptors on the proteasome (see the section titled Ubiquitin Recognition at the Proteasome). An unstructured region of the substrate is then engaged by the AAA+ motor, which subsequently pulls on, translocates, and unfolds the substrate (see the sections titled Substrate Requirements for Proteasome Degradation, and The Proteasome AAA+ ATPase Motor) (46). Substrate engagement is accompanied by a major conformational change that switches the RP into a state ideally suited for processive substrate translocation and translocation-coupled deubiquitination (see the sections titled Proteasome Conformational Changes, and Proteasomal Deubiquitinases) (42). The proteasome’s various conformational states thus appear to differentially facilitate and coordinate the individual steps of substrate processing (37–45, 47). On the basis of these findings, it is now clear that proteasome function depends on complex conformational equilibria, which are influenced by a range of factors, including the nucleotide state of the Rpt subunits, the presence of protein substrate, and the occupancies of DUBs and ubiquitin receptors. In addition, major advances in localizing and characterizing the various ubiquitin receptors and DUBs have provided a first glimpse into how the proteasome may differentially use them to recruit certain substrates and fine-tune degradation activities. The following sections focus on the recent progress in our understanding of the 19S RP, its fascinating dynamics, and versatile functions in substrate selection and processing.
Figure 2.

Model for substrate engagement by the proteasome. (Left) Proteasome is shown in the s1 state (EMDB: 3534) with a model substrate (red) tethered through a tetraubiquitin chain (purple) near the presumed location of the Rpn10 ubiquitin-interacting motif. In this s1 state, Rpn11 (orange) is offset to the right, making the entrance to the central pore accessible for insertion of a substrate’s unstructured initiation region. (Right) Substrate engagement shifts the proteasome to the s3 or s4 state (s4 is shown; EMDB: 3537) (40) in which the N-ring and AAA+ (ATPases associated with various cellular activities) ring are coaxially aligned to facilitate substrate translocation and the 20S gate is open for polypeptide transfer into the internal degradation chamber. Rpn11 is located directly above the entrance to the central pore, where it acts as a gatekeeper and removes ubiquitin chains from substrates during translocation. The initiation region of the substrate must be long enough to bridge the gap between the N-ring and the pore loops of the Rpts.
PROTEASOME CONFORMATIONAL CHANGES
Recent high-resolution cryo-EM work on the 26S proteasome has revealed that the 19S RP has a complex conformational landscape. Two conformations were initially identified and assigned to the substrate-free (s1) and the substrate-processing (s3) states. These two states predominate in in vitro structural studies of purified proteasomes from S. cerevisiae (27, 28, 37–39, 41, 48) and Homo sapiens (44, 49–52), and they are also observed in situ by cryo-electron tomography of rat hippocampal neurons (53). Recent rapid technological advancement in EM has allowed for further subclassification of proteasome states, and now, up to seven different proteasome conformations have been identified (40, 43–45). It is likely that further structural and biochemical studies will continue to reveal additional conformational states. In the current nomenclature, the main states of the yeast proteasome are referred to as s1, s2, s3, and s4 (39, 40), whereas the closely related conformations of the human proteasome are termed SA, SB, SC, and SD(1,2,3) (Table 2) (44, 45). For clarity, we use the yeast nomenclature in this review.
Table 2.
Proteasome conformations and prominent features of each conformation
| Proteasome conformation | Prominent features of conformation | ||||
|---|---|---|---|---|---|
| Saccharomyces cerevisiae | Homo sapiens | Rpn11 position | Relative position of N-ring, AAA+ ring, and 20S core | 20S core particle gate | AAA+ domain configuration |
| s1 (27, 28, 48, 50) | SA (44, 50–52) | Offset from central processing pore | Not aligned | Full density, closed | Steep spiral staircase, Rpt3 at the top |
| s2 (39) | SB (44, 51) | Aligned with central processing pore | Not aligned | Full density, closed | Steep spiral staircase, Rpt3 at the top |
| s3 (37, 38, 41) | SC (44) | Aligned with central processing pore | Aligned | Full density, closed | Shallow spiral staircase, Rpt1 at the top |
| s4 (40) | SD(1, 2, 3) (44, 45) | Aligned with central processing pore | Aligned | Partial density, open | Shallow spiral staircase, Rpt5 at the top |
The order of the four main states is suggested by their structural comparison, as there is a progression of movement from the s1 state through the s2, s3, and s4 states, with s4 being least similar to s1. Throughout all conformations, the general structure of the core particle remains virtually unchanged, whereas the orientation of the base and lid relative to each other and to the core particle varies dramatically (Figure 1b,c; Table 2). Between the s1 and s4 conformations, the lid undergoes an ~30° rotation relative to the base, pivoting around the contact points between its PCI domains and the AAA+ domains of the base. During the transition from s1 to s2, this lid rotation moves the essential DUB Rpn11 from an offset to a coaxially aligned position directly above the central processing pore of the base. Even though this position seems ideal for translocation-coupled deubiquitination, it likely restricts substrate access to the central pore, leading to the proposal that only the s1 state is capable of efficiently engaging an incoming polypeptide with its translocation machinery. The s1 state is therefore assumed to represent the primary substrate-binding conformation (Table 2, Figure 2) (37, 39–41, 44). This conformation is also likely the resting state of the proteasome, as it is the major conformation observed for ATP-bound proteasomes in the absence of added substrate or other factors. The other conformations were induced by trapping the proteasome during substrate degradation or in the presence of ATP analogs (Figure 3), which leaves some uncertainty about their physiological role (37–40, 43, 45).
Figure 3.

Conformational landscape of the proteasome. (a) Qualitative comparison of how the presence of nonhydrolyzable ATP analogs, protein substrate, and ubiquitin-bound Ubp6 affects individual enzymatic activities of the proteasome. Up and down arrows indicate the stimulation and inhibition, respectively, of enzymatic activities of the proteasome compared with their basal activities; two up arrows indicate hyperstimulation. Basal activities are defined here for the proteasome in the presence of ATP and absence of substrate proteins. (b) Data from 17 electron microscopy data sets that directly compare the relative abundances of proteasome conformations under various conditions. The data sets are clustered according to the experimental conditions. Increased ATPase and core-peptidase activities appear to be correlated with greater abundance of s3 and s4 states. (Right) Resolutions of all structures obtained, representing the FSC value at 0.143. An asterisk indicates resolution was attained with an FSC value of 0.3; double asterisks indicate resolution was attained with an FSC value of 0.5. Abbreviations: FSC, Fourier shell correlation; NA, not applicable; NR, not reported; Ub, ubiquitin.
Whereas the transition from s1 to s2 involves mainly the lid subcomplex, the subsequent transition to s3 is accompanied by major rearrangements in Rpt1–Rpt6, the ATPase subunits of the base (39, 44). The N-ring of the Rpts shifts toward Rpn1, and the AAA+ domains move and tilt in the same direction, creating a wider, continuous central channel that is aligned with the axial pore of the core particle (Table 2, Figure 1c) (37, 39, 40, 44). Concurrently, the interfaces between the AAA+ domains of neighboring Rpts become more uniform and adopt a conformation more similar to other AAA+ motors (37, 40).
Finally, during the transition from s3 to s4, the gates to the core particle appear to open up (Figure 1c, Table 2) (40, 44), completing a substrate passage that extends from the N-ring through the AAA+ motor domains into the core. Conformations with more open gates, such as s4, are therefore assumed to reflect the actively translocating states of the RP. The variable opening of the gates is likely induced by differential interactions between the C-terminal tails of Rpt subunits and the 20S core particle (8, 9, 13, 40, 44, 54). The C termini of Rpt2, Rpt3, and Rpt5 contain conserved hydrophobic-Tyr-X (HbYX) motifs, whose docking into hydrophobic pockets on the axial face of the core particle induces gate opening (8, 9, 12, 13, 55). In the s4 state, the C terminus of Rpt6 makes additional contacts with the 20S core, which may trigger the more complete gate opening observed for this state (40).
Other important changes that are coupled with the conformational transitions involve the intrinsic ubiquitin receptors Rpn10 and Rpn1, one of the most flexible subunits of the proteasome complex. Between s1 and s3 or s4, Rpn1 changes its interactions with the N-terminal coiled coil of Rpt1 and Rpt2; thus, it rotates relative to the ATPase ring and the central pore (37, 39, 40, 43, 44). The rotation of Rpn1 brings its binding site for the UBL of Ubp6 closer to the base ATPases, allowing Ubp6 to make stable contacts with the base ATPase site that is responsible for stimulation of Ubp6 DUB activity in non-s1 states. This dependence on the rotation of Rpn1 explains the ability of Ubp6 to sense the conformational state of the proteasome in its DUB activity (47, 56– 58). During the same transitions, Rpn10 moves concomitant with Rpn11 and makes additional contacts with the N-terminal coiled coil of Rpt4 and Rpt5 (Figure 1b) (37, 38, 44). Although the functional importance of these ubiquitin-receptor transitions has yet to be determined, the different positions may influence the proteasome’s affinity for ubiquitin chains or direct substrates to the central pore.
The functional models derived from the EM structural data are supported by biochemical measurements of proteasomal peptide hydrolysis, ATP hydrolysis, and deubiquitination, which suggests an intimate relationship between these activities. Studies using freely diffusible fluorogenic peptides to read out gate opening have shown that the 19S RP finely controls the 20S gates and that this activity is stimulated by multiple factors, including the engagement of a protein substrate, the presence of Ubp6, and binding of nonhydrolyzable nucleotide analogs to the base ATPases (25, 58–62) (Figure 3a, Table 3). Thus, there is consistency between structural and biochemical studies indicating these factors bias the proteasome toward the open-gate s3 and s4 states and influence gate-opening activity (Figure 3b).
Table 3.
