

Abstract
DNA polymerase θ (POLQ) plays an important role in alternative nonhomologous end joining or microhomology-mediated end joining (alt-NHEJ/MMEJ). Here, we show that POLQ is not only required for MMEJ to repair DNA double-strand breaks (DSBs) generated by endonucleases such as I-SceI or Cas9, but is also needed for repair of DSBs derived from DNA nicks generated by Cas9 nickase. Consistently, we found that POLQ deficiency leads to sensitivity to topoisomerase inhibitors that cause DNA single-strand break (SSB) accumulation at replication forks and to ATR inhibitors that induce replication fork collapse. These studies support the function of POLQ in coping with replication stress and repairing DSBs upon fork collapse. POLQ overexpression is present in many cancer types and is associated with poor prognosis, including breast cancer regardless of BRCA1 status. We provide proof-of-concept evidence to support a novel cancer treatment strategy that combines POLQ inhibition with administration of topoisomerase or ATR inhibitors, which induces replication stress and fork collapse. Given the prevalence of POLQ overexpression in tumors, such strategy may have a significant impact on developing targeted cancer treatment.
Keywords: DNA repair, DNA replication stress, genomic instability, breast cancer, inhibitor, ATR kinase, microhomology-mediated end joining, POLQ, DNA double-strand break repair, cancer, genome integrity
Introduction
DNA double-strand breaks (DSBs)3 are a major cause of genome instability. In mammalian cells, nonhomologous end joining (NHEJ) and homologous recombination (HR) are two major pathways for repairing DSBs (1). NHEJ is Ku-dependent, juxtaposing and ligating DSB ends without use of a template (2). HR uses sister chromatids containing identical DNA sequences as repair templates and is considered the most conserved mechanism (3). Other alternative DSB repair pathways also exist. Microhomology-mediated end joining (MMEJ) involves alignment of microhomology sequences at or internal to DSB ends for end joining, which often leads to deletions of DNA sequences flanking the original breaks (4). Studies of the cancer genome have revealed significant microhomology at chromosomal breakpoints, suggesting that MMEJ contributes significantly to chromosomal rearrangements during tumorigenesis (5, 6).
DNA polymerase θ (POLQ) is a unique A-family polymerase, encoding a 290-kDa protein with an N-terminal helicase-like domain and a C-terminal DNA polymerase domain that are separated by a nonstructured central domain (7). POLQ knockout (KO) mice are viable but hypersensitive to DNA-damaging agents that cause DSBs, such as ionizing radiation (IR) and bleomycin (8). POLQ promotes alt-NHEJ, which often uses microhomology at repair junctions (8–10). POLQ overexpression is observed in large numbers of cancers, and up-regulation is associated with poor prognosis (11–13). For example, about 70% of breast cancers show 5-fold or more overexpression of POLQ (12, 13), and such overexpression is observed in both HR-proficient and -deficient breast cancers (12, 14).
Replication stress often induces fork stalling, leading to DSB formation. The ATR-mediated checkpoint is activated to protect replication forks from collapsing and to promote replication restart (15). HR is a primary pathway for replication restart from stalled and collapsed forks and repair of DSBs at replication forks (16). Stalled replication forks undergo remodeling through a process called replication fork reversal, which is achieved through coordinated annealing of two newly synthesized DNA strands to form four-way junction structures resembling Holliday junctions (17). Fork reversal is an important protective mechanism allowing original lesions to be removed before replication restart or bypassed using a template-switching mechanism. The Holliday junction-like structures at regressed forks can also be processed by structure-specific endonucleases to generate one-ended DSBs, and fork restart is achieved by break-induced replication, a specific type of HR (18). Although HR-mediated restart is considered an important mechanism for fork recovery, evidence suggests that prolonged fork stalling in mammalian cells often causes fork collapse, resulting in DSB formation and fork inactivation, which does not allow for replication restart (19). Instead, collapsed replication forks often wait for the arrival of a converging fork so that one-ended DSBs at the collapsed forks can become double-ended DSBs that are then repaired by HR.
In this study, we investigated the role of POLQ in MMEJ. We found that POLQ is not only required for MMEJ in repairing DSBs generated by endonucleases, but is also important for repairing DSBs derived from single-strand DNA nicks using the MMEJ mechanism. This reveals that POLQ plays an important role in repairing DSBs generated upon replication fork collapse and suggests a new function of POLQ in coping with replication stress. Based on observations that inactivation of POLQ results in sensitivity to topoisomerase inhibitors and ATR inhibitors, we have proposed a new strategy to treat POLQ-overexpressing cancers using the combined inhibition of POLQ and fork-damaging agents.
Results
Human POLQ knockout cells are sensitive to topoisomerase inhibitors
POLQ defects in mouse cells lead to IR sensitivity and chromosomal breakage (8, 20–22). To study the role of POLQ in human cells, we used CRISPR/Cas9 to inactivate POLQ in U2OS cells. We first used gRNA1 to target exon 3, which is present at the beginning of the helicase-like domain (Fig. 1A and Fig. S1A). To exclude the possibility of internal translation restart, we targeted POLQ again in the obtained exon 3 frameshift mutants at exon 14 by gRNA2, with the cleavage site situated before multiple putative restart sites. The gRNA2 site is also upstream of the polymerase domain. Because both the helicase-like domain and polymerase domain are required for resistance to IR and for mediating MMEJ (23), a double KO strategy would also ensure inactivation of POLQ function in DSB repair. Indeed, POLQ double KO cells are sensitive to IR (Fig. S2A), confirming that they are truly defective in POLQ activity.
Figure 1.

