

Abstract
mTORC1 regulates protein synthesis and in turn is regulated by growth factors, energy status, and amino acid availability. In kidney cell (HEK293-T) culture, the GAP activity toward RAG (GATOR1) protein complex suppresses activation of the RAG A/B–RAG C/D heterodimer when amino acids are insufficient. During amino acid sufficiency, the RAG heterodimer recruits mTORC1 to the lysosomal membrane where its interaction with Ras homolog enriched in brain (Rheb) stimulates mTORC1's kinase activity. The DEP domain containing 5 (DEPDC5) protein, a GATOR1 subunit, causes familial focal epilepsy when mutated, and global knockout of the Depdc5 gene is embryonically lethal. To study the function of DEPDC5 in skeletal muscle, we generated a muscle-specific inducible Depdc5 knockout mouse, hypothesizing that knocking out Depdc5 in muscle would make mTORC1 constitutively active, causing hypertrophy and improving muscle function. Examining mTORC1 signaling, morphology, mitochondrial respiratory capacity, contractile function, and applied physical function (e.g. rotarod, treadmill, grip test, and wheel running), we observed that mTORC1 activity was significantly higher in knockout (KO) mice, indicated by the increased phosphorylation of mTOR and its downstream effectors (by 118% for p-mTOR/mTOR, 114% for p-S6K1/S6K1, and 35% for p-4E-BP1/4E-BP1). The KO animals also exhibited soleus muscle cell hypertrophy and a 2.5-fold increase in mitochondrial respiratory capacity. However, contrary to our hypothesis, neither physical nor contractile function improved. In conclusion, DEPDC5 depletion in adult skeletal muscle removes GATOR1 inhibition of mTORC1, resulting in muscle hypertrophy and increased mitochondrial respiration, but does not improve overall muscle quality and function.
Keywords: GATOR1 (GTPase-activating protein (GAP) activity toward Rags-1), gene knockout, muscle, muscle hypertrophy, translation control, transgenic mice, mTOR complex (mTORC), amino acid sensing, muscle contractility
Introduction
mTOR complex 1 (mTORC1)2 is a master regulator of protein synthesis that receives inputs from a variety of sources (e.g. energy balance, oxygen concentration, growth factors, contraction/exercise, and amino acid availability). When conditions are favorable, activated mTORC1 phosphorylates downstream targets, such as TFEB (transcription factor EB) and ULK1 (unc-51 like autophagy activating kinase 1), that restrict autophagy, as well as S6K1 (ribosomal protein 6 kinase 1) and 4E-BP1 (eukaryote translation initiation factor 4E–binding protein 1), which promote mRNA translation by the ribosome. The control of mTORC1 signaling is complex, and many review articles describe the process (1–3). Much of this work, particularly research of amino acid sensing and mTORC1 signaling, is based upon findings from cell culture (often HEK-293T immortalized embryonic kidney cells).
Translocation of mTORC1 to the lysosomal membrane stimulates its kinase activity via allosteric activation by Rheb (Ras homolog enriched in brain) (4). The RAG A/B–RAG C/D heterodimer recruits mTORC1 to the lysosomal membrane when sufficient amino acids, particularly leucine and arginine, are available (5). GATOR1 (GAP activity toward RAG 1) is a protein complex consisting of three subunits: NPRL2 (nitrogen permease regulator-like 2), NPRL3 (nitrogen permease regulator-like 3), and DEPDC5 (DEP domain containing 5). GATOR1 is a negative regulator of the RAG heterodimer, preventing translocation of mTORC1 to the lysosome during amino acid insufficiency by acting as a GTPase-activating protein to keep RAG A/B in the inactive GDP state (6). GATOR1 is deactivated by GATOR2 (GAP activity toward RAG 2), if amino acid availability within the cell is increased (7, 8). Interestingly, Depdc5 mRNA expression in human skeletal muscle (obtained from biopsy samples) changes rapidly in response to an increase in intracellular amino acid concentrations during postprandial conditions (9).
Transgenic mouse models have been developed that globally knock out Nprl2 (10), Nprl3 (11), and Depdc5 (12). The common theme of these knockouts is embryonic lethality. Nprl2 knockout reduces methionine synthesis from homocysteine and causes hematopoietic dysregulation (10). Nprl3 knockout results in lethal developmental abnormalities in the cardiovascular system (11). Global knockout of Depdc5 causes severe abnormal embryonic morphology and mTORC1 hyperactivity in mice (12) and rats (13). Neuron-specific knockout of Depdc5 results in an epileptic phenotype similar to the human presentation of DEPDC5 protein mutation familial epilepsy (14).
In skeletal muscle cells, amino acid–sensing pathways control protein synthesis and hypertrophy through mTORC1. DEPDC5, as a component of the GATOR1 complex, is a negative regulator of mTORC1. Given that global or embryonic knockout of DEPDC5 protein is embryonically lethal, we bred an inducible skeletal muscle-specific knockout to study DEPDC5 function in muscle. To accomplish this, we used cre-loxP technology in a transgenic mouse that expresses tamoxifen-inducible cre only in limb skeletal muscle: the Tg(ACTA1-cre/Esr1*)2Kesr (CRE) mouse available commercially from the Jackson Laboratory (stock no. 025750) and crossed the CRE line (either heterozygous CRE+/−, or homozygous CRE+/+) with a transgenic mouse with the mutant homozygous allele Depdc5tm1c(EUCOMM)Hmgu (Depdc5fl/fl for the homozygous allele) (see “Genotyping” under “Method details” for further explanation). We hypothesized that Depdc5 knockout in skeletal muscle would result in a constitutively active mTORC1, promote muscle cell growth (hypertrophy), and result in increased functional performance. To test this hypothesis, we compared non–tamoxifen-treated mice (control or WT) to tamoxifen-treated mice (experimental, denoted as KO for knockout) and used immunohistochemistry to measure muscle cell cross-sectional area by fiber type, immunoblotting to determine mTORC1 pathway activation, and performed functional analysis using in vitro contractile physiology (muscle force production), rotarod (overall motor function), grip test (forelimb strength), treadmill (endurance), and voluntary wheel running (activity). In addition, because mitochondrial activity has been shown to be regulated by 4E-BP1, a downstream effector of mTORC1 (15), we determined mitochondrial respiratory capacity with high-resolution respirometry (Orobouros Instruments Oxygraph-2k), citric acid cycle activity with a citrate synthase enzymatic activity assay, and relative levels of the COX4 protein using Western blotting and performed an enzymatic activity immunohistochemical stain for succinate dehydrogenase.
Results
DEPDC5 depletion in KO muscle
The transgene PCR product (∼749 bp) is expressed only in tamoxifen-treated KO skeletal muscle (tibialis anterior, TA) and not in off-target tissues such as the heart and skin or in nontransgenic or tamoxifen negative mice. DEPDC5 mRNA expression was also reduced in KO when compared with WT. Indeed, most KO samples did not result in PCR replication of DEPDC5 within the maximum 40 cycles. Gel electrophoresis of the PCR amplicons demonstrated little to no expression of DEPDC5 mRNA in skeletal muscle. DEPDC5 protein expression was reduced in a Western blotting comparison of WT versus KO in skeletal muscle (Fig. 1 and Fig. S1).
