

Summary
Microglia and astrocytes modulate inflammation and neurodegeneration in the central nervous system (CNS)1–3. Microglia modulate pro-inflammatory and neurotoxic activities in astrocytes, but the mechanisms involved are not completely understood4,5. Here we report that TGF-α and VEGF-B produced by microglia regulate astrocyte pathogenic activities in the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS). Microglia-derived TGF-α acts via ErbB1 in astrocytes to limit their pathogenic activities and EAE development. Conversely, microglial VEGF-B triggers FLT-1 signaling in astrocytes and worsens EAE. VEGF-B and TGF-α also participate in the microglial control of human astrocytes. Furthermore, TGF-α and VEGF-B expression in CD14+ cells correlates with MS lesion stage. Finally, metabolites of dietary tryptophan (Trp) produced by the commensal flora control microglial activation and TGF-α and VEGF-B production, modulating the transcriptional program of astrocytes and CNS inflammation through a mechanism mediated by the aryl hydrocarbon receptor (AHR). In summary, we identified novel positive and negative regulators that mediate the microglial control of astrocytes. Moreover, these findings define a pathway through which microbial metabolites limit pathogenic activities in microglia and astrocytes, suppressing CNS inflammation. This pathway may guide new therapies for MS and other neurologic disorders.
Microglia are reported to express AHR 6,7. To investigate the role of microglial AHR on CNS inflammation we generated CX3CR1-CreERT2 AHRflox/flox mice (CX3CR1-AHR mice) in which the Cx3cr1 promoter drives the expression of Cre recombinase fused to an estrogen ligand-binding domain 8. Upon treatment of CX3CR1-AHR mice with tamoxifen, AHR-expressing peripheral CX3CR1+ cells are replenished from the bone marrow (BM) while microglia remain AHR deficient without impaired survival (Extended Data 1a-d). Microglial AHR deletion worsened EAE, increasing demyelination and CNS monocyte recruitment (Figs. 1a-c); the T-cell response was unaffected (Extended Data 1e,f). AHR deletion in peripheral CX3CR1+ cells achieved by chronic tamoxifen administration to BM chimeras of WT mice reconstituted with CX3CR1-AHR BM 9 led to earlier EAE onset, without affecting maximal scores and disease recovery (Extended Data 1g). AHR deficiency in both CNS-resident and peripheral CX3CR1+ cells accelerated EAE onset and impaired recovery (Extended Data 1h). Collectively, these data suggest that microglial AHR limits EAE.
Figure 1. AHR limits microglial pro-inflammatory transcriptional responses during EAE.
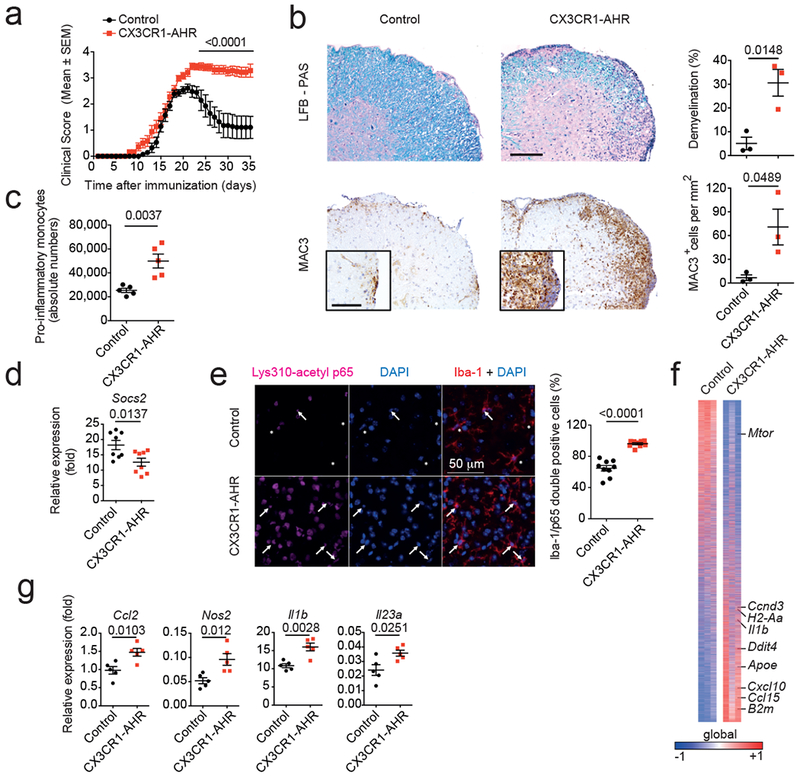
(a) EAE clinical scores in control and CX3CR1-AHR mice (n=10 mice per group). Data are mean ± s.e.m. and representative of two independent experiments. P derived by two-way ANOVA. (b) Representative spinal cord sections from control and CX3CR1-AHR EAE mice stained for Luxol Fast blue (LFB) for demyelination (top), and MAC3 for macrophage infiltration (bottom). Representative of 3 sections of n=3 mice. Right, quantification of demyelination and macrophage infiltration. Data are mean ± s.e.m.; P values determined by two-sided Student’s t-test. (c) Pro-inflammatory monocytes in the CNS of Control and CX3CR1-AHR EAE mice. Data are mean ± s.e.m. and representative of two independent experiments with n = 5 mice per group. P value was determined by two-sided Student’s t-test. (d) Microglial mRNA expression determined by qPCR in control (n = 8) and CX3CR1-AHR (n = 8) EAE mice; P value determined by two-sided Student’s t-test. (e) Lys310-acetyl p65 in Iba-1 positive cells in control and CX3CR1-AHR EAE mice (left). Quantification of Iba-1/p65 double positive cells (right). Data are mean ± s.e.m. representative of two independent experiments with n = 9 mice per group; P value determined by two-sided Student’s t-test. (f) Heat map of 9.957 genes expressed in microglia from control and CX3CR1-AHR mice (n = 3 mice per group). Gene expression is row-centered and log2-transformed, and saturated at levels −0.5 and +0.5 for visualization satisfying a false discovery rate (FDR)<0.1. (g) Micloglial mRNA expression determined by qPCR in control (n = 5) and CX3CR1-AHR (n = 5) EAE mice. Data are mean ± s.e.m. and P values were determined by two-sided Student’s t-test.
NF-κB controls microglial responses during EAE 8, and AHR can limit NF-κB activation in a SOCS2-dependent manner 10,11. The deletion of microglial AHR decreased Socs2 expression and increased NF-κB p65 nuclear localization in spinal cord Iba-1+ myeloid cells during EAE (Figs. 1d,e). Moreover, AHR deletion led to the upregulation of transcripts associated with microglial activation (Apoe, Ddit4 and B2m), inflammation and neurodegeneration (Ccl2, Nos2, Il1b and Il23a) 8,12 (Figs. 1f,g).
