

Abstract
Diabetes mellitus (DM) is an independent risk factor for atrial fibrillation, but the underlying ionic mechanism for this association remains unclear. We recently reported that expression of the small-conductance calcium-activated potassium channel 2 (SK2, encoded by KCCN2) in atria from diabetic mice is significantly down-regulated, resulting in reduced SK currents in atrial myocytes from these mice. We also reported that the level of SK2 mRNA expression is not reduced in DM atria but that the ubiquitin-proteasome system (UPS), a major mechanism of intracellular protein degradation, is activated in vascular smooth muscle cells in DM. This suggests a possible role of the UPS in reduced SK currents. To test this possibility, we examined the role of the UPS in atrial SK2 down-regulation in DM. We found that a muscle-specific E3 ligase, F-box protein 32 (FBXO-32, also called atrogin-1), was significantly up-regulated in diabetic mouse atria. Enhanced FBXO-32 expression in atrial cells significantly reduced SK2 protein expression, and siRNA-mediated FBXO-32 knockdown increased SK2 protein expression. Furthermore, co-transfection of SK2 with FBXO-32 complementary DNA in HEK293 cells significantly reduced SK2 expression, whereas co-transfection with atrogin-1ΔF complementary DNA (a nonfunctional FBXO-32 variant in which the F-box domain is deleted) did not have any effects on SK2. These results indicate that FBXO-32 contributes to SK2 down-regulation and that the F-box domain is essential for FBXO-32 function. In conclusion, DM-induced SK2 channel down-regulation appears to be due to an FBXO-32-dependent increase in UPS-mediated SK2 protein degradation.
Keywords: potassium channel, diabetes, cardiomyocyte, cardiovascular disease, ubiquitin ligase, protein degradation, atrial fibrillation, atrogin-1, diabetes mellitus, HL-1 cells, SK channels
Introduction
DM4 is a global epidemic, and the number of people with diabetes has quadrupled since 1980 (1). According to the American Diabetes Association, in 2015 (http://www.diabetes.org/diabetes-basics/statistics/?loc=db-slabnav),5 30.0 million Americans or 9.4% of the population have DM, and 1.5 million Americans are diagnosed with DM every year, imposing a financial burden of 245 billion dollars annually. DM affects men and women, young and old, and people of all ethnic backgrounds (2). DM is one of the leading causes of death (3) and is associated with increased risks of cardiovascular diseases, including hypertension, dyslipidemia, myocardial infarction, heart failure, stroke, and cardiovascular deaths (4). DM has also been implicated as a risk factor for the development of AF (5, 6). However, very little is known about the mechanism through which DM modulates atrial electrophysiology.
A genome-wide association study (GWAS) study has indicated that there is a strong association between the SK channels and the development of AF (7). Recently, we reported that the expression of SK2 and SK3 channel proteins was significantly down-regulated by 85% and 92% respectively, whereas that of the SK1 channel was not changed in streptozotocin-induced diabetic mouse atria (8). This was associated with a significant reduction in SK currents in diabetic mouse atrial myocytes together with prolongation of the action potential duration (8). In addition, we identified significantly increased oxidative stress in diabetic cardiac atria and in HL-1 cells cultured in high glucose (8), which may link to changes in the SK channel in DM. Further, we identified that the decreased SK channel protein expression was not due to a decrease in mRNA expression (8), suggesting posttranslational regulation, such as increased protein degradation. The UPS is a principal mechanism for cellular protein degradation (9, 10). The integrity of proteome homeostasis is challenged in DM. Evidence for this includes the accumulation of damaged proteins as a consequence of increased oxidative stress with progressive deterioration in the ability to preserve a functional cellular proteome in DM (8, 11, 12). Degradation by the UPS is regulated by protein-specific ubiquitin E3 ligase enzymes that facilitate the ubiquitination of substrate proteins, destining them for degradation in the 26S proteasome (10, 13). To understand the regulation of SK channel degradation in diabetic atria, it is critical to identify and characterize the E3 ligase involved. In this study, we examined the role of muscle-specific E3 ligase in the regulation of SK channels by the UPS and tested the hypothesis that atrial SK channel protein down-regulation in DM is due to accelerated proteolysis as a result of up-regulation in E3 ligase and UPS activities.