POLQ KO cells are sensitive to topoisomerase inhibitors. A, schematic representation of CRISPR/Cas9 targeting sites in POLQ gene. Exons of the POLQ helicase domain, the polymerase domain, and the central domain are marked in red, blue, and green, respectively. Two gRNA sequences, gRNA1 and gRNA2, targeting exon 3 and exon 14, respectively, are marked. The protospacer adjacent motif sequences are in the blue squares. The insertions or deletions at the gRNA sites resulting in POLQ KO of U2OS POLQ KO clone 1 (POLQKO-1) are shown. B, U2OS and U2OS-derived POLQKO-1 cells were treated with the indicated concentrations of etoposide (left) and CPT (right; CPT concentration starting at 0.008 μm with 2- or 4-fold increments) for 48 h, and cell viability assays were performed. C, U2OS cells expressing shRNA-1 for POLQ (shPOLQ-1) or the vector were treated with the indicated concentration of etoposide and CPT for 48 h, and cell viability assays were performed (left). Inhibition of POLQ expression after expressing shRNA-1 was shown by qPCR, and the expression without inhibition was set at 1 (right). In all experiments, error bars represent the S.D. of at least three independent experiments.
To test whether POLQ inactivation in human cells would cause sensitivities to other damaging agents, we treated U2OS POLQ KO cells with camptothecin (CPT), a topoisomerase I inhibitor, and etoposide, a topoisomerase II inhibitor. We found that POLQ KO in U2OS cells leads to enhanced sensitivity to CPT and etoposide (Fig. 1B and Figs. S1B and S2 (B and C)). We also expressed FLAG-POLQ in POLQ KO-1 U2OS cells using the tet-on inducible system. Expression of POLQ suppresses sensitivity of POLQ KO-1 cells to CPT and etoposide (Fig. S3). We further showed that knockdown of POLQ expression by shRNAs also causes increased sensitivity to CPT and etoposide (Fig. 1C and Fig. S4). Because both POLQ KO and POLQ depletion result in sensitivity to CPT and etoposide, our observation is probably not due to an off-target effect of gRNA or shRNA. As inhibition of topoisomerases often leads to an accumulation of single-strand DNA nicks, which would result in DSBs after replication, these data suggest that POLQ is probably involved in repair of DSBs that are generated upon fork collapse.
POLQ is needed to repair DSBs generated from DNA nicks through the MMEJ mechanism
Double-ended DSBs can be repaired by MMEJ, which has been shown using the EGFP-based MMEJ reporter (Fig. 2A) after cleavage by I-SceI (24) or by WT Cas9 endonuclease (Cas9WT) with the gRNA site situated in the middle of the I-SceI site on the MMEJ reporter (Fig. 2B, left). Interestingly, when we used Cas9 nickase (Cas9D10A) (25) to generate a nick between the two 9-bp microhomologies, MMEJ could also be induced (Fig. 2B, right). The lower MMEJ efficiency by Cas9D10A compared with Cas9WT is probably due to the efficient repair of DNA nicks by single-strand break (SSB) repair mechanisms before DNA nicks are converted to DSBs during replication.
Figure 2.

POLQ is important for using MMEJ in repairing DSBs generated from DNA nicks. A, schematic drawing of the EGFP-MMEJ reporter. The positions of the I-SceI cleavage site and gRNA site are indicated. 9bp, microhomology sequences flanking the cleavage sites. B, U2OS (EGFP-MMEJ) cells were transfected with or without Cas9WT or Cas9D10A, and MMEJ efficiency was determined 5 days later by FACS analysis. C, U2OS (EGFP-MMEJ) cells were infected with or without lentiviruses encoding POLQ shRNA-1, and MMEJ was assayed 5 days after Cas9WT or Cas9D10A transfection (left). The efficiency of POLQ knockdown was determined by qPCR (right). D, model to illustrate POLQ-mediated MMEJ in repairing single-strand nicks on replication forks (see “POLQ is needed to repair DSBs generated from DNA nicks through the MMEJ mechanism” details). In all experiments, error bars represent the S.D. of at least three independent experiments.
Consistent with the notion that POLQ is required for MMEJ in repairing DSBs, depletion of POLQ by shRNAs impairs MMEJ when DSBs are generated by Cas9WT (Fig. 2C (left) and Fig. S5). When we used Cas9D10A nickase, SSB-induced MMEJ is also reduced when POLQ is silenced (Fig. 2C (right) and Fig. S5). This is consistent with observations that POLQ deficiency leads to sensitivities to CPT and etoposide. We propose that SSBs present at replication forks are converted to single-ended DSBs upon replication, which would result in double-ended DSBs when converging forks arrive from the other side, and POLQ-mediated MMEJ is involved in the repair of such double-ended DSBs generated upon fork collapse (Fig. 2D). These data suggest that POLQ is important for repairing DSBs that result from DNA replication and thus is needed in dealing with replication stress.
POLQ is important for repairing DSBs that have accumulated due to replication stress or ATR inactivation
Because our results suggest that POLQ is important for repairing DSBs that have been generated from DNA nicks on replication forks, we tested whether POLQ is required for repair of DSBs caused by replication stress. We treated cells with hydroxyurea (HU) and monitored γH2AX formation. We observed that POLQ depletion by shRNAs causes increased levels of γH2AX after HU treatment (Fig. 3A), suggesting that POLQ is needed for repairing DSBs when forks have collapsed upon replication stress.
Figure 3.

POLQ suppresses DSB formation upon replication stress. A, U2OS cells with or without POLQ shRNA-1 expression were either treated with HU (2 mm, 24 h) or not and were lysed for Western blot analysis of γH2AX, using Ku70 as the loading control. γH2AX levels were quantified and normalized to the loading control (Ku70), and relative γH2AX levels are shown on the left, with γH2AX levels in the sample post-HU treatment without POLQ shRNA-1 set at 1. The efficiency of POLQ knockdown is shown in Fig. 1C (right). B, POLQWT and POLQKO-1 U2OS cells were infected with lentiviruses encoding ATR shRNA (shATR) or control, and cell lysates were prepared for Western blot analysis of the indicated proteins. The relative levels of γH2AX are shown by quantifying the band density and normalizing to Ku70, with the WT sample expressing ATR shRNA set as 1. C, comet assays were performed with control and POLQKO-1 U2OS cells after infection with lentiviruses encoding ATR-shRNA or vector. Left, images of the comet assays are shown. The length of the tail moment is plotted using dots (right), and tail moments were exported as box-and-whisker plots. The 25th to 75th percentiles and the median are shown in each box. The whisker ends show the minimum and maximum values. Data were analyzed by one-way analysis of variance, and the p value is shown (**, p < 0.01). In all experiments, error bars represent the S.D. of at least three independent experiments.