Microglia modulate astrocyte phenotype and function 13. Indeed, microglial AHR deletion upregulated the expression of genes in astrocytes associated with inflammation and neurodegeneration, such as Ccl2, Il1b and Nos2 (Figs. 2a,b). Bioinformatic analyses aimed to identify candidate cause/effect relationships between dysregulated transcriptional responses in microglia and astrocytes identified two transcriptional modules in astrocytes, potentially controlled by microglia-produced Tgfa and Vegfb during EAE (Extended Data 2a and Fig. 2c). Similar microglial Vegfb expression levels were detected throughout the CNS during EAE; Tgfa expression was slightly decreased in spinal cord microglia (Extended Data 2b).
Figure 2. AHR-regulated microglial TGF-α and VEGF-B control astrocytes during EAE.
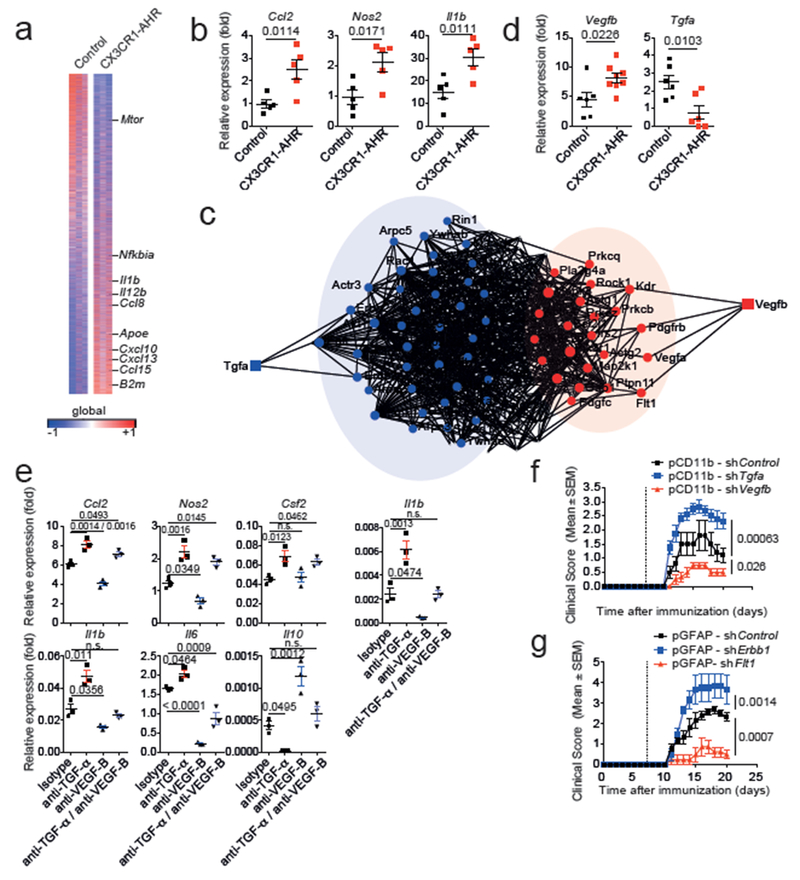
(a) Heat map of 14.823 genes (detected at level 0.1 in at least 2 of 3 samples) expressed in astrocytes from control and CX3CR1-AHR mice. Gene expression is row-centered log2-transformed and saturated at −0.5 and +0.5 levels for visualization satisfying an FDR<0.1; n = 3 independent biological samples per group. (b) mRNA expression determined by qPCR in control and CX3CR1-AHR EAE mice (n = 5 per group). Data are mean ± s.e.m., P values determined by two-sided Student’s t-test. (c) Network diagram of differentially regulated genes in astrocytes and their predicted upstream regulators in microglia (n = 3 independent samples per group). (d) Microglial mRNA expression determined by qPCR in control and CX3CR1-AHR EAE mice. Data are mean ± s.e.m. P values determined by two-sided Student’s t-test; representative of two independent experiments with n = 6 control and n = 8 (Vegfb) or n = 6 (Tgfa) CX3CR1-AHR mice per group. (e) Effect of MCM and blocking antibodies to TGF-α and VEGF-B on gene expression in primary astrocytes determined by pPCR after 24 hrs. Representative of three independent experiments with n = 3 biological replicates. Data are mean ± s.e.m., P values determined by one-way ANOVA followed by Tukey’s post-hoc test. n.s. not significant. (f,g) EAE in C57Bl/6J mice injected with lentiviral knock-down constructs targeting Tgfa, Vegfb or control in microglia (f), or Erbb1, Flt1 or control in astrocytes (g). Data are mean ± s.e.m. and representative of two independent experiments with n=5 mice per group, P values determined by two-way ANOVA.
Microglial AHR deletion decreased Tgfa and increased Vegfb expression during EAE (Fig. 2d). AHR regulates gene expression by direct interactions with target DNA regions, and also by controlling other transcription factors such as NF-κB 14,15. We identified AHR and NF-κB responsive elements (XREs and NREs, respectively) in the Vegfb and Tgfa promoters (Extended Data 2c). AHR deletion increased NF-κB p65 recruitment to NREs in the Vegfb promoter in microglia during EAE (Extended Data 2d). NF-κB p65 transactivated the Vegfb promoter in reporter assays; AHR suppressed this transactivation as well as Vegfb promoter basal activity (Extended Data 2e). AHR was also recruited to XREs in the Tgfa promoter in microglia (Extended Data 2f) and transactivated the Tgfa promoter in reporter studies (Extended Data 2g). These findings suggest that AHR regulates microglial Tgfa and Vegfb expression through its direct effects on the Tgfa and Vegfb promoters, and through its ability to limit NF-κB activation.
We then analyzed the effects of microglial TGF-α and VEGF-B on astrocytes. Microglial AHR inhibition decreased Tgfa and increased Vegfb expression (Extended Data 3a), while it boosted pro-inflammatory Ccl2 and neurotoxic Nos2 expression induced in astrocytes by microglial supernatants (Extended Data 3b-f). Antibody blockade showed that TGF-α and VEGF-B mediated these effects with a relative dominance of TGF-α suppressive effects (Fig. 2e). Recombinant TGF-α decreased pro-inflammatory chemokine (Ccl2, Csf2) and cytokine (Il6) expression induced in murine astrocytes by TNF-α and IL-1β, while it enhanced Il10 expression (Extended Data 3g). Conversely, VEGF-B boosted Ccl2, Csf2 and Nos2 expression in astrocytes. Similarly, VEGF-B pretreatment enhanced the toxicity of astrocyte-conditioned medium (ACM) towards neurons and oligodendrocytes; TGF-α reduced this toxicity (Extended Data 3h,i). VEGF-B pretreatment also enhanced pro-inflammatory monocyte recruitment and microglia activation by ACM; these activities were inhibited by TGF-α (Extended Data 3j,k). These data suggest that TGF-α and VEGF-B control astrocyte functions that contribute to CNS pathology.
