

Summary
Plasma cells are terminally differentiated B lymphocytes that constitutively secrete antibodies. These antibodies can provide protection against pathogens, and their quantity and quality are the best clinical correlates of vaccine efficacy. As such, plasma cell lifespan is the primary determinant of the duration of humoral immunity. Yet dysregulation of plasma cell function can cause autoimmunity or multiple myeloma. The longevity of plasma cells is primarily dictated by nutrient uptake and non-transcriptionally regulated metabolic pathways. We have previously shown a positive effect of glucose uptake and catabolism on plasma cell longevity and function. In this review, we discuss these findings with an emphasis on nutrient uptake and its effects on respiratory capacity, lifespan, endoplasmic reticulum stress, and antibody secretion in plasma cells. We further discuss how some of these pathways may be dysregulated in multiple myeloma, potentially providing new therapeutic targets. Finally, we speculate on the connection between plasma cell intrinsic metabolism and systemic changes in nutrient availability and metabolic diseases.
Keywords: plasma cell, metabolism, glucose, glutamine, multiple myeloma
1. INTRODUCTION
The distinguishing feature of adaptive immunity is “immunological memory”, in which long-lived effector populations persist long after antigen is cleared. In this regard, both B and T lymphocytes exhibit similar characteristics and comparable stages of activation, clonal expansion and contraction, and differentiation. This is advantageous for researchers in lymphocyte biology, as mechanistic insights in one lineage can usually be extended to the analogous populations arising in the other. Plasma cells, however, are an exception to this rule, as they lack a similar counterpart in the T cell lineage. While this presents the difficulty of drawing parallels, it also provides a clean slate to examine and explore unique metabolic properties of these cells.
Plasma cells are terminal effectors within the B cell lineage and devote most of their transcriptional and metabolic resources to synthesize antibodies 1, 2. As a consequence, relatively small numbers of cells can produce large quantities of protective antibodies 3. Thus, the specificity and longevity of plasma cells are the major determinants of durable humoral immunity 4. The resultant antibodies pre-exist and provide sterilizing immunity against pathogens upon secondary exposures to physiological inocula. The longevity of this protection was highlighted in a 26-year longitudinal study in humans, whose aim was to estimate the durability of the antibody response against previously administered vaccines and antecedent infections. The authors estimated the half-life of antibody production to range between 11 years up to an entire lifetime, depending on the specific vaccine or infection 5. This parameter is not to be confused for the half-life of an antibody molecule, which ranges from a few days to several weeks depending on the isotype 6. This sustained level of lifelong immunoglobulin production indicates that plasma cells are either continuously generated through activation of upstream B cells or are a fixed pool of cells that persist without replenishment. Non-specific and periodic activation of memory B cells by toll-like receptor ligands was proposed to generate plasma cells and maintain durable antibody-mediated immunity 7. However, more persuasive evidence for the latter mechanism came from reports in mice showing that antigen-specific plasma cell numbers in the spleen gradually waned post immunization, while the numbers of bone marrow resident plasma cells increased and then remained constant for the entire duration of the assay 8, 9. Further, serum antibody titers were unaffected in humans or mice treated with depleting antibodies against CD20, which is expressed by upstream B cells but not by plasma cells. Moreover, human studies have found no correlation between memory B cell numbers and serum antibody titers 5, 10–12. Finally, other researchers found no evidence that inoculation of mice with TLR ligands led to meaningful activation of memory B cells in vivo 13. These data demonstrate that the durability of antibody production is directly linked to plasma cell lifespan, and not replenishment from re-stimulated memory B cells. Thus, in the context of the findings by Amanna and colleagues, we conclude that the different half-lives of antibody production after vaccination or infection are due to heterogeneity in the lifespan of the plasma cells 5. Defining pathways that promote plasma cell longevity is a major goal for vaccine development, especially for immunizations that lead to very transient protection against infections 14, 15.
Plasma cell lifespan and anatomical location are properties often used to distinguish antibody-secreting cell subsets. The shortest-lived plasma cells persist only for several days after their formation and are mostly localized to the extrafollicular regions of secondary lymphoid organs 16, 17. In contrast, the longest-lived plasma cells are largely bone marrow-resident and persist for the entire lifespan of the organism 8, 9, 18. Many plasma cells of intermediate lifespans have recently been found in the spleen, bone marrow, and intestine. In humans, CD19 expression marks relatively short-lived plasma cells in both the bone marrow and the intestine 19–21. In mice, plasma cell subsets of varying lifespans can be distinguished by expression of B220, B-lymphocyte induced maturation protein 1 (BLIMP1), CD93 (also known as AA4.1), major histocompatibility class 2 (MHCII), CXCR3, and uptake of the fluorescent glucose analog 2-deoxy-2-(7-Nitro-2,1,3-benzoxadiazol-4-yl)amino)-D-glucose (2NBDG) 1, 22–25 (Figure 1). Plasma cells can also be classified based on their non-antibody producing functions, exemplified by Lymphocyte-Activation gene 3 (LAG3) expression. LAG-3+ plasma cells were found to secrete Interleukin-10 (IL-10) and exhibit a more natural regulatory phenotype as compared to LAG3− plasma cells 26. These multiple phenotypes should to be considered and integrated with our explanation of plasma cell longevity and heterogeneity.
Figure 1: Mouse plasma cell subsets and markers.

BM: bone marrow. +/− indicates that a fraction of the cells are positive for the marker.
In addition to neutralization of pathogens, antibodies also perform other effector functions, which include activation of complement cascades, recruitment of phagocytes, triggering of mast cells and basophils during hypersensitivity reactions, passive protection of developing fetuses and neonates, as well as regulating bacterial populations and biofilms in mucosal surfaces such as the gut 27–29. Yet aside from their otherwise protective roles, self-reactive antibodies are associated with many autoimmune disorders. Plasma cells can also become transformed to cause multiple myeloma (MM), a bone marrow-associated malignancy 30. In this review, we discuss the recent findings surrounding the transition from B cells to the plasma cell fate and the metabolic changes that accompany it. We also discuss plasma cell metabolism as a function of both intrinsic and extrinsic signals that shape the function of these cells during health and disease, and finally offer insights and future directions in light of recent work.
2. METABOLIC CHANGES DURING B CELL ACTIVATION AND DIFFERENTIATION
Much of the literature regarding B cell and plasma cell metabolism has been recently reviewed 31–33. We will re-summarize many of these conclusions below and in Figure 2, but we place the majority of our emphasis on the potential meaning and caveats of our own findings, and future directions for the field.
Figure 2: Metabolic cues direct plasma cell longevity and function.
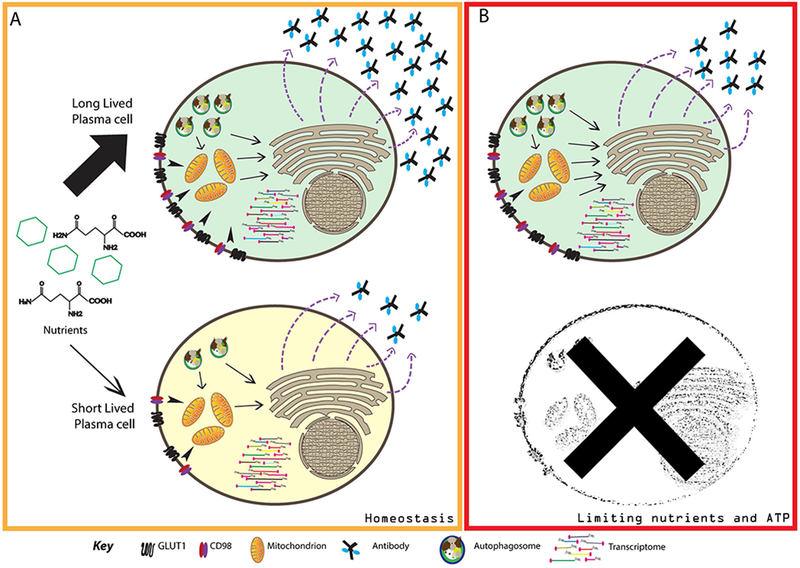
Schematic representation of the metabolic differences between short- and long-lived plasma cells (SLPCs and LLPCs). (A) Despite exhibiting similar transcriptomes, LLPCs take up higher levels of glucose and glutamine than do SLPCs under homeostatic conditions. Nutrients can also be provided through autophagy, which is also higher in LLPCs. These nutrients collectively are catabolized and directed to mitochondria for ATP generation, or used in synthesis and glycosylation of antibodies. ER stress is equivalent between plasma cell subsets, but SLPCs degrade antibody molecules more than LLPCs. As a result, LLPCs secrete more antibody molecules than SLPCs, despite equivalent rates of protein and antibody synthesis. (B) Under limiting nutrient availability and low ATP generation, LLPCs can expand their basal respiratory capacity to compensate. SLPCs are unable to perform this function and as a result, initiate pathways of programmed cell death.
2.1. Antigen-inexperienced B cells
B cell development is a process that initiates in the bone marrow and continues in the spleen across phenotypically distinguishable CD19+ subsets 34. Naïve B cells are composed of antigen-inexperienced follicular (also known as B2), marginal zone, B1a, and B1b subsets. The average lifespan of a naïve B2 B cell is approximately 13 weeks, as compared to marginal zone B cells (22 weeks), the other mature antigen-inexperienced B cell population in the periphery 35. The transcription factor Paired Box 5 (PAX5) is crucial for the maintenance of B cell identity, and by modulating its target genes, is responsible for a large fraction of the transcriptional changes that mediate lineage commitment and maturation 36, 37. Despite this critical role in establishing and maintaining B cell identity, Pax5 is frequently mutated in B cell malignancies 38. To explain the tumor-suppressive role of PAX5, a series of elegant papers demonstrated that in pre-B Acute Lymphoblastic Leukemia (ALL), PAX5 suppresses glucose uptake and ribonucleoside synthesis 39, 40. Mutating PAX5 alleviates some of these metabolic constraints, thereby allowing pre-B ALL cells to divide indefinitely. Presumably PAX5 plays a similar role in naïve B cells, thereby ensuring minimal baseline metabolic activity 41. Naïve B cells also rely on B cell receptor (BCR)- and B-cell activating factor (BAFF)-mediated signals through the mammalian target of rapamycin (mTOR) pathway, which has been shown to contribute to increases in cell size, mitochondrial activity, protein synthesis, and general activity of B cells prior to antigen-induced proliferation 42–44. In addition to naïve B cells, marginal zone B cells are also mature antigen-inexperienced splenic B cells and are crucial participants in responses to T-independent antigens 45, 46. However, little is documented about the metabolic status of these cells, save that B cell-specific ablation of tuberous sclerosis 1, a subunit of the mTORC1 inhibitor complex, led to a dramatic loss of marginal zone B cells and affected downstream T-independent responses 47.
2.2. Activated B cells
Naïve B cell metabolic activity is triggered upon B cell receptor signaling and/or Toll-like receptor activation. The B cell activation program induces changes in cell size, increased uptake of nutrients, and increased rate of transcription and translation 48–50. In addition to B cell receptor engagement, B cells also receive additional signals such as those through the Toll-like receptors, as well as costimulatory and cytokine signals provided through T cell help. This two-signal mode of activation reflects a similarity between T and B lymphocytes, and in the absence of this second signal, B cells undergo mitochondrial dysfunction and superoxide-induced cell death 51. Co-stimulation also favors uptake of nutrients necessary for anabolism to support activation. Glucose uptake increases concomitantly with upregulation of the glucose transporter GLUT1 (SLC2A1) and is catabolized through glycolysis 52. The resultant lactate allows for restoration of NAD+ and redox balance 53. Alternatively, pyruvate derived from glycolysis can be carried into mitochondria to contribute carbons to the tricarboxylic acid cycle 53. Glucose can also be oxidized through the pentose phosphate pathway, which generates ribonucleosides that are critical for subsequent proliferation 52. Through the tricarboxylic acid cycle, glucose-derived pyruvate can be converted into citrate, which further feeds into lipogenic pathways through activity of the enzyme ATP-citrate lyase 49. These pathways feed into the formation of cholesterol, phosphatidylcholine, phosphatidylinositol, ceramides, and other fatty acids, which in turn serve as substrates for endoplasmic reticulum (ER) and plasma membrane expansion as well as organelle biogenesis 49.