Results
Regulation of SK2 by atrogin-1
We have recently reported that the protein expression of SK2 and SK3 is significantly down-regulated in the atria of type 1 diabetic mice, whereas that of SK1 was unchanged (8). We also found that the mRNA expression of these SK channels was not reduced in diabetes (8). These results suggest that the down-regulation of SK channel protein expression in the diabetic mouse atrium may result from enhanced protein degradation. We have reported previously that atrogin-1, a muscle-specific E3 ligase, is up-regulated in diabetic mouse vessels and in vascular smooth muscle cells cultured with high glucose (HG) (11). To determine whether atrogin-1 is involved with SK channel degradation, we conducted our experiments using HL-1 cells (8). Overexpression of atrogin-1 was achieved by transduction of an adenoviral vector carrying the WT atrogin-1 (Ad-atrogin-1). The protein levels of atrogin-1, SK1, SK2, and SK3 were examined 48 h after transduction of atrogin-1 in HL-1 cells. The results showed that there was a 3.4-fold overexpression of atrogin-1 with Ad-atrogin-1 compared with that with a control vector (n = 3, p < 0.05) (Fig. 1A), and this was accompanied by a significant reduction of SK2 protein expression by 78% (n = 3, p < 0.05) (Fig. 1A). Overexpression of atrogin-1 did not produce any significant alterations in SK1 and SK3 protein expression (Fig. 1A). To further evaluate the regulation of SK channel expression by atrogin-1, we performed knockdown of atrogin-1 in HL-1 cells using atrogin-1 siRNAs. Forty-eight hours after transfection, atrogin-1 protein was reduced by 71% in cells transfected with siRNA compared with those with the scrambled RNAi control (n = 3, p < 0.05) (Fig. 1B). Knockdown of atrogin-1 produced a 2.1-fold increase in SK2 protein expression (n = 3, p < 0.05) but no significant change in that of SK1 and SK3 (Fig. 1B). These results indicate that mouse atrial SK2 is a target of atrogin-1 regulation.
Figure 1.

Atrogin-1 regulates SK2 protein expression in HL-1 cells. A, adenovirus-mediated overexpression of atrogin-1 in HL-1 cells was associated with a significant reduction in SK2 protein levels (*, p < 0.05; n = 3) but had no effect on those of SK1 and SK3 (p = N.S., n = 3). B, knockdown of atrogin-1 using atrogin-1 siRNAs achieved a 71% reduction in atrogin-1 protein expression in HL-1 cells, and this was accompanied by a significant 2.1-fold up-regulation of SK2 protein (*, p < 0.05; n = 3). Atrogin-1 knockdown had no significant effect on the levels of SK1 and SK3 protein expression (p = N.S., n = 3).
Down-regulation of SK2 protein expression in diabetic mouse atria is associated with up-regulation of atrogin-1
We examined the levels of atrogin-1 expression in the atrial myocardium of type 1 (streptozotocin-induced) and type 2 db/db diabetic mice. We found that atrogin-1 expression was significantly up-regulated in association with significant down-regulation of SK2 in both type 1 (n = 6 for each group, p < 0.05) and type 2 diabetic mouse atria (n = 5 for each group, p < 0.05) (Fig. 2, A and B). These results indicate that reduction of atrial SK2 channel protein expression is a common feature in the atria of type 1 and type 2 diabetic mice, which are associated with enhanced atrogin-1 expression.
Figure 2.

SK channel expression in control and diabetic mouse atria. A, immunoblots of SK channels and atrogin-1 using homogenates of control and type 1 (streptozotocin-induced) diabetic mouse atria. Group data show significant down-regulation of SK2 and up-regulation of atrogin-1 protein expression in type 1 DM (n = 6 for each group; *, p < 0.05). B, immunoblots of SK channels and atrogin-1 using homogenates from control and type 2 db/db diabetic mouse atria. Group data show significant downregulation of SK2 and up-regulation of atrogin-1 protein expression in type 2 DM (n = 5 for each group; *, p < 0.05).
Proteasomal degradation of SK2
To determine the role of proteasomal degradation in SK2 protein regulation, HL-1 cells were transduced with Ad-atrogin-1 in the presence or absence of the proteasome inhibitor MG-132 (24 h, 10 μm). Treatment with MG-132 abolished the effects of atrogin-1, and the protein expression of SK2 was preserved (n = 6, p = N.S. versus control) (Fig. 3A). In HL-1 cells cultured in HG (25 mm), the protein expression of atrogin-1 was significantly up-regulated (n = 6, p < 0.05), and this was accompanied by significantly reduced SK2 protein expression (n = 6, p < 0.05) (Fig. 3B, left panel). In addition, knockdown of atrogin-1 by siRNA resulted in significant up-regulation of SK2 protein expression in these cells (n = 3, p < 0.05) (Fig. 3B, right panel). These results indicate that the UPS plays a critical role in regulating SK2 protein expression in atrial myocytes cultured in HG.
Figure 3.

Role of the ubiquitin-proteasome system in the regulation of SK2 protein expression. A, incubation with a proteasomal inhibitor, MG132 (24 h, 10 μm), abolished the effects of atrogin-1 on SK2 protein degradation (n = 6 for each group; *, p < 0.05). B, left panel, adenovirus-mediated up-regulation of atrogin-1 resulted in significant downregulation of SK2 in HL-1 cells cultured in HG (n = 6 for each group; *, p < 0.05). Right panel, knockdown of atrogin-1 with siRNA resulted in significant up-regulation of SK2 in HL-1 cells cultured in HG (n = 3 for each group; *, p < 0.05).
The role of the PDZ-binding motif in UPS-mediated SK2 degradation
Further demonstration of the specificity of atrogin-1 in SK2 protein degradation was performed by co-transfection of FLAG-tagged SK2 WT cDNAs (FLAG-SK2) with atrogin-1 WT cDNAs or atrogin-1ΔF cDNAs (a nonfunctional mutant with the F-box domain deleted) in HEK293 cells. There was no significant alteration in the level of atrogin-1ΔF protein expression compared with that of atrogin-1 WT (Fig. 4A). Forty-eight hours after transfection, expression of FLAG-SK2 was significantly lower in cells co-transfected with atrogin-1 WT cDNAs compared with those co-transfected with empty plasmids (control) (0.15 ± 0.06 for atrogin-1 versus 0.71 ± 0.14 for the control, n = 3, p < 0.05) (Fig. 4A). In contrast, co-transfection with atrogin-1ΔF did not reduce the level of FLAG-SK2 protein expression (0.71 ± 0.04 versus 0.71 ± 0.14 for the control, n = 3, p = N.S.) (Fig. 4A).