Figure 1.

Validation of skeletal muscle–specific inactivation of Depdc5. A, DNA. Only in the TA muscle was the recombinant product produced, and not in heart or skin (ear punch). Liver does not highly express the floxed allele at 1349 bp. The plus (+) or minus (−) signs indicate whether tamoxifen has been given (+) or not (−). Mice that have received tamoxifen to induce recombination have a product at 749 bp. Arrows highlight experimental KO mice that have received tamoxifen. B, mRNA. In mRNA isolated from TA, WT, or control mice have the PCR mRNA amplicon at 76 bp, but KO do not. The left graph is randomly mixed males and females. In the right graph, triangles indicate KO, and diamonds indicate WT, with filled shapes indicating females. C, protein. In whole TA muscle homogenate, WT mice show a much greater abundance of DEPDC5 than the KO. PC, positive control (much larger amount of TA from WT mouse: 100 μg protein/well in Western blotting and 4× mRNA amount in RNA electrophoresis); GAS, gastrocnemius; KO, DEPDC5fl/fl × CRE+/− or +/+; EV, empty vector; DEPDC5fl/fl, homozygous for Depdc5tm1c(EUCOMM)Hmgu; CRE+/− or CRE+/+, hetero- or homozygous for Tg(ACTA1-cre/Esr1*)2Kesr.
mTORC1 signaling increased in KO
All mice were fed ad libitum. In KO, when compared with WT, phosphorylation of mTOR at Ser-2448) increased by 118% (p < 0.001), phosphorylation of S6K1 at Thr-389 increased by 114% (p = 0.008), and the phosphorylation of 4E-BP1 Thr-37/46) increased by 35% (p < 0.001) (Fig. 2).
Figure 2.

mTORC1 signaling increased in knockout mice. *, p < 0.05; WT, WT mice; KO, transgenic knockout mice. Each symbol represents an individual muscle sample. Triangle, WT; diamond, KO; filled symbols, females; open symbols, males. To the right of each group: rectangle, means ± S.E.; M, male; F, female. For mTOR and S6K signaling, n = 5 M KO, 2 F KO, 5 M WT, and 4 F WT. For 4E-BP1 signaling, n = 5 M KO, 1 F KO, 5 M WT, and 1 F WT.
Muscle cell hypertrophy in KO
Male KO demonstrated significant muscle fiber hypertrophy (increased cross-sectional area (CSA)) versus WT males in Soleus (SOL) type 1 (+13%, p = 0.034), 2a (+17%, p = 0.048), and 2x (+21%, p = 0.010) fiber types. In the TA, type 2a muscle fibers demonstrated a tendency for hypertrophy (+17%, p = 0.089) in males, with no increase in 2x or 2b cells (data not shown). Female KO animals did not have significant changes in muscle fiber CSA (Fig. 3 and Table S1).
Figure 3.

Muscle cell size increased in male knockout mice. A, SOL MHC 1. B, SOL MHC 2a. C, SOL MHC 2. A–C, SOL CSA +15% overall (p = 0.030). D, TA CSA did not change significantly overall, but there was a trend for an increase in the MHC 2a cells. E, representative image of SOL WT cross-section. F, representative image of SOL KO cross-section. E and F, 1, 2x, 2a, and 2b = MHC type. *, p < 0.05; #, 0.05 < p < 0.10; WT, WT mice; KO, transgenic knockout mice. Each symbol represents an individual muscle sample. Circle, WT; diamond, KO. M, male. To the right of each group: rectangle, means ± S.E. In SOL: n = 5 WT and n = 6 KO. For TA MHC 2a: n = 4 KO and n = 7 WT.
Fiber type in the TA and SOL
In the TA of male KO, there was evidence of reduced numbers of the more oxidative and slower twitch type 1 fibers −79%, p = 0.051; and type 2a fibers −65.8%, p = 0.064, with a concomitant, albeit nonsignificant, shift to a more glycolytic and faster fiber type (type 2B fibers +31%, p = 0.279). There was no significant evidence of a shift in fiber type in SOL. In contrast, the female mice demonstrated no significant change in fiber type isoforms (Table S1).
Function is not improved in KO
Other than the grip test in male KO, which was reduced by 15%, none of the functional tests (e.g. rotarod, treadmill, voluntary wheel running) demonstrated significant change in KO (from pre- to 6-weeks post-tamoxifen treatment as compared with WT), when the mass of the mice was accounted for in both pre- and post-testing (Fig. 4, Table S2, and Figs. S2–S5). Body mass did not significantly change in males. However, female WT mice did gain 6.3% (p = 0.037), over the experimental period, but female KO mice did not change (Table S2). Muscle wet mass was not significantly different between KO and WT for SOL, plantaris, extensor digitorum longus (EDL), or gastrocnemius muscles (p > 0.05) in either males or females. The TA muscle mass demonstrated no change in males, but was significantly lower in KO females (Table S2). Muscle contractile function (i.e. maximal tetanic force in EDL and SOL) was also not changed significantly in KO (see Table S2 for further details of the contractile physiology data). Female KO mice also had no improved function.
Figure 4.

Functional aptitude not improved in knockout mice. Function did not change significantly (p < 0.05) in any of the measurements other than grip test, which was reduced by 15% (p = 0.002). The Pre designation indicates the value obtained before tamoxifen injection, and Post indicates values from testing done 6 weeks after the last tamoxifen injection. All mice in this figure are males; see the supporting information for female mice. WT, WT mice; KO, transgenic knockout mice. Each symbol represents an individual muscle sample from a male mouse. Circle, WT; diamond, KO, In A–C, E, and F, to the right of each group: rectangle, means ± S.E. In D, the error bars show the standard error, and the number inside the graph indicates the mean km/day. Grip, rotarod, treadmill: WT n = 10, KO n = 15; running wheel: WT n = 6, KO n = 11; EDL force: WT n = 7, KO n = 11; SOL force: WT n = 10, KO n = 11.
KO mitochondrial respiratory capacity increases
Respiration coupled to ATP production (state 3 respiration) was significantly greater in KO versus WT (46 ± 6 versus 116 ± 4 pmol/s/mg; p < 0.001). Similarly, maximal uncoupled respiration was significantly greater in KO (67 ± 11 versus 160 ± 0 pmol/s/mg; p < 0.001; Fig. 5), indicating greater respiratory capacity per mg of muscle tissue in KO. Despite this 2.5-fold difference in mitochondrial respiratory capacity, respiratory control in response to ADP did not differ between the two groups, suggesting that mitochondrial quality was similar between KO and WT (Fig. S6). Similarly, the substrate control for succinate and the flux control ratio, calculated by normalizing the leak and coupled respiration to the maximal uncoupled respiration, were all similar between WT and KO (Fig. S6). Collectively, these data suggest that the significant differences in TA mitochondrial respiratory capacity between WT and KO are not the result of alterations in mitochondrial coupling control or quality. This means that KO may have greater mitochondrial protein abundance. This is supported by data in the SOL that demonstrated, at least in male KO mice, COX4 protein was increased ∼20% (although females actually had a similar magnitude of decline) (Fig. 6).