In addition to glucose, glutamine uptake also increases after B cell activation. This is likely achieved by the PDK1- and Akt- dependent induction of Slc3a2 expression, a common subunit of multiple amino acid transporters 54, 55. In addition to glutamine, this transporter is crucial for the uptake of multiple large neutral amino acids, which are substrates for protein synthesis and feed into other metabolic pathways 56. While SLC3A2 pairs with SLC7A5 to form CD98, it can also pair with SLC1A5 to make up the ASCT2 transporter, both of which facilitate the uptake of large neutral amino acids by B cells 57. Glutamine can feed into the TCA cycle as α-ketoglutarate, thereby acting as an anaplerotic substrate to replenish TCA cycle intermediates 53. Through the TCA cycle, glutamine can be used to generate other amino acids such as glutamate and aspartate, citrate for use in lipogenic pathways, and succinate which is oxidized to provide electrons for respiration and ATP generation 23. The uptake of both glucose and glutamine are tightly regulated processes and are controlled by expression of the microRNA let-7, which suppresses expression of Hexokinase-2 and c-Myc 58. In addition to these nutrients, leucine uptake promotes mTORC1 activation in B cells 59. Thus, activation signals promote nutrient uptake to allow B cells to expand and divide.
After exposure to the antigen and initiating activation programs, B cells migrate towards the interface between the T and B cell zones in the secondary lymphoid organ to recruit help from T cells 60. T cells in turn, through recognition of the peptide-MHC-II complex on the surface of B cells, provide help to B cells in the form of costimulatory interactions involving CD154-CD40, ICOS-ICOSL, OX40-OX40L, LFA-2-ICAM-1 as well as through secretion of cytokines and growth factors 61. These initial interactions enable B cells to subsequently undergo proliferate and form foci at the outer edges of the B cell follicles 62. Some of these cells may undergo isotype switching and differentiate into short-lived plasma cells and contribute to the early humoral response while others can form memory B cells 63, 64. Alternatively, some B cells migrate to the centers of B cell follicles and establish germinal centers (GCs) 65.
2.3. Germinal centers
Depending on the infection or immunization, GCs can be detected as early as 3 days post-immunization and can persist for many weeks 66–69. The GC is organized into a dark zone, consisting of highly proliferative B cells, and a light zone comprised of non-dividing B cells 70. Within the germinal centers, B cells express activation-induced cytidine deaminase (AID), which is responsible for both somatic hypermutation and immunoglobulin isotype-switching 71. Dark-zone GC B cells proliferate rapidly while accumulating somatic mutations in antibody receptor-encoding genes 72, 73. These cells then migrate to the light zone where they compete among themselves for antigen, which is endocytosed and subsequently presented through MHCII to T cells in an attempt to procure survival signals 73. Only a small fraction of these cells are selected in the light zone and subsequently return to the dark zone undergo more rounds of proliferation, class switching, and affinity maturation.
Much of the proliferative burst in the dark zone has been shown to rely on c-Myc, as its ablation leads to complete abrogation of GCs 74, 75. c-Myc is induced in GC B cells by the action of BCR and CD40 signals 76. Signals through the B cell receptor and CD40 also induce mTOR activation, thereby permitting B cells to re-enter cycles of proliferation 76, 77. c-Myc also promotes glycolytic activity by upregulating Hexokinase and Pyruvate kinase in activated cells while modestly increasing enzyme expression of the downstream tricarboxylic acid cycle and pentose phosphate pathways 78. In T cells, c-Myc also leads to CD98 upregulation and upregulation of Glutaminase 2 (Gls2), suggesting therefore that it also participates in glutamine metabolism 78. It is possible that a similar metabolic regulation is also at play in GC B cells. Intriguingly though, c-Myc is inhibited by the GC-promoting transcription factor B cell lymphoma 6 (BCL6) and is undetectable in DZ B cells 74, 75, 79. To mediate its effects throughout the dark zone, c-Myc activates the expression of AP4, which maintains expression of many c-Myc targets 79. c-Myc downregulation is a crucial event leading to the expression of Glycogen synthase kinase 3 (GSK-3), which in turn promotes glycolysis, mitochondrial biogenesis, and ROS formation in proliferating B cells 80.
The rapid proliferation and nutrient consumption within GC B cells potentially render them susceptible to starvation-induced cell death. Thus, autophagy is another key pathway upregulated in GC B cells. Interestingly though, B cell activation transiently downregulates mTORC1-dependent canonical autophagy while promoting upregulation of the non-canonical pathway 81. The large numbers of proliferating cells may also induce a state of low oxygen availability within the GC, observed by hypoxia-marking dyes and expression of HIF1α. Genetic deletion of VHL, an inhibitor of HIF1 transcription factors, reduces cellular proliferation, AID expression, and dampens mTOR signaling in GCs 82.
GCs can output memory B lymphocytes and plasma cells. Early hapten studies reported that GC-derived memory B cells tend to express antibodies of lower affinity than do plasma cells, suggesting the existence of an affinity threshold that distinguishes these fates within the GC 83. Our own studies have found that the isotype-switched memory B cell compartment is significantly more diverse than long-lived plasma cells 3. This allows for memory B cells, but not long-lived plasma cells to mount recall responses to divergent and heterologous pathogens. Mechanistically, GC B cells fated for the memory B cell lineage, identified by expression of several markers, may be actively selected for low affinity 84–86. This is mediated in part by BACH2, a critical transcription factor necessary for the memory B cell lineage, which is repressed when cells receive strong T Follicular helper (Tfh) cell-mediated signals 86. Reciprocally, GC B cells that receive Tfh signals induce high levels of Interferon Regulatory Factor 4 (IRF4) to promote the plasma cell fate (discussed more below). The active selection of low affinity GC cells into the memory compartment is puzzling, as it is unclear how such cells would be selected without also recruiting autoreactive or irrelevant B cells. In an elegant genetic system in which isotype switching is mediated by Cre recombinase but AID-mediated somatic hypermutation is prevented, memory B and plasma cell formation is largely unperturbed 87. These data argue against an absolute final affinity threshold that must be reached. Since each GC is an autonomous unit with different clonal compositions, it remains possible that a relative affinity threshold still exists 88. In this case, cells with relatively high antibody affinities within a given GC might be recruited into the plasma cell compartment, irrespective of the absolute on- and off-rates. Regardless, long-lived plasma cells derived from GCs are formed relatively late in the response, whereas memory B cells are formed early 66. Thus, distinct signals must be provided in early versus late GCs. In helminth infections, Tfh cells within GCs express IL-21 initially, but gradually lose IL-21 and express IL-4 instead 89. Of note, IL-4 promotes glucose uptake by B cells, suggesting that part of the signaling differences between early and late GCs may be metabolic in nature 90. Indeed, many of the transcriptional differences between early and late germinal centers are in genes involved in metabolism 66.
2.4. Memory B cells
Memory B cells are composed of several subsets that differ in their ability to generate new GCs and plasma cells. In both mice and humans, IgM+ memory B cells tend to generate GCs preferentially, whereas IgG+ memory cells are skewed toward the plasma cell fate 67, 91, 92. These lineage biases are likely intrinsic to the memory cell transcriptional program rather than the strength of BCR signals, as CD80 and PDL2 expression can functionally separate subsets irrespective of the antibody isotype 93. While metabolic pathways in memory T cells have been studied extensively, memory B cell metabolism remains largely unexplored with a few recent exceptions 94. Like their GC and plasma cell counterparts, memory B cell responses depend on autophagy. Memory B cells deficient in the autophagy gene ATG7 are present in normal numbers initially after immunization or infection with influenza, but then wane rapidly 95, 96. This diminution results from oxidative stress rather than apoptosis or necrosis. Another recent study showed that toll-like receptor ligands and Interferon-α caused human IgD+ memory B cells to engage glycolysis and the mTORC1 pathway in vitro. Compared with naïve B cells, this led to enhanced differentiation by memory B cells into plasmablasts 97. It is possible that these functional differences are caused by enhanced sensitivity to toll-like receptor ligands by memory B cells.
2.5. Plasma cells
The exit signals from the GC reaction that promote plasma cell formation are not fully known. Recent studies have suggested that high-affinity B cells in the light zone receive strong CD40 signals, downregulate BCL6, and exit the GC reaction as IRF4+ CD69+ cells that eventually differentiate into plasmablasts 98. Expression of IRF4 is important for the formation and function of GCs 99–101. Its subsequent upregulation then enables IRF4 to associate with low affinity DNA binding sites and promote expression of BLIMP-1 99, 100, 102. BLIMP-1 expression then silences GC B cell transcriptional programs while simultaneously promoting expression of genes required for plasma cell differentiation and function 59, 103. Initially, plasmablasts are short-lived, immature, proliferating cells, but gradually acquire a more mature plasma cell phenotype based on BLIMP-1 expression 24. Plasma cells formed in the earliest stages of the response tend to be largely short-lived and persist only for a few days in the periphery, though there is substantial heterogeneity in turnover rates 16, 22, 23. Some plasma cells produced towards the end of the GC response migrate to the bone marrow and persist there in survival niches, durably secreting high-affinity antibodies 8, 9, 18, 66, 104.
Once a plasma cell is formed, much of its metabolism is diverted from organelle biogenesis and anabolism towards antibody synthesis and glycosylation. Through much of B cell activation and its subsequent stages, glucose is taken up via the transporter GLUT1, and it is likely that it remains the chief glucose transporter in these cells 52. Genetic ablation of GLUT1, however, does not fully ablate plasma cell formation, suggesting that glucose uptake might depend on additional as yet unidentified sugar transporters 52. GLUT6 (Slc2a6) and the solute carrier SLC50A1 are candidates to perform this function as they are highly expressed in both murine splenic and bone marrow plasma cells 23, 105. Yet GLUT6 remains largely uncharacterized, whereas SLC50A1 has primarily been implicated in sugar efflux rather than import 106, 107. Primary human plasma cells represent even more of a mystery, as none of the known glucose transporters are highly expressed at the transcript level, despite clear evidence of glucose import and catabolism 19, 105. Irrespective of its mode of entry, much of the glucose taken up by plasma cells is shuttled into hexosamine biosynthetic pathways in plasma cells, which provides sugars for protein glycosylation 48, 105.
Glycosylation patterns influence how antibodies interact with Fc receptors, thereby conferring distinct effector functions 108. Glycosylation primarily occurs at Asn297 as an N-linked glycan in the constant region of the heavy chain, while a secondary site also exists in some antibodies in the variable region of the heavy chain. While mutating this residue to a glutamine has little impact on antibody formation, inhibiting glycosylation completely using tunicamycin results in plasma cell death en masse by promoting protein misfolding and an excessive unfolded protein response 109, 110. It is intriguing to note that conditions of inflammation or priming antigen can modulate glycosylation patterns on the surface of the antibody molecule 111–113. The signals to plasma cells and downstream mechanisms of differential glycosylation remain unknown.
Plasma cells secrete thousands of antibody molecules per second, and this depends upon an expanded ER. This expansion is primarily driven by the Inositol-requiring enzyme 1α (IRE1α) pathway, the most well-studied of the unfolded protein response (UPR) signals in plasma cells 114. IRE1α is a ribonuclease and splicing factor, responsible for the removal of 26 internal nucleotides in XBP-1 transcripts, leading to the formation of spliced X-Box Binding Protein 1 (sXBP1) 115. In turn, sXBP1 promotes mitochondrial and ER biogenesis, and its deletion in B cells leads to reduced ER expansion and antibody secretion 114, 116, 117. This deletion however does not impact plasma cell numbers, suggesting that antibody secretion is not essential for differentiation 118, 119. sXBP-1 expression also shuttles glucose towards hexosamine biosynthesis by regulating expression of key enzymes like Glutamine Fructose-6-phosphate transferase 1 (GFAT1), Glucosamine-phosphate N-acetyltransferase 1 (GNPNAT1) and phosphoglucomutase 3 (PGM3) 120. In plasma cells, the IRE1α pathway and sXBP1 targets were found to be equivalent across long-lived 2NBDG+ and short-lived 2NBDG− subsets, suggesting that this mode of UPR is important for antibody secretion but does not correlate with longevity 23. A second pathway downstream of the UPR is cleavage of ATF6α, leading to a functional transcription factor 121. Expression of ATF6α targets was also similar across plasma cell subsets, and its genetic deletion had no effect on plasma cell longevity or antibody secretion 23, 122. The third pathway activated by the UPR is phosphorylation of eif2α 121. Based on in vitro cultures, this pathway was proposed to be inactive in plasma cells 123–125. However, analysis of plasma cells directly ex vivo showed robust activation of this pathway, with slightly elevated p-eif2α levels in the B220+ subsets 23. Accordingly, ablation of EIF2AK3/PERK, the kinase that phosphorylates eukaryotic translation initiation factor 2α (eif2α) during ER stress, led to a selective loss of B220+ plasma cells 23. However, within the B220+ plasma cells, 2NBDG+ cells were similarly sensitivity to PERK deletion as were shorter-lived 2NDBG− subsets 23. Moreover, B220− plasma cells were unaffected by the loss of PERK 23. Thus, despite previous findings demonstrating that ER stress-dependent pathways effect in vitro plasma cell death, the differences in lifespan between plasma cell subsets in vivo are not explained by alterations in the UPR 126–128
Interestingly, long-lived plasma cells show equivalent rates of antibody expression as do their short-lived counterparts but have a greater rate of immunoglobulin secretion 23. These differences are again unlikely to be explained by ER stress responses, given that UPR pathways are activated similarly across all subsets 23. Against our predictions, short-lived plasma cell antibody secretion could not be consistently enhanced by provision of exogenous glycosylation sugars. This suggests that a shortage of these intermediary metabolites do not explain diminished secretion rates, though we cannot be certain that these glycosylation sugars were properly incorporated via the hexosamine salvage pathway 129. Instead, provision of additional extracellular amino acids enhanced mitochondrial respiration, intrinsic amino acid biosynthesis, and antibody secretion. This is partly driven by glutamine, which is used for mitochondrial anaplerotic reactions and for glutamate and aspartate synthesis 23. Stable isotope tracing experiments demonstrated that 20-30% of succinate, which is oxidized to fumarate to generate electrons for respiration, is contributed by glutamine 23. Yet it is unlikely that glutamine is responsible for the entire effect of extracellular amino acids, as we have preliminarily observed no survival or secretion defects in glutaminase-deficient plasma cells. Mechanistically, it seems unlikely that these additional amino acids promote antibody secretion by enhancing the rate of translation, as immunoglobulin transcripts and proteins are produced similarly across plasma cell subsets 23. Instead, it may be that enhanced energy production, mitochondrial anaplerosis, or effects on the secretory apparatus indirectly facilitate antibody release 130.