To investigate the interplay between microglia and astrocytes in vivo, we knocked down Tgfa and Vegfb expression in microglia using lentivirus-delivered shRNAs expressed under the control of the Itgam (CD11b) promoter. The knock-down did not affect astrocyte numbers or morphology, nor Tgfa and Vegfb expression in CNS-infiltrating monocytes (Extended Data 4a-f). Microglial Tgfa knock-down worsened EAE, while Vegfb knock-down ameliorated it (Fig. 2f). Similar observations were made when the TGF-α or VEGF-B receptors ErbB1 or Flt-1, respectively, were knocked down in astrocytes (Fig. 2g, Extended Data 4a,b,e,g). Of note, VEGF-B administration did not induce demyelination in naive mice (Extended Data 4h), suggesting that VEGF-B synergizes with other factors to boost EAE pathology. Moreover, the knock-down of Tgfa and Vegfb in astrocytes or of their receptors in microglia did not affect EAE (Extended Data 5), supporting a microglia to astrocyte directionality in their effects.
Transcriptional analyses suggested that TGF-α/ErbB1 and VEGF-B/Flt-1 regulate NF-κB in astrocytes (Extended Data 6a-d), known to drive their pathogenic activities during CNS inflammation 10,16–18. Indeed, NF-κB signaling in astrocytes was increased following microglial AHR deletion (Extended Data 2a). Interestingly, VEGF-B boosted astrocytic NF-κB activation; this boost was inhibited by TGF-α (Extended Data 6e). Moreover, NF-κB blockade suppressed the increase in pro-inflammatory gene expression induced by VEGF-B in astrocytes (Extended Data 6f,g). Collectively, these findings suggest that by controlling NF-κB signaling, VEGF-B and TGF-α modulate astrocyte pathogenic activities.
The microbial metabolism of dietary Trp generates AHR agonists such as I3S, which limits astrocyte pathogenic activities and EAE development 10,19,20. To investigate the role of microglial AHR in the control of CNS inflammation by dietary Trp metabolites, we subjected control and CX3CR1-AHR mice to a Trp-depleted diet initiated 21 days after EAE induction (TDD). TDD interfered with disease recovery in control mice (Fig. 3a). Trp or I3S administration ameliorated EAE in control but not in CX3CR1-AHR TDD-fed mice (Fig. 3a), suggesting that microglial AHR participates in EAE amelioration by Trp metabolites. In addition, TDD initiated 14 days after EAE induction worsened disease in control mice and also in mice with AHR-deficient microglia or astrocytes (Extended Data 7a-c), suggesting that Trp metabolites limit CNS inflammation via both, microglial and astrocytic AHR. Indeed, I3S administration initiated 14 days after EAE induction ameliorated disease via AHR in astrocytes and microglia (Extended Data 7b,c). Similar results were obtained when AHR was knocked down in astrocytes or microglia (Extended Data 7d-g). Collectively, these findings suggest that microglial AHR deletion renders astrocytes unresponsive to the anti-inflammatory effects of AHR ligands at later EAE stages.
Figure 3. Trp metabolites control microglia/astrocyte interactions and CNS inflammation.

(a) Clinical scores in control and CX3CR1-AHR mice treated with TDD, TDD+Trp or TDD+I3S from day 21 after EAE induction (n=10 mice per group). Data are mean ± s.e.m. representative of two independent experiments. P values were derived by two-way ANOVA. n.s. not significant. (b) Microglia were isolated and subjected to RNA-sequencing. Heatmap of expressed genes of normalized reads of n = 2 independent samples per group. (c) t-distributed stochastic neighbor embedding (tSNE) plot of RNA-Sequencing data isolated from microglia of mice as in (b). (d) microglial mRNA expression determined by qPCR in EAE mice as in (a). Data are mean + s.e.m. representative of two independent experiments with n = 3 replicates. P values determined by one-way ANOVA followed by Tukey’s post-hoc test. (e) mRNA expression determined by qPCR in microglia from EAE mice as in (a). Data are mean + s.e.m. and representative of two independent experiments with n = 3 replicates. P values determined by one-way ANOVA followed by Tukey’s post-hoc test. (f) Heatmap depicting mRNA expression in astrocytes from EAE mice as in (a), as determined by RNA-seq of normalized reads of n = 2 independent samples per group.
The transcriptional response of microglia in TDD control mice resembled that of AHR-deficient microglia (Fig. 3b,c). Indeed, the microglial expression of Ahr and its target gene Cyp1b1 was suppressed in TDD-fed and CX3CR1-AHR mice, and could be restored by Trp or I3S supplementation in control but not in CX3CR1-AHR mice (Fig. 3d). Moreover, dietary Trp metabolites promoted Socs2 expression, associated to NF-κB regulation by AHR 10,11, and suppressed the microglial expression of NF-κB dependent transcripts such as Tnfa in an AHR-dependent manner (Figs. 3b and Extended Data 8a). In addition, dietary Trp metabolites also regulated microglial Tgfa and Vegfb expression via AHR (Fig. 3e). Accordingly, astrocytes from CX3CR1-AHR and TDD-treated control mice showed increased expression of genes linked to EAE pathogenesis such as Ccl2 and Nos2 (Extended Data 8b and Fig. 3f). Collectively, these findings suggest that dietary Trp metabolites such as I3S limit NF-κB driven pro-inflammatory programs in microglia and suppress their ability to promote pro-inflammatory activities in astrocytes.
We validated our observations using human samples. AHR was activated by I3S and inhibited by the antagonist CH223191 in primary human microglia, as indicated by the expression of AHR and its target CYP1B1 (Fig. 4a). Microglial AHR activation suppressed pro-inflammatory and neurotoxic gene expression (TNFA, IL6, IL12A, NOS2) and boosted anti-inflammatory IL10 expression; AHR activation also promoted TGFA and suppressed VEGFB expression in human microglia (Figs. 4b,c). More importantly, TGF-α and VEGF-B suppressed and boosted, respectively, pro-inflammatory gene expression in primary human astrocytes (Fig. 4d).
Figure 4. VEGF-B and TGF-α control human astrocytes and are expressed by CD14+ cells in MS lesions.
