

Abstract
G protein–coupled receptors (GPCRs) are currently the target of more than 30% of the marketed medicines. However, there is an important medical need for ligands with improved pharmacological activities on validated drug targets. Moreover, most of these ligands remain poorly characterized, notably because of a lack of pharmacological tools. Thus, there is an important demand for innovative assays that can detect and drive the design of compounds with novel or improved pharmacological properties. In particular, a functional and screening-compatible GPCR–G protein interaction assay is still unavailable. Here, we report on a nanoluciferase-based complementation technique to detect ligands that promote a GPCR–G protein interaction. We demonstrate that our system can be used to profile compounds with regard to the G proteins they activate through a given GPCR. Furthermore, we established a proof of applicability of screening for distinct G proteins on dopamine receptor D2 whose differential coupling to Gαi/o family members has been extensively studied. In a D2–Gαi1 versus D2–Gαo screening, we retrieved five agonists that are currently being used in antiparkinsonian medications. We determined that in this assay, piribedil and pergolide are full agonists for the recruitment of Gαi1 but are partial agonists for Gαo, that the agonist activity of ropinirole is biased in favor of Gαi1 recruitment, and that the agonist activity of apomorphine is biased for Gαo. We propose that this newly developed assay could be used to develop molecules that selectively modulate a particular G protein pathway.
Keywords: G protein–coupled receptor (GPCR), G protein, high-throughput screening, drug screening, pharmacology, biased signaling, complementation assay, dopamine, functional selectivity, Nanoluciferase
Introduction
G protein–coupled receptors (GPCRs)4 are a large family of membrane proteins that have pivotal functions in physiology and are directly targeted by more than 30% of our therapeutic arsenal (1). Given their successes in drug discovery, it is common sense to postulate that uncharacterized members hold great potential in terms of innovative therapeutic strategies (2–5). Several authors recently proposed that the paucity of adequate pharmacological tools was precluding research on elusive receptors (5, 6). In addition, there is still an unmet medical need in various diseases for drugs with improved properties such as increased potency, higher selectivity, or refined efficacy for existing validated drug targets. Thus, the drug discovery process would benefit from more sophisticated assays able to detect with maximal accuracy the ligands with a desired pharmacological profile.
GPCR signaling has been extensively studied and is notoriously complex. The current paradigm states that the binding of a ligand to its receptor stabilizes active conformations that in turn triggers through allosteric effects the formation of an active complex bound with intracellular partners (7). It is generally accepted that the prime event following ligand binding is the interaction of the active receptor with heterotrimeric G proteins composed of α and βγ subunits (8). These elements dissociate upon activation, and each of them has the capacity to promote distinct signaling pathways (9). Following G protein activation, the receptor undergoes desensitization and internalization through diverse processes involving phosphorylation by GPCR-specific kinases (GRK) and scaffolding by proteins such as arrestins (10).
In humans, the G protein family comprises 16 members that are classified according to the identity of their α subunits into four families (11). The Gαs family (Gαslong, Gαsshort, and Gαolf) increases and the Gαi/o family (Gαi1, Gαi2, Gαi3, Gαo, and Gαz) decreases adenylate cyclase activity. Hence, Gαs/olf and Gαi/o oppositely regulate levels of cAMP in the cell (9). The Gαq/11 family (Gαq, Gα11, Gα15, and Gα16) notably triggers the activation of the phospholipase C-β and calcium mobilization through the release of diacylglycerol and inositol triphosphate (9). Gα12 and Gα13 (Gα12/13) activate the small GTPase regulators Rho-GEF (9). Although individual receptors were initially seen as being selective for a given pathway and thus coupled to a single G protein, it has been observed that most receptors display at least some level of promiscuousness toward different G proteins or G protein families (9). Another layer of complexity exists in GPCR signaling with the observation of functional selectivity, which can be defined as the ability of a ligand to stabilize a specific receptor conformation leading to a unique and ligand-determined profile of signaling pathway activations (7).
A large array of pharmacological assays has been developed for the identification of substances able to modulate G proteins through GPCRs. Most of them focus on the downstream events triggered by G protein activation such as the generation of cAMP (Gαs/olf and Gαi/o) (12, 13), Ca2+ (14) and inositol monophosphate (Gαq/11), the downstream activation of gene promoters, or more recently, the shedding of transforming growth factor α (Gαq/11 and Gα12/13) (15). More generic and holistic assays measuring the accumulation of the nonhydrolysable GTPγS (reflecting G protein activation) (16) or cell morphology (17) have also been extensively used. To extend the scope of an assay to more than one or two pathways, promiscuous G proteins linking most receptors to common second messengers are generally added to the system (18). Collectively, these assays are limited in the way that they cannot give direct information on the kind of G protein, or even on the identity of the GPCR, that has been activated by a given ligand. Thus, during screening campaigns, they are prone to deliver a high rate of false positives (13).
More recent techniques based on resonance energy transfer (BRET and FRET) have given unprecedented access to the study of individual interaction between G protein and their receptors in living cells (19). However, these approaches suffer from important drawbacks such as limited sensitivity (BRET) or high background noise (FRET) that limit their use, for instance in high-throughput screenings (20). In addition, they require the presence of bulky donor and/or acceptors, although reduced-size donors such as NanoLuc (BRET (21)) or FlasH (FRET (22)) are now routinely used.
Here, we describe a simple and flexible NanoLuc-based complementation assay that overcomes these issues and gives access to the profiling of ligands for their ability to induce GPCR interaction with individual G protein. In addition, the procedure is compatible with the settings of high-throughput screening. We applied this methodology to several prototypical class A receptors and their cognate G proteins. The D2 receptor is a well-validated drug target in psychosis and Parkinson's disease and is coupled to the Gαi/o family. We used the D2 receptor as a proof-of-concept to perform a screening of a library containing known drugs and active compounds against the D2 receptor.
Results
NanoLuc complementation can monitor receptor–G protein interaction in living cells
To detect the real-time interaction between receptor and G proteins, we selected the NanoLuc Binary Technology (NanoBiT) that is based on NanoLuc, an engineered luciferase from the deep sea shrimp Oplophorus gracilirostris (23). We reasoned that the reported increased brightness of the enzyme would overcome sensitivity issues of other systems, such as firefly luciferase complementation, BRET or FRET. In addition, we expected the small size of the NanoBiT partners (a small subunit (SmBiT, 11 AA)) and a larger subunit ((LgBiT, 158 AA), Fig. 1A (24)) could minimize perturbations of GPCR pharmacology that have been outlined with large fluorescent proteins (19).
Figure 1.

NanoLuc complementation system. A, constructs for the NanoLuc complementation assay between a GPCR linked to SmBiT, NP, or HiBiT peptides and a Gα subunit linked to LgBiT. Upon stimulation (yellow circle), the coupling of heterotrimeric G protein to the receptor triggers the exchange of a GDP (purple star) by a GTP (green star) followed by dissociation of Gα from Gβγ subunits. The interaction between GPCR and Gα subunit induces the formation of a complete NanoLuciferase and light emission in the presence of its substrate. B, amino acid sequence and KD value of the different small peptides (SmBiT, NP, and HiBiT). Red amino acids represent mutated amino acids compared to NP.