A major unresolved question is how these metabolic programs are initiated and maintained in long-lived plasma cells. A number of signals, such as TLR ligands, BCR ligation, and cytokines such as IL-4 promote glucose uptake when B cells become activated 48, 52, 77, 80, 90. Yet whether these same signals instruct a metabolic program as plasma cells are generated is unknown. Moreover, it is unclear whether extrinsic niche factors maintain this metabolic program in plasma cells, or whether a self-sustaining metabolic feedback loop is initiated during ontogeny. As these questions are resolved, new strategies to enhance or antagonize plasma cells for therapeutic purposes should become clear.
3. A METABOLIC EXPLANATION FOR PLASMA CELL LONGEVITY
Recent studies have demonstrated surprising transcriptional similarity between short- and long-lived plasma cells, both in humans and in mice. In human cells, unbiased comparisons of short-lived tonsillar plasma cells with long-lived bone marrow plasma cells revealed only 7 transcriptional differences 131. Comparisons of refined populations of short-lived CD19+ plasma cells with long-lived CD19− plasma cells in the human bone marrow also revealed relatively few differences, only a fraction of which were consistent across studies 19, 23. In the mouse, transcriptional profiles were again similar when splenic (ostensibly) short-lived plasma cells were compared with bone marrow plasma cells 1. Our own studies using bulk and single-cell RNA-seq demonstrated that except for genes involved in proliferation and, unexpectedly, neutrophil effector pathways, the transcriptomes between 2NBDG+ and 2NBDG− mouse plasma cells were surprisingly similar 23, 105. We were able to corroborate differences in cellular proliferation across subsets with incorporation of the thymidine analog 5-Bromo-2’-deoxyuridine (BrdU), and it may be possible that neutrophil degranulation pathways may play longevity-independent effector roles in the plasma cell subsets 23. Genes such as BLIMP-1, Myeloid cell leukemia 1 (MCL1), CD28, and B-cell maturation antigen (BCMA), which are essential for the long-term survival of plasma cells, were similarly expressed transcriptionally across all mature mouse plasma cells irrespective of their longevity 1, 23, 105, 132–135. In previous work we and others demonstrated that the transcription factor ZBTB20 promotes plasma cell longevity 136, 137. Reciprocally, ZBTB32, a related family member, antagonizes plasma cell longevity 138. In light of the lack of obvious transcriptional differences between short- and long-lived plasma cells, the direct effects of ZBTB20 might actually occur in upstream germinal center reactions, perhaps to impose a stable metabolic program in plasma cell precursors 139. Similarly, ZBTB32 is expressed in upstream memory B cells rather than in plasma cells themselves 93, 138. We certainly cannot exclude the possibility that very subtle alterations in transcription below our limit of detection cause large changes in plasma cell lifespan. However, if such functionally important and consistent transcriptional changes do exist, it will be difficult to uncover these differences using currently available methods. It is likely, therefore that differences in post- or non-transcriptional regulation, such as metabolism, explain the variability of plasma cell lifespan across subsets.
3.1. Mitochondrial spare respiratory capacity as a determinant of plasma cell longevity
Previous studies in other cell types demonstrated mitochondrial spare respiratory capacity as a metabolic correlate of longevity 140, 141. SRC is the difference between the maximum possible ability of a cell to synthesize ATP through the mitochondrial pathway and its basal rates. Spare respiratory capacity is by definition revealed by treatment with uncouplers such as 2,4-Dinitrophenol (DNP) or Carbonyl cyanide-4-trifluoromethoxyphenylhydrazone (FCCP), which are ionophores that permeabilize the inner mitochondrial membrane. These ionophores thus force cells to respire maximally in a futile effort to restore the electrochemical gradient, thereby uncoupling ATP synthesis from respiration 142. The property of spare respiratory capacity was shown in long-lived cerebrocortical neurons and was proposed to act as a bioenergetic buffer to promote longevity 140. The physiological importance of spare respiratory capacity, however, has been difficult to demonstrate for several reasons. First, spare respiratory capacity is defined in vitro after addition of uncouplers such as FCCP. Because cells would never be exposed to such treatment in vivo, the pathways exposed by such pharmacological manipulations ex vivo might never be used under physiological conditions. Second, genetic perturbations remained to be discovered that would allow for a specific ablation of spare respiratory capacity while leaving basal respiration intact. Thus, when our initial observations demonstrated much greater spare respiratory capacity in long-lived plasma cells than in short-lived plasma cells, much work remained to prove the importance of this property in vivo.
Fortuitously, we discovered that treatment of long-lived plasma cells with UK5099, a drug that prevents pyruvate from entering the mitochondria, specifically ablated spare respiratory capacity but left basal respiration intact 105, 143. Even more fortuitously, our colleagues had recently generated mice conditionally deficient in MPC2, an essential subunit of the mitochondrial pyruvate carrier 144. The heterodimeric complex of MPC1 and MPC2 is the target of UK5099, and each subunit is critical for carrying cytoplasmic pyruvate into the inner matrix of the mitochondria 145, 146. Deletion of Mpc2 led to a progressive loss of bone marrow plasma cells in vivo as well as antigen-specific antibodies, but not upstream B cells 105. Thus, to our knowledge, this study was the first to formally demonstrate that the ex vivo property of spare respiratory capacity was important in vivo for cellular longevity.
Based on available evidence, apoptosis is likely the dominant mechanism that leads to short-lived plasma cell death upon bioenergetics crises. Mice carrying transgenes of Bcl2 maintain plasma cell numbers after immunization 147. Reciprocally, plasma cells that lack Mcl1 die rapidly 133. Yet despite the dominance of apoptosis as a death effector mechanism in plasma cells, other pathways may also contribute. Indeed, plasma cells lacking pro-apoptotic Bim are not fully protected from death 148. Several studies have proposed ER stress-dependent death, though these pathways are equally active in short- and long-lived plasma cell subsets 23, 126, 127. Aside from apoptosis, the most widely accepted forms of intentional cell death are necroptosis, pyroptosis, and ferroptosis 149–151. External methods of clearance mediated by phagocytes are another mechanism of clearance, which is used to remove aging red blood cells 152. An unusual form of oxidative death has been described in Atg7−/− memory B cells, but to our knowledge these alternate death effector mechanisms have not been studied in primary plasma cells 95.
3.2. Nutrient uptake by plasma cells
Our findings with UK5099 and genetic knockouts demonstrated that mitochondrial pyruvate import is the basis for spare respiratory capacity in long-lived plasma cells. The spare respiratory capacity differences between short- and long-lived plasma cells could not obviously be linked to intrinsic differences in mitochondrial function or numbers, yet this issue still needs to be explored further. Mitochondria from long-lived plasma cells are larger and appear more fused than those in short-lived plasma cells, and mitochondrial fusion has been shown to be critical for mediating memory T cell function 105, 153. Regardless, pyruvate is derived from glycolysis, as inhibiting glycealdehyde-3-phosphate dehydrogenase (GAPDH) dramatically reduced spare respiratory capacity 105. Consistently, halving the glucose concentrations in the assay media led to a decrease in spare respiratory capacity. These data implicate increased glucose uptake by long-lived plasma cells relative to short-lived plasma cells as the primary basis for spare respiratory capacity.
Consistent with this conclusion, in vivo uptake of the fluorescent glucose analog 2NBDG correlates well with plasma cell longevity. Bone marrow long-lived plasma cells imported high amounts of 2NBDG whereas splenic plasma cells were more heterogeneous 23, 105. BrdU pulse-chase experiments clearly demonstrated enhanced longevity by 2NBDG+ plasma cell subsets relative to their 2NBDG− counterparts 23. 2NBDG+ subsets of plasma cells showed higher spare respiratory capacity than did their 2NBDG− counterparts, further supporting the conclusion that longevity is linked to the ability to import extracellular glucose 105. Moreover, 2NBDG+ subsets in the spleen exhibited high rates of antibody secretion, higher levels of autophagosomes, as well as longer half-lives as compared to their 2NBDG− counterparts 23. We also found that each plasma cell subset had mostly distinct immunoglobulin repertoires, were generated contemporaneously through the course of an NP-Ovalbumin (OVA) immunization, and showed similar expression of CD93, a marker of mature plasma cells, indicating that these subsets are formed independently of each other 23, 25. When long- and short-lived plasma cells were isolated and cultured ex vivo, 2NBDG uptake by long-lived plasma cells remained higher than in short-lived plasma cells. These data suggest an intrinsic ability of long-lived plasma cells to robustly import nutrients, rather than changes in the nutrient content in the local microenvironments of long- and short-lived plasma cells 23, 105. Moreover, the data suggest that the decision to commit to longevity (or not) may be determined sometime before or during plasma cell differentiation, and in part involves establishment of a sustainable metabolic program. One caveat to these studies is the concern over the specificity of 2NBDG and its transporters 154. In preliminary observations, we have found that while radioactive glucose uptake is much higher by bone marrow long-lived plasma cells relative to other plasma cell subsets, similar trends are not readily apparent between splenic 2NBDG+ and 2NBDG− subsets. One technical caveat we have noticed anecdotally is that plasma cells assayed immediately after having been fluorescence activated cell sorted display very poor glucose import rates, perhaps due to stresses imposed by the process 155. These cells recover after a rest period ex vivo. Given the minimal expression of known glucose transporters in mouse and human plasma cells, as-yet unknown genes and pathways likely promote extracellular sugar import.
3.3. Methods to assess plasma cell metabolism
Chemical inhibitors that disrupt anabolic pathways and dyes that mark nutrient uptake and mitochondrial function have proven very useful in perturbing and quantifying metabolic pathways. These dyes provide an essential bridge between classical metabolism experiments performed on bulk populations in vitro and single-cell resolution studies that can be performed in vivo. Yet as highlighted by our preliminary 2NBDG results, some degree of caution is warranted when using dyes with unclear specificities and inhibitors with problematic off-target effects. The precise targets of most of the widely used Mitotracker and MitoSox dyes have not been defined, and the use of these dyes can be confounded by non-specific over-labelling or by cellular drug efflux pumps. To highlight these issues, hematopoietic stem cells were thought to possess very little mitochondrial mass based on mitochondrial dye stains 156–162. Yet recent studies demonstrated instead that hematopoietic stem cells have substantial mitochondrial mass as visualized by electron microscopy and show robust labeling with Mitotracker dyes when pre-treated with verapamil, an inhibitor of ABC drug transporters 163. As another example of how drug manipulations should be treated with caution, we performed pharmacological experiments with plasma cell subsets and etomoxir, an inhibitor of fatty acid oxidation, to define the carbon sources that drive basal respiration. Our results suggested that approximately 50% of basal respiration is derived from long-chain fatty acids 105. New information in the field, however, has demonstrated clear off-target effects of etomoxir. In addition to acting on Carnitine palmitoyltransferase 1 (CPT1) proteins, which allow long-chain fatty acids to pass the outer mitochondrial membrane, etomoxir can also inhibit 1,2-Diacyl-sn-glycerol acyl transferase, a key enzyme in the de novo synthesis of glycolipids and at high concentrations, it inhibits the electron transport chain 164, 165. High concentrations of etomoxir also inhibited mitochondrial adenine nucleoside transporter and depleted intracellular Coenzyme A levels in macrophages 166. Further, genetic ablation of CPT2, which carries long-chain fatty acids into the inner matrix of the mitochondria, was unable to recapitulate findings obtained on etomoxir treatment of cells, implying therefore an inconsistency between pharmacological and genetic data 165–169. Preliminary data from our group also points towards no defects in mouse plasma cell frequencies upon genetic ablation of CPT2, suggesting therefore that long-chain fatty acids may not have as pronounced a role in plasma cell biology as we had initially anticipated. These findings thus serve as a needed warning in the use of pharmacological inhibitors alone to dissect biological phenomena.