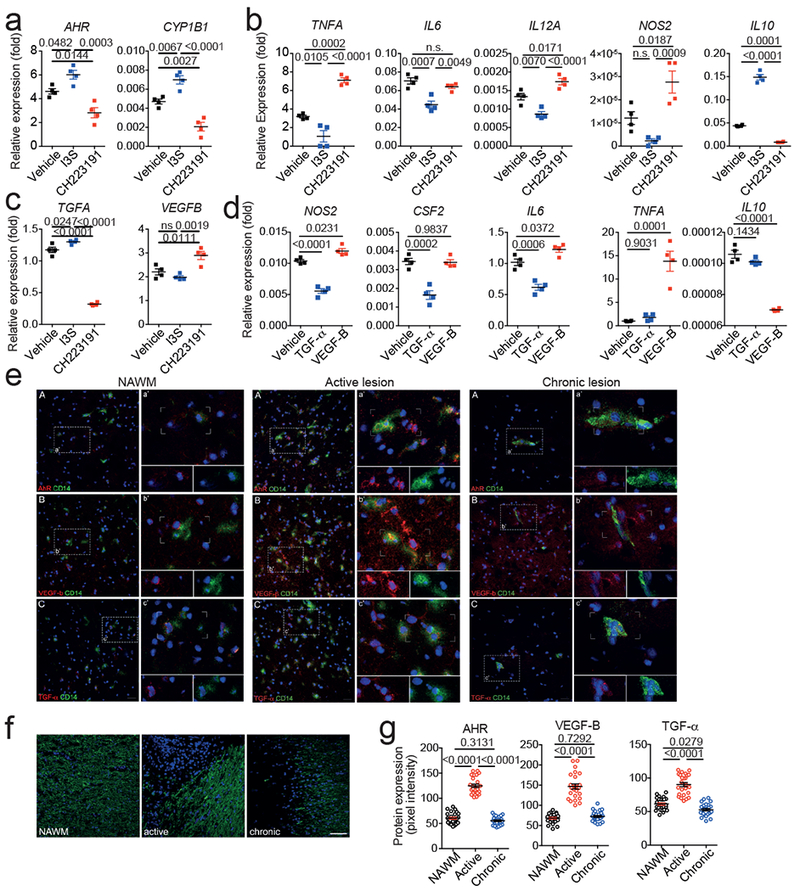
(a-c) mRNA expression determined by qPCR in human microglia activated in the presence of I3S or the AHR antagonist CH223191 24 hours after activation. n=4 biological replicates. Data are mean + s.e.m. representative of three independent experiments. P values derived by one-way ANOVA followed by Tukey’s post-hoc test. (d) mRNA expression determined by qPCR in primary human astrocytes activated in the presence of TGF-α or VEGF-B. n=4 biological replicates. Data are mean + s.e.m. representative of three independent experiments. P values derived by one-way ANOVA followed by Tukey’s post-hoc test. (e) Immunofluorescence staining for AHR (A, red), VEGF-B (B, red), or TGF-α (C, red), CD14 (green), and DAPI (blue) in human brain samples corresponding to normal appearing white matter (NAWM), active, and chronic MS lesions Data are representative of n = 12 fields from three distinct MS brains. Inserts highlight co-expression of AHR and CD14, VEGF-B and CD14, and TGF-α and CD14. (f) Myelin oligodendrocyte glycoprotein (MOG, green) staining in MS tissues. Nuclear staining was done using Hoescht (blue). Representative sections of NAWM, active and chronic lesions from MS patients (n=3). (g) Quantification of AHR, VEGF-B and TGF-α expression in NAWM, active, or chronic lesions in MS tissue. Data shown are representative of n = 25 fields from three distinct MS brains. Data are mean + s.e.m. P values derived by one-way ANOVA followed by Tukey’s post-hoc test.
Finally, we analyzed AHR, TGF-α and VEGF-B expression on MS brain samples. We detected AHR, TGF-α and VEGF-B expression in CD14+ cells (microglia and recruited monocytes) in the normally appearing white matter (NAWM), demyelinated active and chronic MS lesions; the highest AHR, VEGF-B and TGF-α expression was detected in CD14+ cells in MS active lesions (Figs. 4e,f and Extended Data 8c). In chronic MS lesions VEGF-B and AHR expression decreased to NAWM levels, while TGF-α expression was decreased to levels below those detected in NAWM, resulting in a higher VEGF-B/TGF-α ratio than in NAWM (Fig. 4g and Extended Data 8d). These findings suggest that VEGF-B and TGF-α participate in the control of astrocytes by microglia in humans and contribute to MS pathogenesis.
In summary, we found that AHR-controlled microglial VEGF-B and TGF-α regulate astrocyte pathogenic activities during EAE. VEGF-A promotes CNS pathology by multiple mechanisms including angiogenesis induction 21, and microglia 22 and T cell 23 stimulation, but less is known about VEGF-B which does not promote angiogenesis in the CNS 24 and shows neuroprotective effects in some models 25. Our data suggest that Flt-1 activation in astrocytes by VEGF-B produced by microglia and other sources 26 promotes CNS inflammation, identifying VEGF-B/Flt-1 signaling inhibitors as candidate therapeutics for CNS inflammation. Conversely, TGF-α induces astrogliosis and neuroprotective factor production, and increases neuronal survival and axonal growth in multiple contexts, including spinal cord injury (SCI) models 27,28. Indeed, based on the promotion of axon regeneration by reactive astrocytes in SCI models 29, it is tempting to speculate that microglial TGF-α promotes these beneficial astrocyte activities. Future studies should address whether the control of TGF-α/ErbB1 signaling via AHR contributes to the beneficial effects of commensal bacteria on SCI 30. In conclusion, our findings define a gut/brain axis by which metabolites of dietary Trp controlled by the commensal flora act directly on CNS-resident microglia and astrocytes 10 to limit inflammation and neurodegeneration via AHR.
Methods:
Animals.
C57BL/6J mice were obtained from the Jackson Laboratory and were all female. CX3CR1-CreERT2 mice 8 were a kind gift from Steffen Jung (Weizmann Institute of science) and were bred to AHRfl/fl mice. To delete microglial AHR, 4 to 5 week old mice were injected subcutaneously with 4 mg tamoxifen (Sigma) in 200 µl warm corn oil at two time points 48 hours apart. CX3CR1-CreERT2 negative AHRfl/fl mice were used as controls. Four weeks later, EAE was induced. To delete AHR in all CX3CR1-expressing cells, Control and CX3CR1-AHR mice were gavaged weekly with tamoxifen starting from 5 weeks of age. EAE was induced and weekly tamoxifen gavages were continued after EAE induction.