Figure 4.

Mutations in the PDZ-binding motif of SK2 prevented atrogin-1-mediated-ubiquitination and degradation of SK2. A, left panel, there was no alteration in atrogin-1ΔF protein expression compared with that of the WT atrogin-1. Right panel, co-transfection of atrogin-1 and FLAG-SK2 resulted in significant down-regulation of SK2 protein expression compared with the control (FLAG-SK2 co-transfected with empty plasmids) (*, p < 0.05, n = 3). However, co-transfection with atrogin-1ΔF had no effect on the level of SK2 expression compared with control (p = N.S., n = 3). B, top panel, immunoprecipitation with anti-ubiquitin antibody against HEK293 cell lysates with each combination of co-transfection. First lane, SK2 WT co-transfected with ubiquitin only; second lane, SK2 WT co-transfected with ubiquitin and atrogin-1; third lane, the SK2 PDZ binding motif mutant co-transfected with atrogin-1 and ubiquitin; fourth lane, SK2 WT co-transfected with atrogin-1ΔF and ubiquitin; fifth lane, lysate control; sixth lane, nonimmune IgG negative control. Ubiquitination of SK2 WT (second lane) was more substantially enhanced than all other combinations and controls. Nonimmune IgG control on the lysate of the second lane was negative (sixth lane). The experiment was independently repeated three times and produced similar results (shown in C). Bottom panel, the inputs of the tested samples as indicated in the top panel. SK2 and its mutant expression were detected by anti-FLAG antibody against the FLAG tag on the channels, and atrogin-1 and its mutant were detected by anti-myc antibody to the Myc tag fused with atrogin-1 and its mutant. C, group data for the experiments in B presented in dot scatterplots showing that significant SK2 ubiquitination occurred when SK2 WT and atrogin-1 WT were present (n = 3, p < 0.05). The positions of the lanes correspond to the position and composition of table columns in B. Data represent all anti-FLAG-reactive bands in densitometry arbitrary units (AU).
Co-transfection with atrogin-1 cDNAs and ubiquitin cDNAs with FLAG-SK2 WT cDNAs or with FLAG-SK2-PDZ binding motif mutant cDNAs showed that the level of protein ubiquitination was significantly higher in FLAG-SK2 WT than that in FLAG-SK2-mutant (Fig. 4B, second and third lanes). In contrast, co-transfection with atrogin-1ΔF cDNAs and ubiquitin cDNAs had no significant effect on SK2 ubiquitination (Fig. 4B, fourth lane). Group data are presented in Fig. 4C, showing significant SK2 ubiquitination when SK2 WT and atrogin-1 WT are present (n = 3, p < 0.05). These results suggest that the FBXO domain in atrogin-1 and the PDZ binding motif of SK2 are both required to facilitate SK2 ubiquitination and degradation.
Enhanced expression of atrogin-1 reduced SK2 channel activities
We determined the functional consequences of enhanced atrogin-1 expression on SK2 channel activity. FLAG-SK2 WT cDNAs were co-transfected with atrogin-1 WT cDNAs or atrogin-1ΔF cDNAs in HEK293 cells. Patch clamp studies confirmed that SK2 current densities were significantly reduced by 3- to 5-fold (n = 4∼5, p < 0.05) in cells with FLAG-SK2 and atrogin-1 WT co-transfection compared with FLAG-SK2 and atrogin-1ΔF co-transfection (Fig. 5). These results are consistent with the findings from molecular biological experiments.
Figure 5.
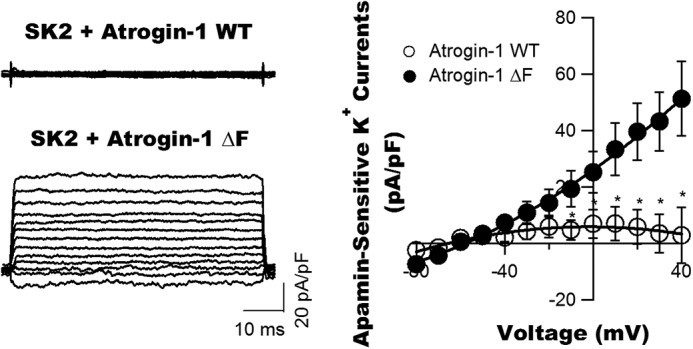
Down-regulation of SK current densities in HEK293 cells by atrogin-1 co-transfection. SK2 WT cDNAs were transfected into HEK293 cells with atrogin-1 WT or atrogin-1ΔF cDNAs. Forty-eight hours after transfection, the apamin-sensitive currents were elicited from a holding potential of −60 mV to different testing potentials of −80 mV to +40 mV in +10-mV increments in the presence and absence of apamin (100 pm). The current–voltage relationships showed a significant reduction in apamin-sensitive K+ components (defined as SK2 currents in picoamperes/picofarads) in cells with co-expression of atrogin-1 WT compared with those with atrogin-1ΔF expression (n = 4 for both groups; *, p < 0.05).