ATP analogs used in proteasome studies
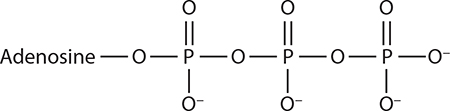
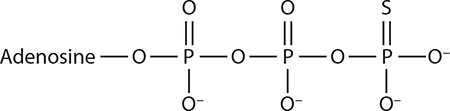
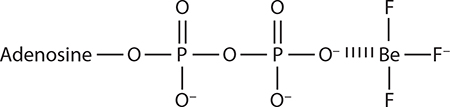
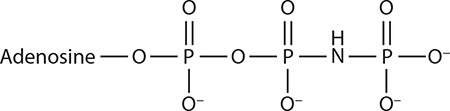
It is unclear whether beryllium fluoride binds as BeF2 or BeF3−. Shown here is the BeF3− form that best resembles PO4 −.
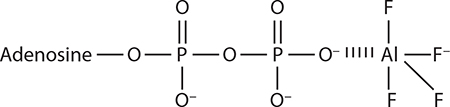
It is not known whether aluminum fluoride binds as AlF3 or AlF3−. Again, the form resembling PO4− (AlF4−) is shown here.
Another major difference between conformations is the arrangement of the AAA+ domains, and here too, biochemical and structural studies converge. Substrate engagement, for instance, induces the transition from s1 to s3 or s4 (37), where the contacts between neighboring AAA+ subdomains are more fully formed. Correspondingly, substrate engagement increases the ATPase activity of the Rpt motor (25, 47, 58, 63) (Figure 3a). Similarly, Ubp6 stimulates proteasomal ATPase activity, and it also shifts the conformational equilibrium of the RP away from the s1 state (47, 51, 56, 63). These data are consistent with a model where the s1 conformation is the ground state of the proteasome in which the base hydrolyzes ATP slowly. Factors that stimulate ATPase activity do so by biasing the proteasome toward different conformations that hydrolyze ATP more quickly. This assumption is further corroborated by the fact that nonhydrolyzable nucleotide analogs that likely stabilize uniform Rpt-subunit interfaces also bias the proteasome conformational landscape away from the s1 conformer (38–40, 43, 45) (Figure 3b, Table 3).
Most EM structural studies conclude that the ATP-bound proteasome in the absence of nucleotide analogs, effector proteins, substrates, or inhibitors is found predominantly in the s1 state, but multiple other conformations are still present in varying proportions (37, 39–41, 44, 49) (Figure 3b). In particular, the s2 and s3 conformations appear with different frequencies in comparable EM data sets (Figure 3b). Differences in these conformational distributions may be attributable to variable ATP/ADP ratios in sample preparations, as high concentrations of ADP affect the proteasome conformation (40) and weaken interactions between the 19S RP and the 20S core (61, 64, 65). Furthermore, it is possible that substrates copurify with proteasomes and bias the conformational landscape toward the s3 state (37, 41). Although much effort has been dedicated to defining different proteasome conformations, the biochemical activities are only correlative at this point, and the relevance of any conformer to on-pathway proteasome degradation has not yet been assessed. Moreover, posttranslational modifications regulate proteasome function in vivo (66, 67), but their effects on the conformational landscape and biochemical activities have not yet been studied. Modification of the proteasome is a burgeoning aspect of the proteasome field, but it is not covered in this review (67, 68).
THE PROTEASOME AAA+ ATPASE MOTOR
Unfolding and translocation of protein substrates into the proteolytic core is driven by the Rpt1-Rpt6 heterohexameric motor, which functions as the engine of the proteasome, converting the chemical energy of ATP binding and hydrolysis into mechanical work. The architecture of the Rpt ring resembles a trimer of dimers, with the N-terminal helices forming coiled coils between pairs of neighboring subunits (Rpt1/Rpt2, Rpt6/Rpt3, and Rpt4/Rpt5) that contribute to proper arrangement of subunits during assembly (69–71) (Figure 4a,b). Downstream of the coiled coils, the OB-fold domains of Rpt1–Rpt6 form the N-ring, which is essential for the structural stability of the hexamer and acts as a bottleneck against which the AAA+ motor likely pulls the protein substrates during mechanical threading to induce their unfolding.
Figure 4.

AAA+ motor architecture, nucleotide binding, and conformational changes. (a) Side (left) and top (right) views of the EM density for the Rpt1–Rpt6 hexamer in the s3 state (EMDB: 3536) (40). The small AAA+ subdomain of each Rpt forms a rigid body with the large AAA+ subdomain of the clockwise-next neighboring subunit. The rigid body formed between Rpt3 (red) and Rpt6 (dark blue) is shown with a dashed loop. In the top view (right), the EM densities for Rpt3 and Rpt6 are shown in transparent grey, and the molecular models for both Rpts are fitted into the density, with the large AAA+ subdomain of Rpt3 in red and the small AAA+ subdomain of Rpt6 in blue, to highlight the rigid-body interaction. (b, left) Molecular model of Rpt3 (PDB: 5mpb) (40); (right) representation of the relative positions of the large and small AAA+ subdomains as well as the pore loop responsible for mechanical substrate translocation. (c) Schematic of the Rpt1–Rpt6 hexamer, showing the reported nucleotide occupancy of each AAA+ binding pocket for EM reconstructions in the presence of ATP (s1 state) (innermost circle in dark gray) or different ATP analogs (ADP-BeFx, ADP-AlFx, and two distinct ATPγS structures): Black dots indicate the presence of nucleotide density, dots are absent from reported empty sites, and outlined gray dots denote sites with lower probability occupancy or lower affinity. For the s1 state, all available data sets show nucleotide density in every pocket (white dots), although Rpt6 exhibits a smaller density and a lack of arginine-finger contacts, suggesting that the bound nucleotide is ADP (white star) (40, 43, 44, 50, 51). The two ATPγS data sets reflect the higher-certainty assignments from Zhu et al. (45), the results for ADP-AlF4-bound proteasomes were taken from Ding et al. (43), and the ADP-BeFx data set reflects the assignments of the s4 state described in Wehmer et al. (40). (d) Representations of the splayed-out Rpt subunits in the steep spiral arrangement of the s1 state and the more planar staircase conformation in s3. All subunits are oriented with the channel-facing pore loops pointing to the right. The dashed lines serve to highlight the apparent tilt of the AAA+ large domains. Abbreviations: AAA+, ATPases associated with various cellular activities; EM, electron microscopy; OB, oligonucleotide/oligosaccharide binding.
In the s3 and s4 conformations of the base, the small AAA+ subdomain of every Rpt subunit forms static contacts and thus a rigid body with the large AAA+ subdomain of the clockwise-next neighboring subunit (37, 38, 40, 44) (Figure 4a). Therefore, the AAA+ hexamer can be thought of as six rigid bodies that are connected by linkers between the large and small subdomains. They are expected to move in response to ATP hydrolysis and change their vertical arrangement in a topologically constrained fashion. The conserved pore loops that sterically interact with substrate in the central pore (Figure 4b) originate from the large AAA+ subdomains and are linked to movements of the rigid bodies, enabling the translation of nucleotide-dependent changes into mechanical pulling for substrate translocation. The AAA+ domain interfaces that form rigid-body contacts are similar to those seen for other polypeptide translocating AAA+ motors, including the archaeal proteasome homolog PAN (proteasome-activating nucleotidase) and the bacterial translocase ClpX (72–74). In addition, a number of structures have been recently solved of AAA+ motors actively translocating substrates, and they all exhibited rigid-body movements of their AAA+ domains as well (75–79).
An emergent feature common to these AAA+ motors is a spiral staircase (often termed lockwasher) arrangement of their rigid bodies, in which neighboring large AAA+ subdomains tilt progressively downward, with a break in the arrangement between the first and last units (see Figure 4d) (73, 75–81). This staircase arrangement may be tied to the hydrolysis cycle of the motors, with each position in the staircase corresponding to a particular hydrolysis event (80). For the proteasome, cryo-EM studies have revealed that the Rpt ring is highly dynamic, with the most pronounced structural rearrangement occurring between the s1 and s3 states. In the s1 state, rigid-body interactions between neighboring Rpt subunits are largely absent; the large AAA+ domains are differentially lifted out of the plane of the ring; and their pore loops are arranged in an extended spiral, with Rpt3 in the highest position (Figure 4d) (27, 48, 52). This arrangement is not typical of actively translocating AAA+ hexamers and may represent an “off” state for other motors (75). In the proteasome, this motor state may be stabilized in the s1 conformation by interactions between the base, lid, and core. During the transition from the s1 to the s3 and s4 states, the ring of ATPases flattens out and adopts a shallower staircase arrangement analogous to that of other active AAA+ translocases (37–41, 43, 44). In these conformations, a number of different Rpts have been observed at the top of the staircase, but these different states have not yet been assigned to particular steps in the hydrolysis or translocation cycle (27, 37–41, 43–45, 48, 52).
Mutational analyses of the base have shown that the Rpts are functionally nonredundant and may have differential roles according to their vertical position in the spiral arrangement of the hexamer. Active site mutations for each Rpt subunit in vivo showed that nucleotide binding by Rpt1 or Rpt5 is not required for viability, and the effects of pore loop mutations in vivo were similarly varied (26, 82). In a heterologous expression system that allows for detailed biochemical investigation of otherwise-lethal mutations in the proteasome ATPases, eliminating the ATP hydrolysis activity in the Rpt subunits that adopt the top positions in the s1-state spiral (Rpt3 and 4) leads to the most severe defects in substrate processing, but not necessarily the strongest defect in ATP hydrolysis (25). These experiments suggest a particular role of those subunits closest to the pore entrance in facilitating substrate engagement and the transition out of the s1 state.