Bone marrow chimera were generated as previously described to minimize irradiation-induced artifacts 31,32. Briefly, 7 weeks old wild type recipient mice were lethally radiated with a dose of 9.5 Gy. 1 day later, mice were administered 5×106 bone marrow cells isolated from donor femora and tibiae by intravenous injection. Donors were CX3CR1Cre neg AHRfl/fl and CX3CR1Cre pos AHRfl/fl mice. Bone marrow recipients were then rested for 3 weeks and thereafter treated with weekly tamoxifen gavages for another 3 weeks; after a total of 6 weeks, EAE was induced as described below. Tamoxifen administration was continued weekly during EAE. All mice were on the C57Bl/6 background and were kept in a pathogen-free facility at the Harvard Institutes of Medicine. All experiments were carried out in accordance with guidelines prescribed by the Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School.
EAE induction and treatment.
EAE was induced in 8 week old mice by subcutaneous immunization with 150 µg MOG35–55 peptide (Genemed Synthesis Inc., San Antonio, Texas) emulsified in complete Freund’s adjuvant (CFA, Difco Laboratories) per mouse, followed by administration of 200 ng pertussis toxin (PTX, List biological Laboratories, Inc.) on days 0 and 2 as described10. Clinical signs of EAE were assessed as follows: 0, no signs of disease; 1, loss of tone in the tail; 2, hind limb paresis; 3, hind limb paralysis; 4, tetraplegia; 5, moribund. All agents were purchased from Sigma-Aldrich.
Isolation of cells from adult mouse CNS.
Mononuclear cells were isolated from the CNS as previously described and astrocytes, monocytes, and microglia were sorted as described before 10,16,17. Isolated CNS cells were stained with fluorochrome-conjugated antibody to CD11b (M1/70, 1:100), CD45 (90, 1:100), Ly6C1 (HK1.4, 1:100), CD105 (N418, 1:100), CD140a (APA5, 1:100), CD11c (N418, 1:100), F4/80 (BM8, 1:50), O4 (O4, Miltenyi Biotec, 1:10), and CD19 (eBio1D3, 1:100). All antibodies were from eBioscience or BD Pharmingen, unless otherwise mentioned. Microglia were sorted as CD11b+ cells with low CD45 expression and low LY6C1 (CD11b+CD45lowLy6C1low), inflammatory monocytes were considered as CD45hiCD11b+Ly6C1hi. Astrocytes were sorted as CD11blowCD45low Ly6C1lowCD105lowCD140alowCD11blowF4/80lowO4lowCD19low after the exclusion of lymphocytes, microglia, oligodendrocytes, and monocytes.
Flow cytometry staining and acquisition.
Mononuclear cell suspensions were prepared as previously described 10,16,17. Antibodies for flow cytometry were purchased from eBioscience or BD Pharmingen and used at a concentration of 1:100 unless recommended otherwise by the manufacturer. Mouse AHR antibody (IC6697G) and mouse FLT-1 antibody (FAB4711A) were from R&D Systems, VEGF-B (RM0008–6E72) and TGF-α (MF9) from Novus Biologicals, EGF Receptor (D38B1) and p-p65 (93H1) from Cell Signaling. Cells were then analyzed on a LSRII or MACSQuant flow cytometer (BD Biosciences and Miltenyi Biotec, respectively). As outlined in the individual figures, Th1 cells were defined as CD3+CD4+IFN-γ+IL-17−IL-10−Foxp3−, Th17 cells as CD3+CD4+IFN-γ−IL-17+IL-10−Foxp3−, Treg cells as CD3+CD4+IFN-γ−IL-17−Foxp3+, microglia as CD11b+CD45lowLy6Clow, and pro-inflammatory monocytes as CD45hiCD11b+Ly6Chi.
RNA-sequencing.
Mice were sacrificed at day 25 after disease induction and astrocytes isolated as described above. RNA was sequenced using the strand-specific TruSeq protocol. High coverage (>50M) strand-specific paired-end 76bp reads were aligned to the mm10/GRCm38 mouse reference genome using TopHat v2.0.11 33. Gene expression levels were estimated for 38922 GenCode Release M2 (GRCm38.p2) mouse gene annotations using Cuffquant and Cuffnorm v2.2.1 quartile normalized FPKMs 33.
Data availability.
RNA sequencing data have been uploaded and is accessible under the access code https://figshare.com/s/c109f251b149b7a843b3.
nCounter gene expression.
50 ng of total RNA was hybridized with reporter and capture probes in custom-made astrocyte-targeted nCounter Gene Expression code set according to manufacturer’s instructions (NanoString Technologies). Data were analyzed using nSolver Analysis software.
qPCR.
RNA was extracted with RNAeasy kit (Qiagen), cDNA was prepared and used for qPCR with the results normalized to gapdh. All primers and probes were from Applied Biosystems. Mouse: Ahr Mm00478932_m1, Aldh1a1 Mm00657317_m1, Aqp4 Mm00802131_m1, Ccl20 Mm01268754_m1, Ccl2 Mm00441242_m1, Ccl8 Mm01297183_m1, Cxcl3 Mm01701838_m1, Cyp1b1 Mm00487229_m1, Erbb1 Mm01187858_m1, Flt1 Mm00438980_m1, Gapdh Mm99999915_g1, Gfap Mm01253033_m1, Il10 Mm00439614_m1, Il12a Mm00434165_m1, Il23a Mm01160011_g1, Il6 Mm00446190_m1, Itgam Mm00434455_m1, Nos2 Mm00440502_m1, Tgfa Mm00446232_m1, Tnfa Mm00443258_m1, Vegfb Mm00442102_m1. Human: AHR Hs00169233_m1, CCL2 Hs00234140_m1, CYP1B1 Hs02382916_s1, ERBB1, FLT1, IL6 Hs00985639_m1, NOS2 Hs01075529_m1, TGFA Hs00608187_m1, TNFA Hs01113624_g1, VEGFB Hs00173634_m1.
T-cell proliferation.
Splenocytes were cultured in X-VIVO 15 medium (Lonza) and were plated for 72 h at a density of 5 × 105 cells per well with increasing concentrations of MOG35–55 peptide. During the final 16 h, cells were pulsed with 1 Ci [3H]thymidine (PerkinElmer), followed by collection on glass fiber filters and analysis of incorporated [3H]thymidine in a beta-counter (1450 MicroBeta TriLux; PerkinElmer). For intracellular cytokine staining, cells were stimulated for 4 h with PMA (phorbol 12-myristate 13-acetate; 50 ng/ml; Sigma), ionomycin (1 μg/ml; Sigma) and monensin (GolgiStop; 2 μM BD Biosciences). After staining of surface markers, cells were fixed and made permeable according to the manufacturer’s instructions (BD Cytofix/Cytoperm Kit (BD Biosciences) or Foxp3 Fixation/Permeabilization (eBioscience).