As proof of concept for such an approach, we first selected three receptors coupled to the Gαi/o family: the long isoform of the dopamine receptor subtype 2 (D2) (25); the histamine receptor H3 (26); and the succinate receptor (SUCNR1) (27). The SmBiT was attached to the C terminus of the receptors, and the LgBiT was introduced in the loop connecting helices A and B of the Gαi1 protein (Gαi1—LgB91) (Fig. 1A). The interaction between labeled receptor and Gαi1–LgB91 was estimated by measuring the emitted light upon agonist stimulation. All the tested receptors induced a rapid increase of luminescent signal upon stimulation (Fig. 2, A–C). However, the amplitude and stability of the obtained signal was relatively weak. We reasoned that the transient nature of the GPCR–G protein interaction could, to some extent, explain the low level of the observed signal. SmBiT has been optimized to have a low affinity for LgBiT (KD = 190 μm (24)) compared with the native sequence (natural peptide or NP, KD = 0.9 μm (24)) to minimize the perturbation of the physiological interaction induced by the presence of complementing partners (24). A third peptide with high affinity (HiBiT, KD = 0.7 nm (24)) has also been described (Fig. 1B). To improve the signal-to-noise ratio of our assay, we hypothesized that the affinity of the small complementing peptide could be a critical parameter for the sensitivity of the detection system. Therefore, we tested the other two peptides, NP and HiBiT, fused at the same location as SmBiT on the tested receptors. For the three receptors, the amplitude and the stability of the signal were markedly increased when NP was used as the complementation peptide (Fig. 2, D–F). For both these peptides, the signal remained stable for at least 10 min (Fig. 2, D–I), which is consistent with the literature describing real-time GPCR–G protein interaction (28, 29).
Figure 2.

NanoLuc complementation measurement of interaction between Gαi1 and GPCRs linked to SmBiT, NP, or HiBiT peptides in living cells. NanoLuc activity kinetics measured in HEK293 cells co-expressing Gαi1–LgB91 and GPCRs linked to three different small peptides are indicated in each panel. A–C, D2, H3, and SUCNR1 linked to SmBiT; D–F, D2, H3, and SUCNR1 linked to NP; G–I, D2, H3, and SUCNR1 linked to HiBiT, before and after injection of their respective ligand (dopamine 1 μm, imetit 100 nm, and succinate 1 mm). Results are expressed as the normalization of the NanoLuc signal in the presence of agonist to the signal in the absence of agonist. Data are representative of the mean ± S.E. of at least three independent experiments.
NanoLuc complementation is completely reversible when natural peptide or SmBiT are fused to the receptor
Next, we envisaged the possibility that the use of complementing partners with different affinities for each other could completely distort the GPCR–G protein interaction and increase the risk of detecting nonpharmacological interactions and the accumulation of irreversible receptor–G protein complexes. Thus, we tested the reversibility of the complementation between the three small peptides (SmBiT, NP, and HiBiT) and LgBiT in our system. Following stimulation with dopamine, a competitive D2 receptor antagonist (sulpiride) was injected. For the D2 receptor fused with the SmBiT, a rapid decrease of the NanoLuc signal that reached basal level was observed upon antagonist addition (Fig. 3A). When the experiment was repeated with the constructs containing NP, a similar pattern was observed, although the signal-to-noise ratio was increased (Fig. 3B). When the HiBiT was used as a partner, the rate of dissociation was markedly decreased, and the signal did not reach the basal level, suggesting an incomplete dissociation of the receptor–G protein complex (Fig. 3C). In light of such results, we decided to select the system using the NP for further investigations.
Figure 3.
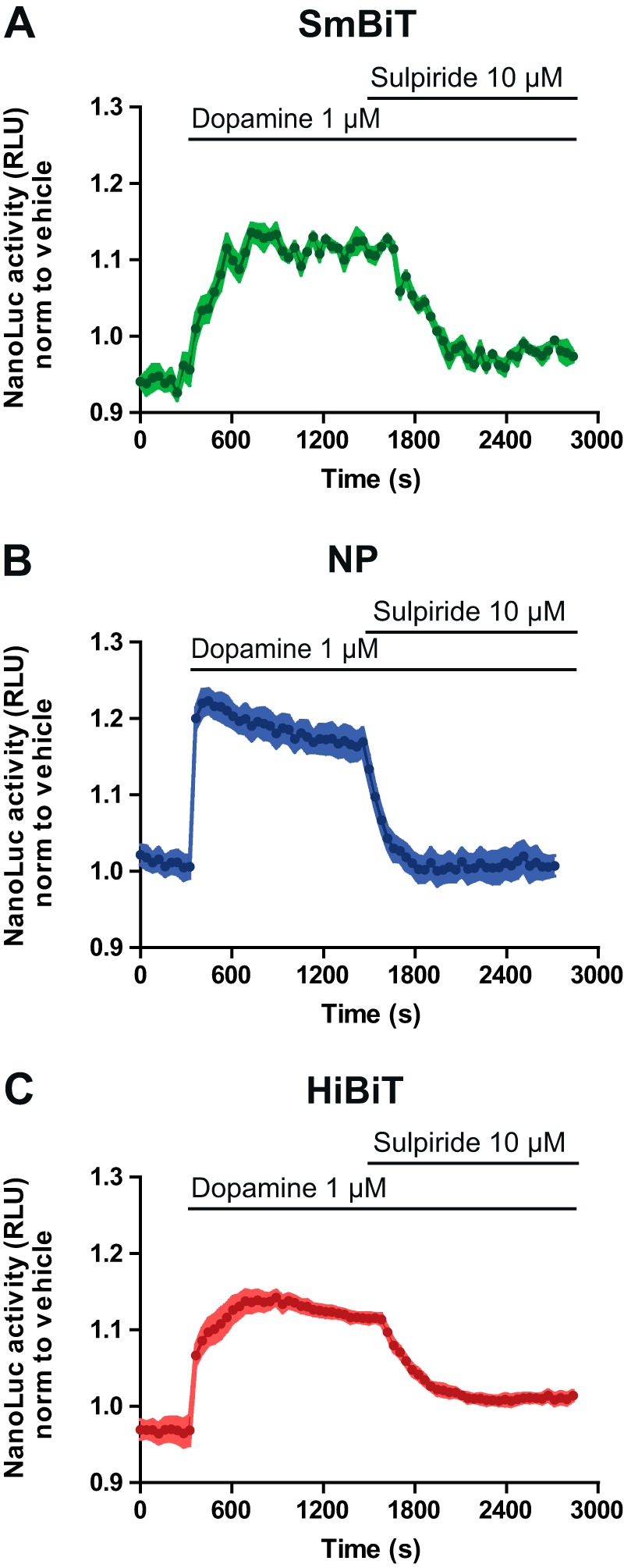
Reversibility of the SmBiT, NP, and HiBiT peptides. NanoLuc activity was measured in HEK293 cells co-expressing Gαi1–LgB91 and D2 linked to SmBiT (A), NP (B), or HiBiT (C), before and after stimulation with the receptor ligand dopamine (1 μm). Twenty minutes after addition of agonist, a selective-receptor antagonist sulpiride (10 μm) was injected. Data represent stimulated cells normalized to nonstimulated cells. Data are representative of the mean ± S.E. of at least three independent experiments.
GPCR–G protein NanoLuc complementation can be applied to all G protein subtypes
We questioned the possibility of implementing the detection system to other G protein subtypes. Thus, we also expanded our detection system to other families of Gα proteins. We introduced the LgBiT in the loop connecting helices A and B of the other Gα proteins and tested the long isoform of Gαs with the β2-adrenergic receptor (β2AR) (Fig. 4A), Gαq and Gα11 with the histamine receptor H1 (H1) (Fig. 4B), and Gα12 and Gα13 with the thromboxane A2 receptor (TPα) (Fig. 4C). Upon stimulation with their respective ligands, a significant interaction between the receptor and the Gα subunit was recorded in cells co-transfected with β2AR-NP/Gαs–LgB113, H1-NP/Gαq/11–LgB97, and TPα-NP/Gα12/13–LgB115/106 (Fig. 4, A–C). We then aimed at performing a complete profiling of G protein recruitment on a test receptor. We chose the well-characterized D2 dopaminergic receptor as a model because it has been previously reported to couple differently to a diversity of G proteins from the Gαi/o family (30). We designed additional sensors for the other α subunits of the Gi/o family. We inserted the LgBiT at the same topological location in Gαi2 and Gαi3. For Gαo (the “a” isoform), the initial constructs gave a poor signal, and after different rounds of optimization, the LgBiT was placed just after helix C, and a deletion of the first 52 amino acids at the N-terminal side (Gαo–LgB143d) was performed. We stimulated with dopamine the HEK293 cells transiently transfected with D2–NP and each of these Gα subunits fused with LgBit. Exposure of D2–NP to its endogenous agonist, dopamine, induced a significant increase of luminescence signal when the receptor was co-expressed in HEK293 cells with either Gαi1–LgB91, Gαi2–LgB91, Gαi3–LgB91, or Gαo–LgB143d but no activity when the D2–NP was co-transfected with plasmids containing the validated constructs Gαs–LgB113, Gαq–LgB97, Gα11–LgB97, Gα12–LgB115, or Gα13–LgB106 (Fig. 4D). The interaction profile of the D2 with Gα subunits that was obtained with the present system was consistent with the ones already described in the literature (31–33).