Figure 5.
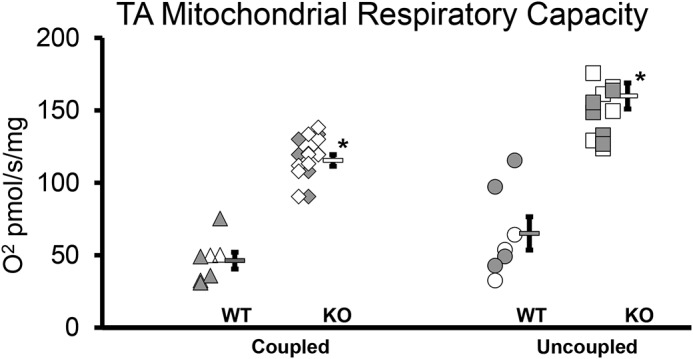
Mitochondrial TA respiration increased in knockout mice. Both coupled and uncoupled mitochondrial respiration increased in KO. *, p < 0.001. WT, WT mice (n = 7); KO, transgenic knockout mice (n = 13). Each symbol indicates a sample from an individual mouse, with filled symbols indicating female mice. To the right of each group is the group mean, and the error bar is the standard error.
Figure 6.

COX4. A, SOL COX4/GAPDH ratio is 19. 0% higher in male KO (open triangles, n = 5, p = 0.047) than in WT (combined sexes, n = 4 M and 4 F) and the ratio is 19.3% lower in female KO (filled triangles, n = 3, p = 0.021). Male KO ratio is 47.5% higher than the female KO (p = 0.004). Different letters indicate significant difference between groups. WT M and WT F were not different; thus, the groups were combined. Each symbol represents an individual muscle sample. Filled symbol, female; open symbol, male; diamond, WT; triangle, KO. To the right of each group: rectangle, means ± S.E. WT, WT mice; KO, knockout mice. B, representative images. 10 μg of protein loaded per well on a 4–15% gradient acrylamide gel. M, males; F, females; PC, positive control.
Furthermore, we showed that there was increased citric acid cycle (TCA) activity in both male and female KO SOL of ∼158% (Fig. 7). In addition, when we stained for succinate dehydrogenase (SDH) activity in the SOL, we uncovered a positive correlation between higher SDH activity and muscle size (Fig. S7), with SDH+ fiber frequency 13% greater in male KO mice than WT on average.
Figure 7.
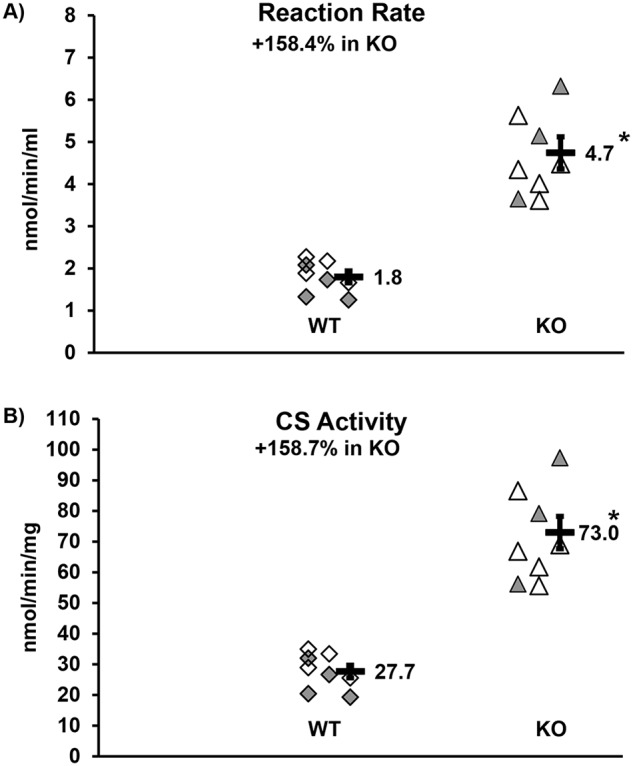
SOL citrate synthase activity. Both the WT control (diamonds) and KO experimental (triangles) groups are made of mixed gender: n = 4 males (open) and n = 4 females (shaded). There were no gender differences; thus we combined the groups. Each symbol indicates the mean value of two independent assays of an individual mouse soleus muscle sample. The bar is the mean of each group (number to the right) with error bar indicating standard error. *, p < 0.001. A, reaction rate. B, citrate synthase (CS) activity.
Discussion
We set out to determine the impact of Depdc5 knockout on skeletal muscle metabolism and function. As expected, the muscle-specific knockout of DEPDC5 in adult mice induced mTORC1 to become constitutively active in muscle tissue as demonstrated by increased phosphorylation of mTOR and its downstream effectors in KO animals as compared with WT. Male, but not female mice, had an increase in muscle cell size (i.e. hypertrophy) with the loss of the DEPDC5 protein. Six weeks following tamoxifen treatment, there was no change in maximal isometric force (i.e. in vitro EDL and SOL contractile physiology), activity (wheel running), overall motor ability (rotarod), or endurance (treadmill). However, evidence for a reduction in forelimb strength (grip test) in KO animals was observed. The knockout of DEPDC5 in adult muscle resulted in hyperactive mTORC1 signaling in fed mice, underscoring a critical role for muscle DEPDC5 in mTORC1 activation in vivo.
These data are in line with those reported in the brain of animals with global knockout (12, 13) or neuron-specific knockout (14). Similarly, knockout of NPRL2 in mouse embryonic fibroblasts resulted in the inability of starvation to repress mTORC1 signaling (10), whereas knocking out NPRL3 resulted in changes in mRNA expression consistent with down-regulation of mTORC1 signaling (11). When NPRL2 was knocked out in muscle, mTORC1 was activated despite fasted conditions (16). Whether muscle DEPDC5 knockout is sufficient to preserve mTORC1 signaling in a fasted state, in vivo, remains an unanswered question that warrants further study.
In the current study, we found evidence of cellular hypertrophy following the loss of DEPDC5 in skeletal muscle. However, this hypertrophy was not consistent across all muscles, fiber types, or sexes. We suspect that the male mice may have exhibited greater hypertrophy because of a higher level of aggressive activity (i.e. wrestling/fighting), whereas the females were observed to be more sedentary/relaxed than the males in our study. Indeed, male mice can be aggressive (17) and have bouts of fighting when group housed, which may provide an anabolic stimulus (18). In contrast, aggression is not a behavior associated with female mice. Researchers sometimes choose to use only female mice to avoid dealing with aggressive behaviors (19). Additionally, the male physiology may have been permissive of hypertrophy in the absence of a dedicated exercise stimulus, because young male mice would have increased levels of testosterone when compared with young female mice (20). However, there was no percentage increase in overall body mass in the KO compared with WT over the same period, nor were significant increases in muscle wet mass observed. Future work examining whether traditional hypertrophy models (e.g. synergist ablation), exercise training, or disuse models (e.g. hindlimb suspension or casting) in inducible Depdc5 muscle-specific KO animals will be useful in understanding the interaction of physical activity and mTORC1 activation in regulating muscle size and function.