Primary astrocyte and microglia cultures.
Cerebral cortices from neonatal mice (1–3 days) were dissected, carefully stripped of their meninges, digested with 0.25% trypsin-EDTA and DNAse I (1 mg/ml) for 15 mins, and dispersed to single-cell level by passing through a cell strainer (70 µm). The cell suspension was then cultured at 37 °C in humidified 5% CO2, 95% air on poly-L-Lysine (Sigma) precoated 75 cm2 cell culture flasks. Medium was replaced every 4–5 d. After 7–10 d cells reached confluence and astrocytes and microglia were isolated by mild trypsinization with Trypsin-EDTA (0.06%) as previously described 10,16,17. Cells were >95% astrocytes as determined by staining with GFAP or GLAST, with less than 5% contamination of CD11b+ microglia cells (not shown). Conversely, microglia cultures stained CD11b+CD45lowLy6C1low >95%. After the isolation procedure, cells were further plated as required for the specific experiments. Concentrations of agents were 100 ng/ml for LPS (Sigma), 50 µg/ml for poly(I:C), 100 ng/ml for IL-1β, 50 ng/ml for TNF-α, 0.1 ng/ml TGF-α, 10 ng/ml VEGF-B (all R&D Systems), 50 µg/ml 3-Indoxyl-sulfate (Sigma), 100 nM NF-kB Blocker Bengamide B (Tocris). Unless otherwise indicated, RNA was isolated 24 hours after start of treatment. For Western Blot, cells were pretreated with I3S or vehicle for 24 h, thereafter LPS was added and protein prepared after 2 h.
Plasmids.
Constructs encoding p65, AHR, pTgfa-Luc, pVegfb-Luc, as well as pTK-Renilla were obtained from Addgene. The pLenti-GFAP-EGFP-mir30-shAct1 vector was a gift from Guang-Xian Zhang 34. The pLenti-CD11b-EGFP-mir30-shRNA was also provided by Guang-Xian Zhang, who generated it by exchanging the GFAP promoter with the CD11b promoter.
In vivo knockdown with shRNA lentivirus.
shRNA sequences against ahr, tgfa, vegfb, erbb1 or flt1 and a non-targeting control shRNA were cloned into pLenti-GFAP-EGFP-mir30-shRNA or pLenti-CD11b-EGFP-mir30-shRNA using the following validated shRNA sequence against Ahr (5’-CCGGCATCGACATAACGGACGAAATCTCGAGATTTCGTCCGTTATGTCGATGTTTTTG-3’), Tgfa (5′-CCGGTCGTCAGGATGCGTGTCTTATCTCGAGATAAGACACGCATCCTGACGATTTTTG-3’), Vegfb (5′-CCGGGCCAATGTGAATGCAGACCAACTCGAGTTGGTCTGCATTCACATTGGCTTTTTG-3’), Erbb1 (5′-CCGGGCTGGATGATAGATGCTGATACTCGAGTATCAGCATCTATCATCCAGCTTTTTG-3’) or Flt1 (5’-CCGGCGTGACCTTTAATCGTGCTTTCTCGAGAAAGCACGATTAAAGGTCACGTTTTT-3’) as described 16. Lentivirus particles were generated by transfecting HEK-293FT cells (Invitrogen) with the pLenti-GFAP-EGFP-mir30-shRNA or pLenti-CD11b-EGFP-mir30-shRNA vector and the ViraPower Packaging mix (helper plasmids pLP1, pLP2, pLP/VSV-G, Invitrogen). Supernatants were collected, filtered through a 0.45-μm PVDF filter, and concentrated overnight with the Lenti-X concentrator kit (Clontech). The viral titer was determined using the Lenti-X qRT-PCR titration kit (Clontech). For in vivo knock-down immunized mice were anesthetized at indicated time points, positioned in a Kopf Stereotaxic Alignment System and injected with 107 IU of respective virus using a Hamilton syringe 0.44 mm posterior to the bregma, 1.0 mm lateral to it and 2.3 mm below the skull surface. The injection system was retracted slowly, skin incisions closed carefully by surgical sutures, mice allowed to wake up in a cage pre-warmed with a red light and mice checked twice daily thereafter.
Assessment of toxicity towards neurons and oligodendrocytes.
N2A Neuronal cells (ATCC CCL-131, ATCC, Manassas) or mouse oligodendrocytes (Celprogen, 11004–02) were grown in 96 Well-plates and pre-activated with mIFN-γ (100 ng/ml, R&D Systems) for 24 h. Thereafter, medium was supplemented after extensive PBS washes with ACM. Cytotoxicity was determined by quantifying LDH release (CytoTox 96® Non-Radioactive Cytotoxicity Assay, Promega) after 24 h as suggested by manufacturer’s protocol.
Monocyte migration assay.
Splenic monocytes from WT mice were pre-enriched by CD11b beads (Miltenyi) and sorted for F4/80+SSClowLy6Chi. These monocytes were seeded in the upper chamber of a 24-well cell culture insert with 5 μm pore-size (Corning) containing ACM. Migrating monocytes were quantified in the lower chamber after 3 h.
Microglia polarization assays.
WT microglia were co-cultured with astrocytes pre-treated with TGF-α or VEGF-B and extensively washed. After 24 hours, microglia were re-isolated, RNA was isolated, transcribed and subjected to qPCR analysis.
Subcellular fractionation and immunoblot analysis.
In vitro microglia cultures were treated as indicated in specific experiments, subcellular fractions generated using Cell Fractionation kit (Cell Signaling) and 10 µg of nuclear and cytoplasmic fractions were separated by 4–12% Bis-Tris Nupage gels (Invitrogen, USA) and transferred onto PVDF membranes (Millipore). As primary antibodies rabbit anti-GAPDH mAb (14C10, Cell Signaling), anti-Histone H3 rabbit polyclonal Ab (EMD Millipore), anti-NF-κB p65 rabbit mAb (D14E12, Cell Signaling) were used, followed by goat anti-rabbit IgG HRP linked AB (7074S, Cell Signaling). All antibodies were used at a dilution of 1:1.000. Blots were developed using the SuperSignal West Femto Maximum sensitivity kit (Thermo Scientific / Life Technologies). Data quantification was done using Image J software 1.48v (NIH) and specific signals normalized to GAPDH (cytoplasm) or Histone 3 (nucleus).
Chromatin immunoprecipitation (ChIP).