ATR plays a critical role in protecting replication forks, and in its absence, DSBs are accumulated due to fork collapse (15). We tested whether POLQ is needed to repair DSBs that are generated upon ATR inactivation. We found that ATR depletion by shRNAs leads to more γH2AX accumulation in POLQ KO cells compared with POLQ WT U2OS cells (Fig. 3B). To directly demonstrate DSB formation, we performed a neutral comet assay. The comet tail moment is significantly increased in POLQ KO cells when ATR is inactivated by shRNAs, providing direct evidence for DSB formation (Fig. 3C).
Collectively, these data support the model that POLQ is important for repairing DSBs that are associated with fork collapse due to replication stress or a failure of ATR-mediated fork protection.
Inactivation of POLQ causes increased cell death in ATR-deficient cells
Because POLQ is important for repair of DSBs that have accumulated due to compromised fork protection in ATR-deficient cells, we reasoned that inactivation of POLQ would cause more cell death in ATR-deficient cells. In support of this, depletion of ATR by shRNAs results in greater cell death in POLQ KO U2OS cells compared with that in POLQ WT U2OS cells (Fig. 4A). U2OS POLQ KO cells are also more sensitive to ATR inhibitor VE822 compared with POLQ WT cells (Fig. 4B). Similarly, depletion of POLQ by shRNAs in U2OS results in greater sensitivity to ATR inhibitors (Fig. 4C). These data indicate that inhibition of POLQ activity leads to more cell death when ATR activity is compromised and suggest a synthetic lethality interaction between POLQ and ATR.
Figure 4.
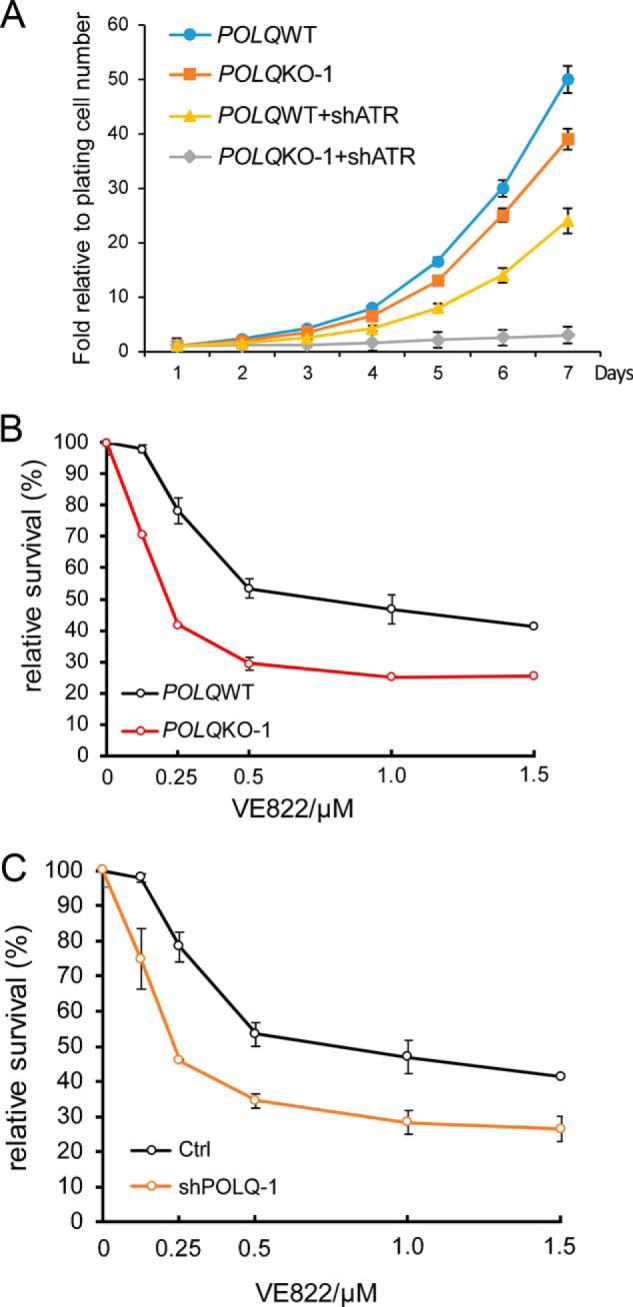
POLQ-deficient cells are compromised in viability when ATR is inhibited. A, POLQWT and POLQKO-1 U2OS cells were infected with lentiviruses encoding ATR shRNA or the vector, and growth curves were plotted. B, POLQWT and POLQKO-1 U2OS cells were treated with ATR inhibitor VE-822 for 48 h at the indicated concentrations, and cell viability assays were performed. C, U2OS cells with or without POLQ shRNA-1 expression were treated with VE-822 for 48 h at the indicated concentrations, and cell viability assays were performed. The efficiency of POLQ knockdown is shown in Fig. 1C (right). In all experiments, error bars represent the S.D. of at least three independent experiments.