While the use of genetic models potentially offers a more specific solution to dissecting pathways crucial for plasma cell function, they too present their own intrinsic challenges. For example, deletion of CPT1A is readily compensated by endogenous expression of the functionally redundant gene products CPT1B and CPT1C 168. Moreover, germline knockouts of essential genes often result in embryonic lethality of the strain, as is the case with the Mpc2-knockout mouse 170. Plasma cells present their own unique challenges, as no lineage-specific Cre recombinase lines exist. Using the estrogen receptor to the Cre recombinase provides a tamoxifen inducible murine model of gene deletion. The latter system was beneficial for our studies in which we were able to genetically ablate Mpc2 in mixed bone marrow chimeras months after reconstitution and subsequent vaccination against West Nile Virus (WNV) 105. Alternative approaches have used mice in which the Cre expression cassette has been driven by B cell-specific promoters, such as those of CD79α, CD23, CD19, and the γ1-heavy chain promoter 171–174. A shortcoming of these Cre-mediated strategies is that deletion of genes of interest occurs ubiquitously (as in the Rosa26-CreERT2 system) or in the lineages preceding plasma cell differentiation (in the B cell promoter-Cre systems). An experimental challenge then becomes how to distinguish between defects in upstream B cell activation and plasma cell formation versus maintenance. A needed solution is to generate new mouse lines in which Cre recombinase expression is restricted to plasma cells. Resolving this dilemma would provide an important tool for plasma cell research.
3.4. Cell extrinsic factors
While cell intrinsic metabolic differences functionally distinguish short-lived from long-lived plasma cells, cell-extrinsic differences also play a role. A Proliferation Inducing Ligand (APRIL) enhances plasma cell survival in vitro, and genetic ablation of the APRIL receptor BCMA leads to a loss of plasma cells in vivo 132, 175, 176. These signals promote the expression of MCL1, the critical BCL2-family anti-apoptotic factor expressed in plasma cells 132, 133. A number of other extrinsic stimuli, such as IL-6 and Tumor Necrosis Factor α, and engagement of CD28 and CD44 also promote plasma cell survival ex vivo 134, 177, 178. Genetic knockout studies, however, have provided mixed results, suggesting that plasma cell-intrinsic signaling by these factors might not all be essential in vivo 133, 179. A number of accessory cell types such as eosinophils, basophils, megakaryocytes, regulatory T cells, and mesenchymal cells have all been proposed to enhance plasma cell survival 178, 180–184. The necessity of some of these cell types and secreted cytokines has been disputed, but a common proposed feature of these populations is their ability to secrete pro-survival cytokines and in turn support plasma cell longevity 185–189. The integration of these signals with primary plasma cell metabolism, however, has been understudied with a notable recent exception. Human plasma cells co-cultured with primary mesenchymal stromal cells (MSCs) survived in in vitro cultures for up to 8 weeks, versus those cultured in media alone, that died within one day 190. Functional secretome analyses revealed that fibronectin-1 and the 14-3-3 zeta/delta proteins promote plasma cell survival, suggesting a role for the extracellular matrix proteins 190. Culturing these cells under physiological oxygen tension and with MSC supernatants further enhanced survival, suggesting an as yet unexplored role for hypoxia in plasma cell survival. A deeper understanding of the complex interactions between these cells and systems will undoubtedly provide a more complete basic understanding of the factors that promote plasma cell longevity, as well as malignancies.
4. MULTIPLE MYELOMA
Multiple myeloma (MM) is a genetically heterogeneous cancer driven by malignant plasma cells in the bone marrow. MM is marked by immunodeficiency, anemia, renal failure, and bone lytic lesions. The use of drugs like Thalidomide, Lenalidomide, Bortezomib, and Dexamethasone in combination or coupled with autologous hematological cell transfers or stem cell therapy have reported some amelioration of the disease and its symptoms, but MM remains incurable 191, 192. While it is difficult to culture myeloma cells from patients in routine cell culture media, a model for studying this disease exists in mice, which involves transplantation of the spontaneously arising 5TMM cells from aged C57BL/KaLwRij mice into syngeneic younger recipients 193, 194 (discussed more recently in 195). Human xenograft models for multiple myeloma do exist, but these models are complex and most work is performed in vitro and on cell lines 196, 197.
While extracellular survival factors and niches may be limiting for normal long-lived plasma cell survival and numbers, MM growth and maintenance becomes independent of such constraints. It is possible that to achieve this independence, MM adopts distinct survival signals from their normal counterparts. In addition to cytokines such as IL-6 and BAFF that promote normal plasma cell survival, other factors such as Receptor Activator of NF-κB (RANK), Vascular Endothelial Growth factor (VEGF), Colony stimulating factor-1 (Csf1), Fibroblast Growth factor-2 (FGF-2), angiopoietin-1, and IL-8 can promote MM survival 198, 199. MM may also intrinsically engage iNOS-cGMP signaling, c-Myc, and Src and ERK kinases to persist and grow 200–202. Through secretion of IL-10 and Transforming Growth Factor β (TGF-β), regulatory T cells may also promote MM persistence either directly or by preventing rejection 203. Many of these signals and cytokines effect metabolic changes in other systems. Whether they mediate similar changes in MM remains to be fully explored.
As the critical metabolic pathways of plasma cells become defined, it will be important to determine if these same pathways can be targeted to eliminate MM. Long-lived plasma cells support durable immunity, but their bystander ablation as part of MM therapies would not be immediately life-threatening and likely an acceptable side effect. Nonetheless, an ideal therapy would target MM selectively while sparing non-malignant plasma cells. As noted above, spare respiratory capacity is a distinguishing feature of long-lived plasma cells, but such properties to our knowledge have not yet been reported for MM. In unpublished data, we have found that the 5TGM1 MM cell lines exhibit similar spare respiratory capacity and fluorescent glucose analog uptake as do primary plasma cells, but it remains unclear whether these MM cells are dependent on this pathway to form tumors in vivo. MM cells in general do exhibit enhanced nutrient uptake over non-transformed primary cells, which is a hallmark of cancerous cells. These cells are heavily reliant on glucose, and consequently are susceptible to chemotherapeutic strategies involving 3-Bromopyruvate and 2-Deoxyglucose, known inhibitors of key glycolytic enzymes 204–206. MM is characterized by high expression of Hexokinase-II and Pyruvate kinase M2, both of which are key enzymes in the breakdown of glucose leading to the formation of pyruvate 207, 208. This tendency towards aerobic glycolysis, or the Warburg effect, is a unique feature of highly proliferative cells and is likely to shunt the excess of pyruvate towards lactate formation, which in turn is excreted through the monocarboxylate transporters MCT1 and MCT4 209. Under normal circumstances, long-lived plasma cells use glucose mainly for antibody glycosylation, and to a lesser extent for ATP 23, 105. Thus, the dependence on aerobic glycolysis in MM is a marked shift from the normal metabolic phenotype of long-lived plasma cells and is likely to promote the malignant transformation of these cells. Further, glycolysis can activate MCL1, an anti-apoptotic factor promoting myeloma survival 210. Despite the importance of glucose in the metabolism of MM cells, the transporter responsible for its uptake is inconsistent between human and mouse cell lines. GLUT1 and GLUT6 are the chief glucose transporters in primary and MM murine cell lines, but human MM lines show elevated levels of the glucose transporters GLUT4, GLUT8, and GLUT11 with minimal expression of GLUT1 105, 210.
The importance of glutamine in plasma cell metabolism was drawn from studies carried out in MM cell lines. Inhibition of glutaminolysis results in MM death 211. This may be another key difference between MM and normal long-lived plasma cells, since as mentioned above, genetic ablation of glutaminolysis has not revealed obvious defects in plasma cell numbers or secretion thus far. c-Myc is an important factor contributing to the tumorigenic phenotype of MM cells and enhances expression of glutamine transporters as well as favoring glutaminolysis 211, 212. In addition to being a key source of fuel, glutamine uptake also triggers the mTOR pathway and stimulates cell proliferation through a STAT3-dependent mechanism 213. Glutamine also can upregulate HIF1α and can possibly mediate survival of the tumor cell under conditions of hypoxia, a condition typically associated with the MM bone marrow 213. In human MM cell lines, glutamine was preferentially shuttled through the tricarboxylic acid cycle as compared to glucose, and led to the synthesis of the oncometabolite 4-hydroxyglutarate 214. In MM cells, glutamine uptake was also shown to prevent dissociation of the pro-apoptotic BIM from its complex with BCL2, thus preventing cell-intrinsic apoptotic pathways 215. To summarize, it appears that MM cells are more heavily dependent on glycolysis and glutaminolysis than are their normal long-lived plasma cell counterparts, and these pathways may represent Achilles heels to target the disease.
5. NUTRITIONAL INFLUENCES ON PLASMA CELL FUNCTION
As mentioned earlier, the metabolic phenotype and longevity of plasma cells likely results from a complex interplay of cell-intrinsic and -extrinsic factors. There is a growing body of literature suggesting that plasma cell generation and function may be impacted by metabolic changes that occur at the organismal and systemic levels. To highlight this point, consider the property of spare respiratory capacity in long-lived plasma cells. It remains unclear under what physiological conditions this spare respiratory capacity is engaged. Despite many efforts to define specific plasma cell survival factors, such as APRIL, IL-6, CD28 engagement, and many others, we have yet to identify an extrinsic stimulus that requires spare respiratory capacity to promote plasma cell survival. Therefore, we have instead speculated that the fluctuations in systemic nutrients over the course of a day, or even a lifespan, place metabolic stresses on cells 105. Perhaps spare respiratory capacity protects against these stresses to prevent bioenergetic crises, especially for cells as metabolically active as long-lived plasma cells. Nonetheless, this explanation remains speculative and not entirely satisfying in the absence of experimental evidence. Therefore, a key direction will be to connect metabolic changes at the systemic level to the specific plasma cell metabolic pathways and their functional outcomes. We highlight some possible directions and relevant literature below.
5.1. Caloric restriction
A restriction in dietary calories positively correlates with longevity across many organisms. Yet these studies are invariably carried out in controlled, sterile environments, and negative impacts on immunological responses should be weighed. One of the first reports of the effects of caloric restriction on the immune system involved sheep red blood cell immunizations of rats restricted to 25% of their ad libitum diet. It was found that rats on the 6-week caloric restriction regimen exhibited marked reductions in lymphocyte numbers as well as reduced serum IgM and IgG titers in response to immunization 216. Further, offspring of pregnant dams on caloric restriction exhibited similar reduced antibody responses, even if they were weaned on a normal diet. These diminished responses to T-dependent antigens also reflected poorer activation and proliferation in mouse B cell cultures and had similar effects on macrophage and T cell function in mice on caloric restriction 217. In a model of WNV infection, mice on caloric restriction had fewer Interferon-γ-producing T cells and exhibited higher mortality as compared to mice fed ad libitum diets 218. A recent study involving non-obese human volunteers monitored over two years recapitulated the data from mice, where volunteers on caloric restriction showed reduced inflammation as well as markedly low circulating white blood cell and lymphocyte counts 219. The caloric restriction-induced lymphopenia, however, is limited to peripheral organs as bone marrow lymphocyte numbers were unaffected in aged mice on caloric restriction 220. The effects of caloric restriction on the immune system may be dependent on the age of the individual too, as old mice on caloric restriction showed restored lymphocyte cellularity when fed ad libitum, in contrast to the findings reported by Chandra on neonates 216, 220. In models of autoimmunity such as the (NZMxNZB) F1 model of lupus and the MRL/Mp-lpr/lpr model of glomerular nephritis, caloric restriction delayed the onset in disease symptoms and enhanced the lifespan of the mice 221, 222. Thus, the available evidence suggests that caloric restriction induces a systemic inhibition of immune cell function. As infectious disease remains one of the major causes of death worldwide, caloric restriction may not be effective in enhancing lifespan in the real world 223.