Another common feature of other related AAA+ motors is allosteric interactions that constrain subunits to bind at most four nucleotides per hexamer at any one time (79, 83–85). Similar allosteric networks have also been identified in the proteasome homolog PAN and in the 26S proteasome (60, 64, 86, 87). Recent higher-resolution EM studies have now begun to reveal the nucleotide occupancies of individual Rpt subunits in the proteasome, though in many cases the limited resolution of the nucleotide prevented distinguishing between ADP and ATP (40, 43–45, 50, 51) (Figure 4c). Another challenge is that averaging large numbers of particles for cryo-EM reconstructions may obscure the underlying heterogeneous arrangement of nucleotides. One intriguing study used extensive subclassifications to show that a planar s4 conformation of the Rpt ring has only four bound nucleotides, which is in agreement with scenarios observed for other motors (Figure 4c) (45). Thus, even though AAA+ hexamers can explore a large conformational landscape, they may share common mechanisms of translocation. In ClpX, for instance, ATP hydrolysis in one subunit can drive the motion of all other subunits owing to their rigid-body contacts and conformational coupling in the hexamer (88, 89). AAA+ motor subunits arranged in a spiral staircase may thus advance in a coordinated fashion by one register after each ATP hydrolysis event (77–80). The arrangements of Rpt pore loops in the s3, s4, and recently reported ADP-AlFx-bound states of the proteasome also suggest a downward-directed paddling motion of individual subunits to propel substrate through the central pore (Figure 4d) (37–41, 43–45).
In addition, the substrate polypeptide seems to play a role in the allosteric control of AAA+ motor activity. For most AAA+ protein unfoldases, engagement and threading of a polypeptide stimulate ATPase activity, likely because subunits exert force on each other through substratemediated contacts between pore loops (88, 89). This bridging of pore loops by the substrate and the reciprocal application of force between ATPase subunits may also be responsible for the transition of the RP from the s1 to the s3 or s4 conformations upon substrate engagement. Moreover, an intriguing consequence of the proteasome’s complexity and architecture is a novel mode of long-range allosteric communication that is mediated by the ubiquitin-conjugated substrate polypeptide. Through mechanical tension on the substrate, the proteasomal AAA+ motor appears to induce a conformational switch in the Rpn11 DUB located ~40 Å away, above the entrance to the central pore, thus stimulating cotranslocational deubiquitination (42).
SUBSTRATE REQUIREMENTS FOR PROTEASOME DEGRADATION
Even though the proteasome processes a large variety of proteins in the cell (4), all of its substrates must possess two essential parts: a targeting signal and an unstructured initiation region (90, 91) (Figure 2). Various recent studies using different model substrates have provided more clarity on the precise nature of both requirements. The predominant targeting signals in vivo are polyubiquitin chains, which are formed by first covalently attaching the C terminus of ubiquitin via an isopeptide bond to a substrate lysine and then repeatedly linking additional moieties to lysines in ubiquitin (1). Depending on the lysine-linkage type, these chains exhibit variable compactness and distinct conformations, which serve as a ubiquitin code to control substrate targeting. Proteins modified with one or several K11-, K48-, K29-, and, at least in vitro, also K63-linked chains are recruited to the proteasome by interacting with the intrinsic ubiquitin receptors Rpn1, Rpn10, and Rpn13, or the transiently bound ubiquitin receptors Rad23 and Dsk2 (18, 92–101). A single tetraubiquitin chain is a more effective targeting signal than a single monoubiquitin, but multiple short (mono- or di-) ubiquitin modifications can also lead to efficient degradation (102, 103). Some natural substrates have intrinsic affinity for the proteasome (104, 105), but proteins can also be targeted by artificial ubiquitin-independent tethering systems or fusions to proteasome components such as Rad23 and Rpn10 (47, 91, 106–109). These numerous delivery strategies imply sufficient flexibility provided by the ubiquitin chain, linkers in ubiquitin receptors such as Rpn10 and Rad23, or flexible segments in artificial recruitment systems to allow the substrate to properly orient itself for insertion of its initiation region into the pore (see Figure 5).
Figure 5.

Ubiquitin receptors and DUBs (deubiquitinases) at the proteasome. (a) Cryo-electron microscopy (EM) reconstruction of the 26S proteasome in complex with ubiquitin-bound Ubp6 (EM: 3034; PDB: 5a5b) (56), with EM density for the lid shown in yellow, the base in light blue, the core particle in gray, Rpn11 in orange, Ubp6 in lighter orange, and individual ubiquitin receptors in darker blue. A schematic version highlighting the DUBs and ubiquitin receptors labeled by figure panel is shown in the inset (lower right). The essential DUB Rpn11 (orange) sits just above the N-ring pore with its active site 35 Å away from the active site of Ubp6 (lighter orange, with bound ubiquitin density in pink). Bridging EM density links Ubp6’s ATPase-contacting ubiquitin-specific protease (USP) domain to the UBL (ubiquitin-like) domain bound at the T2 site of Rpn1. The UBL domain of Mus musculus Ubp6 (PDB: 1wgg) (orange ribbon) is fit into the EM density seen at the T2 site of Rpn1, using Chimera’s Fit in Map tool. The USP domain of Ubp6 (PDB: 5a5b) is also depicted as an orange ribbon within the EM density. Each ubiquitin receptor on the proteasome is shown with the ribbon diagram of the EM-based atomic model docked into the EM density. Proteasome-bound ubiquitin (pink ribbon) is modeled by docking existing ubiquitin-receptor costructures into the EM density for each ubiquitin receptor. K48-linked diubiquitin bound to the dual UIMs (ubiquitin-interacting motifs) of human S5a/PSMD4 (Rpn10) is shown in a possible location on the proteasome, placed by confining the most N-terminal residue of the UIM structure 17 Å from the most C-terminal residue of the EM docked Rpn10 VWA (von Willebrand factor type A) domain. This constraint is determined by the five amino acid linker unresolved between the UIM and VWA domains in the available structures of the Homo sapiens Rpn10 homolog (PDB: 2kde) (134). (b) The side view of the human S5a/PSMD4 (Rpn10) with bound diubiquitin highlights the gap (dashed line) between the VWA domain and the UIMs. In organisms containing more than a single UIM in Rpn10, UBLs such as hHR23 (dark green) can bind to the UIM2 site in a similar manner as ubiquitin, as illustrated by overlaying the hHR23 S5a structure with the ubiquitin dimer-bound structure of S5a (PDB: 1p9d) (173). Here, the C terminus of ubiquitin (pink sphere) is shown 76 Å from the active site of Rpn11, but this distance is only a single possibility owing to the flexibility in the linker between the UIMs and the VWA domain anchoring Rpn10 to the proteasome. (c) The T1 site of Rpn1 is shown bound to the K48-linked ubiquitin dimer, with the free ubiquitin C terminus (pink sphere) sitting 93 Å from the active site of Rpn11 (distance indicated as green dashed line) (PDB: 2n3v) (18). Side view compares the interaction of UBLs or ubiquitin with Rpn1. Rad23 (dark green) interacts with the portion of the T1 Rpn1 site that is bound by the distal ubiquitin (the ubiquitin without a free C terminus in the dimer) in the ubiquitin dimer-bound structure (PDB: 2nbw) (131). The free C terminus of ubiquitin is highlighted with a pink sphere, whereas the most C-terminal residue of Rad23 is highlighted with a smaller green sphere. The M. musculus Ubp6 UBL is placed at the T2 site, which is distinct from the T1 ubiquitin binding site. The structure of the human Rpn13 homolog ADRM1 (dark blue) (PDB: 5v1z) (174) in complex with ubiquitin and the respective Rpn2 peptide is placed into the EM density, using the orientation defined by Wehmer et al. (40) (PDB: 5mpd). This orientation places the C terminus of ubiquitin 91 Å from the active site of Rpn11. (d) The UBL of Dsk2 binds to Rpn13 in a similar manner as ubiquitin, highlighted by the overlay of the ADRM1 bound to hPLIC2, a Dsk2 homolog (dark green) (PDB: 2nbv) (131), with the C-terminal residues indicated by same-colored spheres.
The presence of an unstructured region, either at the terminus or as an internal flexible loop, is a strict prerequisite for degradation by the proteasome (110, 111). Intrinsic subunits of the proteasome complex or transiently bound factors lack flexible segments suitable for engagement and thus avoid degradation. As a proof of principle, the addition of a flexible initiation region at the terminus of Rad23 leads to efficient degradation, whereas an internal unstructured region inherent to Rad23 is not long enough to engage with the proteasomal motor (108). The minimum length requirement for a terminal initiation region is ~20–30 amino acids (the requirements for an internal initiation region are more strict), and its distance from the targeting signal, e.g., the substrate-attached ubiquitin chain, influences the rate of proteasomal processing (46, 102, 108). It remains unclear how the length requirement for the initiation region is affected by certain characteristics of the ubiquitin chain, for instance, linkage type or branching, that may determine which of the ubiquitin receptors on the proteasome is preferentially used for substrate delivery (46) (see the section titled Ubiquitin Recognition at the Proteasome and Figure 5).
The requirements for a sufficiently long handle on the substrate are not surprising, given the structural features of the proteasome, with the static N-ring above the AAA+ domain hexamer forming the only entrance to the central processing channel (Figure 2). Thus, there is a 30–40-Å gap between this constriction point at the entrance and the pore loops of the motor that must engage the substrate polypeptide to drive translocation and unfolding. For proteasome substrates that do not contain an intrinsic, flexible initiation region, another hexameric AAA+ unfoldase, Cdc48 in yeast and p97 or VCP in higher eukaryotes, may completely or at least partially unfold them to create a handle for proteasomal processing (112, 113). Cdc48 does not depend on extended flexible regions in its substrates, possibly because it lacks an N-ring above the AAA+ motor and the central channel with its translocating pore loops is more easily accessible even for well-folded proteins (114, 115). However, the detailed mechanisms for substrate recognition and engagement by Cdc48 remain elusive.
An unstructured region not only is required for reaching the translocation machinery deep in the proteasome central pore but also plays an important role in committing substrates to proteasomal processing (116). This commitment step is critical for direct coupling between substrate degradation and deubiquitination at the proteasome (42). The sequence composition of this region has large effects on the rate at which a substrate is degraded both in vivo and in vitro (117–119). Compared with diverse regions, those with lower sequence complexity and small side chains (i.e., stretches of serines or glycines) lead to much less efficient degradation, and other parameters such as flexibility, hydrophobicity, and charge further modulate such effects (119). One explanation for sequence dependence could be that the AAA+ pore loops need a good grip on the substrate polypeptide chain both to prevent backsliding and escape and to apply a high enough unfolding force when the first folded domain of the substrate arrives at the entrance to the pore. For instance, introducing a low complexity sequence in front of a stably folded domain appears to compromise motor grip, thus encouraging slippage and subsequent release of partially processed substrate (110, 117, 118, 120). Such degradation stop signals allow for partial proteasomal degradation and consequent activation of transcription factors such as NFκB, Spt23, and Mga2, which are engaged at an unstructured but slippery internal loop (120, 121). The biophysical basis for the sequence dependence of motor grip is still unclear, but it is conceivable that the lack of large side chains weakens the steric interaction between the substrate polypeptide and the pore loops of the AAA+ motor.