Figure 4.

NanoLuc complementation measurement of GPCR and G protein interactions in living cells. A–D, NanoLuc activity was measured in HEK293 cells co-expressing Gα subunit linked to LgBiT and GPCR linked to NP as indicated in each panel. Cells were stimulated with vehicle or with their respective agonist (isoproterenol, histamine, and dopamine 10 μm; U46619 10 nm), and the results are expressed as the normalization of agonist-treated cells to untreated cells. Data represent the mean ± S.D. of three independent experiments performed in triplicate. Statistical significance between stimulated and unstimulated cells was assessed using a nonparametric Mann-Whitney test (***, p < 0.001).
GPCR–G protein NanoLuc complementation system is amenable to high-throughput screening
We further examined whether the GPCR–G protein NanoLuc complementation assay could be used for a high-throughput screening campaign. To transpose this assay to a screening protocol, we first optimized the Gαi1 construct. The LgBiT was inserted at different locations of the Gα protein and tested in the presence of D2 receptor tagged with the NP (Fig. S1A). The LgBiT placed at the N terminus of Gαi1 (Gαi1–LgBN-term) showed the strongest signal among all the constructs tested. Similar investigations on Gαo confirmed that Gαo–LgB143d was the construct that gave the best signal-to-noise ratio (Fig. S1B). Next, we performed a flow cytometry experiment to determine the relative expression of the D2–NP receptor at the membrane compared with the intracellular expression of Gαi1–LgBN-term and Gαo–LgB143d in transiently transfected HEK293 cells. We observed a similar expression of receptor and Gα protein constructs for the two conditions of transfection (Fig. S2). To obtain the higher signal-to-noise ratio, we tested a different stoichiometry of D2–NP and Gαi1–LgBN-term/Gαo–LgB143d for the transfection. A ratio 1:1 of receptor and Gα subunit revealed a higher signal-to-noise ratio and was used for the subsequent experiments (Fig. S3A). Next, we reasoned that the Gβγ dimer could affect the amplitude of the signal obtained, as it has been shown for the RET-based systems (28). However, the co-expression of Gαo–LgB143d with its complementary Gβ1γ2 dimer did not modify the recruitment of Gαo–LgB143d to D2–NP. Interestingly, the co-expression of D2–NP and Gαi1–LgBN-term with Gβ1γ2 subunits decreased the signal of NanoLuc complementation upon dopamine stimulation, although it did not affect the pEC50 (Fig. S3B). Thus, the detection of the interaction between NP-tagged GPCR and the LgBiT-tagged Gα subunit does not require the co-expression with the Gβγ dimer.
Concentration-response curves that were determined on the system revealed specific interaction between D2–NP/Gαi1–LgBN-term (EC50 = 2 nm) and D2–NP/Gαo–LgB143d (EC50 = 116 nm) in the presence of dopamine (Fig. 5, A and B). During the kinetic measurement of the D2–NP/Gαi1–LgBN-term or the D2–NP/Gαo–LgB143d interaction induced by dopamine stimulation, we observed an increase of signal that started to decrease immediately upon competitive antagonist (sulpiride and spiperone) addition (Fig. 5, C and D). The affinity of sulpiride for the D2–NP construct that was estimated with a Schild plot (Fig. S4) was not significantly different between the two assays and was consistent with the literature (34). We applied statistical methods to determine whether this assay would be suitable for high-throughput screening. We calculated the Z′ factor, which includes the mean and the standard deviation of the positive and negative control (35). This factor reflects the dynamic range of the assay and its ability to detect active ligands. A Z′ factor that is between 0.5 and 1 is characterized by a large band separation between positive and negative signals and can be considered as an excellent assay for hit identification (35). When cells co-expressing D2–NP with Gαi1–LgBN-term or Gαo–LgB143d were stimulated with either dopamine or the vehicle in a 96-well-plate, the Z′ factors were of 0.61 and 0.69, respectively (Fig. 5, E and F).
Figure 5.

Screening compatible NanoLuc complementation assay for GPCR and G protein interactions. HEK293 cells co-expressing D2–NP/Gαi1–LgBN-term or Gαo–LgB143d are shown. A and B, concentration-response curves of dopamine. C and D, kinetic measurements of NanoLuc activity during stimulation with dopamine and after addition of two different antagonists (sulpiride and spiperone). E and F, assay performance (Z′ factor determination) for D2 untreated and treated with dopamine (10 μm) on transiently co-transfected cells (n = 48). Data are representative of the mean ± S.E. of at least three independent experiments.
Gαi1- and Gαo-based screening of a SOSA library identifies D2 agonists with distinct pharmacological profiles
To further validate our approach, we screened a SOSA library composed of 1200 known active compounds and drugs (Prestwick Chemical Library®) on D2–NP-expressing cells together with the Gαi1- or Gαo-based complementation constructs (Gαi1–LgBN-term or Gαo–LgB143d) in parallel. Several agonists of the D2 receptor were present in this library, and we reasoned that they would serve as internal positive controls. We fixed the threshold for hit identification as the mean of the negative control plus three times the standard deviation. With this criterion, we detected the six agonists (the D2 agonists were listed according to the BJP/IUPHAR database http://www.guidetopharmacology.org)5 (53) of the D2 receptor present in the library (Fig. 6, A and B).
Figure 6.

Screening of Prestwick Chemical Library® on D2–Gαi1 and D2–Gαo. A and B, 1200 compounds were tested at the same concentration (5 μm) on cells co-expressing D2–NP/Gαi1–LgBN-term or Gαo–LgB143d. Results are expressed as the ratio over vehicle (DMSO)-treated (compounds) cells.
Next, we tested a wide range of agonist concentrations and compared the profile of each ligand with regard to dopamine that was defined as the reference ligand. Surprisingly, the pEC50 of dopamine was very different between the Gαi1- and Gαo-based interaction assay (Table 1; pEC50: 8.64 ± 0.10 and 6.60 ± 0.05, respectively). We postulated that the difference between pEC50 could be the consequence of using different constructs. Actually, when we tested several constructs of Gαi1–LgB, we also obtained a difference of pEC50 upon stimulation with dopamine (Fig. S1).
Table 1.
Agonist activity on Gαi1 and Gαo interaction of D2 ligands
Agonist-induced Gαi1 or Gαo interaction with D2 is shown. Efficacy (Emax) is relative to the maximum effect of dopamine (=100%). Bias factor was calculated as described under “Experimental procedures.” Values in parentheses indicate S.E.