While these studies indicate that there is a plasma cell phenotype associated with caloric restriction, their primary readout is serum immunoglobulin levels, which may not be a specific indicator of changes in cellular kinetics or function of antibody-secreting cells. Impacts on T cells, dendritic cells, stroma, and a myriad of other cell types might indirectly lead to altered antibody responses. While a more formal demonstration of the impact of caloric restriction on the B cell response is needed, our findings surrounding the metabolic phenotypes of plasma cells is in accordance with the data presented. As discussed previously, plasma cells exhibit a dependence on glucose and glutamine for their optimal function, and limiting these nutrients therefore, as in caloric restriction, might have a deleterious effect on the longevity and persistence of these cells 23, 105. It is also noteworthy that treating mice with Rapamycin, the inhibitor of the mTOR complex, was shown to extend the lifespan of treated rodents, in a manner akin to CR 224. Given the central role mTOR plays in B cell activation, nutrient uptake, class switch recombination, and the overall transition from B cells to plasma cells, it is possible that the effects of caloric restriction may act through alterations of B cell-intrinsic mTOR signaling. The effects of mTOR signaling in B cell differentiation are stage-dependent, as treatment with rapamycin or through genetic deletion of Raptor resulted in reduced GCs, class switching, and promoted short-lived plasma cell formation 225, 226. In already differentiated plasma cells, mTOR inhibition led to reduced antibody synthesis, but did not affect the longevity of plasma cells 225. The metabolic changes that specifically occur in plasma cells during caloric restriction still remain to be defined and may help provide mechanistic explanations for immune system dysfunction during caloric restriction.
5.2. Obesity
Reciprocally, obesity and diabetes might provide an overabundance of nutrients, leading to different systemic changes in the immune response. While obesity predisposes to many diseases, it has also been shown to impair immune responsiveness, thus creating an obstacle in the treatment of infectious diseases. In the recent 2009 H1N1 Influenza A pandemic, 91% of the reported deaths in the state of California were of individuals who had a body-mass index (BMI) greater than 30, and almost 51% of hospitalized patients were categorized as obese to morbidly obese, indicating a strong correlation between obesity and poor responsiveness to the virus 227. Other reports also correlated higher BMI with poor antibody responses to the Hepatitis B (HBV) vaccine in adults and Tetanus Toxoid (TT) vaccination in children. Obese children actually showed higher anti-influenza antibodies in their serum 4 weeks after vaccination as compared to vaccinated non-obese children 228. At 4 months, however, seroprotection and seroconversion rates against A/H1N1 and A/H3N2 were identical between both groups of children while obese children showed higher antibody titers against the B strain relative to the non-obese vaccinated children 228. Similar findings were recapitulated in obese North American adults, who showed higher seroprotection and seroconversion at a month post vaccination as compared to lean participants. Problematically, though, a greater drop in anti-Influenza antibodies was observed among obese individuals at 12 months post vaccination compared to their non-obese counterparts 229. Further, individuals with type 2 diabetes showed similar antibody kinetics in response to the trivalent-inactivated influenza vaccine as compared to healthy individuals, suggesting that these antibody response kinetics are specific to obesity 230. Thus, obesity correlates with an elevated antibody response at early time points, but also correlates with transient anti-influenza antibody responses. As the measurements in all these studies were serum enzyme-linked immunoassays, we speculate that obesity favors the formation of short-lived plasma cells which are responsible for the heightened antibody response, but by an unknown mechanism does not permit either the formation or establishment of the long-lived plasma cell compartment.
There are several possibilities as to how obesity could influence the formation of the long-lived plasma cell compartment. First, it is likely that obesity and its associated systemic inflammation may alter the composition of the bone marrow, thereby rendering it unsupportive to the establishment of the long-lived plasma cells. Evidence for this possibility has come from experiments in murine models of obesity, including high-fat diet fed mice or leptin-leptin receptor deficient animals. Bone marrow resident mesenchymal stem cells from mice on a high-fat diet as well as from mesenchymal stem cell-specific lepRfl/fl mice expressed gene signatures associated with adipogenesis, suggesting that obesity can induce an altered differentiation route for these multipotent cells 231, 232. Diet-induced obesity also reduced the proliferative and self-renewing capacity of hematopoietic stem and progenitor cells in the bone marrow, in turn gradually impeding B lymphopoiesis 233–235. This gradual lymphopenia may help explain poor immune responses in aged individuals 227, 229, 230. Obesity might thus prevent normal stem cell differentiation pathways in the bone marrow and in turn limit the formation of cell types that provide plasma cells essential survival factors. Consequently, plasma cells may be deprived of essential survival factors, thereby compromising durable immunity. A second possibility for poor long-lived plasma cell formation is that the elevated levels of sugars and lipids due to obesity may influence plasma cell metabolism directly. For example, excessive glucose can be shunted into sorbitol, which is associated with diabetes and cellular toxicities 236. Exercise is widely promoted as a conservative but effective intervention for obesity. Yet limited data suggest that exercise has minimal effects on B cells and plasma cell function in both mice and humans 237–239. As above, measuring the impact of obesity and exercise on plasma cell-intrinsic metabolism will be critical for moving the field from observations of phenomena to mechanistic studies.
6. CONCLUSIONS
While our findings and those from many others in the field of immunometabolism have defined specific metabolic pathways for defining cellular differentiation outcomes and function, these are just stepping stones towards a more comprehensive understanding of the B cell response. Our work highlights the importance of nutrient uptake in the longevity of the plasma cell, and the cell-intrinsic mechanisms required to balance energy uptake and utilization in plasma cells and their subsets. An unexpected set of findings by our group and others demonstrate that these metabolic pathways come with very few transcriptional consequences, suggesting that plasma cell subsets may differ from each other at the level of their proteomes. In vivo, plasma cell behavior is influenced by the local environment, and almost certainly by the systemic metabolic state. To define how these extrinsic cues influence plasma cell metabolism, longevity, and function, approaches that integrate cell-intrinsic, microenvironmental, and systemic parameters are necessary. This approach will be beneficial in our understanding of complex diseases that are based on metabolic dysregulation and provide guidance for the design of vaccines.
Acknowledgements
The authors are supported by National Institutes of Health grants R01AI129945 and R01AI099108 (D.B.). The funding sources had no role in data collection, interpretation, or writing of this manuscript.
Footnotes
Declaration of interests
D.B. is a co-founder of Sana Biotechnology, Inc. and owns significant stock in Forty-Seven Inc. L.D. reports no conflict of interest.
References
- 1.Shi W, Liao Y, Willis SN, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol 2015; 16: 663–673. DOI: 10.1038/ni.3154. [DOI] [PubMed] [Google Scholar]
- 2.Hibi T and Dosch HM. Limiting dilution analysis of the B cell compartment in human bone marrow. European journal of immunology 1986; 16: 139–145. DOI: 10.1002/eji.1830160206. [DOI] [PubMed] [Google Scholar]
- 3.Purtha WE, Tedder TF, Johnson S, et al. Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. The Journal of experimental medicine 2011. 2011/12/14 DOI: 10.1084/jem.20110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zinkernagel RM and Hengartner H. Protective ‘immunity’ by pre-existent neutralizing antibody titers and preactivated T cells but not by so-called ‘immunological memory’. Immunological reviews 2006; 211: 310–319. [DOI] [PubMed] [Google Scholar]
- 5.Amanna IJ, Carlson NE and Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med 2007; 357: 1903–1915. DOI: 10.1056/NEJMoa066092. [DOI] [PubMed] [Google Scholar]
- 6.Vieira P and Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol 1988; 18: 313–316. DOI: 10.1002/eji.1830180221. [DOI] [PubMed] [Google Scholar]
- 7.Bernasconi NL, Traggiai E and Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002; 298: 2199–2202. [DOI] [PubMed] [Google Scholar]
- 8.Manz RA, Thiel A and Radbruch A. Lifetime of plasma cells in the bone marrow. Nature 1997; 388: 133–134. DOI: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 9.Slifka MK, Antia R, Whitmire JK, et al. Humoral immunity due to long-lived plasma cells. Immunity 1998; 8: 363–372. [DOI] [PubMed] [Google Scholar]
- 10.Ahuja A, Anderson SM, Khalil A, et al. Maintenance of the plasma cell pool is independent of memory B cells. Proc Natl Acad Sci U S A 2008; 105: 4802–4807. DOI: 10.1073/pnas.0800555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiLillo DJ, Hamaguchi Y, Ueda Y, et al. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol 2008; 180: 361–371. [DOI] [PubMed] [Google Scholar]
- 12.Cambridge G, Leandro MJ, Edwards JC, et al. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis and rheumatism 2003; 48: 2146–2154. Research Support, Non-U.S. Gov’t 2003/08/09 DOI: 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- 13.Richard K, Pierce SK and Song W. The agonists of TLR4 and 9 are sufficient to activate memory B cells to differentiate into plasma cells in vitro but not in vivo. J Immunol 2008; 181: 1746–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olotu A, Fegan G, Wambua J, et al. Four-year efficacy of RTS,S/AS01E and its interaction with malaria exposure. The New England journal of medicine 2013; 368: 1111–1120. DOI: 10.1056/NEJMoa1207564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wendelboe AM, Van Rie A, Salmaso S, et al. Duration of immunity against pertussis after natural infection or vaccination. The Pediatric infectious disease journal 2005; 24: S58–61. Comparative Study Review 2005/05/07. [DOI] [PubMed] [Google Scholar]
- 16.Sze DM, Toellner KM, Garcia de Vinuesa C, et al. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J Exp Med 2000; 192: 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fagraeus A The plasma cellular reaction and its relation to the formation of antibodies in vitro. J Immunol 1948; 58: 1–13. [PubMed] [Google Scholar]
- 18.McMillan R, Longmire RL, Yelenosky R, et al. Immunoglobulin synthesis by human lymphoid tissues: normal bone marrow as a major site of IgG production. J Immunol 1972; 109: 1386–1394. [PubMed] [Google Scholar]
- 19.Halliley JL, Tipton CM, Liesveld J, et al. Long-Lived Plasma Cells Are Contained within the CD19(−)CD38(hi)CD138(+) Subset in Human Bone Marrow. Immunity 2015; 43: 132–145. DOI: 10.1016/j.immuni.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landsverk OJ, Snir O, Casado RB, et al. Antibody-secreting plasma cells persist for decades in human intestine. J Exp Med 2017; 214: 309–317. DOI: 10.1084/jem.20161590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mei HE, Wirries I, Frolich D, et al. A unique population of IgG-expressing plasma cells lacking CD19 is enriched in human bone marrow. Blood 2015; 125: 1739–1748. DOI: 10.1182/blood-2014-02-555169. [DOI] [PubMed] [Google Scholar]
- 22.Chernova I, Jones DD, Wilmore JR, et al. Lasting antibody responses are mediated by a combination of newly formed and established bone marrow plasma cells drawn from clonally distinct precursors. J Immunol 2014; 193: 4971–4979. DOI: 10.4049/jimmunol.1401264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lam WY, Jash A, Yao CH, et al. Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell Rep 2018; 24: 2479–2492 e2476 DOI: 10.1016/j.celrep.2018.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kallies A, Hasbold J, Tarlinton DM, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med 2004; 200: 967–977. DOI: 10.1084/jem.20040973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chevrier S, Genton C, Kallies A, et al. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc Natl Acad Sci U S A 2009; 106: 3895–3900. DOI: 10.1073/pnas.0809736106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lino AC, Dang VD, Lampropoulou V, et al. LAG-3 Inhibitory Receptor Expression Identifies Immunosuppressive Natural Regulatory Plasma Cells. Immunity 2018; 49: 120–133 e129 DOI: 10.1016/j.immuni.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerutti A The regulation of IgA class switching. Nat Rev Immunol 2008; 8: 421–434. DOI: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell RG. IgE, allergies and helminth parasites: a new perspective on an old conundrum. Immunol Cell Biol 1996; 74: 337–345. DOI: 10.1038/icb.1996.60. [DOI] [PubMed] [Google Scholar]
- 29.Kato LM, Kawamoto S, Maruya M, et al. The role of the adaptive immune system in regulation of gut microbiota. Immunol Rev 2014; 260: 67–75. DOI: 10.1111/imr.12185. [DOI] [PubMed] [Google Scholar]
- 30.Hallek M, Bergsagel PL and Anderson KC. Multiple myeloma: increasing evidence for a multistep transformation process. Blood 1998; 91: 3–21. [PMC free article] [PubMed] [Google Scholar]