Inactivation of POLQ sensitizes POLQ-overexpressing breast cancer cells to topoisomerase and ATR inhibitors
POLQ is overexpressed in a majority of breast cancers (12, 13). We performed quantitative RT-PCR to assess POLQ expression in a panel of breast cancer cell lines compared with the normal breast cancer cell line MCF10A. All tested breast cancer lines expressed higher levels of POLQ than MCF10A, with seven of 11 cell lines expressing POLQ more than 5-fold (Fig. 5A). BT-474 and MDA-MB-436, the two cell lines with the highest POLQ expression, are of luminal and basal origin, respectively (Table 1). BT-474 contains WT alleles for p53 and BRCA1, whereas MDA-MB-436 is triple-negative (negative for estrogen receptors (ER−), progesterone receptors (PR−), and HER2 (HER2−)) and has mutated p53 and BRCA1. Both BT-474 and MDA-MB-436 cell lines are more resistant to CPT and etoposide compared with MCF7 and MDA-MB-231 cell lines, which express relatively low levels of POLQ (Fig. 5B).
Figure 5.

POLQ is frequently overexpressed in breast cancer cell lines. A, POLQ expression levels in a panel of breast cancer cell lines and a normal breast cell line MCF10A were determined by qPCR normalized to HPRT as a reference gene. B, the indicated cancer cell lines were treated with CPT (left) and etoposide (right) at the indicated concentrations, and cell viability assays were performed 48 h after treatment. In all experiments, error bars represent the S.D. of at least three independent experiments.
Table 1.
Breast cancer and normal cell lines
TN, triple negative cell lines; ND, not determined; Mut, mutant.
| Cell lines | POLQ expressiona | Classification | p53 | BRCA1 | TN |
|---|---|---|---|---|---|
| -fold | |||||
| Cancer cell lines | |||||
| BT-474 | 17.2 | Luminal | WT | WT | No |
| MDA-MB-436 | 15.4 | Basal | Mut | Mut | Yes |
| BT-20 | 10.4 | Basal | Mut | WT | Yes |
| MDA-MB-415 | 10.3 | Luminal | WT | WT | No |
| ZR-75-B | 7.4 | Luminal | ND | WT | No |
| MDA-MB-435 | 7.3 | Basal | Mut | WT | Yes |
| BT-549 | 6.1 | Basal | Mut | WT | Yes |
| HBL-100 | 3.5 | Basal | WT | WT | No |
| MCF7 | 3.5 | Luminal | WT | WT | No |
| SUM149PT | 3.0 | Basal | Mut | Mut | Yes |
| MDA-MB-231 | 2.1 | Basal | Mut | WT | Yes |
| Normal cell line | |||||
| MCF10A | 1 | Basal | WT | WT | No |
a -Fold level compared with MCF10A.
We reason that overexpression of POLQ provides an efficient mechanism of repairing DSBs that are generated upon fork collapse. Thus, depletion of POLQ would sensitize breast cancer cells, especially those with POLQ overexpression, to DNA-damaging agents that cause fork collapse. Indeed, depletion of POLQ by shRNAs in BT-474 and MDA-MB-436 breast cancer cell lines, which highly express POLQ, sensitizes these cell lines to CPT and etoposide (Fig. 6A and Fig. S6). We also showed that inactivation of POLQ sensitizes BT-474 and MDA-MB-436 cell lines to ATR inhibitor VE822 (Fig. 6B). Because BT-474 contains the WT BRCA1 allele whereas MDA-MB-436 carries a mutated BRCA1 allele, the combined treatment of POLQ inactivation with inhibitors to topoisomerase or ATR could be used for effective treatment of POLQ-overexpressing breast cancers irrespective of BRCA1 status.
Figure 6.

Silencing POLQ expression sensitizes breast cancer cell lines BT-474 and MDA-MB-436 to topoisomerase and ATR inhibitors. A, POLQ is depleted or not in MDA-MB-436 and BT-474 cells using shRNA-1, and sensitivity to CPT (left) and etoposide (right) was determined through cell viability assays 48 h after treatment. B, BT-474 and MDA-MB-436 cells with or without POLQ shRNA-1 expression were treated with VE-822 at the indicated concentrations, and cell viability assays were performed 48 h after treatment (left). Inhibition of POLQ expression after shRNA expression was shown by qPCR (right). C, BT-474 cells either expressing ATR shRNAs or not were treated with or without VE-822 (2 μm, 48 h), and Western bolt analysis was performed for the indicated proteins. The relative level of γH2AX is shown by quantifying band density and normalizing to Ku70, with the sample treated with VE822 without POLQ shRNA-1 expression set as 1. D, BT-474 cells expressing ATR shRNAs or not were treated with or without VE-822 (2 μm, 48 h), and comet assays were performed. Images of the comet assays are shown (left), and the length of the tail moment is plotted using dots (right). Data were analyzed by one-way analysis of variance, and the p value is shown (*, p < 0.05; **, p < 0.01). For a more detailed description, see Fig. 3C. In all experiments, error bars represent the S.D. of at least three independent experiments.
We further examined γH2AX levels in BT-474 cells after knockdown of POLQ and/or treatment with ATR inhibitor VE822. Whereas inhibiting ATR activity causes an increase in γH2AX levels, inactivation of POLQ and ATR synergistically increases γH2AX accumulation (Fig. 6C). Comet assays confirmed DSB accumulation in BT-474 cells after inactivation of POLQ by shRNA and ATR by VE822 (Fig. 6D). Thus, POLQ inhibition potentiates toxicity of ATR inhibitor by compromising DSB repair in breast cancer cells.