Although recent research has explored the minimum requirements for a substrate, a thorough mechanistic and structural understanding of how the proteasome recognizes a substrate is still lacking. The difficulty lies both in the complexity of substrate recognition by ubiquitin receptors (discussed in the following section) and in the significant challenges of producing large quantities of model substrates with defined ubiquitin modifications. Ideally, a structure with visibly bound substrate would help to fully elucidate the proteasome’s interactions with ubiquitin and the substrate polypeptide.
UBIQUITIN RECOGNITION AT THE PROTEASOME
Before a flexible initiation region can engage with the AAA+ motor, the substrate needs to be recruited to the proteasome through interactions of its ubiquitin modification with intrinsic or extrinsic ubiquitin receptors. The proteasome contains at least three intrinsic receptors: the UIMs (ubiquitin-interacting motifs) of Rpn10/S5a, the Pru (plextrin-like receptor for ubiquitin) domain of Rpn13/ADRM1, and the T1 site of Rpn1/PSMD2 (Figure 5a) (18–21, 122).
Intrinsic and Extrinsic Ubiquitin Receptors
Each ubiquitin receptor is placed approximately the length of a tetraubiquitin from the entrance to the N-ring through which substrates are threaded into the motor (Figure 5a). Placement on the periphery of the proteasomal complex provides these receptors with some flexibility, which may allow the proteasome to accommodate substrates with diverse geometries of ubiquitin chains and folded domains, not only during initial recruitment, but also for engagement, unfolding, and deubiquitination. Accordingly, Rpn13 and Rpn1 consistently show the lowest resolution in EM reconstructions of the proteasome, and even the ubiquitin receptor–containing portion of Rpn10 has yet to be definitively localized. Rpn10 binds the proteasome via a VWA (von Willebrand factor type A) domain, and, depending on the organism, one to three UIMs are attached to this domain through flexible linkers (26, 123). This flexible attachment of UIMs allows them to explore a wide radius around the RP, which may also explain some of the previously observed unexpected crosslinking between ubiquitin and nonreceptor subunits (26, 123–125).
In addition to the intrinsic receptors Rpn1, Rpn10, and Rpn13, several extrinsic receptors can deliver substrates through dynamic interactions with both the proteasome and ubiquitin chains. These receptors act by combining ubiquitin-chain recognition through ubiquitinassociated (UBA) domains with proteasome binding via a flexibly tethered, N-terminal UBL domain (126–130). Whereas S. cerevisiae contains the shuttle receptors Rad23/hHR23 and Dsk2/hPLIC2/ubiquilins, higher eukaryotes contain many more paralogs, along with other UBL/UBA-containing proteins that have not been implicated in direct proteasome delivery. Despite their similar domain architectures, different shuttle-receptor paralogs appear to have distinct preferences for specific intrinsic ubiquitin receptors owing to differences in their individual UBL interfaces (131). The dynamic nature of these interactions has complicated a reliable determination of which intrinsic receptor is bound by individual shuttle receptors, and some UBLs can support proteasome delivery in vitro even if they are unlikely to act in that capacity in vivo (109, 132, 133). Differences in the number of UIMs present in Rpn10 also complicate the assignment of the preferential proteasomal binding site for UBLs. In organisms where Rpn10 contains multiple UIMs, the second UIM acts as a binding site for extrinsic receptors such as the Rad23 homolog hHR23 (Figure 5b) (134). However, recent structural studies on Rad23 and hPLIC2, a homolog of Dsk2, clearly define the molecular basis for their preferences to bind the Rpn1 T1 site and Rpn13/Adrm1, respectively, in a manner similar to ubiquitin binding to those sites (Figure 5c,d) (131).
Utilizing extrinsic receptors with their flexible domain architecture may allow the proteasome to accommodate an even wider variety of substrate geometries and ubiquitin modifications than with the stably bound intrinsic receptors alone. Extrinsic receptors can also provide proteasomal substrate selection upstream of ubiquitination by recognizing and binding disordered regions of substrates, and using their UBL domains to interact directly with E3 ubiquitin ligases, which allows them to shuttle condemned proteins from ubiquitination to proteasomal degradation (100, 135–138). As a result, misfolded or mistargeted proteins that would not be recognized by the proteasome’s intrinsic ubiquitin receptors due to the lack of a permanently unstructured region or ubiquitin chains can still be directed to the proteasome (138).
Specificity of Ubiquitin Receptors
The proteasomal ubiquitin receptors do not exhibit exclusive binding of certain ubiquitin-chain linkages, in agreement with in vivo and in vitro findings that all linkage types can be recognized as a signal for degradation at the proteasome (97, 139). However, each receptor provides unique contributions to linkage-type preferences, with intrinsic receptors preferring K48-linked chains over other linkage types, but the biological relevance of these overall preferences remains unclear (19, 124, 131, 140, 141). In vivo, specific recruitment of substrates with K48-linked ubiquitin chains for proteasomal degradation may be largely determined by the linkage-type specificity of Cdc48-dependent processes upstream of the proteasome, rather than the linkage-type preferences of the proteasomal ubiquitin receptors (141). Disruption of ubiquitin binding to single or several ubiquitin receptors is well tolerated in S. cerevisiae but leads to accumulation of polyubiquitinated species (18, 19, 142), speaking to the overlapping functions of ubiquitin receptors in substrate recruitment. Similarly, known proteasome substrates are stabilized in a variety of different receptor-knockout backgrounds, consistent with largely overlapping substrate pools for individual receptors (141, 143). In higher eukaryotes, deletion of single proteasomal receptors suggests incompletely overlapping roles in substrate recruitment. Deletion of Rpn10’s UIMs, for instance, is embryonically lethal in Mus musculus, whereas deletion of Rpn13 produces viable mice with tissue-specific proteasome defects that can be magnified by codisruption of Rpn10 in those same tissues (144–146). So, even though receptor-specific essential substrates are apparently not present in S. cerevisiae, they seem to exist in higher eukaryotes.
Variations in the location and orientation of substrate when recruited through different binding sites may have consequences for the efficiency of engagement and degradation. For example, the geometries of substrate-attached ubiquitin chains relative to folded domains influences turnover by the proteasome (46). Multiple ubiquitin receptors might also work in tandem on the same substrate, not only to increase the substrate affinity through avidity, but also to funnel a substrate toward the central pore of the base in an ideal orientation for engagement of the unstructured initiation region. Because extrinsic receptors use the same sites on intrinsic receptors as ubiquitin, simultaneous disruption of all intrinsic receptors should prevent ubiquitin-dependent substrate delivery in general; however, current mutations are not lethal despite the essential nature of the proteasome (18). It is possible that these mutations incompletely disrupt the binding of ubiquitin and extrinsic receptors (18), but there may also be additional uncharacterized substrate-delivery mechanisms, as hinted by ubiquitin crosslinking to sites beyond the currently established receptors (125, 147).
Finally, as each ubiquitin receptor is placed in a distinct location on the proteasome, substrates recruited through them may have differential access to other proteasome-associated factors, such as the E3 ligases Hul5/UBE3C and parkin. Parkin interacts with the ubiquitin receptor Rpn13, whereas Hul5 binds to the proteasome through Rpn2 (148, 149), yet neither has been visualized in high-resolution EM reconstructions of the proteasome. Hul5 is thought to act mainly in extending substrate ubiquitin chains, and its location on Rpn2 may make Rpn13-bound chains, for example, more accessible to this ligase. Similarly, the peripherally associated proteasome DUBs Ubp6 and Uch37 are located adjacent to different ubiquitin receptors: Uch37 binds the proteasome through Rpn13, whereas the UBL of Ubp6 is tethered to Rpn1 at the T2 site that is distinct from the T1 ubiquitin binding site (18, 35, 36, 150). Their proximity to ubiquitin receptors may influence the extent to which ubiquitin chains bound to one receptor or another are susceptible to deubiquitination by these proteasome-associated DUBs.
PROTEASOMAL DEUBIQUITINASES
Because ubiquitin receptors guide substrates decorated with ubiquitin and ubiquitin chains to the proteasome, it is not surprising that the proteasome contains DUBs capable of removing or editing the ubiquitin signal. In isolation, these DUBs have fairly poor isopeptidase activity, but their activities are increased upon interaction with the proteasome. This allosteric relationship between the DUBs and the proteasome works both ways: In turn, the various catalytic functions of the proteasome such as ATPase activity, gate opening, and substrate degradation are activated or repressed in the presence of DUBs. EM has identified these DUBs in key locations within the RP, either near ubiquitin receptors or, in the case of Rpn11, directly above the N-ring and the entrance to the central processing channel (Figure 5a). Recent studies have provided structural insights into DUB activation by the proteasome, how these enzymes affect the proteasome conformational states, and what potential role they play in substrate degradation.
Rpn11, the Essential Deubiquitinase
The most important proteasomal DUB is Rpn11, a JAMM metalloprotease closely related to the NEDD8 isopeptidase CSN5 of the COP9 signalosome (151). Rpn11 contains a catalytic zinc ion, coordinated by an EXnHXHX10D metal-binding motif (30, 31), and is absolutely essential for proteasome function and cell viability (152–154). The catalytic-site mutations His109Ala and His111Ala (referred to as the AXA mutation) do not disrupt assembly of the proteasome or its conformational states, but they do inhibit degradation and are lethal in yeast (30). Rpn11 resides just above the N-ring of the AAA+ motor and adjacent to the ubiquitin receptor Rpn10 (27, 123), in a location that is ideally suited for its key role in deubiquitination after the substrate is engaged, but before the AAA+ ATPase would unfold ubiquitin (Figures 5 and 6a). Importantly, Rpn11’s proximity to the N-ring sterically precludes cleavage between folded ubiquitin moieties and, thus, within ubiquitin chains (155). Instead, it removes ubiquitin modifications en bloc by hydrolyzing the isopeptide bond at the very base of the chain between the substrate lysine and the C terminus of the first ubiquitin moiety (31).