- 31.Boothby M and Rickert RC. Metabolic Regulation of the Immune Humoral Response. Immunity 2017; 46: 743–755. DOI: 10.1016/j.immuni.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam WY and Bhattacharya D. Metabolic Links between Plasma Cell Survival, Secretion, and Stress. Trends Immunol 2018; 39: 19–27. DOI: 10.1016/j.it.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egawa T and Bhattacharya D. Regulation of metabolic supply and demand during B cell activation and subsequent differentiation. Current opinion in immunology 2018; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allman D and Pillai S. Peripheral B cell subsets. Curr Opin Immunol 2008; 20: 149–157. DOI: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones DD, Wilmore JR and Allman D. Cellular Dynamics of Memory B Cell Populations: IgM+ and IgG+ Memory B Cells Persist Indefinitely as Quiescent Cells. J Immunol 2015; 195: 4753–4759. DOI: 10.4049/jimmunol.1501365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuxa M and Busslinger M. Reporter gene insertions reveal a strictly B lymphoid-specific expression pattern of Pax5 in support of its B cell identity function. J Immunol 2007; 178: 3031–3037. [DOI] [PubMed] [Google Scholar]
- 37.Revilla IDR, Bilic I, Vilagos B, et al. The B-cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. EMBO J 2012; 31: 3130–3146. DOI: 10.1038/emboj.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446: 758–764. DOI: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 39.Chan LN, Chen Z, Braas D, et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 2017; 542: 479–483. DOI: 10.1038/nature21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao G, Chan LN, Klemm L, et al. B-Cell-Specific Diversion of Glucose Carbon Utilization Reveals a Unique Vulnerability in B Cell Malignancies. Cell 2018; 173: 470–484 e418 DOI: 10.1016/j.cell.2018.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milasta S, Dillon CP, Sturm OE, et al. Apoptosis-Inducing-Factor-Dependent Mitochondrial Function Is Required for T Cell but Not B Cell Function. Immunity 2016; 44: 88–102. DOI: 10.1016/j.immuni.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patke A, Mecklenbrauker I, Erdjument-Bromage H, et al. BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. J Exp Med 2006; 203: 2551–2562. DOI: 10.1084/jem.20060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baracho GV, Cato MH, Zhu Z, et al. PDK1 regulates B cell differentiation and homeostasis. Proc Natl Acad Sci U S A 2014; 111: 9573–9578. DOI: 10.1073/pnas.1314562111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calamito M, Juntilla MM, Thomas M, et al. Akt1 and Akt2 promote peripheral B-cell maturation and survival. Blood 2010; 115: 4043–4050. DOI: 10.1182/blood-2009-09-241638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliver AM, Martin F and Kearney JF. IgMhighCD21high lymphocytes enriched in the splenic marginal zone generate effector cells more rapidly than the bulk of follicular B cells. J Immunol 1999; 162: 7198–7207. [PubMed] [Google Scholar]
- 46.Martin F, Oliver AM and Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity 2001; 14: 617–629. [DOI] [PubMed] [Google Scholar]
- 47.Benhamron S and Tirosh B. Direct activation of mTOR in B lymphocytes confers impairment in B-cell maturation andloss of marginal zone B cells. Eur J Immunol 2011; 41: 2390–2396. DOI: 10.1002/eji.201041336. [DOI] [PubMed] [Google Scholar]
- 48.Doughty CA, Bleiman BF, Wagner DJ, et al. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 2006; 107: 4458–4465. DOI: 10.1182/blood-2005-12-4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dufort FJ, Gumina MR, Ta NL, et al. Glucose-dependent de novo lipogenesis in B lymphocytes: a requirement for atp-citrate lyase in lipopolysaccharide-induced differentiation. J Biol Chem 2014; 289: 7011–7024. DOI: 10.1074/jbc.M114.551051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woodland RT, Fox CJ, Schmidt MR, et al. Multiple signaling pathways promote B lymphocyte stimulator dependent B-cell growth and survival. Blood 2008; 111: 750–760. DOI: 10.1182/blood-2007-03-077222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akkaya M, Traba J, Roesler AS, et al. Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat Immunol 2018. DOI: 10.1038/s41590-018-0156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caro-Maldonado A, Wang R, Nichols AG, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol 2014; 192: 3626–3636. DOI: 10.4049/jimmunol.1302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nelson DL and Cox MM. Lehninger Principles of Biochemistry. 2017. [Google Scholar]
- 54.Cantor J, Browne CD, Ruppert R, et al. CD98hc facilitates B cell proliferation and adaptive humoral immunity. Nat Immunol 2009; 10: 412–419. DOI: 10.1038/ni.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelly AP, Finlay DK, Hinton HJ, et al. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J 2007; 26: 3441–3450. DOI: 10.1038/sj.emboj.7601761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanai Y, Segawa H, Miyamoto K, et al. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). The Journal of biological chemistry 1998; 273: 23629–23632. [DOI] [PubMed] [Google Scholar]
- 57.Le A, Lane AN, Hamaker M, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell metabolism 2012; 15: 110–121. DOI: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang S, Yan W, Wang SE, et al. Let-7 Suppresses B Cell Activation through Restricting the Availability of Necessary Nutrients. Cell Metab 2018; 27: 393–403 e394 DOI: 10.1016/j.cmet.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 59.Tellier J, Shi W, Minnich M, et al. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 2016; 17: 323–330. DOI: 10.1038/ni.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reif K, Ekland EH, Ohl L, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature 2002; 416: 94–99. [DOI] [PubMed] [Google Scholar]
- 61.Bishop GA and Hostager BS. B lymphocyte activation by contact-mediated interactions with T lymphocytes. Current opinion in immunology 2001; 13: 278–285. Review 2001/06/19. [DOI] [PubMed] [Google Scholar]
- 62.Coffey F, Alabyev B and Manser T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity 2009; 30: 599–609. 2009/03/24 DOI: S1074-7613(09)00116-2 [pii] 10.1016/j.immuni.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor JJ, Pape KA and Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. The Journal of experimental medicine 2012; 209: 597–606. DOI: 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toyama H, Okada S, Hatano M, et al. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity 2002; 17: 329–339. [DOI] [PubMed] [Google Scholar]
- 65.Pereira JP, Kelly LM, Xu Y, et al. EBI2 mediates B cell segregation between the outer and centre follicle. Nature 2009; 460: 1122–1126. 2009/07/15 DOI: nature08226 [pii] 10.1038/nature08226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weisel FJ, Zuccarino-Catania GV, Chikina M, et al. A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 2016; 44: 116–130. DOI: 10.1016/j.immuni.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dogan I, Bertocci B, Vilmont V, et al. Multiple layers of B cell memory with different effector functions. Nature immunology 2009; 10: 1292–1299. 2009/10/27 DOI: ni.1814 [pii] 10.1038/ni.1814. [DOI] [PubMed] [Google Scholar]
- 68.Paus D, Phan TG, Chan TD, et al. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. The Journal of experimental medicine 2006; 203: 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rothaeusler K and Baumgarth N. B-cell fate decisions following influenza virus infection. European journal of immunology 2010; 40: 366–377. DOI: 10.1002/eji.200939798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Victora GD and Nussenzweig MC. Germinal centers. Annual review of immunology 2012; 30: 429–457. DOI: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 71.Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 2000; 102: 565–575. [DOI] [PubMed] [Google Scholar]
- 72.Allen CD, Okada T, Tang HL, et al. Imaging of germinal center selection events during affinity maturation. Science 2007; 315: 528–531. 2006/12/23 DOI: 1136736 [pii] 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- 73.Victora GD, Schwickert TA, Fooksman DR, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 2010; 143: 592–605. 2010/11/16 DOI: S0092-8674(10)01236-5 [pii] 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Calado DP, Sasaki Y, Godinho SA, et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol 2012; 13: 1092–1100. DOI: 10.1038/ni.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dominguez-Sola D, Victora GD, Ying CY, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol 2012; 13: 1083–1091. DOI: 10.1038/ni.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luo W, Weisel F and Shlomchik MJ. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity 2018; 48: 313–326 e315 DOI: 10.1016/j.immuni.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ersching J, Efeyan A, Mesin L, et al. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity 2017; 46: 1045–1058 e1046 DOI: 10.1016/j.immuni.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang R, Dillon CP, Shi LZ, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011; 35: 871–882. DOI: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chou C, Verbaro DJ, Tonc E, et al. The Transcription Factor AP4 Mediates Resolution of Chronic Viral Infection through Amplification of Germinal Center B Cell Responses. Immunity 2016; 45: 570–582. DOI: 10.1016/j.immuni.2016.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jellusova J, Cato MH, Apgar JR, et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat Immunol 2017; 18: 303–312. DOI: 10.1038/ni.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martinez-Martin N, Maldonado P, Gasparrini F, et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science 2017; 355: 641–647. DOI: 10.1126/science.aal3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho SH, Raybuck AL, Stengel K, et al. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 2016; 537: 234–238. DOI: 10.1038/nature19334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith KG, Light A, Nossal GJ, et al. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. The EMBO journal 1997; 16: 2996–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suan D, Krautler NJ, Maag JLV, et al. CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity 2017; 47: 1142–1153 e1144 DOI: 10.1016/j.immuni.2017.11.022. [DOI] [PubMed] [Google Scholar]
- 85.Laidlaw BJ, Schmidt TH, Green JA, et al. The Eph-related tyrosine kinase ligand Ephrin-B1 marks germinal center and memory precursor B cells. The Journal of experimental medicine 2017; 214: 639–649. DOI: 10.1084/jem.20161461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shinnakasu R, Inoue T, Kometani K, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nature immunology 2016; 17: 861–869. DOI: 10.1038/ni.3460. [DOI] [PubMed] [Google Scholar]
- 87.Gitlin AD, von Boehmer L, Gazumyan A, et al. Independent Roles of Switching and Hypermutation in the Development and Persistence of B Lymphocyte Memory. Immunity 2016; 44: 769–781. DOI: 10.1016/j.immuni.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tas JM, Mesin L, Pasqual G, et al. Visualizing antibody affinity maturation in germinal centers. Science 2016; 351: 1048–1054. DOI: 10.1126/science.aad3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weinstein JS, Herman EI, Lainez B, et al. TFH cells progressively differentiate to regulate the germinal center response. Nature immunology 2016; 17: 1197–1205. DOI: 10.1038/ni.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dufort FJ, Bleiman BF, Gumina MR, et al. Cutting edge: IL-4-mediated protection of primary B lymphocytes from apoptosis via Stat6-dependent regulation of glycolytic metabolism. J Immunol 2007; 179: 4953–4957. [DOI] [PubMed] [Google Scholar]
- 91.Pape KA, Taylor JJ, Maul RW, et al. Different B cell populations mediate early and late memory during an endogenous immune response. Science 2011; 331: 1203–1207. DOI: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seifert M, Przekopowitz M, Taudien S, et al. Functional capacities of human IgM memory B cells in early inflammatory responses and secondary germinal center reactions. Proceedings of the National Academy of Sciences of the United States of America 2015; 112: E546–555. DOI: 10.1073/pnas.1416276112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zuccarino-Catania GV, Sadanand S, Weisel FJ, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nature immunology 2014; 15: 631–637. DOI: 10.1038/ni.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Buck MD, Sowell RT, Kaech SM, et al. Metabolic Instruction of Immunity. Cell 2017; 169: 570–586. DOI: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen M, Hong MJ, Sun H, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nature medicine 2014; 20: 503–510. DOI: 10.1038/nm.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen M, Kodali S, Jang A, et al. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J Immunol 2015; 194: 2607–2615. DOI: 10.4049/jimmunol.1403001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Torigoe M, Iwata S, Nakayamada S, et al. Metabolic Reprogramming Commits Differentiation of Human CD27(+)IgD(+) B Cells to Plasmablasts or CD27(−)IgD(−) Cells. J Immunol 2017; 199: 425–434. Research Support, Non-U.S. Gov’t 2017/06/20 DOI: 10.4049/jimmunol.1601908. [DOI] [PubMed] [Google Scholar]
- 98.Ise W, Fujii K, Shiroguchi K, et al. T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity 2018; 48: 702–715 e704 DOI: 10.1016/j.immuni.2018.03.027. [DOI] [PubMed] [Google Scholar]
- 99.Sciammas R, Shaffer AL, Schatz JH, et al. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 2006; 25: 225–236. DOI: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 100.Ochiai K, Maienschein-Cline M, Simonetti G, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 2013; 38: 918–929. DOI: 10.1016/j.immuni.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Klein U, Casola S, Cattoretti G, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol 2006; 7: 773–782. DOI: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 102.Willis SN, Good-Jacobson KL, Curtis J, et al. Transcription factor IRF4 regulates germinal center cell formation through a B cell-intrinsic mechanism. J Immunol 2014; 192: 3200–3206. DOI: 10.4049/jimmunol.1303216. [DOI] [PubMed] [Google Scholar]