Discussion
In this study, we made several important observations. First, the expression of SK2 is markedly down-regulated in both type 1 and type 2 diabetic mouse atria. Second, the expression of atrogin-1 is significantly up-regulated in DM atria. Third, we provided compelling evidence that the expression of SK2 is closely regulated by atrogin-1-mediated UPS-dependent degradation. Fourth, both the F-box in atrogin-1 and the PDZ binding motif in SK2 are required for atrogin-1–mediated SK2 protein ubiquitination and degradation. Fifth, down-regulation of SK2 channel expression by atrogin-1 results in loss of channel activity. These findings provide novel insights into the molecular mechanism of cardiac atrial SK channel regulation by atrogin-1 that support our previous work in DM (8).
SK channels are widely expressed in many tissues, including the myocardium, vascular endothelium, and smooth and skeletal muscle and the central nervous system (14, 15). These important potassium-selective ion channels are more abundant in the atria than in the ventricles of the heart (16). SK channels are voltage-independent and are activated by increased intracellular calcium, coupling intracellular calcium homeostasis with cell membrane potentials. SK channels are known to be important in the regulation of excitability in the central nervous system, contributing to action potential after-hyperpolarization and regulating spike frequency adaptation and fundamental parameters of excitability (15, 17). However, the role of SK channels in cardiac electrophysiology remains unclear. Multiple studies suggest that SK channels are important mediators of pro-arrhythmogenic electrical remodeling in the atria (16, 18–20). SK2 knockout mice have been reported to have more frequent early after-depolarizations, action potential duration prolongations, and enhanced AF inducibility compared with WT controls (20). However, inhibition of SK channels in other animal models showed antiarrhythmic effects (14, 21). These results suggest that the electrophysiological consequence of SK channel expression is complex and could be anti-arrhythmic or pro-arrhythmic in different clinical situations. Hence, regulation of SK channel expression is highly important physiologically, with effects on atrial electrophysiology and arrhythmogenesis. We have recently reported that SK channel activity is significantly diminished in type 1 diabetic mouse atria as a result of reduced SK2 protein expression, which contributes to the significant prolongation in action potential duration and early after-depolarization formation (8). These changes may lead to an increased susceptibility to develop atrial arrhythmias. However, the precise molecular mechanism that causes SK channel protein down-regulation in diabetic atria is unclear. One of the novel findings in this study is that the UPS is associated with the degradation of SK2 channel proteins.
More than 80% of cellular proteins are degraded in a highly specific and selective fashion via the UPS (10, 22). A defective UPS may cause devastating conditions such as neurodegeneration (23), cardiac diseases (24, 25), autoimmunity, and inflammatory diseases (26, 27). Proteins targeted for degradation are ubiquitinated through the sequential action of three enzymes: an ubiquitin-activating enzyme (E1), an ubiquitin carrier protein (E2), and an ubiquitin-protein isopeptide ligase (E3) (10, 22). There is one E1, more than 25 E2s, and more than 1000 E3s. Each E3 recognizes a specific motif on the substrate protein; therefore, the specificity of the UPS depends on E3s (28). Proteins destined for degradation by the UPS pathway are covalently ubiquitinated by E3 to signal for breakdown by the 26S proteasome (13). Hence, it is important to identify the specific E3s for target proteins to understand the regulation of their expression. F-box only proteins (FBXOs) are key components of the Skp1-Cullin-F-box (SCF) type ubiquitin ligase complex, functioning as sites for enzyme–substrate interaction (29). Atrogin-1, a member of the FBXO proteins, is reported to be highly expressed during muscle atrophy (13). We have found previously that atrogin-1 expression is up-regulated in vascular smooth muscle cells in diabetic vessels and when cultured in HG. Up-regulation of atrogin-1 under these conditions plays a critical role in the ubiquitin–proteasome–mediated degradation of BK-β1, a subunit of the high-conductance calcium- and voltage-activated potassium (BK) channel, leading to the down-regulation of BK channel function in DM (11). In this study, we found that an increase in the expression of atrogin-1 in HL-1 cells reduces the protein expression of SK2 channels (Fig. 1A). In contrast, knockdown of atrogin-1 by siRNAs in HL-1 cells resulted in up-regulation of SK2 protein (Figs. 1B and Fig. 3B). In comparison, the levels of SK1 and SK3 expression were not affected by that of atrogin-1 (Fig. 1), suggesting that the effects of atrogin-1 on SK channels in DM are isoform-specific. Indeed, we have reported previously that the down-regulation of SK3 in DM was regulated by microRNA-499 (30), indicating that distinct mechanisms are involved in the regulation of the expression of different SK isoforms. Interestingly, SK1 also contains a PDZ-binding motif (31). However, SK1 expression was not affected by atrogin-1, suggesting that the PDZ-binding motif alone may not be sufficient for atrogin-1–mediated regulation of SK1. Channel conformation and splice variants might be important in their regulation by atrogin-1; however, the role of such elements is beyond the scope of this study. These results strongly indicate that atrogin-1 and the UPS are critical regulators of atrial SK2 expression in DM.