Discussion
POLQ plays an important role in alt-NHEJ, which often uses microhomology sequences at the repair junction (26). POLQ-mediated alt-NHEJ has been observed after endonuclease cleavage, dysfunctional telomere fusion, and chromosomal translocation (8–10). Consistently, using our EGFP-based MMEJ reporter, we showed that MMEJ induced by DSBs that were generated by Cas9WT depends on POLQ. Interestingly, we found that when DNA nicks are generated by Cas9D10A nickase, MMEJ is also induced, and such SSB-induced MMEJ depends on POLQ as well. This observation is consistent with results showing that POLQ deficiency results in an increased sensitivity to topoisomerase inhibitors (8) (this study), which causes SSB formation at replication forks. This is also in line with previous studies showing that in C. elegans, POLQ is involved in repairing replication-associated DNA breaks, and in Drosophila, mus308/POLQ maintains normal fork progression during rereplication in follicle cells (27, 28). We propose that POLQ-mediated MMEJ is involved in repairing DSBs that are converted from SSBs present on replication forks and POLQ plays an important role in dealing with replication stress. In support of this conclusion, we showed that POLQ deficiency leads to increased DSB accumulation upon replication stress caused by HU or loss of ATR activity. Previously, targeting POLQ has only been suggested for sensitizing tumor cells to radiation based on its end-joining activity. Our study reveals new utility of targeting POLQ in cancer treatment in combination with agents that cause DNA replication stress and DNA damage on replication forks.
When replication passes through SSBs or when forks have collapsed due to replication stress, it is expected that one-ended DSBs would be generated, but they are not appropriate DNA lesions for MMEJ to repair. Break-induced replication is used to repair such single-ended DSBs and to promote replication restart (18). However, it has been shown that in mammalian cells, collapsed forks derived from prolonged fork stalling are often inactivated for restart (19), and instead, resultant one-ended DSBs become two-ended upon the arrival of converging forks and are then repaired by HR. We showed that in addition to HR, MMEJ is also involved in repairing such double-ended DSBs (Fig. 2D). Because these double-ended DSBs are generated due to fork collapse, ssDNA is expected to be present at DSB ends, and they are not favorable substrates for Ku-dependent NHEJ but would be appropriate for MMEJ substrates to repair.
POLQ overexpression is observed in many cancers, including breast, lung, gastric, and colorectal cancers, and is associated with adverse clinical outcomes (11–13). This overexpression is independent of HR status in cancer cells (11, 12, 14), and the cause is not well understood. Based on our study, we suggest that the role of POLQ in MMEJ is important for coping with replication stress and repairing DSBs generated from collapsed forks. Oncogene expression in the precancerous stage causes replication stress, and POLQ-dependent MMEJ in addition to HR would be involved in repairing the resulting DSBs. Thus, POLQ overexpression potentially provides a growth advantage, allowing cell survival of oncogenic stress.
POLQ is an attractive drug target, because it is not essential for normal cell growth, and its overexpression is widespread in tumors (29). Synthetic lethality interactions between POLQ with ATM and the players in NHEJ and HR, such as Ku70 and BRCA1/2, have previously been reported (9, 10, 30), suggesting that DSB repair defects in other pathways would cause reliance on MMEJ for survival. Inhibition of POLQ in NHEJ-, HR-, or ATM-defective tumors could thus be the basis of a targeted cancer treatment strategy. Our study reveals a new function of POLQ in dealing with replication stress by repairing DSBs caused by fork collapse. These findings suggest another new treatment strategy by combining POLQ inhibition with replication stress or fork-damaging agents. POLQ overexpression is frequently found in tumors, suggesting that stimulation of MMEJ to repair stress-induced DSBs may be a common mechanism to cope with oncogenic replication stress. On the flip side, in those tumors that do not overexpress POLQ, DSB repair mechanisms other than MMEJ may be boosted to deal with replication stress. Thus, inhibition of POLQ may work more effectively in POLQ-overexpressing cancer cells when combined with replication stress–inducing agents.
As a proof-of-principle study, we showed that inhibition of POLQ in breast cancer cell lines with POLQ overexpression effectively sensitizes tumor cells to DNA-damaging agents such as CPT, etoposide, and ATR inhibitors, which induce replication stress and cause fork collapse. Because replication stress is a hallmark of tumor cells and POLQ overexpression is common in many tumors, our proposed strategy combining POLQ inhibition with topoisomerase inhibitors or ATR inhibitors could have a wide range of applications in cancer treatment. For instance, ATR is critical for the survival of tumor cells under high levels of replication stress induced by oncogene expression (31). Based on this, ATR inhibitors have been developed as a new strategy for cancer treatment and have been used as single agents to treat Ewing sarcoma and MLL-rearranged acute myeloid leukemia (32, 33). However, for other types of cancers, ATR inhibitors are effective only when tumors have defects in another cellular pathway, such as loss of ATM, repair proteins XRCC1 and ERCC1, or chromatin-remodeling protein ARID1A (34–37). Our study puts forth a new therapeutic strategy using ATR inhibitors in combination with POLQ inhibition to treat a wide range of tumors that overexpress POLQ.
Poly(ADP-ribose) polymerase (PARP) inhibitors have been successfully used to treat BRCA-deficient breast and ovarian cancers (38). However, only a fraction of breast cancers are defective in BRCA1, BRCA2, or their functionally related HR pathway (“BRCAness”; up to 25% (39)). Acquired resistance to PARP inhibitors also develops rapidly. It is highly desirable to develop alternative treatment strategies that will be effective for a majority of breast cancers, including those with proficient HR activity. POLQ is overexpressed 5-fold or more in about 70% of breast cancer in both HR-proficient and deficient tumors, and overexpression is associated with poor prognosis (12, 14). Our proposed strategy could therefore be used for treating a wide range of aggressive breast cancers, including HR-proficient breast cancers. This strategy could also complement PARP inhibitor treatment for tumors with innate or acquired resistance to PARP inhibitors.
POLQ has been recognized as an attractive anti-cancer target, and potent POLQ inhibitors are being developed by many laboratories and biotech companies. Our study presents a new potential use of POLQ inhibitors for targeted cancer treatment in combination with DNA-damaging agents or inhibitors.
Experimental procedures
Cell cultures and transfection
U2OS, HCT116, and 293T cells were cultured at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium with 10% fetal bovine serum in the presence of antibiotics. BT-474, MDA-MB-436, and other breast cancer cell lines were obtained from ATCC and cultured according to recommended conditions. Transfection of 293T cells and U2OS cells was done using calcium phosphate or polyethyleneimine methods following standard protocols.