- 103.Minnich M, Tagoh H, Bonelt P, et al. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol 2016; 17: 331–343. DOI: 10.1038/ni.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Han S, Hathcock K, Zheng B, et al. Cellular interaction in germinal centers. Roles of CD40 ligand and B7-2 in established germinal centers. J Immunol 1995; 155: 556–567. 1995/07/15. [PubMed] [Google Scholar]
- 105.Lam WY, Becker AM, Kennerly KM, et al. Mitochondrial Pyruvate Import Promotes Long-Term Survival of Antibody-Secreting Plasma Cells. Immunity 2016; 45: 60–73. DOI: 10.1016/j.immuni.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Byrne FL, Olzomer EM, Brink R, et al. Knockout of glucose transporter GLUT6 has minimal effects on whole body metabolic physiology in mice. Am J Physiol Endocrinol Metab 2018; 315: E286–E293. DOI: 10.1152/ajpendo.00082.2018. [DOI] [PubMed] [Google Scholar]
- 107.Chen LQ, Hou BH, Lalonde S, et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010; 468: 527–532. DOI: 10.1038/nature09606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jennewein MF and Alter G. The Immunoregulatory Roles of Antibody Glycosylation. Trends Immunol 2017; 38: 358–372. DOI: 10.1016/j.it.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 109.Tao MH and Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol 1989; 143: 2595–2601. [PubMed] [Google Scholar]
- 110.Hickman S, Kulczycki A Jr., Lynch RG, et al. Studies of the mechanism of tunicamycin in hibition of IgA and IgE secretion by plasma cells. J Biol Chem 1977; 252: 4402–4408. [PubMed] [Google Scholar]
- 111.Wang TT, Maamary J, Tan GS, et al. Anti-HA Glycoforms Drive B Cell Affinity Selection and Determine Influenza Vaccine Efficacy. Cell 2015; 162: 160–169. DOI: 10.1016/j.cell.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang TT, Sewatanon J, Memoli MJ, et al. IgG antibodies to dengue enhanced for FcgammaRIIIA binding determine disease severity. Science 2017; 355: 395–398. DOI: 10.1126/science.aai8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu LL, Chung AW, Rosebrock TR, et al. A Functional Role for Antibodies in Tuberculosis. Cell 2016; 167: 433–443 e414 DOI: 10.1016/j.cell.2016.08.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001; 412: 300–307. DOI: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 115.Yoshida H, Matsui T, Yamamoto A, et al. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107: 881–891. [DOI] [PubMed] [Google Scholar]
- 116.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004; 21: 81–93. DOI: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 117.Sriburi R, Jackowski S, Mori K, et al. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 2004; 167: 35–41. DOI: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McGehee AM, Dougan SK, Klemm EJ, et al. XBP-1-deficient plasmablasts show normal protein folding but altered glycosylation and lipid synthesis. J Immunol 2009; 183: 3690–3699. DOI: 10.4049/jimmunol.0900953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Taubenheim N, Tarlinton DM, Crawford S, et al. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol 2012; 189: 3328–3338. DOI: 10.4049/jimmunol.1201042. [DOI] [PubMed] [Google Scholar]
- 120.Wang ZV, Deng Y, Gao N, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014; 156: 1179–1192. DOI: 10.1016/j.cell.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ron D and Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8: 519–529. DOI: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 122.Aragon IV, Barrington RA, Jackowski S, et al. The specialized unfolded protein response of B lymphocytes: ATF6alpha-independent development of antibody-secreting B cells. Molecular immunology 2012; 51: 347–355. DOI: 10.1016/j.molimm.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gass JN, Jiang HY, Wek RC, et al. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Molecular immunology 2008; 45: 1035–1043. DOI: 10.1016/j.molimm.2007.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ma Y, Shimizu Y, Mann MJ, et al. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones 2010; 15: 281–293. DOI: 10.1007/s12192-009-0142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Goldfinger M, Shmuel M, Benhamron S, et al. Protein synthesis in plasma cells is regulated by crosstalk between endoplasmic reticulum stress and mTOR signaling. European journal of immunology 2011; 41: 491–502. DOI: 10.1002/eji.201040677. [DOI] [PubMed] [Google Scholar]
- 126.Auner HW, Beham-Schmid C, Dillon N, et al. The life span of short-lived plasma cells is partly determined by a block on activation of apoptotic caspases acting in combination with endoplasmic reticulum stress. Blood 2010; 116: 3445–3455. DOI: 10.1182/blood-2009-10-250423. [DOI] [PubMed] [Google Scholar]
- 127.Pelletier N, Casamayor-Palleja M, De Luca K, et al. The endoplasmic reticulum is a key component of the plasma cell death pathway. J Immunol 2006; 176: 1340–1347. [DOI] [PubMed] [Google Scholar]
- 128.Gaudette BT, Iwakoshi NN and Boise LH. Bcl-xL protein protects from C/EBP homologous protein (CHOP)-dependent apoptosis during plasma cell differentiation. The Journal of biological chemistry 2014; 289: 23629–23640. DOI: 10.1074/jbc.M114.569376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wellen KE, Lu C, Mancuso A, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes & development 2010; 24: 2784–2799. DOI: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zacharogianni M, Kondylis V, Tang Y, et al. ERK7 is a negative regulator of protein secretion in response to amino-acid starvation by modulating Sec16 membrane association. The EMBO journal 2011; 30: 3684–3700. DOI: 10.1038/emboj.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Valor LM, Rodriguez-Bayona B, Ramos-Amaya AB, et al. The transcriptional profiling of human in vivo-generated plasma cells identifies selective imbalances in monoclonal gammopathies. PloS one 2017; 12: e0183264 DOI: 10.1371/journal.pone.0183264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.O’Connor BP, Raman VS, Erickson LD, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med 2004; 199: 91–98. DOI: 10.1084/jem.20031330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Peperzak V, Vikstrom I, Walker J, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol 2013; 14: 290–297. DOI: 10.1038/ni.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rozanski CH, Arens R, Carlson LM, et al. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med 2011; 208: 1435–1446. DOI: 10.1084/jem.20110040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shapiro-Shelef M, Lin KI, Savitsky D, et al. Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J Exp Med 2005; 202: 1471–1476. DOI: 10.1084/jem.20051611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang Y and Bhattacharya D. Adjuvant-specific regulation of long-term antibody responses by ZBTB20. The Journal of experimental medicine 2014; 211: 841–856. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t 2014/04/09 DOI: 10.1084/jem.20131821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chevrier S, Emslie D, Shi W, et al. The BTB-ZF transcription factor Zbtb20 is driven by Irf4 to promote plasma cell differentiation and longevity. The Journal of experimental medicine 2014; 211: 827–840. DOI: 10.1084/jem.20131831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jash A, Wang Y, Weisel FJ, et al. ZBTB32 Restricts the Duration of Memory B Cell Recall Responses. J Immunol 2016; 197: 1159–1168. DOI: 10.4049/jimmunol.1600882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sutherland AP, Zhang H, Zhang Y, et al. Zinc finger protein Zbtb20 is essential for postnatal survival and glucose homeostasis. Molecular and cellular biology 2009; 29: 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yadava N and Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. The Journal of neuroscience : the official journal of the Society for Neuroscience 2007; 27: 7310–7317. DOI: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van der Windt GJ, Everts B, Chang CH, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012; 36: 68–78. DOI: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brand MD and Nicholls DG. Assessing mitochondrial dysfunction in cells. The Biochemical journal 2011; 435: 297–312. DOI: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Halestrap AP. The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. The Biochemical journal 1975; 148: 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McCommis KS, Chen Z, Fu X, et al. Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell Metab 2015; 22: 682–694. DOI: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bricker DK, Taylor EB, Schell JC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012; 337: 96–100. DOI: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Herzig S, Raemy E, Montessuit S, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012; 337: 93–96. DOI: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- 147.Smith KG, Weiss U, Rajewsky K, et al. Bcl-2 increases memory B cell recruitment but does not perturb selection in germinal centers. Immunity 1994; 1: 803–813. [DOI] [PubMed] [Google Scholar]
- 148.Fischer SF, Bouillet P, O’Donnell K, et al. Proapoptotic BH3-only protein Bim is essential for developmentally programmed death of germinal center-derived memory B cells and antibody-forming cells. Blood 2007; 110: 3978–3984. DOI: 10.1182/blood-2007-05-091306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Holler N, Zaru R, Micheau O, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature immunology 2000; 1: 489–495. DOI: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 150.Brennan MA and Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol 2000; 38: 31–40. [DOI] [PubMed] [Google Scholar]
- 151.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149: 1060–1072. DOI: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Oldenborg PA, Zheleznyak A, Fang YF, et al. Role of CD47 as a marker of self on red blood cells. Science 2000; 288: 2051–2054. [DOI] [PubMed] [Google Scholar]
- 153.Buck MD, O’Sullivan D, Klein Geltink RI, et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016; 166: 63–76. DOI: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tao J, Diaz RK, Teixeira CR, et al. Transport of a Fluorescent Analogue of Glucose (2-NBDG) versus Radiolabeled Sugars by Rumen Bacteria and Escherichia coli. Biochemistry 2016; 55: 2578–2589. DOI: 10.1021/acs.biochem.5b01286. [DOI] [PubMed] [Google Scholar]
- 155.Llufrio EM, Wang L, Naser FJ, et al. Sorting cells alters their redox state and cellular metabolome. Redox Biol 2018; 16: 381–387. DOI: 10.1016/j.redox.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mantel C, Messina-Graham S, Moh A, et al. Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood 2012; 120: 2589–2599. DOI: 10.1182/blood-2012-01-404004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mohrin M, Shin J, Liu Y, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015; 347: 1374–1377. DOI: 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Romero-Moya D, Bueno C, Montes R, et al. Cord blood-derived CD34+ hematopoietic cells with low mitochondrial mass are enriched in hematopoietic repopulating stem cell function. Haematologica 2013; 98: 1022–1029. DOI: 10.3324/haematol.2012.079244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Simsek T, Kocabas F, Zheng J, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010; 7: 380–390. DOI: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Takubo K, Nagamatsu G, Kobayashi CI, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013; 12: 49–61. DOI: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vannini N, Girotra M, Naveiras O, et al. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Commun 2016; 7: 13125 DOI: 10.1038/ncomms13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xiao N, Jani K, Morgan K, et al. Hematopoietic stem cells lacking Ott1 display aspects associated with aging and are unable to maintain quiescence during proliferative stress. Blood 2012; 119: 4898–4907. DOI: 10.1182/blood-2012-01-403089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.de Almeida MJ, Luchsinger LL, Corrigan DJ, et al. Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell Stem Cell 2017; 21: 725–729 e724 DOI: 10.1016/j.stem.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xu FY, Taylor WA, Hurd JA, et al. Etomoxir mediates differential metabolic channeling of fatty acid and glycerol precursors into cardiolipin in H9c2 cells. J Lipid Res 2003; 44: 415–423. DOI: 10.1194/jlr.M200335-JLR200. [DOI] [PubMed] [Google Scholar]
- 165.Yao CH, Liu GY, Wang R, et al. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation. PLoS Biol 2018; 16: e2003782 DOI: 10.1371/journal.pbio.2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Divakaruni AS, Hsieh WY, Minarrieta L, et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab 2018; 28: 490–503 e497 DOI: 10.1016/j.cmet.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lee J, Ellis JM and Wolfgang MJ. Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell reports 2015; 10: 266–279. DOI: 10.1016/j.celrep.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Raud B, Roy DG, Divakaruni AS, et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell metabolism 2018; 28: 504–515 e507 DOI: 10.1016/j.cmet.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nomura M, Liu J, Rovira, II, et al. Fatty acid oxidation in macrophage polarization. Nature immunology 2016; 17: 216–217. DOI: 10.1038/ni.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vigueira PA, McCommis KS, Schweitzer GG, et al. Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep 2014; 7: 2042–2053. DOI: 10.1016/j.celrep.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hobeika E, Thiemann S, Storch B, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proceedings of the National Academy of Sciences of the United States of America 2006; 103: 13789–13794. DOI: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kwon K, Hutter C, Sun Q, et al. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 2008; 28: 751–762. DOI: 10.1016/j.immuni.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 173.Rickert RC, Roes J and Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic acids research 1997; 25: 1317–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Casola S, Cattoretti G, Uyttersprot N, et al. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proceedings of the National Academy of Sciences of the United States of America 2006; 103: 7396–7401. DOI: 10.1073/pnas.0602353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Benson MJ, Dillon SR, Castigli E, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol 2008; 180: 3655–3659. [DOI] [PubMed] [Google Scholar]