We found that atrogin-1 expression was significantly up-regulated in the atria of both type 1 and type 2 diabetic mice (Fig. 2), and this was accompanied by significantly decreased SK2 channel protein. Molecular biological studies confirmed that the F-box domain of atrogin-1 and the PDZ-binding motif of SK2 are both required for atrogin-1-mediated SK2 degradation. Disruption of the SK2 PDZ-binding motif abolished SK2 protein ubiquitination in the presence of atrogin-1 (Fig. 4B), and deletion of the F-box domain of atrogin-1 abolished its deleterious effects on SK2 channel expression and activity (Fig. 5). Hence, our results delineate a fundamental mechanism that underlies the regulation of SK2 channel protein expression and function by atrogin-1 in diabetes.
In summary, we have reported novel findings showing that the down-regulation of the cardiac SK2 channel in DM is due to enhanced atrogin-1 expression. Our results provide novel insights into the mechanisms of atrial electrophysiology and arrhythmogenesis in DM. To our knowledge, this study is the first report of the molecular regulation of SK channel degradation in DM. These findings may have important clinical implications. Modulation of SK channel turnover by the UPS is a potential therapeutic approach for preserving the integrity of atrial electrophysiology and the prevention of arrhythmia development in DM. These important findings warrant further studies.
Experimental procedures
Diabetic mice
Development of type 1 diabetes was induced in male mice (Harlan Inc., Indianapolis, IN) at 6 to 8 weeks of age by streptozotocin (100 mg/kg body weight, intraperitoneally) as described previously (11). Control mice received vehicle injections. Type 2 diabetic db/db mice (BKS.Cg-Dock7m+/+Leprdb/J) and age-matched control mice (Dock7m+/Dock7m) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice with blood glucose higher than 300 mg/dl were considered diabetic and used for experiments after 8 weeks. Handling and care of animals as well as animal procedures were approved by the Institutional Animal Care and Use Committee of the Mayo Clinic Foundation.
HL-1 cell culture
HL-1 cells, a continuously proliferating cardiomyocyte cell line derived from mouse atrial tumors, were maintained in modified Claycomb medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum, 100 μg/ml penicillin–streptomycin, 0.1 mm norepinephrine, and 2 mm l-glutamine as described previously (32). Cells were seeded onto gelatin/fibronectin-coated plates, replenished daily with 1 ml of medium per 5 cm2 culture area, and enzymatically dissociated when confluent and reseeded at 1:4 dilutions. HL-1 cells were cultured with modified Claycomb medium containing either normal glucose (5 mm) or HG (25 mm) for 2 weeks before the experiments.
Adenoviral expression of atrogin-1 in HL-1 cells
Overexpression of atrogin-1 in HL-1 cells was performed as we described previously (12). Briefly, HL-1 cells cultured in Claycomb medium were transduced with an adenovirus carrying the CMV promoter and human atrogin-1 (Vector BioLabs, Philadelphia, PA) or with a control adenovirus carrying the CMV promoter only (Vector BioLabs) at a multiplicity of infection of 50 for 48 h. Efficacy of infection was confirmed by atrogin-1 Western blot analysis.
Atrogin-1 knockdown by siRNA
Knockdown of atrogin-1 by siRNA was performed as described previously (33). A pool of three different atrogin-1 siRNA duplexes against mouse atrogin-1 mRNA (sc-44807; CUCAGUACUUCCAUCAAGAtt (sense) and UCUUGAUGGAAGUACUGAGtt (antisense), GUACGAUGUUACCCAAGAAtt (sense) and UUCUUGGGUAACAUCGUACtt (antisense), and CGUGUUCUCUGGCAACAUAtt (sense) and UAUGUUGCCAGAGAACACGtt (antisense)) and the scrambled control siRNA (sc-37007; UUCUCCGAACGUGUCACGUtt (sense) and ACGUGACACGUUCGGAGAAtt (antisense)) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). HL-1 cells at 70% confluence were transfected with atrogin-1 siRNA (100 nm) or scrambled control RNAi using Lipofectamine 2000 (Invitrogen). Fresh medium was replenished 5 h after transfection, and the cells were examined 48 h after transfection.
Site-directed mutagenesis and cDNA transfection in HEK293 cells
Mouse SK2 WT cDNAs (accession no. NM_001312905.1) were subcloned into pIRES2-EGFP. Atrogin-1 WT and atrogin-1ΔF cDNAs in pCMC-Tag 3B were provided by Dr. Monte S. Willis (11). The FLAG-SK V475A mutant was created using the QuikChange site-directed mutagenesis kit (Stratagene Co., La Jolla, CA) (35). Orientations of constructs and correctness of the mutation were verified by DNA sequencing at the DNA facility core of the Mayo Clinic. HEK293 cells were cultured in Dulbecco's modified Eagle's medium containing 5 mm glucose. cDNAs were transfected into HEK293 cells using Lipofectamine 2000 (Thermo Fisher Scientific Inc.) and used for experiments 48 h after transfection (35).