Cells were cross-linked with 1% paraformaldehyde and lysed with 350 µl lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1) containing 1× protease inhibitor cocktail (Roche Molecular Biochemicals, USA). Chromatin was sheared by sonication and supernatants were collected after centrifugation and diluted in ChIP incubation buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0). 10 µg of antibody was prebound for 6 h to protein A– and protein G–Dynal magnetic beads (Invitrogen, USA) and washed three times with PBS plus 1% BSA, and then added to the diluted chromatin and immunoprecipitated rotating overnight. The magnetic bead-chromatin complexes were then washed 3 times in RIPA buffer (50 mM HEPES (pH 7.6), 1 mM EDTA, 0.7% Sodiumdeoxycholate, 1% NP-40, 0.5 M LiCl) followed by 2 times with TE buffer. Immunoprecipitated chromatin was then extracted with 1% SDS, 0.1 M NaHCO3 and heated at 65 °C for 8 h to reverse the paraformaldehyde cross-linking. DNA fragments were purified with a QIAquick DNA purification kit (Qiagen, USA) and analyzed using the SYBR Green real-time PCR kit (Takara Bio Inc., USA). Anti-AHR (BML-SA210, Enzo Life Sciences, USA), anti-NF-κB p65 (D14E12) XP Rabbit mAb (Cat. #8242, Cell Signaling Technology, USA), and rIgG isotype control were used as indicated in specific experiments. The following primer pairs were used: VEGFB_NFKb1: forward 5’-TCTGTGGCATAGAAACCCAAAG-3’, reverse 5’-AC CCTAAGTCACTGGCTGTC-3’, VEGFB_AHR1: forward 5’-ACCTTCTTCACAGGACAG CC-3’ and reverse 5’-AGTCTCCGAACTCTGGTGTC-3’, VEGFB_AHR2: 5’-GAGTTA ACTGCAATTCCTTCACA-3’and reverse 5’-CTGGAGGGTGGTGCTGAAG-3’, VEGFB_ NFK2/AHR3: forward 5’-TTCATTGGTCCTCTCCCTGC-3’ and reverse 5’-CAGGGGAA AGGGGACACAC-3’, VEGFB_AHR3+4: forward 5’-GTCCCCTTTCCCCTGCAG-3’ and reverse 5’AGAGGCTCATGTGACCTAAACA-3’, TGFa_AHR1: forward 5’-GCCAAGG GAGCATGAACTAG-3’ and reverse 5’-GATGCTCAAAGTTTCAGAGTTGA-3’, TGFa_ AHR2: forward 5’-AGGAGAGGGGTCAGTCTGAT-3’ and reverse 5’-AGAGGG AAAC ACAAGAAGGGA-3’, TGFa_AHR3: forward 5’-GACTCAGAGTGGGGCCAG −3’ and reverse 5’-GAGTCGCTCAGGATCCAGTC-3’.
Human primary astrocytes and microglia.
Human fetal astrocytes and microglia were isolated as previously described 16,35 from human CNS tissue from fetuses at 17–23 weeks of gestation obtained from the Human Fetal Tissue Repository (Albert Einstein College of Medicine) and from the University of Washington Birth Defects Research Laboratory (BDRL, Project Number 5R24HD000836–51) following Canadian Institutes of Health and NIH Research–approved guidelines. Primary human astrocytes and microglia were treated with hVEGF-B (0.1 ng/ml, R&D Systems), hTGF-α (0.1 ng/ml, R&D systems), or vehicle, poly(I:C) (10 mg/ml) with or without 3-indoxyl-sulfate (50 µM, Sigma) or untreated (control) as indicated in the respective figures. After 24 h, total RNA was isolated, transcribed and subjected to qPCR.
Immunohistochemistry and astrocyte morphometry.
Mice were anesthetized (i.p. 100 mg ketamine and 5 mg xylazine per kg body weight) and transcardially perfused with PBS followed by 4% paraformaldehyde in PBS. CNS and other organs were postfixed for 4–6 h at 4 °C, washed in PBS and incubated in 30% sucrose in PBS at 4 °C until fully enriched. Samples were embedded in OCT (Tissue-Tek) for frozen sectioning on a cryostat (Leica). All the stainings were performed on 30 μm thick transversal spinal cord sections. The sections were permeabilized in blocking solution (0.5% Triton-X 100, 5% bovine serum albumin (BSA) + 5% normal donkey serum + 0,1% NaN3 in PBS) for 1 hr at room temperature. Primary antibodies were dissolved in blocking solution and incubated overnight at 4°C with the following primary antibodies: Lys310-acetyl p65 (ab52175, Abcam), Sox9 (AF3075, R&D, 1:200), GFAP (RBK037, Zytomed, 1:5.000), Iba-1 (ab178846, Abcam, 1:500), DAPI (1:5.000). Conjugated secondary antibodies used were Donkey anti-Rabbit Alexa Fluor 647 (Invitrogen A-31573), Donkey anti-Rabbit Alexa Fluor 568 (Invitrogen A-10042) and Donkey anti-Goat Alexa Fluor 488 (Life Technologies A11055) for 2 hrs at room temperature. TUNEL staining was performed using the In Situ Cell Death Detection Kit, TMR red (Roche, Ref.12156792910). Slices were mounted with ProLong Diamond Antifade Mountant (Life Technologies, P36961).
For immunohistochemistry with DAB, after harvesting as described above, spinal cords were cut transversally, embedded in paraffin blocks, cut in 4 μm thick slices in a microtome, cooked 40 mins in citrate buffer at 98°C using a vapor cooker for antigen retrieval, incubated one hour using 10% normal goat serum (SouthernBiotech, 0060–01) in PBS as blocking buffer, incubated overnight at 4°C using primary rat anti-Mac3 antibody (BD-Pharmingen Biosciences, 553322) diluted 1:200 in blocking buffer, incubated biotin-conjugated goat anti-rat antibody (SouthernBiotech, 3050–08) 45 mins at room temperature diluted 1:1.000 in blocking buffer as secondary antibody and finally incubated for 45 mins at room temperature with streptavidine-HRP (SouthernBiotech, 7100–05) diluted 1:1.000 in PBS. The samples were washed four times in PBS+0.1% Triton X-100 between each step. The DAB reaction was run for 5 minutes using the EnVision Flex kit (Dako). Slides were counterstained with Gill’s Hematoxilin, dehydrated trough an ethanol and xylol gradient and mounted in Vitro-Clud (R. Langenbrinck, 04–0001) using a Leica CV5030 system. All images were taken with a Keyence BZ-9000 microscope. For astrocyte reconstruction a Leica Confocal SP8 microscope was used. The Imaris 9.0.2 Software was used to reconstruct and get the morphometry data.