- 176.Belnoue E, Pihlgren M, McGaha TL, et al. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood 2008; 111: 2755–2764. [DOI] [PubMed] [Google Scholar]
- 177.Cassese G, Arce S, Hauser AE, et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol 2003; 171: 1684–1690. [DOI] [PubMed] [Google Scholar]
- 178.Minges Wols HA, Underhill GH, Kansas GS, et al. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol 2002; 169: 4213–4221. [DOI] [PubMed] [Google Scholar]
- 179.Njau MN, Kim JH, Chappell CP, et al. CD28-B7 interaction modulates short- and long-lived plasma cell function. J Immunol 2012; 189: 2758–2767. DOI: 10.4049/jimmunol.1102728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Glatman Zaretsky A, Konradt C, Depis F, et al. T Regulatory Cells Support Plasma Cell Populations in the Bone Marrow. Cell Rep 2017; 18: 1906–1916. DOI: 10.1016/j.celrep.2017.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chu VT, Frohlich A, Steinhauser G, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol 2011; 12: 151–159. DOI: 10.1038/ni.1981. [DOI] [PubMed] [Google Scholar]
- 182.Rodriguez Gomez M, Talke Y, Goebel N, et al. Basophils support the survival of plasma cells in mice. J Immunol 2010; 185: 7180–7185. DOI: 10.4049/jimmunol.1002319. [DOI] [PubMed] [Google Scholar]
- 183.Winter O, Moser K, Mohr E, et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood 2010; 116: 1867–1875. DOI: 10.1182/blood-2009-12-259457. [DOI] [PubMed] [Google Scholar]
- 184.Mohr E, Serre K, Manz RA, et al. Dendritic cells and monocyte/macrophages that create the IL-6/APRIL-rich lymph node microenvironments where plasmablasts mature. J Immunol 2009; 182: 2113–2123. DOI: 10.4049/jimmunol.0802771. [DOI] [PubMed] [Google Scholar]
- 185.Bortnick A, Chernova I, Spencer SP, et al. No strict requirement for eosinophils for bone marrow plasma cell survival. European journal of immunology 2018; 48: 815–821. DOI: 10.1002/eji.201747229. [DOI] [PubMed] [Google Scholar]
- 186.Cravedi P, Lessman DA and Heeger PS. Eosinophils are not required for the induction and maintenance of an alloantibody response. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2013; 13: 2696–2702. DOI: 10.1111/ajt.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wilmore JR and Allman D. Here, There, and Anywhere? Arguments for and against the Physical Plasma Cell Survival Niche. J Immunol 2017; 199: 839–845. DOI: 10.4049/jimmunol.1700461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Harker JA, Lewis GM, Mack L, et al. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 2011; 334: 825–829. DOI: 10.1126/science.1208421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rawstron AC, Fenton JA, Ashcroft J, et al. The interleukin-6 receptor alpha-chain (CD126) is expressed by neoplastic but not normal plasma cells. Blood 2000; 96: 3880–3886. [PubMed] [Google Scholar]
- 190.Nguyen DC, Garimalla S, Xiao H, et al. Factors of the bone marrow microniche that support human plasma cell survival and immunoglobulin secretion. Nat Commun 2018; 9: 3698 DOI: 10.1038/s41467-018-05853-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cavo M, Tacchetti P, Patriarca F, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet 2010; 376: 2075–2085. DOI: 10.1016/S0140-6736(10)61424-9. [DOI] [PubMed] [Google Scholar]
- 192.Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352: 2487–2498. DOI: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 193.Radl J, Hollander CF, van den Berg P, et al. Idiopathic paraproteinaemia. I. Studies in an animal model--the ageing C57BL/KaLwRij mouse. Clin Exp Immunol 1978; 33: 395–402. [PMC free article] [PubMed] [Google Scholar]
- 194.Radl J, De Glopper ED, Schuit HR, et al. Idiopathic paraproteinemia. II. Transplantation of the paraprotein-producing clone from old to young C57BL/KaLwRij mice. J Immunol 1979; 122: 609–613. [PubMed] [Google Scholar]
- 195.Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers 2017; 3: 17046 DOI: 10.1038/nrdp.2017.46. [DOI] [PubMed] [Google Scholar]
- 196.Das R, Strowig T, Verma R, et al. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nature medicine 2016; 22: 1351–1357. DOI: 10.1038/nm.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Reinisch A, Thomas D, Corces MR, et al. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nature medicine 2016; 22: 812–821. DOI: 10.1038/nm.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Berardi S, Ria R, Reale A, et al. Multiple myeloma macrophages: pivotal players in the tumor microenvironment. J Oncol 2013; 2013: 183602 DOI: 10.1155/2013/183602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Giuliani N, Storti P, Bolzoni M, et al. Angiogenesis and multiple myeloma. Cancer Microenviron 2011; 4: 325–337. DOI: 10.1007/s12307-011-0072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Saini AS, Shenoy GN, Rath S, et al. Inducible nitric oxide synthase is a major intermediate in signaling pathways for the survival of plasma cells. Nat Immunol 2014; 15: 275–282. DOI: 10.1038/ni.2806. [DOI] [PubMed] [Google Scholar]
- 201.Murray ME, Gavile CM, Nair JR, et al. CD28-mediated pro-survival signaling induces chemotherapeutic resistance in multiple myeloma. Blood 2014; 123: 3770–3779. DOI: 10.1182/blood-2013-10-530964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kim J, Denu RA, Dollar BA, et al. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br J Haematol 2012; 158: 336–346. DOI: 10.1111/j.1365-2141.2012.09154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Beyer M, Kochanek M, Giese T, et al. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood 2006; 107: 3940–3949. DOI: 10.1182/blood-2005-09-3671. [DOI] [PubMed] [Google Scholar]
- 204.Niedzwiecka K, Dylag M, Augustyniak D, et al. Glutathione may have implications in the design of 3-bromopyruvate treatment protocols for both fungal and algal infections as well as multiple myeloma. Oncotarget 2016; 7: 65614–65626. DOI: 10.18632/oncotarget.11592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Majkowska-Skrobek G, Augustyniak D, Lis P, et al. Killing multiple myeloma cells with the small molecule 3-bromopyruvate: implications for therapy. Anticancer Drugs 2014; 25: 673–682. DOI: 10.1097/CAD.0000000000000094. [DOI] [PubMed] [Google Scholar]
- 206.Zhang XD, Deslandes E, Villedieu M, et al. Effect of 2-deoxy-D-glucose on various malignant cell lines in vitro. Anticancer Res 2006; 26: 3561–3566. [PubMed] [Google Scholar]
- 207.Nakano A, Miki H, Nakamura S, et al. Up-regulation of hexokinaseII in myeloma cells: targeting myeloma cells with 3-bromopyruvate. J Bioenerg Biomembr 2012; 44: 31–38. DOI: 10.1007/s10863-012-9412-9. [DOI] [PubMed] [Google Scholar]
- 208.Gu Z, Xia J, Xu H, et al. NEK2 Promotes Aerobic Glycolysis in Multiple Myeloma Through Regulating Splicing of Pyruvate Kinase. J Hematol Oncol 2017; 10: 17 DOI: 10.1186/s13045-017-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Walters DK, Arendt BK and Jelinek DF. CD147 regulates the expression of MCT1 and lactate export in multiple myeloma cells. Cell Cycle 2013; 12: 3175–3183. DOI: 10.4161/cc.26193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.McBrayer SK, Cheng JC, Singhal S, et al. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood 2012; 119: 4686–4697. DOI: 10.1182/blood-2011-09-377846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Effenberger M, Bommert KS, Kunz V, et al. Glutaminase inhibition in multiple myeloma induces apoptosis via MYC degradation. Oncotarget 2017; 8: 85858–85867. DOI: 10.18632/oncotarget.20691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008; 105: 18782–18787. DOI: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cacace A, Sboarina M, Vazeille T, et al. Glutamine activates STAT3 to control cancer cell proliferation independently of glutamine metabolism. Oncogene 2017; 36: 2074–2084. DOI: 10.1038/onc.2016.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gonsalves WI, Ramakrishnan V, Hitosugi T, et al. Glutamine-derived 2-hydroxyglutarate is associated with disease progression in plasma cell malignancies. JCI Insight 2018; 3 DOI: 10.1172/jci.insight.94543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bajpai R, Matulis SM, Wei C, et al. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene 2016; 35: 3955–3964. DOI: 10.1038/onc.2015.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chandra RK. Antibody formation in first and second generation offspring of nutritionally deprived rats. Science 1975; 190: 289–290. [DOI] [PubMed] [Google Scholar]
- 217.Christadoss P, Talal N, Lindstrom J, et al. Suppression of cellular and humoral immunity to T-dependent antigens by calorie restriction. Cell Immunol 1984; 88: 1–8. [DOI] [PubMed] [Google Scholar]
- 218.Goldberg EL, Romero-Aleshire MJ, Renkema KR, et al. Lifespan-extending caloric restriction or mTOR inhibition impair adaptive immunity of old mice by distinct mechanisms. Aging Cell 2015; 14: 130–138. DOI: 10.1111/acel.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Meydani SN, Das SK, Pieper CF, et al. Long-term moderate calorie restriction inhibits inflammation without impairing cell-mediated immunity: a randomized controlled trial in non-obese humans. Aging (Albany NY) 2016; 8: 1416–1431. DOI: 10.18632/aging.100994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Contreras NA, Fontana L, Tosti V, et al. Calorie restriction induces reversible lymphopenia and lymphoid organ atrophy due to cell redistribution. Geroscience 2018; 40: 279–291. DOI: 10.1007/s11357-018-0022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fernandes G, Yunis EJ and Good RA. Influence of diet on survival of mice. Proc Natl Acad Sci U S A 1976; 73: 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kubo C, Day NK and Good RA. Influence of early or late dietary restriction on life span and immunological parameters in MRL/Mp-lpr/lpr mice. Proc Natl Acad Sci U S A 1984; 81: 5831–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Organization WH. Report on Infectious Disease. 2000. Geneva: World Health Organization. [Google Scholar]
- 224.Miller RA, Harrison DE, Astle CM, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2011; 66: 191–201. DOI: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Jones DD, Gaudette BT, Wilmore JR, et al. mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest 2016; 126: 4250–4261. DOI: 10.1172/JCI86504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Keating R, Hertz T, Wehenkel M, et al. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat Immunol 2013; 14: 1266–1276. DOI: 10.1038/ni.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Louie JK, Acosta M, Samuel MC, et al. A novel risk factor for a novel virus: obesity and 2009 pandemic influenza A (H1N1). Clin Infect Dis 2011; 52: 301–312. DOI: 10.1093/cid/ciq152. [DOI] [PubMed] [Google Scholar]
- 228.Esposito S, Giavoli C, Trombetta C, et al. Immunogenicity, safety and tolerability of inactivated trivalent influenza vaccine in overweight and obese children. Vaccine 2016; 34: 56–60. DOI: 10.1016/j.vaccine.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 229.Sheridan PA, Paich HA, Handy J, et al. Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes (Lond) 2012; 36: 1072–1077. DOI: 10.1038/ijo.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Sheridan PA, Paich HA, Handy J, et al. The antibody response to influenza vaccination is not impaired in type 2 diabetics. Vaccine 2015; 33: 3306–3313. DOI: 10.1016/j.vaccine.2015.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.da Silva SV, Renovato-Martins M, Ribeiro-Pereira C, et al. Obesity modifies bone marrow microenvironment and directs bone marrow mesenchymal cells to adipogenesis. Obesity (Silver Spring) 2016; 24: 2522–2532. DOI: 10.1002/oby.21660. [DOI] [PubMed] [Google Scholar]
- 232.Yue R, Zhou BO, Shimada IS, et al. Leptin Receptor Promotes Adipogenesis and Reduces Osteogenesis by Regulating Mesenchymal Stromal Cells in Adult Bone Marrow. Cell Stem Cell 2016; 18: 782–796. DOI: 10.1016/j.stem.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 233.van den Berg SM, Seijkens TT, Kusters PJ, et al. Diet-induced obesity in mice diminishes hematopoietic stem and progenitor cells in the bone marrow. FASEB J 2016; 30: 1779–1788. DOI: 10.1096/fj.201500175. [DOI] [PubMed] [Google Scholar]
- 234.Adler BJ, Green DE, Pagnotti GM, et al. High fat diet rapidly suppresses B lymphopoiesis by disrupting the supportive capacity of the bone marrow niche. PLoS One 2014; 9: e90639 DOI: 10.1371/journal.pone.0090639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Chan ME, Adler BJ, Green DE, et al. Bone structure and B-cell populations, crippled by obesity, are partially rescued by brief daily exposure to low-magnitude mechanical signals. FASEB J 2012; 26: 4855–4863. DOI: 10.1096/fj.12-209841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Greene DA, Lattimer SA and Sima AA. Sorbitol, phosphoinositides, and sodium-potassium-ATPase in the pathogenesis of diabetic complications. The New England journal of medicine 1987; 316: 599–606. DOI: 10.1056/NEJM198703053161007. [DOI] [PubMed] [Google Scholar]
- 237.Warren KJ, Olson MM, Thompson NJ, et al. Exercise Improves Host Response to Influenza Viral Infection in Obese and Non-Obese Mice through Different Mechanisms. PLoS One 2015; 10: e0129713 DOI: 10.1371/journal.pone.0129713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Nehlsen-Cannarella SL, Nieman DC, Balk-Lamberton AJ, et al. The effects of moderate exercise training on immune response. Med Sci Sports Exerc 1991; 23: 64–70. [PubMed] [Google Scholar]
- 239.Turner JE, Spielmann G, Wadley AJ, et al. Exercise-induced B cell mobilisation: Preliminary evidence for an influx of immature cells into the bloodstream. Physiol Behav 2016; 164: 376–382. DOI: 10.1016/j.physbeh.2016.06.023. [DOI] [PubMed] [Google Scholar]