Plasmid construction
The Cas9-WT in px459 vector was purchased from Addgene (catalog no. 62988). Cas9-D10A was generated by site-directed mutagenesis of Cas9-WT plasmid. The gRNA sequence CGCGCCGAGTAGGGATAAC, recognizing a site overlapping with the I-SceI site in the MMEJ reporter (Fig. 2A), was inserted into the BbsI site of Cas9-WT and D10A plasmids to produce Cas9-WT/gRNA or Cas9-D10A/gRNA plasmids.
To generate tet-on FLAG-POLQ plasmid (pTRE3G-POLQ-FH), POLQ cDNA with C-terminal FLAG and HA tags from pCDH-EF1-FHC-POLQ (Addgene, catalog no. 64875) (8) was subcloned into the pTRE3G vector between the SalI and FseI sites by replacing GSX2 in the pTRE3G-GSX2 plasmid (Addgene, catalog no. 96964).
shRNA interference
POLQ and ATR silencing was performed using shRNAs through lentiviral infection with the pLKO vector (Addgene, catalog no. 26655). shRNAs for POLQ and ATR were designed by Dharmacon: shPOLQ-1, ACAACAACCCTTATCGTAAAG; shPOLQ-2, CCTTAAGACTGTAGGTACTAT; shATR, CGAGACTTCTGCGGATTGCAG.
Generation of POLQ KO cell lines by CRISPR and reconstitution of POLQ in KO cells
POLQ gene KO in U2OS cells was generated by transfection of the respective gRNA vectors with Lipofectamine 2000 (Invitrogen, catalog no. 11668027). The POLQ gRNA1 (sequence, GTTTTAGCTCCTACAAGTGC) plasmid was purchased from GeneCopoeia (catalog no. HCP20085-CG01-3-B). The POLQ gRNA2 (sequence, AGAAATATGGATGCAATCGT) plasmid was cloned into the px459 vector (Addgene, catalog no. 62988). For gRNA1, mCherry-positive cells were separated by FACS sorting 48 h post-transfection, followed by single clone isolation. KO clones were confirmed by PCR amplification of genomic DNA and sequencing (primer, ATCGGGATCCATCTACTTCCTCAAAGCAGGCCTT and ATCGGTCGACATGAATGAAACCAAGACTGCCA). For gRNA2, transfected cells were selected for 24 h with puromycin, followed by single clone isolation. KO clones were confirmed by PCR amplification and sequencing (primer, AGCAGCAGTTTTGAATATGGGC and GGAGGATAGTCAGCAGGTTGC).
Tet-on FLAG-POLQ reconstitution was achieved by transfection of the pTRE3G-POLQ-FH plasmid into POLQ KO-1 U2OS cells. After puromycin selection, single clones were picked up and POLQ expression was induced by doxycycline (5 μg/ml, 48 h). Reconstituted clones were confirmed by Western blotting using the anti-FLAG antibody.
Immunoblotting
Cells were lysed by NETN buffer (100 mm NaCl, 20 mm Tris-Cl (pH 8.0), 0.5 mm EDTA, 0.5% (v/v) Nonidet P-40) and radioimmune precipitation buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris-Cl (pH 8.0), 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS). Western blot analysis was performed as described previously (40, 41). The commercial antibodies used were anti-ATR (Bethyl Laboratories A300-138), anti-Ku-70 (Santa Cruz Biotechnology, sc-17789), and anti-H2AX Ser139p (BioLegend, catalog no. 613402).
Real-time quantitative RT-PCR
Total RNA was extracted from cell lines using the RNeasy Mini Kit (Qiagen, catalog no. 74104). cDNA was synthesized through reverse transcription using the iScriptTM cDNA synthesis kit (Bio-Rad, catalog no. 1708890). The SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, catalog no. 1725271) was used for real-time PCR, and primers were as follows: POLQ-F1, 5′-CACACTGCTACAGGACGAATAA; POLQ-R1, 5′-AGGTGGGCTTTCTCCTACTA; POLQ-F2, 5′-GCCAGGGTTCTCTATGCTTC; POLQ-R2, 5′-TCTTCAACTGCTTCCTCTTCC; HPRT-F, 5′-CTGGCGTCGTGATTAGTGAT; HPRT-R, 5′-CTCGAGCAAGACGTTCAGTC; ACTB-F, 5′-CAAGGCCAACCGCGAGAAGATGAC; ACTB-R, 5′-GCCAGAGGCGTACAGGGATAGCACA.
MMEJ assay after Cas9WT and Cas9D10A induction
The EGFP-MMEJ reporter was described previously (24). To induce DSBs or single-strand nicks, MMEJ reporter cells were transfected with Cas9-WT/gRNA or Cas9-D10A/gRNA plasmids, respectively. EGFP-positive events were scored by FACS analysis 5 days later. FACS analysis was performed using a BD Accuri C6 flow cytometer and accompanying data analysis software (CFlow, BD Biosciences).
Growth curve
U2OS and POLQ KO U2OS cell lines were infected with shRNAs and plated. Cells were trypsinized every 24 h and counted by hemocytometer for 6 days. The cell number on day 1 was set to 1.
Drug sensitivity assay
Cells were plated in 96-well plates (5000 cells/well) and treated with etoposide, CPT, and VE822 at various concentrations for 48 h. After treatment, cell survival was measured using the MTS assay (Promega, G3582) following the manufacturer's instructions. Absorbance was read using a microplate spectrophotometer (μQuant, BioTek) at 490 nm.