Other knockout models of mTORC1 pathway genes have demonstrated effect variability. Knocking out RAPTOR in muscle results in repressed mTORC1 signaling and TSC1 knockout in muscle results in up-regulated mTORC1 signaling (21). Both knockouts result in younger mice being both resistant to high-fat diets and leaner than WT mice, but there were differing metabolic implications for each knockout, with severe myopathy developing in both models at older ages. Both young male and female TSC1 knockouts were lighter than age-matched WT, mainly because of decreased fat mass, but the RAPTOR knockouts were only lighter in males, primarily because of lower lean mass. Compared with nontransfected cells, knockdown of TSC1 via electroporation of shRNA was sufficient to promote hypertrophy in the short term, in both normally innervated and denervated muscle, but resulted in atrophy long term in all but soleus muscles (22). Because our KO animals did not change in overall mass compared with WT, we hypothesize that they may have gained lean mass and lost fat mass in equal proportions.
Even though there was evidence of muscle hypertrophy, at least in the male KO, in contrast to our hypothesis, muscle-specific loss of DEPDC5 did not improve function overall, and grip strength was reduced. This result went contrary to our original hypothesis, where we predicted that in the short term that function would be increased. Interestingly, although we show increased mitochondrial respiratory capacity in the KO, this did not translate into increased endurance, perhaps because only skeletal muscle and not cardiac muscle is affected by the knockout. The loss of grip strength detected in the KO could be interpreted as a beginning of muscle myopathy at 6 weeks following the depletion of DEPDC5. Whether further myopathy follows with a more prolonged loss of DEPDC5 in muscle is an interesting avenue for future studies to explore.
Hypertrophic muscle does not always accompany improvements in contractile or physical performance. There are numerous examples from different systems. Hypertrophy in a muscle cell can involve expansion of the sarcoplasm and/or myofibrillar expansion. Without the addition of force producing machinery in parallel (i.e. actin/myosin chains, myofibrils), force output cannot increase. However, the addition of myofibrils can also increase the pennation angle of muscle fibers, thus lowering force output per unit of muscle (23). Weight training can result in hypertrophy without isometric strength gain (24). Myofibrils can be added to lengthen the muscle, which might contribute to muscle mass and velocity of contraction (and thus increased power output) but would not improve force production, per se. In addition, increased capillarization, fibrosis and lipid deposition could increase muscle size and mass without increasing force output (25). For example, in myostatin null mice force output is not increased in ratio to an increase in muscle size (26, 27). Another example would be the super-sized muscle cells observed in Duchene muscular dystrophy models that are actually weaker than their overall muscle size would indicate potentially because of pseudohypertrophy (i.e. hypertrophy of cellular components in muscles unrelated to force production), fibrotic intrusion, and cellular disruption (28).
Another potential side effect of increased mTORC1 signaling is a reduction in autophagy, because mTORC1 is a negative regulator of autophagy through the phosphorylation of ULK-1 (Unc-51 like kinase-1) and TFEB (transcription factor EB) (29). A reduction in autophagy might result in an accumulation of cellular debris, potentially resulting in a reduction of muscle quality. This muscle loss in quality could contribute to a lack of functional benefit from hypertrophy, although we did not find evidence of reduced specific force in the EDL or SOL. Reduced mitophagy could potentially increase reactive oxygen species production, resulting in protein or DNA damage also leading to loss of muscle quality and potential nuclear accretion or apoptosis (30).
Because muscle wet mass and the fiber CSA data in the soleus were not consistent, we compared the fiber type shift to the CSA data and noticed that, although not significant one could speculate that because the muscles tend to switch fiber type from smaller, slower twitch, more oxidative fibers to larger, faster, more glycolytic fibers, the overall volume of the fiber could be maintained in the face of concomitant apoptosis, which would lead to fewer total cells in the muscle. Thus, we hypothesize that, in the context of chronic mTORC1 activation, inhibition of autophagy accompanied by hyperactive mitochondria releasing damaging reactive oxygen species creates an environment with limited ability for the cell to clear damaged organelles and protein complexes, including dysfunctional mitochondria. This condition likely further exacerbates oxidative stress and increases apoptosis, reducing the overall number of cells. Conversely, increased proteasome activity might also occur as the muscle fights to maintain homeostasis in response to chronic mTORC1 activation and mitochondrial hyperactivity. Future studies examining the long-term effects of DEPDC5 depletion in muscle are needed to address this hypothesis.
Based on our findings of increased mitochondrial respiratory capacity (in both male and females), increased citrate synthase activity (in both male and females), and greater relative abundance of COX4 (in male mice), and a trend for increased succinate dehydrogenate activity, coupled with increased mTORC1 signaling, we suspect that the DEPDC5 mice may be hypermetabolic. This result has been observed in other models of altered mTORC1 signaling. Knockdown of TSC1 with shRNA (22) was sufficient to increase oxidative capacity of muscle and increase whole-body oxygen consumption as measured via indirect calorimetry. mTORC1 has been shown to regulate mitochondrial biogenesis and activity via 4E-BP1 (15). Increased mTORC1 signaling may increase protein synthesis, which is thought to contribute ∼25% of the resting metabolic rate in mammals (31). Mice may compensate for increased caloric need simply by eating more food. Thus, increased metabolic rate to fuel this protein production may not have been noticeable in the short term, in the context of body mass. The muscle-specific knockout of the GATOR1 protein subunit NPRL2 results induces aerobic glycolysis and stimulates TCA cycle anaplerotic pathways, presumably to produce biosynthetic intermediates induced by chronic activation of mTORC1 (16). Our findings are supportive of that conclusion, but we also provide evidence for increased electron transport chain activity, signaling a distinct role for DEPDC5 in muscle metabolic regulation. In the long term, we suspect that chronic mTORC1 activation and accompanying hypermetabolism may result in muscle and adipose tissue cachexia.
Conclusion
Skeletal muscle-specific induced knockout of DEPDC5 in adult mice increased mTORC1 signaling, promoted hypertrophy, and increased mitochondrial respiratory capacity. However, these adaptations did not improve muscle function. The next step will be to determine whether long-term DEPDC5 depletion in skeletal muscle inhibits autophagy and induces myopathy.
Experimental procedures
Experimental model and animal details
All animal experiments by were approved the University of Texas Medical Branch Institutional Animal Care and Use Committee and experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Experimental Animals.