Western blot analysis
Isolated mouse atria and HL-1 cells were homogenized or lysed, and the proteins were separated by PAGE, transferred to a nitrocellulose membrane, and immunoblotted against the following antibodies: anti-SK1 (1:200, Alomone Laboratories, Jerusalem, Israel), anti-SK2 (1:200, Alomone Laboratories), and anti-SK3 (1:200, Alomone Laboratories). Blots were probed with anti-glyceraldehyde-3-phosphate dehydrogenase (1:200, Santa Cruz Biotechnology) as a loading control. After extensive washing, blots were exposed to horseradish peroxidase–conjugated secondary antibodies, and signals were developed by the SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific, Rockford, IL). The band optical density was analyzed using Scion Image software (Scion, Frederick, MD). Protein expression was expressed as relative abundance normalized to that of glyceraldehyde-3-phosphate dehydrogenase (36).
Co-immunoprecipitation analysis
Co-immunoprecipitation was performed as we reported previously (36). In brief, HL-1 cells were harvested after two washes with PBS. The collected cells were incubated with 200 μl of radioimmune precipitation assay buffer: 50 mm Tris, 150 mm NaCl, 2 mm NaF, 1 mm EDTA, 1 mm EGTA, 1 mm NaVO4, 1% Triton X-100, and 1 μl of protease inhibitor on ice for 1 h, followed by homogenization and then centrifugation at 8,000 × g at 4 °C for 10 min. The supernatant (about 600 μg of protein in 600 μl) was incubated overnight at 4 °C with anti-ubiquitin antibodies (Santa Cruz Biotechnology) at a final concentration of 5 μg/ml. The samples were then incubated with 35 μl og protein G Plus–agarose (Santa Cruz Biotechnology) at 4 °C for 4 h with rotation. After centrifugation at 2,000 rpm for 3 min and three washes with radioimmune precipitation assay/protease inhibitor buffer, the immunoprecipitates were collected and eluted from agarose with 30 μl of SDS-PAGE loading buffer. The immunoprecipitates were resolved by SDS-PAGE and blotted against anti-FLAG antibodies (1:1,000, Sigma-Aldrich) to detect SK2 expression.
Whole-cell SK2 current recordings
SK2 cDNAs in pIRES2-EGFP were co-transfected with atrogin-1 WT cDNAs in pCMV-Tag3B or atrogin-1ΔF cDNAs in pCMV-Tag3B in HEK293 cells using Effectene transfection reagent (Qiagen Inc., Valencia, CA) as reported previously (36). After 48 h of transfection, the cells were examined for the presence of GFP expression using an UV microscope (Olympus, IX70). SK2 currents were recorded with a holding potential of −60 mV and testing potentials of −80 mV to +40 mV in 10-mV increments using an Axopatch 200B integrating amplifier (Molecular Devices, San Jose, CA). The signals were filtered at 2 kHz and digitized at 50 kHz (16-bit resolution). The glass pipette had a typical resistance of 0.5 to 1 megaohm after filling with the pipette solution, which contained 140 mm KCl, 1.0 mm MgCl2, 0.5 mm Na2GTP, 5.0 mm Na2ATP, 1.0 mm EGTA, 10 mm Hepes, and 0.96 mm CaCl2 (free Ca2+ concentration, 1 μm) (pH 7.35) with KCl (36). The bath solution contained 140 mm N-methylglucamine, 4.0 mm KCl, 1.0 mm MgCl2, 5.0 mm glucose, and 10 mm Hepes (pH 7.4). The SK currents were determined as the K+ current component sensitive to 100 pm apamin (8, 20, 34). All experiments were performed at room temperature (22 °C).
Statistical analysis
Data are presented as mean ± S.E. One-way analysis of variance followed by Tukey test was used to compare data from multiple groups. Statistical significance was defined as p < 0.05.
Author contributions
T.-Y. L., F. Y., T. L., X.-L. W., J. P. A., and H.-C. L. conceptualization; T.-Y. L., F. Y., T. L., X.-L. W., and X. S. data curation; T.-Y. L., F. Y., T. L., X.-L. W., and X. S. formal analysis; T.-Y. L., F. Y., T. L., X.-L. W., J. P. A., and H.-C. L. validation; T.-Y. L., F. Y., T. L., X.-L. W., M. S. W., L.-Q. W., W.-K. S., J. P. A., and H.-C. L. investigation; T.-Y. L., F. Y., T. L., X.-L. W., X. S., M. S. W., J. P. A., and H.-C. L. methodology; T.-Y. L., F. Y., T. L., and H.-C. L. writing-original draft; T.-Y. L., T. L., X.-L. W., J. P. A., and H.-C. L. project administration; T. L. and H.-C. L. funding acquisition; X.-L. W., J. P. A., and H.-C. L. supervision; X.-L. W., X. S., M. S. W., L.-Q. W., W.-K. S., J. P. A., and H.-C. L. writing-review and editing; M. S. W., L.-Q. W., W.-K. S., J. P. A., and H.-C. L. resources; H.-C. L. visualization.
This study was supported by funding from the National Natural Science Foundation of China (81400248 and 81470479); Ruijin Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, China (DLY201604); Xijing Hospital, Fourth Military Medical University, Xi'an, China; the Mayo Clinic Foundation; the American Diabetes Association (ADA 1-12-BS-119, ADA 1-16-IBS-195, and ADA 1-18-IBS-210), and the NHLBI, National Institute of Health (HL74180 and HL080118). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- DM
- diabetes mellitus
- UPS
- ubiquitin–proteasome system
- HG
- high glucose
- N.S.