Neutral comet assay
Cell number was adjusted to 106/ml, and 20 μl of cell suspension was added into 0.8% LMP-agarose in PBS. 60 μl of the cell suspension was plated on a slide and lysed using lysis buffer (2.5 m NaCl, 100 mm EDTA, 10 mm Tris, 10% DMSO, 1% Triton X-100, pH 10) overnight at 4 °C. After incubation in cold electrophoresis buffer (300 mm sodium acetate, 100 mm Tris, pH 8.3), electrophoresis was performed at 25 V and 4 °C for 30 min. Slides were placed in cold 70% ethanol for 30 min, air-dried for 1 h, and stained using SYBR Green (1 μl in 30 ml of TE buffer) for 30 min. Tail moments were quantified using CometScore version 2.0.
Author contributions
Z. W., T. C., and X. W. conceptualization; Z. W. and X. W. resources; Z. W., Y. S., S. L., and S. K. data curation; Z. W. and Y. S. software; Z. W., Y. S., and S. L. formal analysis; Z. W., R. X., T. C., and X. W. supervision; Z. W., R. X., T. C., and X. W. funding acquisition; Z. W., Y. S., S. L., R. X., and X. W. validation; Z. W., Y. S., S. L., R. X., T. C., and X. W. investigation; Z. W., Y. S., and S. L. methodology; Z. W., Y. S., and X. W. writing-original draft; Z. W., Y. S., S. L., and X. W. writing-review and editing; T. C. and X. W. project administration.
Supplementary Material
Acknowledgments
The shRNA vectors pLKO.1-TRC (catalog no. 10878) and pLKO.1-blast (catalog no. 26655) and the Cas9 px459 vector (catalog no. 62988) were obtained from Addgene. pCDH-EF1-FHC-POLQ was a gift from Richard Wood (Addgene catalog no. 64875). We thank the Susan G. Komen Foundation San Diego and their advocates for helpful discussions.
This work is supported by California Breast Cancer Research Program (CBCRP) Grant 23IB and National Institutes of Health Grants CA187052, CA197995, and GM080677 (to X. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6.
- DSB
- double-strand break
- NHEJ
- nonhomologous end joining
- HR
- homologous recombination
- MMEJ
- microhomology-mediated end joining
- KO
- knockout
- IR
- ionizing radiation
- CPT
- camptothecin
- gRNA
- guide RNA
- SSB
- single-strand break
- HU
- hydroxyurea
- PARP
- poly(ADP-ribose) polymerase
- qPCR
- quantitative PCR.
References
- 1. Symington L. S., and Gautier J. (2011) Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 10.1146/annurev-genet-110410-132435 [DOI] [PubMed] [Google Scholar]
- 2. Lieber M. R. (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 10.1146/annurev.biochem.052308.093131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jasin M., and Rothstein R. (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 5, a012740 10.1101/cshperspect.a012740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seol J. H., Shim E. Y., and Lee S. E. (2018) Microhomology-mediated end joining: good, bad and ugly. Mutat. Res. 809, 81–87 10.1016/j.mrfmmm.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiarle R., Zhang Y., Frock R. L., Lewis S. M., Molinie B., Ho Y. J., Myers D. R., Choi V. W., Compagno M., Malkin D. J., Neuberg D., Monti S., Giallourakis C. C., Gostissa M., and Alt F. W. (2011) Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119 10.1016/j.cell.2011.07.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stephens P. J., McBride D. J., Lin M. L., Varela I., Pleasance E. D., Simpson J. T., Stebbings L. A., Leroy C., Edkins S., Mudie L. J., Greenman C. D., Jia M., Latimer C., Teague J. W., Lau K. W., et al. (2009) Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462, 1005–1010 10.1038/nature08645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seki M., Marini F., and Wood R. D. (2003) POLQ (Pol θ), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 31, 6117–6126 10.1093/nar/gkg814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yousefzadeh M. J., Wyatt D. W., Takata K., Mu Y., Hensley S. C., Tomida J., Bylund G. O., Doublié S., Johansson E., Ramsden D. A., McBride K. M., and Wood R. D. (2014) Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 10, e1004654 10.1371/journal.pgen.1004654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mateos-Gomez P. A., Gong F., Nair N., Miller K. M., Lazzerini-Denchi E., and Sfeir A. (2015) Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 10.1038/nature14157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ceccaldi R., Liu J. C., Amunugama R., Hajdu I., Primack B., Petalcorin M. I., O'Connor K. W., Konstantinopoulos P. A., Elledge S. J., Boulton S. J., Yusufzai T., and D'Andrea A. D. (2015) Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 518, 258–262 10.1038/nature14184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawamura K., Bahar R., Seimiya M., Chiyo M., Wada A., Okada S., Hatano M., Tokuhisa T., Kimura H., Watanabe S., Honda I., Sakiyama S., Tagawa M., and O-Wang J. (2004) DNA polymerase theta is preferentially expressed in lymphoid tissues and upregulated in human cancers. Int. J. Cancer 109, 9–16 10.1002/ijc.11666 [DOI] [PubMed] [Google Scholar]
- 12. Lemée F., Bergoglio V., Fernandez-Vidal A., Machado-Silva A., Pillaire M. J., Bieth A., Gentil C., Baker L., Martin A. L., Leduc C., Lam E., Magdeleine E., Filleron T., Oumouhou N., Kaina B., et al. (2010) DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. U.S.A. 107, 13390–13395 10.1073/pnas.0910759107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Higgins G. S., Harris A. L., Prevo R., Helleday T., McKenna W. G., and Buffa F. M. (2010) Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget 1, 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goullet de Rugy T., Bashkurov M., Datti A., Betous R., Guitton-Sert L., Cazaux C., Durocher D., and Hoffmann J. S. (2016) Excess Polθ functions in response to replicative stress in homologous recombination-proficient cancer cells. Biol. Open 5, 1485–1492 10.1242/bio.018028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeman M. K., and Cimprich K. A. (2014) Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petermann E., and Helleday T. (2010) Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 11, 683–687 10.1038/nrm2974 [DOI] [PubMed] [Google Scholar]