Mouse lines
We crossed the HSA-MerCreMer (either heterozygous CRE+/− or homozygous Cre+/+) mouse McCarthy et al. (38), officially Tg(ACTA1-cre/Esr1*)2Kesr/J from the Jackson Laboratory (stock no. 025750) (we refer to as CRE) with a homozygote floxed Depdc5fl/fl mouse donated by David Sabatini of MIT (officially, Depdc5tm1c(EUCOMMHmgu)) and then backcrossed the F1 hybrid (double heterozygote) with the DEPDC5fl/fl to produce the experimental genotype, an inducible muscle-specific knockout of DEPDC5:DEPDC5fl/fl × CRE+/− (noted as KO). Our experimental transgenic mice (homozygous for the floxed Depdc5 and was either homo- or heterozygous for CRE) are officially designated as: Depdc5tm1c(EUCOMMHmgu)/Depdc5tm1c(EUCOMMHmgu) Tg(ACTA1-cre/Esr1*)2Kesr/Tg(ACTA1-cre/Esr1*)2Kesr, or Depdc5tm1c(EUCOMMHmgu)/Depdc5tm1c(EUCOMMHmgu) Tg(ACTA1-cre/Esr1*)2Kesr/0. The control mice were mice not treated with tamoxifen (noted as WT) and were carriers of genes from both parental strains, although not necessarily CRE-positive.
Prior to receiving tamoxifen (at ∼6 months of age), the mice had 1 week of activity testing (voluntary wheel running) followed by 1 week of functional testing (rotarod, grip test, and treadmill). Our experimental mice then received one 170-μl injection of tamoxifen (concentration of 0.08 μg/μl tamoxifen dissolved in ethanol and then mixed in PBS) every day for 5 days. After the mice received tamoxifen, we waited 4 weeks and then retested for function over 2 weeks (1 week on the activity wheel and then 1 week for the other measures), followed by sacrifice and downstream testing at ∼6 weeks post-tamoxifen. The control mice underwent identical procedures apart from the tamoxifen injections. We confirmed genotype of the mice and confirmed genetic recombination following the procedures outlined under “Method details” (detailed in Fig. 1 and Fig. S1).
Method details
Genotyping
The mice were genotyped by PCR. We used tissue derived during the ear punch identification procedure to isolate DNA with the Qiagen DNeasy Blood and Tissue kit (p/n: 69506) using the manufacturer's instructions. The DNA concentration was measured using a Nanodrop 2000, and DNA was diluted to a standardized 10 ng/μl for heart and TA, 5 ng/μl for skin (ear punch), and 40 ng/μl for liver. A Bio-Rad CFX96 was used to run RT-PCRs (primers in Table 1).
Table 1.
Genotyping primer sequences
Depdc5 transgene primers detect the presence of the floxed allele (Depdc5tm1c(EUCOMMHmgu)). See Fig. S1 for more details (and a gene map of what the primers detect) or the wildtype allele. The CRE primers detect the presence or absence of the transgene Tg(ACTA1-cre/Esr1*)2Kesr but cannot detect homo- or heterozygosity. LoxPn, Lox primer n; TGfwd, transgene forward; TgRev, transgene reverse; ICFwd, internal control forward; ICRev, internal control reverse.
| Transgene | Primer | Primer sequences (5′ → 3′) |
|---|---|---|
| Depdc5 | LoxP1 | TCC GCA AAG GTT AGG AGC TAT G |
| LoxP2 | CCC TCA TGC CAG CTC AAA CT | |
| LoxP3 | TTG GTT CCC CTG AAA CTG GG | |
| CRE | TGFwd | AGG TGG ACC TGA TCA TGG AG |
| TGRev | ATA CCG GAG ATC ATG CAA GC | |
| ICFwd | CTA GGC CAC AGA ATT GAA AGA TCT | |
| ICRev | GTA GGT GGA AAT TCT AGC ATC ATC C |
The presence of the Depdc5 floxed transgene was determined using a PCR, followed by gel electrophoresis on a 1.5% agarose gel made with 0.0015% ethidium bromide run at 75 V for 2 h. We assured that genetic recombination was occurring in a subset of mice (Fig. 1) (transgene allele shows a band at ∼749 bp). The floxed allele (Depdc5fl/fl) could produce the 1,349-bp (LoxP3+LoxP2 product) band and the 431-bp band (loxP1+LoxP2 product); recombinant allele (KO, with Depdc5 ablated) could produce the 749-bp band only (LoxP3+LoxP2 product); WT allele (WT) could produce the 1,069-bp band (LoxP3+LoxP2 product) and the 368-bp band (loxP1+LoxP2 product). For each PCR, we used 10 μl of Sybergreen, 1 μl of each primer (LoxP1, LoxP2, and LoxP3 for genotyping; or LoxP2 and LoxP3 to detect recombination), 4 μl of DNA standardized to 10 ng/μl, and QC to 20 μl with DEPDC-treated RNase/DNase-free H2O. The PCR protocol was 3 min at 95 °C; then 35 rounds of 30 s at 90 °C → 30 s at 60 °C → 1 min 20 s at 72 °C; then 3 min at 72 °C; and indefinitely at 4 °C. Further details are in Fig. S1.
The presence of the CRE transgene was determined following the PCR protocol published by the Jackson Laboratory, where the transgene melt curve was at ∼86 °C and the mutant band showed up at ∼440 bp (internal control at 324 bp) on a gel (1.5% agarose gel made with 0.0015% ethidium bromide run at 75 V for 2 h). This protocol for the CRE mice did not distinguish between heterozygous or homozygous genotypes.
Tissue collection
Tissue handling procedures have been previously published (32). We carefully removed the EDL and one SOL at sacrifice, keeping them viable for in vitro contractile physiology. One TA and one SOL were both quickly mounted on cork using optimal temperature cutting compound (Tissue-Tek catalog no. 4583) and were then pinned close to optimal length, followed by freezing the muscles in liquid nitrogen–cooled 2-methylbutane (isopentane). Additional muscles were flash-frozen with liquid nitrogen, including the other TA, the gastrocnemius (Gastroc), and the plantaris. All samples were stored at −80 °C.
We homogenized the Gastroc and TA muscles in buffer (50 mm Tris, 250 mm mannitol, 50 mm NaF, 5 mm sodium pyrophosphate (NaO7P4), 1 mm EDTA, 1 mm EGTA, 1% (v/v) Triton X-100, 1 mm DTT, 1 mm benzamidine, 0.1 mm phenylmethylsulfonyl fluoride, and 5 μg/ml soybean trypsin inhibitor (pH 7.4) at a 1:10 ratio (mass, mg: volume, μl) of muscle to buffer. We determined the protein concentration with the Bradford assay (Bio-Rad catalog no. 500-0006) using a 96-well plate reader (Bio-Rad iMARK).
A subset of TA muscles was used to isolate mRNA as detailed previously (9). In brief, we homogenized frozen muscle tissue in Tri-Reagent (Molecular Research catalog no. TR118) and isolated following manufacturer's instruction. The RNA was quantified on a Nanodrop 2000 (Thermo Scientific), with the mean concentration at 330.8 ± 24.3 ng/μl, the mean 260/280 ratio at 1.69 ± 0.020, and the mean 260/230 ratio at 1.98 ± 0.09. Following the manufacturer's instructions, endogenous leftover DNA was removed (DNA-free kit, Applied Biosystems catalog no. AM1906) and cDNA produced using the Bio-Rad iScript kit (catalog no. 170-8891). cDNA was stored at −80 °C until used.