Figure 6.

Crystal structures of proteasomal DUBs. (a) Crystal structure of Saccharomyces cerevisiae Rpn11 (orange) bound to ubiquitin (green) (PDB: 5u4p) (42) and docked into the electron microscopy density with Ubp6 removed for clarity (EM: 3034; PDB: 5a5b) (56). The central pore is highlighted by a red sphere. (b) Arrangement of the Ins-1 loop of Rpn11 (cyan) in (top) the unbound apo state (PDB: 4o8x) (155) and in (bottom) the ubiquitin-bound state (PDB: 5u4p) (42), with the C-terminal tail of ubiquitin in green. The catalytic zinc ion (gray sphere) is shown coordinated by the catalytic residues (stick representation). (c) Crystal structure of activated Uch37 (purple) bound to the DEUBAD of Rpn13 (yellow) and ubiquitin (green) (PDB: 4wlr) (168). Abbreviations: DEUBAD, deubiquitinase adaptor; DUB, deubiquitinase; Ins-1, Insert-1; Ub, ubiquitin.
Rpn11 and the neighboring lid subunit Rpn8 form an obligate heterodimer that can be expressed and purified in isolation. Crystal structures of this Rpn11/Rpn8 dimer elucidated that Rpn11 does not interact with the substrate moiety on the proximal side (the site closest to the substrate) of the scissile isopeptide bond (155, 156), explaining its high promiscuity and ability to remove ubiquitin chains from the wide variety of proteasome substrates. Crystal structures also revealed that the Insert-1 (Ins-1) region of Rpn11 forms a loop that covers the catalytic groove, restricts access to the active site, and inhibits DUB activity (155, 156). Within the isolated lid, Rpn5 further stabilizes this inhibited state of the Ins-1 loop, preventing Rpn11 from acting as an efficient DUB until it is incorporated into the proteasome (157). This is in contrast to related DUBs, such as Sst2 and AMSH-LP, that act in isolation and whose Ins-1 region adopts a beta-hairpin conformation with an accessible active site (158–160). As shown by a recent ubiquitin-bound structure of Rpn11/Rpn8, interaction with ubiquitin induces the conformational switch of Rpn11’s Ins-1 loop from the closed state to the active beta-hairpin structure (Figure 6b). Biochemical studies also uncovered how this switch and a tight regulation of Rpn11’s DUB activity are critical for efficient substrate degradation by the proteasome (42). Disrupting the inhibitory closed state of Ins-1 increases DUB activity but leads to surprising degradation defects and substrate escape from the proteasome owing to premature ubiquitin-chain removal prior to engagement (42).The inhibitory closed state of Rpn11’s Ins-1 loop enables the essential coupling between degradation and deubiquitination in which substrate translocation by the AAA+ motor accelerates the conformational switch of Rpn11 and deubiquitination only for committed substrates (42).
Ubp6, the Allosteric Regulator of the Proteasome
Ubp6 (Usp 14 in human) was first identified as a proteasomal DUB by using the inhibitor ubiquitinvinylsulfone, which specifically targets the catalytic cysteine of this USP (161). Deleting Ubp6 from the proteasome accelerates the degradation of model substrates in vitro (57) and is not lethal in S. cerevisiae (153). However, this deletion leads to a growth defect owing to increased degradation of proteasome substrates, aberrant ubiquitin turnover, and depletion of free ubiquitin (150). Mouse embryonic fibroblasts with a Usp14 knockout show increased presence of Rpn13 and Uch37 on the proteasome, suggesting overlapping roles for Usp14 and Uch37 (162).
Interaction with the proteasome activates Ubp6 ~300-fold (150), and shifting the RP conformation toward s3 by trapping Rpts with the slowly hydrolyzed ATPγS leads to an additional twofold increase in cleaving the ubiquitin-AMC model substrate (47). An N-terminal UBL domain tethers Ubp6 to Rpn1, but contacts of its catalytic USP domain with the coaxially aligned N-ring and the AAA+ ring of the base are required to displace inhibitory loops, expose the active site, and stimulate DUB activity (18, 47, 51, 56, 150). Owing to this specific interaction with the base in the engaged states, Ubp6’s DUB activity acts as a sensor of the proteasome conformation (47, 57). At the same time, ubiquitin-bound Ubp6 stimulates the ATPase activity and 20S gate opening and inhibits substrate engagement by destabilizing the s1 state or preventing proteasome conformational switching back to s1 (Figure 3a) (47, 56, 58, 150). The interaction with the base places the active sites of Ubp6 and Rpn11 just 35 Å apart, allowing Ubp6 to also interfere with ubiquitin binding to Rpn11 (Figure 5a) (47, 57). Taken together, these findings indicate that Ubp6 plays a key role in allosterically regulating the proteasome, in part depending on its own occupancy with ubiquitin.
Interestingly, when the USP domain is docked against the base and thus fully activated, the N-ring of the ATPase hexamer sterically overlaps with the proximal ubiquitin binding site of Ubp6, similar to the N-ring clash that prevents binding of a proximal ubiquitin moiety to Rpn11.Therefore, the biochemical behavior of Rpn11 and Ubp6 is similar: With poor cleavage of free ubiquitin chains and a preference for cleavage at the base of substrate-attached chains, they release ubiquitin modifications en bloc (32). However, Ubp6 cleaves long, unanchored K48-linked chains better than does Rpn11 (163), and it can cleave ubiquitin chains only when more than one chain is attached to a substrate (32). Furthermore, it cannot substitute for Rpn11 in complete deubiquitination of substrates. Ubp6 has thus been implicated in removing supernumerary ubiquitin chains (32), but the underlying mechanisms remain unknown.
Uch37, the Editing Deubiquitinase
The cysteine-dependent DUB Uch37 was identified as a component within the RP of the H. sapiens and Drosophila melanogaster proteasomes. It is found in Schizosaccharomyces pombe (uch2) but not S. cerevisiae (33, 164). In addition to its role in the proteasome, it also functions as a member of the INO80 chromatin-remodeling complex (165). Similar to Ubp6, Uch37 is activated upon proteasome binding. An active-site crossover loop blocks the catalytic cysteine of Uch37 in isolation (166) but, upon interaction with the N-terminal deubiquitinase adaptor (DEUBAD) domain of Rpn13, is stabilized in a single state that leads to ~fivefold increased affinity for ubiquitin (34–36, 167, 168) (Figure 6c).In isolated Rpn13, the DEUBAD domain intramolecularly interacts with the PRU domain and reduces the ubiquitin affinity of this receptor subunit. Rpn13 binding to the proteasome releases this interaction and makes the domains available for ubiquitin binding and activation of Uch37 (169). These findings suggest that Rpn13 activates Uch37 in the proteasome context and not as a free Uch37/Rpn13 complex (34, 167, 168). In the proteasome, Uch37 can cleave distal K48-, K6-, and K11-linked ubiquitin chains, suggesting that it may edit substrateattached ubiquitin modifications and allow the release of inadequately ubiquitinated proteins from the proteasome (33). Alternatively, Uch37 may remove regulatory ubiquitin modifications from proteasome subunits (170), or it might cleave and thus release unanchored ubiquitin chains from proteasomal receptors (171). Despite its location distant from the AAA+ motor (44, 50, 51, 164), Uch37 also stimulates the gate opening and ATPase activity of the proteasome, which suggests it, like Ubp6, may influence the conformational state of the proteasome (63). These results further highlight the tight cooperation and allosteric communication between components of the proteasome. However, it is still unclear how Uch37 may affect the proteasome conformational state, what its substrates are, and how its activity is tied to the catalytic cycle of the proteasome.
CONCLUSION
The past few years have provided us with a wealth of exciting new insights into the structure, molecular mechanisms, and regulation of the 26S proteasome. Based on its architectural and functional complexity, the proteasome can certainly be seen as the destructive counterpart of the ribosome. Whereas the ribosome utilizes an intricate machinery to synthesize the entire multifarious pool of cellular proteins with high fidelity, the proteasome combines strict substrate selectivity with extreme promiscuity and nondiscriminative processing to degrade hundreds of different proteins with various structural, chemical, and biophysical characteristics. This balancing act is enabled by the complex architecture of the proteasome and a series of degradation steps that are well coordinated, in part through significant conformational changes of the RP.
Since the first subnanometer reconstruction of the proteasome in 2010 (172), cryo-EM and other structural techniques have yielded atomic-resolution models for most proteasomal subunits and revealed a whole series of conformational states that represent a critical structural framework for mechanistic studies of proteasome function and regulation. One of the important challenges lying ahead of us now is to correlate these proteasome conformations with individual steps of substrate processing and reconstruct the entire degradation pathway. Furthermore, we are just starting to understand how the ubiquitin code contained in the position, length, and linkage type of ubiquitin chains may affect substrate recognition and turnover. Recent progress in creating new model substrates with defined ubiquitin chains and biophysical characteristics, combined with advancing knowledge about proteasomal ubiquitin receptors and DUBs, will allow important investigations into the proteasome’s selection of appropriate substrates and the fine-tuning of degradation activities. Such fine-tuning and preferential protein degradation may be prerequisites for the proteasome’s ability to orchestrate many vital processes that depend on rapid turnover of regulatory proteins, while also fulfilling critical housekeeping functions in protein homeostasis.
FUTURE ISSUES.
What is the structure of a substrate-bound 26S proteasome? The field is particularly lacking a high-resolution structure that visualizes interactions between the polypeptide chain and the AAA+ motor and between the ubiquitin chain and ubiquitin receptors or DUBs.
How are the individual steps of substrate degradation correlated with the conformational landscape of the proteasome complex observed by cryo-EM, and what are the allosteric networks controlling conformational changes in the RP?