| Ligand | Gi1 |
Go |
Bias factor, ΔΔlog (Emax/EC50) | ||
|---|---|---|---|---|---|
| pEC50 | Emax | pEC50 | Emax | ||
| % | % | ||||
| Dopamine | 8.64 (0.10) | 100 | 6.60 (0.05) | 100 | 0 (0.12) |
| Apomorphine | 8.25 (0.04) | 103 | 6.62 (0.04) | 63 | −0.27 (0.03) |
| Piribedil | 8.28 (0.04) | 112 | 6.76 (0.14) | 42 | −0.12 (0.18) |
| Pramipexole | 8.59 (0.07) | 119 | 6.80 (0.09) | 77 | 0.04 (0.17) |
| Ropinirole | 8.23 (0.06) | 106 | 5.89 (0.08) | 73 | 0.41 (0.12) |
| Pergolide | 10.13 (0.07) | 100 | 8.26 (0.07) | 54 | 0.09 (0.18) |
We compared all data obtained with both Gαi1 and Gαo assays relative to dopamine. We observed that the Emax of the D2 agonists were similar when we measured the interaction between D2–NP and Gαi1–LgBN-term (Table 1). However, when cells expressing D2–NP and Gαo–LgB143d were stimulated with apomorphine, piribedil, and pramipexole, we detected lower efficacies compared with the one of dopamine as these compounds behaved as a partial agonist for the initiation of interaction between D2 and Gαo. For example, piribedil exhibited a decrease of efficacy up to 3-fold compared with the one measured for dopamine (Fig. 7, A and B). Furthermore, although pergolide was more potent than dopamine as an inducer of both the D2–NP/Gαi1–LgBN-term and D2–NP/Gαo–LgB143d interactions (Table 1; pEC50 of 10.13 ± 0.07 and 8.26 ± 0.07 for Gαi1–LgBN-term and Gαo–LgB143d, respectively), we observed a lower efficacy for pergolide when measuring the interaction between D2–NP and Gαo–LgB143d. Ropinirole-promoted D2–NP/Gαi1–LgBN-term and D2–NP/Gαo–LgB143d interactions were both less potent compared with dopamine (Fig. 7, A and B). In addition, relative to dopamine, ropinirole showed a higher potency for inducing the D2–NP/Gαi1–LgBN-term compared with the D2–NP/Gαo–LgB143d interaction (Table 1; pEC50 of 8.23 ± 0.06 and 5.89 ± 0.08 for Gαi1 and Gαo, respectively).
Figure 7.

Pharmacological characterization of D2 agonists on Gαi1 and Gαo coupling. A and B, concentration-response curves of five D2 agonists on Gαi1 and Gαo interaction. Emax is expressed as the percentage of dopamine maximal activity. Estimation of pEC50 values (±S.D.) for each ligand and agonist efficacy measurements relative to dopamine are provided in Table 1. Correlation between the pEC50 (±S.D.) (C) and Emax (±S.D.) (D) values determined in Gαi1 and Gαo assays. E, bias factors (ΔΔlog(Emax/EC50)) (±S.D.) calculated for tested D2 agonists; positive values indicate bias for Gαi1- over Gαo-dependent pathway, whereas negative values indicate bias for Gαo-dependent pathway. Unpaired Student's t tests were performed on the bias factors to determine the significance of ligand biases between Gαi1 and Gαo pathways (*, p < 0.05). Data points are representative of at least three independent experiments performed in triplicate.
For the different tested ligands, there were no marked differences (when compared with dopamine) between the pEC50 for D2–NP and Gαi1–LgBN-term or D2–NP and Gαo–LgB143d interactions (Fig. 7C). However, when we considered the Emax, partial agonism was recorded when measuring the interaction between D2–NP and Gαo–LgB143d (Fig. 7D). This difference in measured Emax was more notable for piribedil and pergolide (Fig. 7D). For each tested ligand, Δlog(Emax/EC50) was determined for both Gα constructs using dopamine as the reference ligand, followed by the calculation of the bias factor as ΔΔlog(Emax/EC50) = Δlog(Emax/EC50)Gαi1 pathway − Δlog(Emax/EC50)Gαo pathway according to a previously described method (36). A bias factor of 0 corresponds to absence of bias (relative to the reference ligand, dopamine), whereas a bias factor of 1 would mean a 10-fold preference for inducing the Gαi1 interaction. On the contrary, a negative value would mean a preference for the Gαo-dependent pathway. A bias factor of −0.27 (±0.03) (apomorphine), −0.12 (±0.18) (piribedil), 0.04 (±0.17) (pramipexole), 0.41 (±0.12) (ropinirole), and 0.09 (±0.18) (pergolide) was computed for D2 agonists, demonstrating a significant bias for ropinirole and apomorphine through Gαi1–LgBN-term and Gαo–LgB143d, respectively (Fig. 7E). Thus, the GPCR–G protein NanoLuc complementation assay is able to identify biased agonists for certain G proteins.
Furthermore, we tested the possibility of an extension of this assay to detect β-arrestin recruitment. Thus, we also evaluated these five D2 agonists using dopamine as a reference ligand in a β-arrestin 2–based NanoLuc complementation assay. We observed a partial activity for apomorphine, piribedil, ropinirole, and pergolide, whereas pramipexole was a full agonist for β-arrestin 2 recruitment (Fig. S5). This assay was based on the same D2–NP construct and a β-arrestin 2 tagged with the LgBiT at the N terminus. Thereby, we were able to monitor G protein or β-arrestin interaction using the same readout.
Discussion
In this study, our first goal was to develop a simple, robust, and sensitive GPCR assay for the real-time detection of receptor–G protein interaction that would be amenable to high-throughput screening.
The protein-fragment complementation assays have been devised on the principle that two complementary parts of a reporter protein fused to two putative partners would be detectable upon interaction (37). These strategies have greatly advanced our understanding of cellular biology, molecular biology, or pharmacology. However, although they have a relatively high signal-to-noise ratio, they have been so far less popular compared with RET to monitor GPCR–G protein interactions. The main reason for this is that the complementation may perturb the natural interaction between the partners under scrutiny because of a possible intrinsic affinity between the two split parts of the reporter proteins or the irreversibility of the complementation once formed. For example, systems based on the fluorescent protein of the GFP family or some β-galactosidases are unable to dissociate once re-formed and to accumulate in the system (38). The firefly and Gaussia luciferases seem to have a reduced propensity to form stable complexes, but the split parts have affinity for each other, and once reformed they are not very bright (39, 40).
Recently, a novel protein fragment complementation based on a brighter and smaller luciferase called NanoLuc has been described (24) and was already applied in the GPCR field to detect receptor–arrestin association (24, 39, 41). NanoLuc-based complementation presents several advantages compared with other RET- or complementation-based techniques. First of all, the system does not suffer from sensitivity issues because the protein that detects the interaction is a brighter luciferase (42). A second improvement is the small size of both the reconstituted luciferase and one of the complementing partners (13 AA only for the SmBiT, 11 AA for NP) that minimizes the risk for artifacts that would be induced by the bulky nature of the system constituents (24). A third advantage that we noticed is the dynamic reversibility of the system, at least when the SmBiT and NP were used on H3, D2, and SUCNR1 (see Figs. 3B and 5, C and D). It is important to note that we did not formally demonstrate that the true dynamic of the GPCR–G protein interaction was preserved. Therefore, we cannot exclude that the rate of association/dissociation is impaired as a consequence of the complementation. Notwithstanding, the basal affinity of the complementing partners could still promote artifactual constitutive activity by bringing the G protein and the receptor in close vicinity. However, this assay was not designed to study these dynamics but to detect active ligands for uncharacterized receptors. Thus, the possible increase of sensitivity may actually be seen as beneficial.
Several strategies have been successfully applied in the past to specifically monitor the G protein that interacted with or was activated by a receptor bound by an agonist. Historically, the first reported attempts used cellular backgrounds such as the yeast or insect cells that are devoid of most of the G proteins or GPCR present in human cell lines such as HEK293 (43). Another screening-compatible assay that is able to monitor the activation of each G protein individually has been described recently (33). This elegant approach detects the interaction of the activated G protein (Gβγ subunits) with GRK3 with a NanoBRET system and was applied to profile the coupling between receptors and a large set of G proteins (33). Our approach differs in several aspects compared with the one described by Masuho et al. (33). First, some G proteins, such as Gα12, were not detectable by their system (33). Second, although the sensitivity and signal-to-noise ratio is relatively high, the authors did not test the possibility of using that assay for a screening campaign. Third, the nature of the activating receptor giving rise to the recorded signal is not identifiable in a system solely based on G protein activation. Thus, a screening campaign on an elusive receptor with the assay described by Masuho et al. (33) would give a high rate of false-positive ligands that are activating the endogenous GPCRs present on the cell surface. Another putative way to monitor single G protein interaction to a receptor was published during the preparation of this manuscript. Using G proteins truncated at their N-terminal parts that were initially developed to facilitate crystallographic studies (called mini G protein), Wan et al. (44) reported that they could monitor G protein–GPCR interactions but did not challenge the assay in screening conditions. Interestingly, we applied independently a reduction of the size of the G protein to optimize our Gαo construct, further validating this strategy to improve the performance of engineered G proteins. Actually, all the constructs we developed did not give the same results in terms of sensitivity or signal-to-noise ratio. It should be taken into account when implementing the system for other receptors that several constructs should be tested to identify the best ones. This aspect is of particular importance for orphan receptors because the confidence in assay performance is critical if no positive control exists.