- not significant
- cDNA
- complementary DNA
- HEK
- human embryonic kidney
- CMV
- cytomegalovirus
- BK
- large-conductance calcium-activated potassium channel
- SK
- small-conductance calcium-activated potassium channel.
References
- 1. World Health Organization (2016) Global report on diabetes. World Health Organization, Geneva, http://www.who.int/diabetes/global-report/en/ [Google Scholar]
- 2. Black S. A. (2002) Diabetes, diversity, and disparity: what do we do with the evidence? Am. J. Public Health 92, 543–548 10.2105/AJPH.92.4.543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stokes A., and Preston S. H. (2017) Deaths attributable to diabetes in the United States: comparison of data sources and estimation approaches. PLoS ONE 12, e0170219 10.1371/journal.pone.0170219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Resnick H. E., and Howard B. V. (2002) Diabetes and cardiovascular disease. Annu. Rev. Med. 53, 245–267 10.1146/annurev.med.53.082901.103904 [DOI] [PubMed] [Google Scholar]
- 5. Movahed M. R., Hashemzadeh M., and Jamal M. M. (2005) Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int. J. Cardiol. 105, 315–318 10.1016/j.ijcard.2005.02.050 [DOI] [PubMed] [Google Scholar]
- 6. Kannel W. B., and Benjamin E. J. (2008) Status of the epidemiology of atrial fibrillation. Med. Clin. North Am. 92, 17–40, ix 10.1016/j.mcna.2007.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ellinor P. T., Lunetta K. L., Glazer N. L., Pfeufer A., Alonso A., Chung M. K., Sinner M. F., de Bakker P. I., Mueller M., Lubitz S. A., Fox E., Darbar D., Smith N. L., Smith J. D., Schnabel R. B., et al. (2010) Common variants in KCNN3 are associated with lone atrial fibrillation. Nat. Genet. 42, 240–244 10.1038/ng.537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yi F., Ling T. Y., Lu T., Wang X. L., Li J., Claycomb W. C., Shen W. K., and Lee H. C. (2015) Down-regulation of the small conductance calcium-activated potassium channels in diabetic mouse atria. J. Biol. Chem. 290, 7016–7026 10.1074/jbc.M114.607952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jackman R. W., and Kandarian S. C. (2004) The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 287, C834–843 10.1152/ajpcell.00579.2003 [DOI] [PubMed] [Google Scholar]
- 10. Ciechanover A. (2006) The ubiquitin proteolytic system: from an idea to the patient bed. Proc. Am. Thorac. Soc. 3, 21–31 10.1513/pats.200510-106JH [DOI] [PubMed] [Google Scholar]
- 11. Zhang D. M., He T., Katusic Z. S., Lee H. C., and Lu T. (2010) Muscle-specific F-box only proteins facilitate bk channel β1 subunit downregulation in vascular smooth muscle cells of diabetes mellitus. Circ. Res. 107, 1454–1459 10.1161/CIRCRESAHA.110.228361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu T., Chai Q., Yu L., d'Uscio L. V., Katusic Z. S., He T., and Lee H. C. (2012) Reactive oxygen species signaling facilitates FOXO-3a/FBXO-dependent vascular BK channel β1 subunit degradation in diabetic mice. Diabetes 61, 1860–1868 10.2337/db11-1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gomes M. D., Lecker S. H., Jagoe R. T., Navon A., and Goldberg A. L. (2001) Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U.S.A. 98, 14440–14445 10.1073/pnas.251541198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qi X. Y., Diness J. G., Brundel B. J., Zhou X. B., Naud P., Wu C. T., Huang H., Harada M., Aflaki M., Dobrev D., Grunnet M., and Nattel S. (2014) Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 129, 430–440 10.1161/CIRCULATIONAHA.113.003019 [DOI] [PubMed] [Google Scholar]
- 15. Adelman J. P., Maylie J., and Sah P. (2012) Small-conductance Ca2+-activated K+ channels: form and function. Annu. Rev. Physiol. 74, 245–269 10.1146/annurev-physiol-020911-153336 [DOI] [PubMed] [Google Scholar]
- 16. Mahida S. (2014) Expanding role of SK channels in cardiac electrophysiology. Heart Rhythm 11, 1233–1238 10.1016/j.hrthm.2014.03.045 [DOI] [PubMed] [Google Scholar]
- 17. Stocker M. (2004) Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurosci. 5, 758–770 10.1038/nrn1516 [DOI] [PubMed] [Google Scholar]