How do the ubiquitin code and geometric or structural features of the substrate influence recognition and turnover by the proteasome? Are multivalent interactions with several ubiquitin receptors used to boost affinities or direct substrates toward subsequent processing steps?
What are the principles for the mechanochemical coupling between ATP hydrolysis and forceful protein unfolding by AAA+ ATPases? The heterohexameric architecture of the proteasomal ATPase motor promises unique insights into the basic mechanisms of AAA+ protein translocases.
How do extrinsic factors, proteasome interacting proteins, or posttranslational modifications regulate the various proteasome activities? Site-specific proteasome phosphorylation, for instance, has been implicated in controlling cell proliferation and tumorigenesis, but the underlying mechanisms remain unknown.
ACKNOWLEDGMENTS
The authors acknowledge support from the US National Institutes of Health (R01-GM094497 to A.M.), the US National Science Foundation CAREER Program (NSF-MCB-1150288 to A.M.), the US National Science Foundation Graduate Research Fellowship Program (to J.A.M.B.), and the Howard Hughes Medical Institute (E.J., K.C.D., and A.M.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Komander D, Rape M. 2012. The ubiquitin code. Annu. Rev. Biochem 81:203–29 [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharyya S, Yu H, Mim C, Matouschek A. 2014. Regulated protein turnover: snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol 15:122–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collins GA, Goldberg AL. 2017. The logic of the 26S proteasome. Cell 169:792–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finley D 2009. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem 78:477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldberg AL. 2007. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem. Soc. Trans 35:12–17 [DOI] [PubMed] [Google Scholar]
- 6.Budenholzer L, Cheng CL, Li Y, Hochstrasser M. 2017. Proteasome structure and assembly. J. Mol. Biol 429:3500–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadre-Bazzaz K, Whitby FG, Robinson H, Formosa T, Hill CP. 2010. Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol. Cell 37:728–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian G, Park S, Lee MJ, Huck B, McAllister F, et al. 2011. An asymmetric interface between the regulatory and core particles of the proteasome. Nat. Struct. Mol. Biol 18:1259–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillette TG, Kumar B, Thompson D, Slaughter CA, DeMartino GN. 2008. Differential roles of the COOH termini of AAA subunits of PA700 (19 S regulator) in asymmetric assembly and activation of the 26 S proteasome. J. Biol. Chem 283:31813–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glickman MH, Rubin DM, Fried VA, Finley D. 1998. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell. Biol 18:3149–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, et al. 2000. A gated channel into the proteasome core particle. Nat. Struct. Biol 7:1062–67 [DOI] [PubMed] [Google Scholar]
- 12.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. 2008. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell 30:360–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. 2007. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s α ring opens the gate for substrate entry. Mol. Cell 27:731–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finley D, Chen X, Walters KJ. 2016. Gates, channels, and switches: elements of the proteasome machine. Trends Biochem. Sci 41:77–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, et al. 1998. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 94:615–23 [DOI] [PubMed] [Google Scholar]
- 16.Saeki Y, Tanaka K. 2012. Assembly and function of the proteasome. Methods Mol. Biol 832:315–37 [DOI] [PubMed] [Google Scholar]
- 17.He J, Kulkarni K, da Fonseca PC, Krutauz D, Glickman MH, et al. 2012. The structure of the 26S proteasome subunit Rpn2 reveals its PC repeat domain as a closed toroid of two concentric α-helical rings. Structure 20:513–21 [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, et al. 2016. Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351:aad9421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, et al. 2008. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453:481–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Nocker S, Sadis S, Rubin DM, Glickman M, Fu H, et al. 1996. The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol. Cell. Biol 16:6020–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. 1994. A 26 S protease subunit that binds ubiquitin conjugates. J. Biol. Chem 269:7059–61 [PubMed] [Google Scholar]
- 22.Martin A, Baker TA, Sauer RT. 2008. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat. Struct. Mol. Biol 15:1147–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maillard RA, Chistol G, Sen M, Righini M, Tan J, et al. 2011. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145:459–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. 2011. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145:257–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beckwith R, Estrin E, Worden EJ, Martin A. 2013. Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat. Struct. Mol. Biol 20:1164–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erales J, Hoyt MA, Troll F, Coffino P. 2012. Functional asymmetries of proteasome translocase pore. J. Biol. Chem 287:18535–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. 2012. Complete subunit architecture of the proteasome regulatory particle. Nature 482:186–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lasker K, Förster F, Bohn S, Walzthoeni T, Villa E, et al. 2012. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. PNAS 109:1380–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Poot SAH, Tian G, Finley D. 2017. Meddling with fate: the proteasomal deubiquitinating enzymes. J. Mol. Biol 429:3525–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma R, Aravind L, Oania R, McDonald WH, Yates JR 3rd, et al. 2002. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298:611–15 [DOI] [PubMed] [Google Scholar]
- 31.Yao T, Cohen RE. 2002. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 419:403–7 [DOI] [PubMed] [Google Scholar]
- 32.Lee B-H, Lu Y, Prado MA, Shi Y, Tian G, et al. 2016. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 532:398–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lam YA, Xu W, DeMartino GN, Cohen RE. 1997. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature 385:737–40 [DOI] [PubMed] [Google Scholar]
- 34.Yao T, Song L, Xu W, DeMartino GN, Florens L, et al. 2006. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat. Cell Biol 8:994–1002 [DOI] [PubMed] [Google Scholar]
- 35.Hamazaki J, Iemura S-I, Natsume T, Yashiroda H, Tanaka K, Murata S. 2006. A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBOJ. 25:4524–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiu X-B, Ouyang S-Y, Li C-J, Miao S, Wang L, Goldberg AL. 2006. hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J. 25:5742–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matyskiela ME, Lander GC, Martin A. 2013. Conformational switching of the 26S proteasome enables substrate degradation. Nat. Struct. Mol. Biol 20:781–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sledz P, Unverdorben P, Beck F, Pfeifer G, Schweitzer A, et al. 2013. Structure of the 26S proteasome with ATP-γS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. PNAS 110:7264–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unverdorben P, Beck F, Sledz P, Schweitzer A, Pfeifer G, et al. 2014. Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. PNAS 111:5544–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wehmer M, Rudack T, Beck F, Aufderheide A, Pfeifer G, et al. 2017. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. PNAS 114:1305–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luan B, Huang X, Wu J, Mei Z, Wang Y, et al. 2016. Structure of an endogenous yeast 26S proteasome reveals two major conformational states. PNAS 113:2642–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Worden EJ, Dong KC, Martin A. 2017. An AAA motor-driven mechanical switch in Rpn11 controls deubiquitination at the 26S proteasome. Mol. Cell 67:799–811 [DOI] [PubMed] [Google Scholar]
- 43.Ding Z, Fu Z, Xu C, Wang Y, Wang Y, et al. 2017. High-resolution cryo-EM structure of the proteasome in complex with ADP-AlFx. Cell Res. 27:373–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen S, Wu J, Lu Y, Ma YB, Lee BH, et al. 2016. Structural basis for dynamic regulation of the human 26S proteasome. PNAS 113:12991–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Y, Wang WL, Yu D, Ouyang Q, Lu Y, Mao Y. 2017. Nucleotide-drive triple-state remodeling of the AAA-ATPase channel in the activated human 26S proteasome. bioRxiv 10.1101/132613 [DOI] [PMC free article] [PubMed]
- 46.Inobe T, Fishbain S, Prakash S, Matouschek A. 2011. Defining the geometry of the two-component proteasome degron. Nat. Chem. Biol 7:161–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bashore C, Dambacher CM, Goodall EA, Matyskiela ME, Lander GC, Martin A. 2015. Ubp6 deubiquitinase controls conformational dynamics and substrate degradation of the 26S proteasome. Nat. Struct. Mol. Biol 22:712–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beck F, Unverdorben P, Bohn S, Schweitzer A, Pfeifer G, et al. 2012. Near-atomic resolution structural model of the yeast 26S proteasome. PNAS 109:14870–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haselbach D, Schrader J, Lambrecht F, Henneberg F, Chari A, Stark H. 2017. Long-range allosteric regulation of the human 26S proteasome by 20S proteasome-targeting cancer drugs. Nat. Commun 8:15578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, et al. 2016. Structure of the human 26S proteasome at a resolution of 3.9 Å. PNAS 113:7816–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang X, Luan B, Wu J, Shi Y. 2016. An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol 23:778–85 [DOI] [PubMed] [Google Scholar]
- 52.da Fonseca PC, He J, Morris EP. 2012. Molecular model of the human 26S proteasome. Mol. Cell 46:54–66 [DOI] [PubMed] [Google Scholar]
- 53.Asano S, Fukuda Y, Beck F, Aufderheide A, Forster F, et al. 2015. A molecular census of 26S proteasomes in intact neurons. Science 347:439–42 [DOI] [PubMed] [Google Scholar]
- 54.Park S, Li X, Kim HM, Singh CR, Tian G, et al. 2013. Reconfiguration of the proteasome during chaperone-mediated assembly. Nature 497:512–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu Y, Smith DM, Kim HM, Rodriguez V, Goldberg AL, Cheng Y. 2010. Interactions of PAN’s C-termini with archaeal 20S proteasome and implications for the eukaryotic proteasome-ATPase interactions. EMBO J. 29:692–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aufderheide A, Beck F, Stengel F, Hartwig M, Schweitzer A, et al. 2015. Structural characterization of the interaction of Ubp6 with the 26S proteasome. PNAS 112:8626–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanna J, Hathaway NA, Tone Y, Crosas B, Elsasser S, et al. 2006. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell 127:99–111 [DOI] [PubMed] [Google Scholar]