Functional analysis
One researcher assessed all functional outcomes measurements (rotarod, grip test, and treadmill) roughly 1–3 h prior to the dark cycle of the vivarium.
Overall motor function: balance, coordination, and stamina
Mice acclimated on a Panlab LE 8205 rotarod over two practice sessions (32). The practices occurred on consecutive days and consisted of three varied types of trials per session (33). On day 3, three testing trials were completed with the rotarod accelerating from 4 to 40 rpm over 5 min. The best trial was reported. The mice rested for 15 min minimum between trials.
Fore-limb strength
Using a Bioseb GT3 grip tester with a trapeze grip, we tested the mice for upper-limb strength. We have previously published details (32). We reported the best of five attempts.
Endurance/volitional fatigue
Utilizing a PanLab LE 8710 treadmill, we determined endurance level. We have previously published details of this test (32). After 2 days of acclimation training, the mice were tested on a single run on which the starting velocity was 5 cm/s and increased by 1 cm/s every 20 s until volitional fatigue was reached.
Activity level
A subset of mice was singly housed in a cage containing a running wheel (Columbus Instruments) for 7 days to test voluntary wheel running as a proxy for activity level. The number of revolutions per day were counted using the software, then converted into kilometers per day, and averaged over the week as the outcome measure.
Muscle contractile function
We have previously published specific details of this test (32–34). Briefly, the SOL and EDL were carefully extracted and kept viable in oxygenated (95% O2, 5% CO2) Krebs–Ringer buffer held at 25 °C. Suture line (no. 4 gauge) was tied at the myotendinous junctions, with the origin side attached to a force transducer and the distal side to a static clamp. The muscles were suspended between two platinum electrodes.
We stimulated (with hardware: Aurora Scientific: model 6650LR force transducer, dual-mode lever system, hi-power bi-phase stimulator, signal interface; and software: Dynamic Muscle Control version 5.500 and Dynamic Muscle Analysis version 5.300) the muscle with electrical pulses to obtain peak twitch force (Pt), determined optimal sarcomere length (L0, i.e. using the force tension curve to find the muscle length from myotendinous junction to myotendinous junction at Pt) and then peak isometric force (P0) using the force-frequency method. Specific force was determined as P0/PCSA (physiological cross-sectional area). PCSA was determined using the standard formula: PCSA = muscle mass/(muscle length × muscle density); muscle density = 1.06 g/cm3.
Muscle mass
Muscles were blotted dry and weighed prior to being frozen.
Immunohistochemistry
We have previously published these techniques (32). In brief, we sliced muscles into 7-micron sections with a cryostat (Thermo Scientific HM525-NX) and then mounted the sections on slides. To determine fiber type, unfixed slides incubated at room temperature with anti-MHC (1, 2a, and 2b; catalog nos. BA.D5, SC.71, and BF.F3 from Developmental Studies Hybridoma Bank) and anti-laminin (catalog no. L9393, Sigma–Aldrich) antibodies overnight. On the following day, the slides were incubated with isotype- and species-specific fluorescent secondary antibodies (catalog nos. A21242, A21121, A21426, and A21068, Thermo Fisher) and then fixed in methanol. A single blinded researcher assessed fiber type (2× being unstained). To determine muscle fiber type–specific cross-sectional area, mean myofiber, and binned distribution CSA were determined through semi-automated analysis using AxioVision (Zeiss version 4.9.1).
For the succinate dehydrogenase enzymatic staining, we used a previously published protocol (32). In brief, slides with slices of SOL were unfixed and incubated for 1 h of incubation at 37 °C in the following solution: nitro blue tetrazolium (Sigma) and succinate acid disodium (Sigma), dissolved in 0.2 m of PBS. Fibers were determined to be either positive, + (high level of activity), or negative − (lower level of activity), based upon the intensity of the staining.
Microscopy
Images were captured using a Zeiss upright M1 Imager microscope at room temperature 100×, 200×, or 400× total magnification. Blinded image analysis was performed using AxioVision Rel software (version 4.9.1).
Immunoblotting
We have previously published details of these experiments (32, 35). Standard immunoblotting techniques were used to detect protein expression (dilution, antibody) of DEPDC5 (1:750, ABCAM catalog no. ab185565); p-mTOR (S2448) (1:1000, Cell Signaling catalog no. 2971L), mTOR (1:1000, Cell Signaling catalog no. 2972S), p-s6k (Thr-389) (1:1000, Cell Signaling catalog no. 9205), s6k (1:1000, Cell Signaling catalog no. 9202L), p-4EBP1 (Thr-37/46) (1:500, Cell Signaling catalog no. 2855S), EBP1 (1:1000, Cell Signaling catalog no. 9452L), GAPDH (1:2000, Cell Signaling catalog no. 2118), and COX4 (1:1000, Cell Signaling catalog no. 4844). We used a donkey anti-rabbit horseradish peroxidase-conjugated IgG secondary antibody (1:2000 for most, 1:4000 for GAPDH, and 1:8000 for COX4; GE Healthcare ECL catalog no. NA934V) and Super Signal Dura West (Thermo Scientific, catalog no. 80196) was the substrate developing agent.
Quantitative RT-PCR
We performed PCR as previously described (9). Briefly, the primers for mouse Depdc5, with GAPDH and B2M (β2-microgobulin) as the reference genes, were obtained from Bio-Rad PrimerPCR (primer validations in the PrimePCR section at the company website). A Bio-Rad CFX Connect was used to perform the PCR, in triplicate, and the 2−ΔΔCt method was used to calculate the fold change of the KO mRNA expression referenced to the WT. Because the mRNA did not replicate at all via PCR in most KO samples, we arbitrarily set the Ct value at 44 for such runs to calculate the 2−ΔΔCt. Gel electrophoresis was run on a 1.5% agarose gel (0.0015% ethidium bromide) with DEPDC5 PCR product at ∼76 bp.
Mitochondrial function
High resolution respirometry
Mitochondrial respiratory capacity and coupling control were determined in saponin permeabilized myofiber bundles by high resolution respirometry as described previously (36, 37). Briefly, muscle fiber bundles were prepared under magnification in an ice-cold preservation buffer before being weighed and transferred into an Oxygraph-2K high resolution respirometer (Oroboros Instruments, Innsbruck, Austria). Mitochondrial respiration rate was quantified during the sequential titration of substrates (5 mm pyruvate, 2 mm malate, 10 mm glutamate), ADP (5 mm), succinate (10 mm), cytochrome c (10 μm), and finally, the ionphore CCCP (5 μm).
Citrate synthase enzymatic activity assay
Individual soleus samples from control and experimental male and female mice were homogenized in a buffer containing Tris-HCl (125 mm), urea (8 m), and 5% SDS, pH 7. Protease and phosphatase inhibitors (Thermo Scientific no. 78442) were added to the buffer right before homogenization. Protein concentration was measured by the BCA method (Pierce catalog no. 23225). Absorbance measurements were taken at 595-nm wavelength using a microplate reader (iMark, Bio-Rad).