BRET and FRET approaches have been extensively used to monitor G protein recruitment. In principle, a donor (a luciferase in the case of BRET or fluorescent protein in the case of FRET) is fused to one of the interacting partners, and an acceptor is fused to another. In case of sufficient proximity (the donor and acceptor must be at a distance below 10 Å with a correct orientation (19)), the fluorescence of the acceptor can be detected at a unique wavelength, whereas the recorded emission of the donor will be reduced (19). Although these elegant RET approaches shed light on exquisite aspects of receptors, pharmacology such as G protein–GPCR association/dissociation rate or G protein recruitment fingerprints for individual ligands (29, 45, 46). They were never applied to library screenings probably because of their limited sensitivity. In addition, it should be noted that for RET systems to work, the presence of Gβγ is required with precise stoichiometry, which further limit their broad use, especially for screening campaigns. Here, we have demonstrated that the NanoLuc complementation applied to Gα does not necessitate the co-transfection of additional Gβγ subunits (Fig. S3B). For the ease of comparison, we have listed in Table 2 the advantages and limitations of different methods available to monitor G protein–GPCR interactions in living cells.
Table 2.
Reported techniques for the real-time profiling of GPCR-G protein coupling
| Technique | Design | Advantages | Limitations | Amenable to HTS | Refs. |
|---|---|---|---|---|---|
| FRET | CFP and YFP variants | Labeling possible on intra- and extracellular side | Excitation and emission cross-talk | No | 19, 29 |
| No perturbation of the natural interaction | High background (autofluore-scence) | ||||
| Large size of the fluorescent proteins (27 kDa) | |||||
| Addition of Gβγ dimer | |||||
| BRET | RLuc and GFP variants | No perturbation of the natural interaction | Low signal-to-noise ratio | Not determined | 19, 28 |
| No excitation by an external light source | Large size (27 kDa) | ||||
| Low background | Addition of Gβγ dimer | ||||
| NanoBRET | Gβγ-venus and GRK3-NanoLuc | Improved signal compared to BRET | No tagged-receptor or Gα subunit | Yes | 33 |
| Greater light output | Large size of the venus (27 kDa) | ||||
| Medium size of NanoLuc (19 kDa) | Not applicable to Gα12 | ||||
| NanoBiT “Mini G proteins” | LgBiT(1–158) and SmBiT(159–169) | Brighter than BRET methods | Nonideal emission for in vivo applications | Not determined | 24, 44 |
| Low background | Modified G proteins | ||||
| Small size of the SmBiT (1.3 kDa) | Perturbation of the natural interaction | ||||
| Medium size of LgBiT (18 kDa) | |||||
| NanoLuc complementation | NLuc1(1–97) and Nl-Luc2(98–171) | Brighter than BRET methods | Nonideal emission for in vivo applications | Not determined | 52 |
| High signal-to-noise ratio | Perturbation of the natural interaction | ||||
| Low background | |||||
| Medium size of the complementing partners (∼9.5 kDa each) | |||||
| Modified NanoBiT | LgBiT(1–158) and NP(159–171) | Brighter than BRET methods | Nonideal emission for in vivo applications | Yes | This study |
| High signal-to-noise ratio | Perturbation of the natural interaction | ||||
| Low background | |||||
| Small size of the NP (1.5 kDa) | |||||
| Medium size of LgBiT (18 kDa) |
The dopaminergic system is composed of five dopamine receptors (D1 and D5 principally coupled to Gαs/olf and D2-4 coupled mainly to Gαi/o family) of which several are validated therapeutic targets for debilitating conditions such as Parkinson's disease (PD) or psychotic disorders (30). PD is a degenerative disorder marked by tremor, rigidity, and slowness of movement due to a decline of dopaminergic neurons in the substantia nigra (47). In general, the dopaminergic drugs used to treat PD aim at restoring dopamine signaling in affected areas such as the striatum, notably through the activation of the receptor from the D2 family (48). The screening of a library of known drugs on the D2 receptor with a novel GPCR–G protein interaction assay detected five D2 receptor agonists that are currently in use to treat patients suffering from PD (49). The full concentration-response curves that we determined for D2–Gαi1 and -Gαo interactions showed that although there were no relative differences (compared with dopamine) with regard to the potencies of the agonists (except for ropinirole), their Emax displayed signs of partial agonism when recruiting Gαo, in particular for piribedil and pergolide (see Fig. 7 and Table 1). The bias factors that we calculated indicate a different pharmacological profile for the different compounds, especially ropinirole and apomorphine. At this stage, the demonstration that the partial agonism on Gαo is linked to the therapeutic effect would be premature, but the assay presented here has the potential to facilitate the discovery of compounds with a more pronounced bias that could be evaluated for a beneficial therapeutic effect.
Furthermore, the differences in the pharmacological profile we observed between Gαi1 and Gαo with the NanoLuc complementation on the D2 agonist are consistent with the literature. Pioneering work by Strange and co-workers (32, 50) in insect cells demonstrated that different agonists had the ability to elicit different responses depending on the G protein subtype. Using G protein mutants resistant to pertussis toxin treatment, Milligan and co-workers (31) managed to show similar behavior of the D2 receptor toward different G proteins of the Gαi/o family.
The detection of biased agonism is currently based on the differences between activation of G proteins- versus arrestins-based signaling. Usually, the assays to monitor G protein coupling rely on second messengers generation while the arrestin recruitment is estimated with a complementation assay (51). However, we think that it would be preferable to estimate bias with a common strategy instead of different assays because the assays are an important source of artifacts when determining bias. We demonstrated here that the nanoluciferase complementation could also be applied to the estimation of the arrestin recruitment (Fig. S5). Thus, a NanoLuc complementation can be applied to a more robust analysis of biased agonism between arrestin (or any other intracellular partner, in theory) and a given G protein.
In conclusion, this study describes the development and usefulness of a novel system for the detection of direct interactions between GPCR and single G protein. This assay has a dynamic range that is compatible with high-throughput screening. It opens new avenues for programs aimed at the identification in large libraries and optimization of original scaffolds characterized by biased agonism at the level of G protein subtype, a feature that would be impractical with current technologies. Exquisite pharmacological tools such as biased agonists selectively promoting the interaction of a receptor with a restricted set of G proteins should ease our understanding of the physiological rationale for multiple coupling and apparently redundant G proteins.
Experimental procedures
Materials
Dopamine, isoproterenol, succinate, histamine, imetit, sulpiride, piribedil, pramipexole, and pergolide were from Sigma. Ropinirole and U46619 were from Santa Cruz Biotechnology (Dallas, TX); spiperone was from Abcam (Cambridge, UK). Prestwick Chemical Library® was from Prestwick Chemical (Illkirch, France).