- 17. Quinet A., Lemaçon D., and Vindigni A. (2017) Replication fork reversal: players and guardians. Mol. Cell 68, 830–833 10.1016/j.molcel.2017.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anand R. P., Lovett S. T., and Haber J. E. (2013) Break-induced DNA replication. Cold Spring Harb. Perspect. Biol. 5, a010397 10.1101/cshperspect.a010397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petermann E., Orta M. L., Issaeva N., Schultz N., and Helleday T. (2010) Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 37, 492–502 10.1016/j.molcel.2010.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goff J. P., Shields D. S., Seki M., Choi S., Epperly M. W., Dixon T., Wang H., Bakkenist C. J., Dertinger S. D., Torous D. K., Wittschieben J., Wood R. D., and Greenberger J. S. (2009) Lack of DNA polymerase θ (POLQ) radiosensitizes bone marrow stromal cells in vitro and increases reticulocyte micronuclei after total-body irradiation. Radiat. Res. 172, 165–174 10.1667/RR1598.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shima N., Munroe R. J., and Schimenti J. C. (2004) The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol. Cell Biol. 24, 10381–10389 10.1128/MCB.24.23.10381-10389.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shima N., Hartford S. A., Duffy T., Wilson L. A., Schimenti K. J., and Schimenti J. C. (2003) Phenotype-based identification of mouse chromosome instability mutants. Genetics 163, 1031–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mateos-Gomez P. A., Kent T., Deng S. K., McDevitt S., Kashkina E., Hoang T. M., Pomerantz R. T., and Sfeir A. (2017) The helicase domain of Polθ counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol. 24, 1116–1123 10.1038/nsmb.3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Truong L. N., Li Y., Shi L. Z., Hwang P. Y., He J., Wang H., Razavian N., Berns M. W., and Wu X. (2013) Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 110, 7720–7725 10.1073/pnas.1213431110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., and Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Black S. J., Kashkina E., Kent T., and Pomerantz R. T. (2016) DNA polymerase θ: a unique multifunctional end-joining machine. Genes (Basel) 7, E67 10.3390/genes7090067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roerink S. F., van Schendel R., and Tijsterman M. (2014) Polymerase θ-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 24, 954–962 10.1101/gr.170431.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexander J. L., Beagan K., Orr-Weaver T. L., and McVey M. (2016) Multiple mechanisms contribute to double-strand break repair at rereplication forks in Drosophila follicle cells. Proc. Natl. Acad. Sci. U.S.A. 113, 13809–13814 10.1073/pnas.1617110113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Higgins G. S., and Boulton S. J. (2018) Beyond PARP-POLθ as an anticancer target. Science 359, 1217–1218 10.1126/science.aar5149 [DOI] [PubMed] [Google Scholar]
- 30. Wyatt D. W., Feng W., Conlin M. P., Yousefzadeh M. J., Roberts S. A., Mieczkowski P., Wood R. D., Gupta G. P., and Ramsden D. A. (2016) Essential roles for polymerase θ-mediated end joining in the repair of chromosome breaks. Mol. Cell 63, 662–673 10.1016/j.molcel.2016.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karnitz L. M., and Zou L. (2015) Molecular pathways: targeting ATR in cancer therapy. Clin. Cancer Res. 21, 4780–4785 10.1158/1078-0432.CCR-15-0479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morgado-Palacin I., Day A., Murga M., Lafarga V., Anton M. E., Tubbs A., Chen H. T., Ergan A., Anderson R., Bhandoola A., Pike K. G., Barlaam B., Cadogan E., Wang X., Pierce A. J., et al. (2016) Targeting the kinase activities of ATR and ATM exhibits antitumoral activity in mouse models of MLL-rearranged AML. Sci. Signal. 9, ra91 10.1126/scisignal.aad8243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nieto-Soler M., Morgado-Palacin I., Lafarga V., Lecona E., Murga M., Callen E., Azorin D., Alonso J., Lopez-Contreras A. J., Nussenzweig A., and Fernandez-Capetillo O. (2016) Efficacy of ATR inhibitors as single agents in Ewing sarcoma. Oncotarget 7, 58759–58767 10.18632/oncotarget.11643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reaper P. M., Griffiths M. R., Long J. M., Charrier J. D., Maccormick S., Charlton P. A., Golec J. M., and Pollard J. R. (2011) Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 7, 428–430 10.1038/nchembio.573 [DOI] [PubMed] [Google Scholar]
- 35. Mohni K. N., Kavanaugh G. M., and Cortez D. (2014) ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 74, 2835–2845 10.1158/0008-5472.CAN-13-3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sultana R., Abdel-Fatah T., Perry C., Moseley P., Albarakti N., Mohan V., Seedhouse C., Chan S., and Madhusudan S. (2013) Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS One 8, e57098 10.1371/journal.pone.0057098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Williamson C. T., Miller R., Pemberton H. N., Jones S. E., Campbell J., Konde A., Badham N., Rafiq R., Brough R., Gulati A., Ryan C. J., Francis J., Vermulen P. B., Reynolds A. R., Reaper P. M., et al. (2016) ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 7, 13837 10.1038/ncomms13837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Livraghi L., and Garber J. E. (2015) PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med. 13, 188 10.1186/s12916-015-0425-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Turner N., Tutt A., and Ashworth A. (2004) Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 4, 814–819 10.1038/nrc1457 [DOI] [PubMed] [Google Scholar]
- 40. Wang H., Shao Z., Shi L. Z., Hwang P. Y., Truong L. N., Berns M. W., Chen D. J., and Wu X. (2012) CtIP dimerization is critical for its recruitment to chromosomal DNA double-strand breaks. J. Biol. Chem. 287, 21471–21480 10.1074/jbc.M112.355354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen L., Nievera C. J., Lee A. Y., and Wu X. (2008) Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 283, 7713–7720 10.1074/jbc.M710245200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.