Citrate synthase specific activity was determined using a 96-well plate and a final reaction volume of 100 μl/well/sample. 10 mm stocks of acetyl CoA, DTNB, and oxaloacetate were prepared previously. Acetyl CoA (Sigma–Aldrich catalog no. 10101893001) was dissolved in PBS buffer, pH 7, aliquoted, and stored at −20 °C as recommended by the manufacturer. Stock solutions for DTNB (Thermo Scientific catalog no. 22582) and oxaloacetate (EMD Millipore catalog no. 5000-5) were prepared fresh for the assay in 100% ethanol and running buffer (0.1 m Tris-HCl, pH 8), respectively.
Homogenate samples containing 65 μg of protein were adjusted to a 30-μl volume with running buffer. A reagent mix was prepared by adding the following (per sample): acetyl-coA (3 μl), DTNB (1 μl), and running buffer (61 μl). Samples and reagent mix were loaded onto the wells. To start the reaction 5 μl of oxaloacetate were added to each well with a multichannel pipette. A 30-μl sample from homogenization and running buffers along with a positive control (Cayman catalog no. 701045) were also included in the assay to detect any possible interference with the reagents and test validation. Absorbance measurements were taken every 30 s for 15 min at 415-nm wavelength using the kinetic mode with a microplate reader (Bio-Rad). Citrate synthase specific activity results are expressed as nmol/min/mg and reaction rate results as nmol/min/ml. Corrections for optical path and extinction coefficient were included in the calculations. Two independent experiments were performed using the same homogenates.
Statistical analysis
The data are reported as percentages of change ± standard error or as means ± S.E., where appropriate. We report trends at 0.05 < p < 0.10, and significance was designated as p < 0.05. Statistical tests were run separately for males and females. A 2 × 2 repeated measures analysis of covariance (controlling for body mass at pre- and post-tests) compared functional (rotarod, grip test, treadmill, voluntary wheel running) data changes pre- to post-treatment period between KO and WT. Post hoc testing was the least significant difference test. Student's two-tailed t test was used to determine differences in means for nonrepeated measurements. We used SPSS v24 and v23 (IBM) to analyze the statistic.
Author contributions
T. G. G., C. S. F., and B. B. R. conceptualization; T. G. G., C. S. F., C. P., and B. B. R. resources; T. G. G. formal analysis; T. G. G., C. S. F., and B. B. R. supervision; T. G. G., C. S. F., and B. B. R. funding acquisition; T. G. G. and M. W. validation; T. G. G., C. S. F., C.R.B., T. M., R. M., and N. B. investigation; T. G. G., C. S. F., R. M., C. P., and M. W. methodology; T. G. G., C. S. F., and C. P. writing-original draft; T. G. G., C. S. F., and B. B. R. project administration; T. G. G., C. S. F., C. R. B., T. M., R. M., N. B., C. P., M. W., and B. B. R. writing-review and editing.
Supplementary Material
Acknowledgments
We thank the David Sabatini lab at the Whitehead Institute at the Massachusetts Institute of Technology for the generous donation of the Depdc5 floxed mouse line.
This work was supported by National Institutes of Health NCATS CTSA TL1TR001440 NSRA Fellowship and NIA P30 AG024832 Pilot/Developmental grants (to T. G. G.), NIA R56 AG051267 (to B. B. R.), NIAMS R01 AR072061 (to C. S. F.), NIA T32 AG000270 (to C. R. B.), and NCATSS CTSA KL2 scholar award KL2TR001441 (to C. P.) and by Shriners Hospitals for Children Grant 84090 (to C. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S7.
- mTORC1
- mTOR complex 1
- KO
- knockout
- TA
- tibialis anterior
- CSA
- cross-sectional area
- SOL
- soleus
- EDL
- extensor digitorum longus
- SDH
- succinate dehydrogenase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- PCSA
- physiological cross-sectional area
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic acid).
References
- 1. Dibble C. C., and Cantley L. C. (2015) Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555 10.1016/j.tcb.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bar-Peled L., and Sabatini D. M. (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol. 24, 400–406 10.1016/j.tcb.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kimball S. R. (2014) Integration of signals generated by nutrients, hormones, and exercise in skeletal muscle. Am. J. Clin. Nutr. 99, 237S–242S 10.3945/ajcn.113.068387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang H., Jiang X., Li B., Yang H. J., Miller M., Yang A., Dhar A., and Pavletich N. P. (2017) Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373 10.1038/nature25023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., and Sabatini D. M. (2010) Ragulator–rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bar-Peled L., Chantranupong L., Cherniack A. D., Chen W. W., Ottina K. A., Grabiner B. C., Spear E. D., Carter S. L., Meyerson M., and Sabatini D. M. (2013) A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106 10.1126/science.1232044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wolfson R. L., Chantranupong L., Saxton R. A., Shen K., Scaria S. M., Cantor J. R., and Sabatini D. M. (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway, Science 351, 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chantranupong L., Scaria S. M., Saxton R. A., Gygi M. P., Shen K., Wyant G. A., Wang T., Harper J. W., Gygi S. P., and Sabatini D. M. (2016) The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165, 153–164 10.1016/j.cell.2016.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Graber T. G., Borack M. S., Reidy P. T., Volpi E., and Rasmussen B. B. (2017) Essential amino acid ingestion alters expression of genes associated with amino acid sensing, transport, and mTORC1 regulation in human skeletal muscle. Nutr. Metab. (Lond.) 14, 35 10.1186/s12986-017-0187-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dutchak P. A., Laxman S., Estill S. J., Wang C., Wang Y., Wang Y., Bulut G. B., Gao J., Huang L. J., and Tu B. P. (2015) Regulation of hematopoiesis and methionine homeostasis by mTORC1 inhibitor NPRL2. Cell Reports 12, 371–379 10.1016/j.celrep.2015.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kowalczyk M. S., Hughes J. R., Babbs C., Sanchez-Pulido L., Szumska D., Sharpe J. A., Sloane-Stanley J. A., Morriss-Kay G. M., Smoot L. B., Roberts A. E., Watkins H., Bhattacharya S., Gibbons R. J., Ponting C. P., Wood W. G., et al. (2012) Nprl3 is required for normal development of the cardiovascular system. Mamm. Genome 23, 404–415 10.1007/s00335-012-9398-y [DOI] [PubMed] [Google Scholar]
- 12. Hughes J., Dawson R., Tea M., McAninch D., Piltz S., Jackson D., Stewart L., Ricos M. G., Dibbens L. M., Harvey N. L., and Thomas P. (2017) Knockout of the epilepsy gene Depdc5 in mice causes severe embryonic dysmorphology with hyperactivity of mTORC1 signalling. Sci. Rep. 7, 12618 10.1038/s41598-017-12574-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marsan E., Ishida S., Schramm A., Weckhuysen S., Muraca G., Lecas S., NingLiang N., Treins C., Pende M., Roussel D., Le Van Quyen M., Mashimo T., Kanekoi T., Yamamoto T., Sakuma T., et al. (2016) Depdc5 knockout rat: A novel model of mTORopathy. Neurobiol. Dis. 89, 180–189 10.1016/j.nbd.2016.02.010 [DOI] [PubMed] [Google Scholar]