Plasmids
Human D2 long, Gαi2, and Gαi3 were amplified from human ORFeome (version 7.1, http://horfdb.dfci.harvard.edu/hv7/).5 Human H1, H3, TPα, Gαoa, and Gαs were amplified from pcDNA3.1+ coding for each protein (cDNA Resource Center, Bloomsburg, PA). The SUCNR1-coding sequence was amplified from genomic DNA of human embryonic kidney 293 (HEK293) cells. β2AR, Gαi1, Gαq, Gα11, Gα12, and Gα13 were amplified from the cDNA of HEK293 cell mRNA. All receptors were cloned into the pcDNA3.1+ (Invitrogen) after addition of the FLAG epitope (DYKDDDDK) at the N terminus, preceded by the signal sequence KTIIALSYIFCLVFA (54) for D2 and β2AR. After cloning into the pcDNA3.1+, the SmBiT (VTGYRLFEEIL), NP (GVTGWRLCERILA), and HiBiT (VSGWRLFKKIS) were added with a flexible linker (GNSGSSGGGGSGGGGSSG) in-frame to the C terminus of the receptor by polymerase chain reaction (PCR). All G proteins were cloned into the pcDNA3.1+ (Invitrogen) after addition of the HA (YPYDVPDYA) epitope at the N terminus. Then, EcoRV and SacII sites (except BamHI and SacII for Gα12) were inserted by PCR between specific amino acid residues of the Gα protein to insert the LgBiT. The coding sequence of LgBiT (VFTLEDFVGDWEQTAAYNLDQVLEQGGVSSLLQNLAVSVTPIQRIVRSGENALKIDIHVIIPYEGLSADQMAQIEEVFKVVYPVDDHHFKVILPYGTLVIDGVTPNMLNYFGRPYEGIAVFDGKKITVTGTLWNGNKIIDERLITPDGSMLFRVTINS) was amplified by PCR and inserted, flanked by a flexible linker (SGGGGS) and the respective restriction sites, into the Gα subunit sequence. The location of the LgBiT was topologically identical for each of the following Gα subunits: between residues 91 and 92 of Gαi (Gαi1–LgB91, Gαi2–LgB91, and Gαi3–LgB91) and Gαoa (Gαo–LgB91); residues 97 and 98 of Gαq/11 (Gαq–LgB97 and Gα11–LgB97); residues 113 and 114 of Gαs (Gαs–LgB113); residues 115 and 116 of Gα12 (Gα12–LgB115); or residues 106 and 107 of Gα13 (Gα13–LgB106). The Gαi1 constructs used for the screening have been obtained as follows: the LgBiT was added at the N terminus of Gαi1 by cloning its coding sequence into the pNBe3 vector (Promega Corp., Madison, WI) with XhoI and SacI sites, and a HA tag was added at the C-terminal sequence by PCR (Gαi1–LgBN-term). The Gαoa construct used for the screening was obtained by insertion of the LgBiT and flanked by a flexible linker (SGGGGS) and EcoRV/SacII sites, between residues 143 and 144 of Gαoa with a HA tag at the N terminus cloned into the pcDNA3.1+ (Invitrogen). Then, the deletion of the first 52 amino acids was performed using the Q5 site-directed mutagenesis kit (New England Biolabs) (Gαo–LgB143d). For the β-arrestin 2–based NanoLuc complementation assay, β-arrestin 2 was cloned into pNBe3 vector (Promega Corp., Madison, WI) with XhoI and EcoRI sites. All constructs were verified by sequencing.
Cell culture and transfection
HEK293 cells were from American Type Culture Collection (Manassas, VA) and grown in Dulbecco's modified Eagle's medium (Lonza, Verviers, Belgium) containing 10% fetal bovine serum (International Medical Products, Brussels, Belgium), 1% penicillin/streptomycin (Lonza), and 1% l-glutamine (Lonza) at 37 °C with 5% CO2. At 80% confluency, cells were transfected with XtremeGene 9 (Promega Corp., Madison, WI) in a 3:1 (reagent/DNA) ratio according to the manufacturer's recommendations. A 1:1 ratio (GPCR/Gα subunit) was used with a solution of plasmids diluted 1:10 with empty pcDNA3.1+.
Measurement of GPCR–G protein interaction by NanoLuc complementation assay
Twenty four hours after transfection of HEK293 cells with a 1:1 ratio of GPCR and Gα subunit plasmid solutions diluted 1:10 with empty pcDNA3.1+, cells were detached from 20- or 55-cm2 dishes with trypsin. After one wash with PBS, cells were resuspended into Hanks' balanced salt solution (HBSS: 120 mm NaCl, 5.4 mm KCl, 0.8 mm MgSO4, 10 mm HEPES, pH 7.4) supplemented with the NanoLuc substrate (Promega Corp., Madison, WI), seeded into a white 96-well plate (50,000 cells/well), and incubated for 45 min at 37 °C. Cells were stimulated by adding 1 μl of 100× ligand solutions, and luminescence was recorded for several minutes depending on each experiment (Centro XS3 LB960; Berthold Technologies). The same protocol was used for β-arrestin 2 recruitment after a transient transfection of receptor and β-arrestin 2–encoding plasmids diluted 1:10 with empty pcDNA3.1+ into HEK293 cells in a 1:1 ratio.
To determine the Z′ factor, cells co-transfected with D2–NP and Gαi1–LgBN-term or D2–NP and Gαo–LgB143d were resuspended into HBSS supplemented with NanoLuc substrate and seeded in a white 96-well plate (50,000 cells/well) at 37 °C for 45 min. Half of the plate was stimulated with vehicle and the other half with dopamine 100× concentrated, and the luminescence was recorded for 10 min. The screening of the Prestwick Chemical Library® was performed at 5 μm on HEK293 transiently transfected with the pair of D2–NP and Gαi1–LgBN-term or Gαo–LgB143d (1:1 ratio with a solution of plasmids diluted 1:10 with empty pcDNA3.1+). The cells were detached in parallel for Gαi1 and Gαo, resuspended into HBSS supplemented with the NanoLuc substrate (Promega Corp.), and incubated for 45 min at 37 °C. Then, the compounds of the library (dissolved in DMSO) were added into a D2–Gαi1 plate, and luminescence was recorded for 10 min. Next, the compounds of the same plate of the library were added to the D2–Gαo plate, and luminescence was recorded for 10 min.
Data analysis and hit selection criteria
All data show a representative result from three independent experiments, except data from Fig. 4, which represent the mean ± S.D. of three independent experiments. Data were collected as RLU during 10 min after stimulation, and the areas under the curve were calculated and then normalized to the vehicle, considered as 0% activity. Statistical significance of the NanoLuc activity in the presence of ligand from basal activity was assessed using the nonparametric Mann-Whitney test (***, p < 0,001). Concentration-response curves were fitted to the four-parameter Hill equation using the least-squares method (GraphPad Prism, version 5.0 for Windows; GraphPad, La Jolla, CA). To combine the results obtained for all tested ligands, we considered the Emax of dopamine as equal to 100% and represented the activity of each ligand compared with the activity of dopamine.
The Z′ factor was calculated as follows: Z′ = 1 −((3σc+ + 3 σc−)/(|μc+ − μc−|)), where σ represents the standard deviation; μ represents the mean; c+, positive control; and c− negative control. For the screening of the Prestwick Chemical Library®, compounds were considered as hits when the ratio > (mean ratio(VEH) + 3 S.D.(VEH)). Ratio represents RLU(compound)/RLU(VEH), and VEH is vehicle. Bias factor was calculated in two steps. First, we determined the Δlog(Emax/EC50) for the Gαi1 and Gαo pathway, which represents the difference between D2 agonist values and dopamine values used as reference ligand obtained with the Gαi1- and Gαo-based assay. Then, the bias factor corresponding to ΔΔlog(Emax/EC50) was calculated as follows: Δlog(Emax/EC50)Gαi1 pathway − Δlog(Emax/EC50)Gαo pathway.
Author contributions
C. L., N. D., and J. H. conceptualization; C. L. and J. H. data curation; C. L. and J. H. formal analysis; C. L. and J. H. writing-original draft; C. L., N. D., and J. H. writing-review and editing; N. D. resources; J. H. supervision; J. H. funding acquisition; J. H. investigation; J. H. methodology; J. H. project administration.
Supplementary Material
Acknowledgments
The Prestwick Chemical Library® used for the screening was a generous gift from Dr. Jean-Claude Twizere, Laboratory of Protein Signaling and Interactions, GIGA-Molecular Biology of Diseases, University of Liège. We acknowledge the technical assistance of Céline Piron, and we thank Dr. Andy Chevigné for fruitful discussions and critical reading of the manuscript. We thank the GIGA Imaging and Flow Cytometry Platform.