- 18. Chang P. C., and Chen P. S. (2015) SK channels and ventricular arrhythmias in heart failure. Trends Cardiovasc. Med. 25, 508–514 10.1016/j.tcm.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang X. D., Lieu D. K., and Chiamvimonvat N. (2015) Small-conductance Ca2+-activated K+ channels and cardiac arrhythmias. Heart Rhythm 12, 1845–1851 10.1016/j.hrthm.2015.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li N., Timofeyev V., Tuteja D., Xu D., Lu L., Zhang Q., Zhang Z., Singapuri A., Albert T. R., Rajagopal A. V., Bond C. T., Periasamy M., Adelman J., and Chiamvimonvat N. (2009) Ablation of a Ca2+-activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J. Physiol. 587, 1087–1100 10.1113/jphysiol.2008.167718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haugaard M. M., Hesselkilde E. Z., Pehrson S., Carstensen H., Flethøj M., Præstegaard K. F., Sørensen U. S., Diness J. G., Grunnet M., Buhl R., and Jespersen T. (2015) Pharmacologic inhibition of small-conductance calcium-activated potassium (SK) channels by NS8593 reveals atrial antiarrhythmic potential in horses. Heart Rhythm 12, 825–835 10.1016/j.hrthm.2014.12.028 [DOI] [PubMed] [Google Scholar]
- 22. Hershko A. (2005) The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture). Angew. Chem. Int. Ed. Engl. 44, 5932–5943 10.1002/anie.200501724 [DOI] [PubMed] [Google Scholar]
- 23. Ciechanover A., and Brundin P. (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446 10.1016/S0896-6273(03)00606-8 [DOI] [PubMed] [Google Scholar]
- 24. Powell S. R. (2006) The ubiquitin-proteasome system in cardiac physiology and pathology. Am. J. Physiol. Heart Circ. Physiol. 291, H1–H19 10.1152/ajpheart.00062.2006 [DOI] [PubMed] [Google Scholar]
- 25. Zolk O., Schenke C., and Sarikas A. (2006) The ubiquitin-proteasome system: focus on the heart. Cardiovasc. Res. 70, 410–421 10.1016/j.cardiores.2005.12.021 [DOI] [PubMed] [Google Scholar]
- 26. Liu Y. C. (2004) Ubiquitin ligases and the immune response. Annu. Rev. Immunol. 22, 81–127 10.1146/annurev.immunol.22.012703.104813 [DOI] [PubMed] [Google Scholar]
- 27. Malynn B. A., and Ma A. (2010) Ubiquitin makes its mark on immune regulation. Immunity 33, 843–852 10.1016/j.immuni.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang Y. H., and Beaudet A. L. (2004) Human disorders of ubiquitination and proteasomal degradation. Curr. Opin. Pediatr. 16, 419–426 10.1097/01.mop.0000133634.79661.cd [DOI] [PubMed] [Google Scholar]
- 29. Kipreos E. T., and Pagano M. (2000) The F-box protein family. Genome Biol. 1, REVIEWS3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ling T. Y., Wang X. L., Chai Q., Lau T. W., Koestler C. M., Park S. J., Daly R. C., Greason K. L., Jen J., Wu L. Q., Shen W. F., Shen W. K., Cha Y. M., and Lee H. C. (2013) Regulation of the SK3 channel by microRNA-499: potential role in atrial fibrillation. Heart Rhythm 10, 1001–1009 10.1016/j.hrthm.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shmukler B. E., Bond C. T., Wilhelm S., Bruening-Wright A., Maylie J., Adelman J. P., and Alper S. L. (2001) Structure and complex transcription pattern of the mouse SK1 K(Ca) channel gene, KCNN1. Biochim. Biophys. Acta 1518, 36–46 10.1016/S0167-4781(01)00166-X [DOI] [PubMed] [Google Scholar]
- 32. Claycomb W. C., Lanson N. A. Jr, Stallworth B. S., Egeland D. B., Delcarpio J. B., Bahinski A., and Izzo N. J. Jr. (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A. 95, 2979–2984 10.1073/pnas.95.6.2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X. L., Ye D., Peterson T. E., Cao S., Shah V. H., Katusic Z. S., Sieck G. C., and Lee H. C. (2005) Caveolae targeting and regulation of large conductance Ca2+-activated K+ channels in vascular endothelial cells. J. Biol. Chem. 280, 11656–11664 10.1074/jbc.M410987200 [DOI] [PubMed] [Google Scholar]
- 34. Xu Y., Tuteja D., Zhang Z., Xu D., Zhang Y., Rodriguez J., Nie L., Tuxson H. R., Young J. N., Glatter K. A., Vázquez A. E., Yamoah E. N., and Chiamvimonvat N. (2003) Molecular identification and functional roles of a Ca2+-activated K+ channel in human and mouse hearts. J. Biol. Chem. 278, 49085–49094 10.1074/jbc.M307508200 [DOI] [PubMed] [Google Scholar]
- 35. Yi F., Wang H., Chai Q., Wang X., Shen W. K., Willis M. S., Lee H. C., and Lu T. (2014) Regulation of large conductance Ca2+-activated K+ (BK) channel β1 subunit expression by muscle RING finger protein 1 in diabetic vessels. J. Biol. Chem. 289, 10853–10864 10.1074/jbc.M113.520940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu T., Zhang D. M., Wang X. L., He T., Wang R. X., Chai Q., Katusic Z. S., and Lee H. C. (2010) Regulation of coronary arterial BK channels by caveolae-mediated angiotensin II signaling in diabetes mellitus. Circ. Res. 106, 1164–1173 10.1161/CIRCRESAHA.109.209767 [DOI] [PMC free article] [PubMed] [Google Scholar]