- 14. Yuskaitis C. J., Jones B. M., Wolfson R. L., Super C. E., Dhamne S. C., Rotenberg A., Sabatini D. M., Sahin M., and Poduri A. (2018). A mouse model of DEPDC5-related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis. 111, 91–101 10.1016/j.nbd.2017.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morita M., Gravel S.-P., Chénard V., Sikström K., Zheng L., Alain T., Gandin V., Avizonis D., Arguello M., Zakaria C., McLaughlan S., Nouet Y., Pause A., Pollak M., Gottlieb E., et al. (2013). mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18, 698–711 10.1016/j.cmet.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 16. Dutchak P. A., Estill-Terpack S. J., Plec A. A., Zhao X., Yang C., Chen J., Ko B., Deberardinis R. J., Yu Y., and Tu B. P. (2018). Loss of a negative regulator of mTORC1 induces aerobic glycolysis and altered fiber composition in skeletal muscle. Cell Rep. 23, 1907–1914 10.1016/j.celrep.2018.04.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weber E. M., Dallaire J. A., Gaskill B. N., Pritchett-Corning K. R., and Garner J. P. (2017) Aggression in group-housed laboratory mice: why can't we solve the problem? Lab Anim. (NY) 46, 157–161 10.1038/laban.1219 [DOI] [PubMed] [Google Scholar]
- 18. Kappel S., Hawkins P., and Mendl M. T. (2017) To group or not to group?: good practice for housing male laboratory mice. Animals (Basel) 7, E88 10.3390/ani7120088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Loo P. L., Van de Weerd H. A., Van Zutphen L. F., and Baumans V. (2004) Preference for social contact versus environmental enrichment in male laboratory mice. Lab. Anim. 38, 178–188 10.1258/002367704322968867 [DOI] [PubMed] [Google Scholar]
- 20. Haizlip K. M., Harrison B. C., and Leinwand L. A. (2015) Sex-based differences in skeletal muscle kinetics and fiber-type composition. Physiology 30, 30–39 10.1152/physiol.00024.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guridi M., Kupr B., Romanino K., Lin S., Falcetta D., Tintignac L., and Rüegg M. A. (2016) Alterations to mTORC1 signaling in the skeletal muscle differentially affect whole-body metabolism. Skeletal Muscle 6, 13 10.1186/s13395-016-0084-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bentzinger C. F., Lin S., Romanino K., Castets P., Guridi M., Summermatter S., Handschin C., Tintignac L. A., Hall M. N., and Rüegg M. A. (2013). Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skeletal Muscle 3, 6 10.1186/2044-5040-3-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maughan R. J. (1984) Relationship between muscle strength and muscle cross-sectional area: implications for training. Sports Med. 1, 263–269 10.2165/00007256-198401040-00002 [DOI] [PubMed] [Google Scholar]
- 24. Sale D. G., Martin J. E., and Moroz D. E. (1992) Hypertrophy without increased isometric strength after weight training. Eur. J. Appl. Physiol. Occup. Physiol. 64, 51–55 10.1007/BF00376440 [DOI] [PubMed] [Google Scholar]
- 25. Alnaqeeb M. A., Al Zaid N. S., and Goldspink G. (1984) Connective tissue changes and physical properties of developing and ageing skeletal muscle. J. Anat. 139, 677–689 [PMC free article] [PubMed] [Google Scholar]
- 26. Matsakas A., Macharia R., Otto A., Elashry M. I., Mouisel E., Romanello V., Sartori R., Amthor H., Sandri M., Narkar V., and Patel K. (2012) Exercise training attenuates the hypermuscular phenotype and restores skeletal muscle function in the myostatin null mouse. Exp. Physiol. 97, 125–140 10.1113/expphysiol.2011.063008 [DOI] [PubMed] [Google Scholar]
- 27. Amthor H., Macharia R., Navarrete R., Schuelke M., Brown S. C., Otto A., Voit T., Muntoni F., Vrbóva G., Partridge T., Zammit P., Bunger L., and Patel K. (2007) Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Natl. Acad. Sci. U.S.A. 104, 1835–1840 10.1073/pnas.0604893104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen C. N., Chen C. J., Graber T. G., Bratten W. M., Ferrington D. A., and Thompson L. V. (2014) Immunoproteasome in animal models of Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 35, 191–201 10.1007/s10974-014-9385-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong P. M., Puente C., Ganley I. G., and Jiang X. (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9, 124–137 10.4161/auto.23323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ábrigo J., Elorza A. A., Riedel C. A., Vilos C., Simon F., Cabrera D., Estrada L., and Cabello-Verrugio C. (2018) Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxid. Med. Cell. Longev. 2018, 2063179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rolfe D. F., and Brown G. C. (1997) Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 77, 731–758 10.1152/physrev.1997.77.3.731 [DOI] [PubMed] [Google Scholar]
- 32. Graber T. G., Rawls B. L., Tian B., Durham W. J., Brightwell C. R., Brasier A. R., Rasmussen B. B., and Fry C. S. (2018) Repetitive TLR-3 activation in the lung induces skeletal muscle adaptations and cachexia. Exp. Gerontol. 106, 88–100 10.1016/j.exger.2018.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Graber T. G., Ferguson-Stegall L., Kim J.-H., and Thompson L. V. (2013) C57BL/6 neuromuscular healthspan scoring system. J. Gerontol. A Biol. Sci. Med. Sci. 68, 1326–1336 10.1093/gerona/glt032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Graber T. G., Kim J.-H., Grange R. W., McLoon L. K., and Thompson L. V. (2015) C57BL/6 life span study: age-related declines in muscle power production and contractile velocity. Age 37, 9773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fry C. S., Lee J. D., Mula J., Kirby T. J., Jackson J. R., Liu F., Yang L., Mendias C. L., Dupont-Versteegden E. E., McCarthy J. J., and Peterson C. A. (2015) Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 21, 76–80 10.1038/nm.3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Porter C., Herndon D. N., Chondronikola M., Chao T., Annamalai P., Bhattarai N., Saraf M. K., Capek K. D., Reidy P. T., Daquinag A. C., Kolonin M. G., Rasmussen B. B., Borsheim E., Toliver-Kinsky T., and Sidossis L. S. (2016) Human and mouse brown adipose tissue mitochondria have comparable UCP1 function. Cell Metab. 24, 246–255 10.1016/j.cmet.2016.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Porter C., Hurren N. M., Cotter M. V., Bhattarai N., Reidy P. T., Dillon E. L., Durham W. J., Tuvdendorj D., Sheffield-Moore M., Volpi E., Sidossis L. S., Rasmussen B. B., and Børsheim E. (2015) Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Endocrinol Metab. 309, E224–E232 10.1152/ajpendo.00125.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCarthy J. J., Srikuea R., Kirby T. J., Peterson C. A., and Esser K. A. (2012) Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skelet. Muscle 2, 8 10.1186/2044-5040-2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.