This work was supported by the Fonds pour la Recherche Scientifique (F.R.S.-FNRS) Incentive Grant F.4510.14 for Scientific Research, University of Liège (Fonds Spéciaux), and the Léon Fredericq Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- GPCR
- G protein–coupled receptors
- GRK
- GPCR kinase
- D2
- long isoform of the dopamine receptor 2
- NanoBiT
- NanoLuciferase Binary Technology
- BRET
- bioluminescence resonance energy transfer
- FRET
- fluorescence resonance energy transfer
- LgB
- large BiT
- H3
- histamine receptor 3
- SUCNR1
- succinate receptor
- NP
- natural peptide
- NanoLuc
- nanoluciferase
- β2AR
- β2-adrenergic receptor
- H1
- histamine receptor 1
- TPα
- thromboxane A2 receptor α
- RLU
- relative luminescence unit
- RET
- resonance energy transfer
- PD
- Parkinson's disease
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- AA
- amino acid.
References
- 1. Hauser A. S., Attwood M. M., Rask-Andersen M., Schiöth H. B., and Gloriam D. E. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 10.1038/nrd.2017.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sriram K., and Insel P. A. (2018) G protein–coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258 10.1124/mol.117.111062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davenport A. P., Alexander S. P., Sharman J. L., Pawson A. J., Benson H. E., Monaghan A. E., Liew W. C., Mpamhanga C. P., Bonner T. I., Neubig R. R., Pin J.-P., Spedding M., and Harmar A. J. (2013) International union of basic and clinical pharmacology. LXXXVIII. G protein–coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol. Rev. 65, 967–986 10.1124/pr.112.007179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ngo T., Kufareva I., Coleman J. L. j., Graham R. M., Abagyan R., and Smith N. J. (2016) Identifying ligands at orphan GPCRs: current status using structure-based approaches. Br. J. Pharmacol. 173, 2934–2951 10.1111/bph.13452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laschet C., Dupuis N., and Hanson J. (2018) The G protein–coupled receptors deorphanization landscape. Biochem. Pharmacol. 153, 62–74 10.1016/j.bcp.2018.02.016 [DOI] [PubMed] [Google Scholar]
- 6. Roth B. L., and Kroeze W. K. (2015) Integrated approaches for genome-wide interrogation of the druggable non-olfactory G protein–coupled receptor superfamily. J. Biol. Chem. 290, 19471–19477 10.1074/jbc.R115.654764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kenakin (2012) Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br. J. Pharmacol. 165, 1659–1669 10.1111/j.1476-5381.2011.01749.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oldham W. M., and Hamm H. E. (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71 10.1038/nrm2299 [DOI] [PubMed] [Google Scholar]
- 9. Wettschureck N., and Offermanns S. (2005) Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204 10.1152/physrev.00003.2005 [DOI] [PubMed] [Google Scholar]
- 10. Rajagopal S., and Shenoy S. K. (2018) GPCR desensitization: acute and prolonged phases. Cell. Signal. 41, 9–16 10.1016/j.cellsig.2017.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Milligan G., and Kostenis E. (2006) Heterotrimeric G-proteins: a short history. Br. J. Pharmacol. 147, S46–S55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Williams C. (2004) cAMP detection methods in HTS: selecting the best from the rest. Nat. Rev. Drug Discov. 3, 125–135 10.1038/nrd1306 [DOI] [PubMed] [Google Scholar]
- 13. Gilissen J., Geubelle P., Dupuis N., Laschet C., Pirotte B., and Hanson J. (2015) Forskolin-free cAMP assay for Gi-coupled receptors. Biochem. Pharmacol. 98, 381–391 10.1016/j.bcp.2015.09.010 [DOI] [PubMed] [Google Scholar]
- 14. Ma Q., Ye L., Liu H., Shi Y., and Zhou N. (2017) An overview of Ca2+ mobilization assays in GPCR drug discovery. Expert Opin. Drug Discov. 12, 511–523 10.1080/17460441.2017.1303473 [DOI] [PubMed] [Google Scholar]
- 15. Inoue A., Ishiguro J., Kitamura H., Arima N., Okutani M., Shuto A., Higashiyama S., Ohwada T., Arai H., Makide K., and Aoki J. (2012) TGFα shedding assay: an accurate and versatile method for detecting GPCR activation. Nat Methods 9, 1021–1029 10.1038/nmeth.2172 [DOI] [PubMed] [Google Scholar]
- 16. Milligan G. (2003) Principles: extending the utility of [35S]GTPγS binding assays. Trends Pharmacol. Sci. 24, 87–90 10.1016/S0165-6147(02)00027-5 [DOI] [PubMed] [Google Scholar]
- 17. Schröder R., Janssen N., Schmidt J., Kebig A., Merten N., Hennen S., Müller A., Blättermann S., Mohr-Andrä M., Zahn S., Wenzel J., Smith N. J., Gomeza J., Drewke C., Milligan G., et al. (2010) Deconvolution of complex G protein–coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat. Biotechnol. 28, 943–949 10.1038/nbt.1671 [DOI] [PubMed] [Google Scholar]
- 18. Kostenis E., Waelbroeck M., and Milligan G. (2005) Techniques: promiscuous Gα proteins in basic research and drug discovery. Trends Pharmacol. Sci. 26, 595–602 10.1016/j.tips.2005.09.007 [DOI] [PubMed] [Google Scholar]
- 19. Lohse M. J., Nuber S., and Hoffmann C. (2012) Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling. Pharmacol. Rev. 64, 299–336 10.1124/pr.110.004309 [DOI] [PubMed] [Google Scholar]
- 20. Boute N., Jockers R., and Issad T. (2002) The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol. Sci. 23, 351–354 10.1016/S0165-6147(02)02062-X [DOI] [PubMed] [Google Scholar]
- 21. Machleidt T., Woodroofe C. C., Schwinn M. K., Méndez J., Robers M. B., Zimmerman K., Otto P., Daniels D. L., Kirkland T. A., and Wood K. V. (2015) NanoBRET—a novel BRET platform for the analysis of protein–protein interactions. ACS Chem. Biol. 10, 1797–1804 10.1021/acschembio.5b00143 [DOI] [PubMed] [Google Scholar]
- 22. Hoffmann C., Gaietta G., Bünemann M., Adams S. R., Oberdorff-Maass S., Behr B., Vilardaga J.-P., Tsien R. Y., Ellisman M. H., and Lohse M. J. (2005) A FlAsH-based FRET approach to determine G protein–coupled receptor activation in living cells. Nat. Methods 2, 171–176 10.1038/nmeth742 [DOI] [PubMed] [Google Scholar]
- 23. Hall M. P., Unch J., Binkowski B. F., Valley M. P., Butler B. L., Wood M. G., Otto P., Zimmerman K., Vidugiris G., Machleidt T., Robers M. B., Benink H. A., Eggers C. T., Slater M. R., Meisenheimer P. L., et al. (2012) Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 7, 1848–1857 10.1021/cb3002478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dixon A. S., Schwinn M. K., Hall M. P., Zimmerman K., Otto P., Lubben T. H., Butler B. L., Binkowski B. F., Machleidt T., Kirkland T. A., Wood M. G., Eggers C. T., Encell L. P., and Wood K. V. (2016) NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 10.1021/acschembio.5b00753 [DOI] [PubMed] [Google Scholar]
- 25. Beaulieu J.-M., Espinoza S., and Gainetdinov R. R. (2015) Dopamine receptors–IUPHAR review 13. Br. J. Pharmacol. 172, 1–23 10.1111/bph.12906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Panula P., Chazot P. L., Cowart M., Gutzmer R., Leurs R., Liu W. L., Stark H., Thurmond R. L., and Haas H. L. (2015) International union of basic and clinical pharmacology. XCVIII. Histamine receptors. Pharmacol. Rev. 67, 601–655 10.1124/pr.114.010249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gilissen J., Jouret F., Pirotte B., and Hanson J. (2016) Insight into SUCNR1 (GPR91) structure and function. Pharmacol. Ther. 159, 56–65 10.1016/j.pharmthera.2016.01.008 [DOI] [PubMed] [Google Scholar]