- 58.Peth A, Besche HC, Goldberg AL. 2009. Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol. Cell 36:794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li X, Demartino GN. 2009. Variably modulated gating of the 26S proteasome by ATP and polyubiquitin. Biochem. J 421:397–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith DM, Fraga H, Reis C, Kafri G, Goldberg AL. 2011. ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell 144:526–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu CW, Li X, Thompson D, Wooding K, Chang TL, et al. 2006. ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol. Cell 24:39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peth A, Nathan JA, Goldberg AL. 2013. The ATP costs and time required to degrade ubiquitinated proteins by the 26 S proteasome. J. Biol. Chem 288:29215–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peth A, Kukushkin N, Bosse M, Goldberg AL. 2013. Ubiquitinated proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. J. Biol. Chem 288:7781–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL. 2005. ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol. Cell 20:687–98 [DOI] [PubMed] [Google Scholar]
- 65.Kleijnen MF, Roelofs J, Park S, Hathaway NA, Glickman M, et al. 2007. Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol 14:1180–88 [DOI] [PubMed] [Google Scholar]
- 66.Guo X, Wang X, Wang Z, Banerjee S, Yang J, et al. 2016. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol 18:202–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.VerPlank JJS,Goldberg AL. 2017. Regulating protein breakdown through proteasome phosphorylation. Biochem. J 474:3355–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo X, Dixon JE. 2016. The 26S proteasome: a cell cycle regulator regulated by cell cycle. Cell Cycle 15:875–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tomko RJ Jr., Funakoshi M, Schneider K, Wang J, Hochstrasser M. 2010. Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: implications for proteasome structure and assembly. Mol. Cell 38:393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomko RJ Jr., Hochstrasser M. 2011. Order of the proteasomal ATPases and eukaryotic proteasome assembly. Cell Biochem. Biophys 60:13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Inobe T, Genmei R. 2015. N-terminal coiled-coil structure of ATPase subunits of 26S proteasome is crucial for proteasome function. PLOS ONE 10:e0134056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang F, Hu M, Tian G, Zhang P, Finley D, et al. 2009. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol. Cell 34:473–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. 2009. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 139:744–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Glynn SE, Nager AR, Baker TA, Sauer RT. 2012. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat. Struct. Mol. Biol 19:616–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gates SN, Yokom AL, Lin J, Jackrel ME, Rizo AN, et al. 2017. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357:273–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ripstein ZA, Huang R, Augustyniak R, Kay LE, Rubinstein JL. 2017. Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 6:e25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han H, Monroe N, Sundquist WI, Shen PS, Hill CP. 2017. The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. eLife 6:e31324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Monroe N, Han H, Shen PS, Sundquist WI, Hill CP. 2017. Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. eLife 6:e24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Puchades C, Rampello AJ, Shin M, Giuliano CJ, Wiseman RL, et al. 2017. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358:eaao0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomsen ND, Berger JM. 2009. Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139:523–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zehr E, Szyk A, Piszczek G, Szczesna E, Zuo X, Roll-Mecak A. 2017. Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat. Struct. Mol. Biol 24:717–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rubin DM, Glickman MH, Larsen CN, Dhruvakumar S, Finley D. 1998. Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J. 17:4909–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT. 2005. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell 121:1017–27 [DOI] [PubMed] [Google Scholar]
- 84.Yakamavich JA, Baker TA, Sauer RT. 2008. Asymmetric nucleotide transactions of the HslUV protease. J. Mol. Biol 380:946–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nyquist K, Martin A. 2014. Marching to the beat of the ring: polypeptide translocation by AAA+ proteases. Trends Biochem. Sci 39:53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Horwitz AA, Navon A, Groll M, Smith DM, Reis C, Goldberg AL. 2007. ATP-induced structural transitions in PAN, the proteasome-regulatory ATPase complex in Archaea. J. Biol. Chem 282:22921–29 [DOI] [PubMed] [Google Scholar]
- 87.Kim YC, Snoberger A, Schupp J, Smith DM. 2015. ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat. Commun 6:8520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iosefson O, Nager AR, Baker TA, Sauer RT. 2015. Coordinated gripping of substrate by subunits of a AAA+ proteolytic machine. Nat. Chem. Biol 11:201–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Iosefson O, Olivares AO, Baker TA, Sauer RT. 2015. Dissection of axial-pore loop function during unfolding and translocation by a AAA+ proteolytic machine. Cell Rep. 12:1032–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. 2004. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat. Struct. Mol. Biol 11:830–37 [DOI] [PubMed] [Google Scholar]
- 91.Takeuchi J, Chen H, Coffino P. 2007. Proteasome substrate degradation requires association plus extended peptide. EMBO J. 26:123–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chau V, Tobias JW, Bachmair A, Marriott D. 1989. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243:1576–83 [DOI] [PubMed] [Google Scholar]
- 93.Jin L, Williamson A, Banerjee S, Philipp I, Rape M. 2008. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133:653–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, et al. 2009. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137:133–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Johnson ES, Ma PC, Ota IM, Varshavsky A. 1995. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J. Biol. Chem 270:17442–56 [DOI] [PubMed] [Google Scholar]
- 96.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, et al. 2011. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44:325–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saeki Y, Kudo T, Sone T, Kikuchi Y, Yokosawa H, et al. 2009. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J. 28:359–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang D, Chen T, Ziv I, Rosenzweig R, Matiuhin Y, et al. 2009. Together, Rpn10 and Dsk2 can serve as a polyubiquitin chain-length sensor. Mol. Cell 36:1018–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Riedinger C, Boehringer J, Trempe J-F, Lowe ED, Brown NR, et al. 2010. Structure of Rpn10 and its interactions with polyubiquitin chains and the proteasome subunit Rpn12. J. Biol. Chem 285:33992–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Elsasser S, Chandler-Militello D, Müller B, Hanna J, Finley D. 2004. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem 279:26817–22 [DOI] [PubMed] [Google Scholar]
- 101.Mayor T, Graumann J, Bryan J, MacCoss MJ, Deshaies RJ. 2007. Quantitative profiling of ubiquitylated proteins reveals proteasome substrates and the substrate repertoire influenced by the Rpn10 receptor pathway. Mol. Cell Proteom. 6:1885–95 [DOI] [PubMed] [Google Scholar]
- 102.Lu Y, Lee BH, King RW, Finley D, Kirschner MW. 2015. Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science 348:1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shabek N, Herman-Bachinsky Y, Buchsbaum S, Lewinson O, Haj-Yahya M, et al. 2012. The size of the proteasomal substrate determines whether its degradation will be mediated by mono- or polyubiquitylation. Mol. Cell 48:87–97 [DOI] [PubMed] [Google Scholar]
- 104.Zhang M, Pickart CM, Coffino P. 2003. Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J. 22:1488–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Erales J, Coffino P. 2014. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta 1843:216–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Janse DM, Crosas B, Finley D, Church GM. 2004. Localization to the proteasome is sufficient for degradation. J. Biol. Chem 279:21415–20 [DOI] [PubMed] [Google Scholar]
- 107.Wilmington SR, Matouschek A. 2016. An inducible system for rapid degradation of specific cellular proteins using proteasome adaptors. PLOS ONE 11:e0152679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fishbain S, Prakash S, Herrig A, Elsasser S, Matouschek A. 2011. Rad23 escapes degradation because it lacks a proteasome initiation region. Nat. Commun 2:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu H, Kago G, Yellman CM, Matouschek A. 2016. Ubiquitin-like domains can target to the proteasome but proteolysis requires a disordered region. EMBO J. 35:1522–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kraut DA, Matouschek A. 2011. Proteasomal degradation from internal sites favors partial proteolysis via remote domain stabilization. ACS Chem. Biol 6:1087–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Prakash S, Inobe T, Hatch AJ, Matouschek A. 2009. Substrate selection by the proteasome during degradation of protein complexes. Nat. Chem. Biol 5:29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ye Y, Tang WK, Zhang T, Xia D. 2017. A mighty “protein extractor” of the cell: structure and function of the p97/CDC48 ATPase. Front. Mol. Biosci 4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bodnar N, Rapoport T. 2017. Toward an understanding of the Cdc48/p97 ATPase. F1000Research 6:1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Blythe EE, Olson KC, Chau V, Deshaies RJ. 2017. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP*NPLOC4*UFD1L is enhanced by a mutation that causes multisystem proteinopathy. PNAS 114:E4380–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bodnar NO, Rapoport TA. 2017. Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell 169:722–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Peth A, Uchiki T, Goldberg AL. 2010. ATP-dependent steps in the binding of ubiquitin conjugates to the 26S proteasome that commit to degradation. Mol. Cell 40:671–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fishbain S, Inobe T, Israeli E, Chavali S, Yu H, et al. 2015. Sequence composition of disordered regions fine-tunes protein half-life. Nat. Struct. Mol. Biol 22:214–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kraut DA, Israeli E, Schrader EK, Patil A, Nakai K, et al. 2012. Sequence- and species-dependence of proteasomal processivity. ACS Chem. Biol 7:1444–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yu H, Singh Gautam AK, Wilmington SR, Wylie D, Martinez-Fonts K, et al. 2016. Conserved sequence preferences contribute to substrate recognition by the proteasome. J. Biol. Chem 291:14526–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tian L, Holmgren RA, Matouschek A. 2005. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-κB. Nat. Struct. Mol. Biol 12:1045–53 [DOI] [PubMed] [Google Scholar]