- 28. Galés C., Rebois R. V., Hogue M., Trieu P., Breit A., Hébert T. E., and Bouvier M. (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods 2, 177–184 10.1038/nmeth743 [DOI] [PubMed] [Google Scholar]
- 29. Hein P., Frank M., Hoffmann C., Lohse M. J., and Bünemann M. (2005) Dynamics of receptor/G protein coupling in living cells. EMBO J. 24, 4106–4114 10.1038/sj.emboj.7600870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beaulieu J. M., and Gainetdinov R. R. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217 10.1124/pr.110.002642 [DOI] [PubMed] [Google Scholar]
- 31. Lane J. R., Powney B., Wise A., Rees S., and Milligan G. (2007) Protean agonism at the dopamine D2 receptor: (S)-3-(3-hydroxyphenyl)-N-propylpiperidine is an agonist for activation of Go1 but an antagonist/inverse agonist for Gi1,Gi2, and Gi3. Mol. Pharmacol. 71, 1349–1359 10.1124/mol.106.032722 [DOI] [PubMed] [Google Scholar]
- 32. Gazi L., Nickolls S. A., and Strange P. G. (2003) Functional coupling of the human dopamine D2 receptor with Gαi1, Gαi2, Gαi3, and Gαo G proteins: evidence for agonist regulation of G protein selectivity. Br. J. Pharmacol. 138, 775–786 10.1038/sj.bjp.0705116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Masuho I., Ostrovskaya O., Kramer G. M., Jones C. D., Xie K., and Martemyanov K. A. (2015) Distinct profiles of functional discrimination among G proteins determine the actions of G protein–coupled receptors. Sci. Signal. 8, ra123–ra123 10.1126/scisignal.aab4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Freedman S. B., Patel S., Marwood R., Emms F., Seabrook G. R., Knowles M. R., and McAllister G. (1994) Expression and pharmacological characterization of the human D3 dopamine receptor. J. Pharmacol. Exp. Ther. 268, 417–426 [PubMed] [Google Scholar]
- 35. Zhang J., Chung T. D., and Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high-throughput screening assays. J. Biomol. Screen. 4, 67–73 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 36. Kenakin T., Watson C., Muniz-Medina V., Christopoulos A., and Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203 10.1021/cn200111m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Michnick S. W., Ear P. H., Manderson E. N., Remy I., and Stefan E. (2007) Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discov. 6, 569–582 10.1038/nrd2311 [DOI] [PubMed] [Google Scholar]
- 38. Magliery T. J., Wilson C. G., Pan W., Mishler D., Ghosh I., Hamilton A. D., and Regan L. (2005) Detecting protein-protein interactions with a green fluorescent protein fragment Reassembly trap: scope and mechanism. J. Am. Chem. Soc. 127, 146–157 10.1021/ja046699g [DOI] [PubMed] [Google Scholar]
- 39. Dupuis N., Laschet C., Franssen D., Szpakowska M., Gilissen J., Geubelle P., Soni A., Parent A.-S., Pirotte B., Chevigné A., Twizere J.-C., and Hanson J. (2017) Activation of the orphan G protein–coupled receptor GPR27 by surrogate ligands promotes β-Arrestin 2 recruitment. Mol. Pharmacol. 91, 595–608 10.1124/mol.116.107714 [DOI] [PubMed] [Google Scholar]
- 40. Hattori M., Tanaka M., Takakura H., Aoki K., Miura K., Anzai T., and Ozawa T. (2013) Analysis of temporal patterns of GPCR-β-arrestin interactions using split luciferase-fragment complementation. Mol. BioSyst. 9, 957–964 10.1039/c2mb25443c [DOI] [PubMed] [Google Scholar]
- 41. Szpakowska M., Nevins A. M., Meyrath M., Rhainds D., D'huys T., Guité-Vinet F., Dupuis N., Gauthier P.-A., Counson M., Kleist A., St-Onge G., Hanson J., Schols D., Volkman B. F., Heveker N., and Chevigné A. (2018) Different contributions of chemokine N-terminal features attest to a different ligand binding mode and a bias towards activation of ACKR3/CXCR7 compared with CXCR4 and CXCR3. Br. J. Pharmacol. 175, 1419–1438 10.1111/bph.14132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. England C. G., Ehlerding E. B., and Cai W. (2016) NanoLuc: a small luciferase is brightening up the field of bioluminescence. Bioconjug. Chem. 27, 1175–1187 10.1021/acs.bioconjchem.6b00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu R., Wong W., and IJzerman A. P. (2016) Human G protein–coupled receptor studies in Saccharomyces cerevisiae. Biochem. Pharmacol. 114, 103–115 10.1016/j.bcp.2016.02.010 [DOI] [PubMed] [Google Scholar]
- 44. Wan Q., Okashah N., Inoue A., Nehmé R., Carpenter B., Tate C. G., and Lambert N. A. (2018) Mini G protein probes for active G protein–coupled receptors (GPCRs) in live cells. J. Biol. Chem. 293, 7466–7473 10.1074/jbc.RA118.001975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Galés C., Van Durm J. J., Schaak S., Pontier S., Percherancier Y., Audet M., Paris H., and Bouvier M. (2006) Probing the activation-promoted structural rearrangements in preassembled receptor–G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786 10.1038/nsmb1134 [DOI] [PubMed] [Google Scholar]
- 46. Saulière A., Bellot M., Paris H., Denis C., Finana F., Hansen J. T., Altié M.-F., Seguelas M.-H., Pathak A., Hansen J. L., Sénard J.-M., and Galés C. (2012) Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat. Chem. Biol. 8, 622–630 10.1038/nchembio.961 [DOI] [PubMed] [Google Scholar]
- 47. Obeso J. A., Rodriguez-Oroz M. C., Goetz C. G., Marin C., Kordower J. H., Rodriguez M., Hirsch E. C., Farrer M., Schapira A. H., and Halliday G. (2010) Missing pieces in the Parkinson's disease puzzle. Nat. Med. 16, 653–661 10.1038/nm.2165 [DOI] [PubMed] [Google Scholar]
- 48. Oertel W., and Schulz J. B. (2016) Current and experimental treatments of Parkinson disease: a guide for neuroscientists. J Neurochem. 139, Suppl. 1, 325–337 10.1111/jnc.13750 [DOI] [PubMed] [Google Scholar]
- 49. De Keyser J., De Backer J. P., Wilczak N., and Herroelen L. (1995) Dopamine agonists used in the treatment of Parkinson's disease and their selectivity for the D1, D2, and D3 dopamine receptors in human striatum. Prog. Neuropsychopharmacol. Biol. Psychiatry 19, 1147–1154 10.1016/0278-5846(95)00232-4 [DOI] [PubMed] [Google Scholar]
- 50. Cordeaux Y., Nickolls S. A., Flood L. A., Graber S. G., and Strange P. G. (2001) Agonist regulation of D(2) dopamine receptor/G protein interaction. Evidence for agonist selection of G protein subtype. J. Biol. Chem. 276, 28667–28675 10.1074/jbc.M008644200 [DOI] [PubMed] [Google Scholar]
- 51. Winpenny D., Clark M., and Cawkill D. (2016) Biased ligand quantification in drug discovery: from theory to high-throughput screening to identify new biased μ opioid receptor agonists. Br. J. Pharmacol. 173, 1393–1403 10.1111/bph.13441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yano H., Cai N. S., Javitch J. A., and Ferré S. (2018) Luciferase complementation based-detection of G-protein-coupled receptor activity. BioTechniques 65, 9–14 10.2144/btn-2018-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harding S. D., Sharman J. L., Faccenda E., Southan C., Pawson A. J., Ireland S., Gray A. J. G., Bruce L., Alexander S. P. H., Anderton S., Bryant C., Davenport A. P., Doerig C., Fabbro D., Levi-Schaffer F., et al. (2018) The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Res. 46, D1091–D1106 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guan X. M., Kobilka T. S., and Kobilka B. (1992) Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J. Biol. Chem. 267, 21995–21998 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.