- 121.Piwko W, Jentsch S. 2006. Proteasome-mediated protein processing by bidirectional degradation initiated from an internal site. Nat. Struct. Mol. Biol 13:691–97 [DOI] [PubMed] [Google Scholar]
- 122.Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, et al. 2008. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 453:548–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sakata E, Bohn S, Mihalache O, Kiss P, Beck F, et al. 2012. Localization of the proteasomal ubiquitin receptors Rpn10 and Rpn13 by electron cryomicroscopy. PNAS 109:1479–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang Q, Young P, Walters KJ. 2005. Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. J. Mol. Biol 348:727–39 [DOI] [PubMed] [Google Scholar]
- 125.Lam YA, Lawson TG, Velayutham M, Zweier JL, Pickart CM. 2002. A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature 416:763–67 [DOI] [PubMed] [Google Scholar]
- 126.Elsasser S, Gali RR, Schwickart M, Larsen CN, Leggett DS, et al. 2002. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat. Cell Biol 4:725–30 [DOI] [PubMed] [Google Scholar]
- 127.Funakoshi M, Sasaki T, Nishimoto T, Kobayashi H. 2002. Budding yeast Dsk2p is a polyubiquitinbinding protein that can interact with the proteasome. PNAS 99:745–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, et al. 1998. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 391:715–18 [DOI] [PubMed] [Google Scholar]
- 129.Kaplun L, Tzirkin R, Bakhrat A, Shabek N, Ivantsiv Y, Raveh D. 2005. The DNA damage-inducible UbL-UbA protein Ddi1 participates in Mec1-mediated degradation of Ho endonuclease. Mol. Cell. Biol 25:5355–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hofmann K, Bucher P. 1996. The UBA domain: a sequence motif present in multiple enzyme classes of the ubiquitination pathway. Trends Biochem. Sci 21:172–73 [PubMed] [Google Scholar]
- 131.Chen X, Randles L, Shi K, Tarasov SG, Aihara H, Walters KJ. 2016. Structures of Rpn1 T1:Rad23 and hRpn13:hPLIC2 reveal distinct binding mechanisms between substrate receptors and shuttle factors of the proteasome. Structure 24:1257–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gomez TA, Kolawa N, Gee M, Sweredoski MJ, Deshaies RJ. 2011. Identification of a functional docking site in the Rpn1 LRR domain for the UBA-UBL domain protein Ddi1. BMC Biol. 9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Walters KJ, Kleijnen MF, Goh AM, Wagner G, Howley PM. 2002. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 41:1767–77 [DOI] [PubMed] [Google Scholar]
- 134.Zhang N, Wang Q, Ehlinger A, Randles L, Lary JW, et al. 2009. Structure of the S5a:K48-linked diubiquitin complex and its interactions with Rpn13. Mol. Cell 35:280–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen L, Madura K. 2002. Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell. Biol 22:4902–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim I, Mi K, Rao H. 2004. Multiple interactions of Rad23 suggest a mechanism for ubiquitylated substrate delivery important in proteolysis. Mol. Biol. Cell 15:3357–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hänzelmann P, Stingele J, Hofmann K, Schindelin H, Raasi S. 2010. The yeast E4 ubiquitin ligase Ufd2 interacts with the ubiquitin-like domains of Rad23 and Dsk2 via a novel and distinct ubiquitin-like binding domain. J. Biol. Chem 285:20390–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Itakura E, Zavodszky E, Shao S, Wohlever ML, Keenan RJ, Hegde RS. 2016. Ubiquilins chaperone and triage mitochondrial membrane proteins for degradation. Mol. Cell 63:21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sims JJ, Haririnia A, Dickinson BC, Fushman D, Cohen RE. 2009. Avid interactions underlie the K63-linked polyubiquitin binding specificities observed for UBA domains. Nat. Struct. Mol. Biol 16:883–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Raasi S, Pickart CM. 2003. Rad23 ubiquitin-associated domains (UBA) inhibit 26 S proteasome-catalyzed proteolysis by sequestering lysine 48-linked polyubiquitin chains. J. Biol. Chem 278:8951–59 [DOI] [PubMed] [Google Scholar]
- 141.Tsuchiya H, Ohtake F, Arai N, Kaiho A, Yasuda S, et al. 2017. In vivo ubiquitin linkage-type analysis reveals that the Cdc48-Rad23/Dsk2 axis contributes to K48-linked chain specificity of the proteasome. Mol. Cell 66:488–502 [DOI] [PubMed] [Google Scholar]
- 142.Saeki Y, Saitoh A, Toh-e A, Yokosawa H. 2002. Ubiquitin-like proteins and Rpn10 play cooperative roles in ubiquitin-dependent proteolysis. Biochem. Biophys. Res. Commun 293:986–92 [DOI] [PubMed] [Google Scholar]
- 143.Verma R, Oania R, Graumann J, Deshaies RJ. 2004. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell 118:99–110 [DOI] [PubMed] [Google Scholar]
- 144.Hamazaki J, Sasaki K, Kawahara H, Hisanaga S, Tanaka K, Murata S. 2007. Rpn10-mediated degradation of ubiquitinated proteins is essential for mouse development. Mol. Cell. Biol 27:6629–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hamazaki J, Hirayama S, Murata S. 2015. Redundant roles of Rpn10 and Rpn13 in recognition of ubiquitinated proteins and cellular homeostasis. PLOS Genet. 11:e1005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Al-Shami A, Jhaver KG, Vogel P, Wilkins C, Humphries J, et al. 2010. Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLOS ONE 5:e13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chojnacki M, Mansour W, Hameed DS, Singh RK, El Oualid F, et al. 2017. Polyubiquitinphotoactivatable crosslinking reagents for mapping ubiquitin interactome identify Rpn1 as a proteasome ubiquitin-associating subunit. Cell Chem. Biol 24:443–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Crosas B, Hanna J, Kirkpatrick DS, Zhang DP, Tone Y, et al. 2006. Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell 127:1401–13 [DOI] [PubMed] [Google Scholar]
- 149.Aguileta MA, Korac J, Durcan TM, Trempe J-F, Haber M, et al. 2015. The E3 ubiquitin ligase parkin is recruited to the 26S proteasome via the proteasomal ubiquitin receptor Rpn13. J. Biol. Chem 290:7492–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, et al. 2002. Multiple associated proteins regulate proteasome structure and function. Mol. Cell 10:495–507 [DOI] [PubMed] [Google Scholar]
- 151.Cope GA, Suh GSB, Aravind L, Schwarz SE, Zipursky SL, et al. 2002. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 298:608–11 [DOI] [PubMed] [Google Scholar]
- 152.Maytal-Kivity V, Reis N, Hofmann K, Glickman MH. 2002. MPN+, a putative catalytic motif found in a subset of MPN domain proteins from eukaryotes and prokaryotes, is critical for Rpn11 function. BMC Biochem. 3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Guterman A, Glickman MH. 2004. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J. Biol. Chem 279:1729–38 [DOI] [PubMed] [Google Scholar]
- 154.Rinaldi T, Pick E, Gambadoro A, Zilli S, Maytal-Kivity V, et al. 2004. Participation of the proteasomal lid subunit Rpn11 in mitochondrial morphology and function is mapped to a distinct C-terminal domain. Biochem. J 381:275–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Worden EJ, Padovani C, Martin A. 2014. Structure of the Rpn11–Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat. Struct. Mol. Biol 21:220–27 [DOI] [PubMed] [Google Scholar]
- 156.Pathare GR, Nagy I, Sledz P, Anderson DJ, Zhou HJ, et al. 2014. Crystal structure of the proteasomal deubiquitylation module Rpn8–Rpn11. PNAS 111:2984–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dambacher CM, Worden EJ, Herzik MA, Martin A, Lander GC. 2016. Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. eLife 5:e13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sato Y, Yoshikawa A, Yamagata A, Mimura H, Yamashita M, et al. 2008. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature 455:358–62 [DOI] [PubMed] [Google Scholar]
- 159.Davies CW, Paul LN, Kim MI, Das C. 2011. Structural and thermodynamic comparison of the catalytic domain of AMSH and AMSH-LP: nearly identical fold but different stability. J. Mol. Biol 413:416–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shrestha RK, Ronau JA, Davies CW, Guenette RG, Strieter ER, et al. 2014. Insights into the mechanism of deubiquitination by JAMM deubiquitinases from cocrystal structures of the enzyme with the substrate and product. Biochemistry 53:3199–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, Ploegh HL. 2001. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 20:5187–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kim HT, Goldberg AL. 2017. The deubiquitinating enzyme Usp14 allosterically inhibits multiple proteasomal activities and ubiquitin-independent proteolysis. J. Biol. Chem 292:9830–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mansour W, Nakasone MA, von Delbrück M, Yu Z, Krutauz D, et al. 2015. Disassembly of Lys11 and mixed linkage polyubiquitin conjugates provides insights into function of proteasomal deubiquitinases Rpn11 and Ubp6. J. Biol. Chem 290:4688–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hölzl H, Kapelari B, Kellermann J, Seemüller E, Sümegi M, et al. 2000. The regulatory complex of Drosophila melanogaster 26S proteasomes. J. Cell Biol 150:119–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yao T, Song L, Jin J, Cai Y, Takahashi H, et al. 2008. Distinct modes of regulation of the Uch37 deubiquitinating enzyme in the proteasome and in the Ino80 chromatin-remodeling complex. Mol. Cell 31:909–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Burgie SE, Bingman CA, Soni AB, Phillips GN. 2012. Structural characterization of human Uch37. Proteins 80:649–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sahtoe DD, van Dijk WJ, El Oualid F, Ekkebus R, Ovaa H, Sixma TK. 2015. Mechanism of UCH-L5 activation and inhibition by DEUBAD domains in RPN13 and INO80G. Mol. Cell 57:887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.VanderLinden RT, Hemmis CW, Schmitt B, Ndoja A, Whitby FG, et al. 2015. Structural basis for the activation and inhibition of the UCH37 deubiquitylase. Mol. Cell 57:901–11. Correction. 2016. Mol. Cell 61:487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen X, Lee B-H, Finley D, Walters KJ. 2010. Structure of proteasome ubiquitin receptor hRpn13 and its activation by the scaffolding protein hRpn2. Mol. Cell 38:404–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jacobson AD, MacFadden A, Wu Z, Peng J, Liu CW. 2014. Autoregulation of the 26S proteasome by in situ ubiquitination. Mol. Biol. Cell 25:1824–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang NY, Jacobson AD, MacFadden A, Liu CW. 2011. Ubiquitin chain trimming recycles the substrate binding sites of the 26 S proteasome and promotes degradation of lysine 48-linked polyubiquitin conjugates. J. Biol. Chem 286:25540–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bohn S, Beck F, Sakata E, Walzthoeni T, Beck M, et al. 2010. Structure of the 26S proteasome from Schizosaccharomyces pombe at subnanometer resolution. PNAS 107:20992–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mueller TD, Feigon J. 2003. Structural determinants for the binding of ubiquitin-like domains to the proteasome. EMBO J. 22:4634–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.VanderLinden RT, Hemmis CW, Yao T, Robinson H, Hill CP. 2017. Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J. Biol. Chem 292:9493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]