In vivo demyelination assay.
Naive mice were anaesthesized and injected stereotaxically into the corpus callosum (1.4 anterior, 1.0 lateral, 2.1 mm deep) with 2 µl of Lysolecithin (1% w/v, Sigma), VEGF-B (500 ng, R&D Systems), or PBS. Mice were sacrificed 6 days later and subjected to myelin staining as described below.
MS tissue and Immunofluorescence.
Brain tissue was obtained from untreated individuals with clinically diagnosed and neuropathologically confirmed MS, and healthy controls as previously described 10,16. All MS individuals and controls, or their next of kin, had given informed consent for autopsy and use of their brain tissue for research purposes. All the procedures were performed in accordance with local Institutional Review Board guidelines. MS samples were processed and immunostained as previously described2. Briefly, sections were thawed, fixed, washed and blocked with donkey serum 10%. Sections were then incubated overnight at 4°C with antibodies against AHR (rabbit anti AHR, Enzo Life Sciences), CD14 (mouse anti human CD14, 325602, Biolegend), TGF-α (ab112030, Abcam) and VEGF-β (ab185696, Abcam). After washes the samples were incubated at room temperature for 40 minutes with the secondary antibodies (Donkey anti rabbit RRX and donkey anti mouse Alexa 488, Jackson ImmunoResearch). Imaging was performed using a Leica SP5 confocal microscope and the Leica LAS AF software. Images were processed using Adobe Photoshop CS2. For imaging analysis all the data were acquired using the same settings, which were originally standardized on NAWM sections. The degree of co-localization of CD14 with AHR, TGF-α, and VEGF-B was determined using the Volocity software from Perkin Elmer. The overlap coefficient is expressed in percentage where 100% represents the maximum degree of co-localization and 0% denotes no co-localization.
RNA-Seq Data Processing.
RNA-Seq data was analyzed using DESeq2. Gene expression with 0 counts and low expression were removed before differential analysis. Low expressed were filtered out by DESeq’s independent filtering, which removes genes in the lowest 40% quantile of mean normalized counts. Differential genes were selected with FDR < 0.1.
Heatmaps.
Heatmaps were generated with Gene-E program, and the z-scores were calculated for each gene-row using the mean expression of biological replicates.
tSNE plots.
tSNE plot was created using R and the Rtsne package, with parameters perplexity = 1, max iterations = 3000. The mean average of replicates and the top 500 ranked genes for TDD +TRP and TDD in Control mice (n = 3) was taken and the final plot was generated using ggplot.
Ingenuity pathway analysis:
To determine significant pathways, differentially expressed genes that passed FDR < 0.1 were uploaded and analyzed using Ingenuity® Pathway Analysis (IPA) tool. P-values of canonical signaling pathways were calculated using Fischer’s Exact Test. The NF-κB network diagram was generated using IPA.
Network Diagram.
Network diagram for protein-protein interactions was visualized with NetworkAnalyst, (http://www.networkanalyst.ca), using the STRING Interactome database (Confidence score cutoff = 900). Minimum network displaying interacting mediators and molecules were color-coded based on associated pathways for VEGF-B and TGF-α.
Statistical analysis.
Statistical analyses were performed with Prism software (GraphPad), using the statistical tests indicated in the individual figure legends. No samples were excluded. The investigators were blinded as to the treatment of mice in individual experiments. P values of <0.05 were considered significant. All error bars represent s.e.m. or s.d. as noted in the individual figure legends. Unless otherwise stated, 3 independent experiments were used for all assays, and displayed figures are representative.
Extended Data
Extended Data 1. Contribution of AHR in CNS resident and infiltrating immune cells during EAE.

(a) qPCR of indicated genes from microglia, splenic macrophages, and astrocytes from control and CX3CR1-AHR mice on day 28 after EAE induction. Data are mean ± s.e.m. of n = 8 independent samples per group. P values were determined by two-sided Student’s t-test. (b) Flow cytometry analysis of AHR expression in microglia, monocytes and astrocytes from Control and CX3CR1-AHR mice 21 days after EAE induction. Thin line depicts isotype control, thick line AHR staining, and numbers indicate percentage of AHR positive cells. Representative of stainings of n=3 mice per group. (c) Spinal cord samples from naïve Control and CX3CR1-AHR mice were stained for Iba-1 and DAPI and Iba-1+ microglia/mm2 were determined. n=5 mice per group. Data are mean ± s.e.m. and P value was determined by two-sided Student’s t-test. n.s. not significant. (d) TUNEL staining in Iba-1+ microglia in spinal cord sections of Control and CX3CR1-AHR mice as in (c). For the positive control, slides were cooked at 98°C in Citrate buffer during 60 minutes using a vapor cooker. Solid arrows show TUNEL positive microglia. Representative of n = 5 independent experiments. (e) Number of CNS-infiltrating (top) and splenic T-cells (bottom), and splenic pro-inflammatory monocytes (bottom) as determined by flow cytometry. n = 5 samples per group for CNS, n = 4 samples per group for spleen. Data are mean ± s.e.m. and P values were determined by two-sided Student’s t-test. (f) Proliferation assay from splenocytes isolated on day 28 of the experiment (Data are mean ± s.e.m. of n=4 biologically independent samples per group, representative of two independent experiments). (g) Bone marrow chimera were generated using WT mice irradiated as recipients, reconstituted with Control or CX3CR1-AHR bone marrow. Recipients of bone marrow were then rested for 3 weeks and thereafter treated with weekly tamoxifen gavages (4 mg) for another 3 weeks; after a total of 6 weeks, EAE was induced and tamoxifen administration continued weekly during EAE. Left, flow cytometry analysis of AHR expression in microglia and monocytes 21 days after EAE induction. Thin line depicts isotype control, thick line AHR staining, and numbers indicate percentage of AHR positive cells. Representative of stainings of n=3 independent mice per group. Right, EAE clinical course in bone marrow chimera mice. Data are mean ± s.e.m. and P values were determined by two-way ANOVA of n = 4 mice per group. (h) Control and CX3CR1-AHR mice were treated with with oral tamoxifen weekly starting from 5 weeks of age. EAE was induced at 8 weeks under continuation of weekly tamoxifen administration. Left, intracellular FACS staining for AHR in microglia and monocytes from at day 21 of EAE. Representative of stainings of n=3 independent mice per group. Right, clinical course of control and CX3CR1-AHR bone marrow chimera mice. Data are mean ± s.e.m. and P values were determined by two-way ANOVA of n = 4 mice per group.
Extended Data 2. Topical and molecular regulation of TGF-α and VEGF-B.
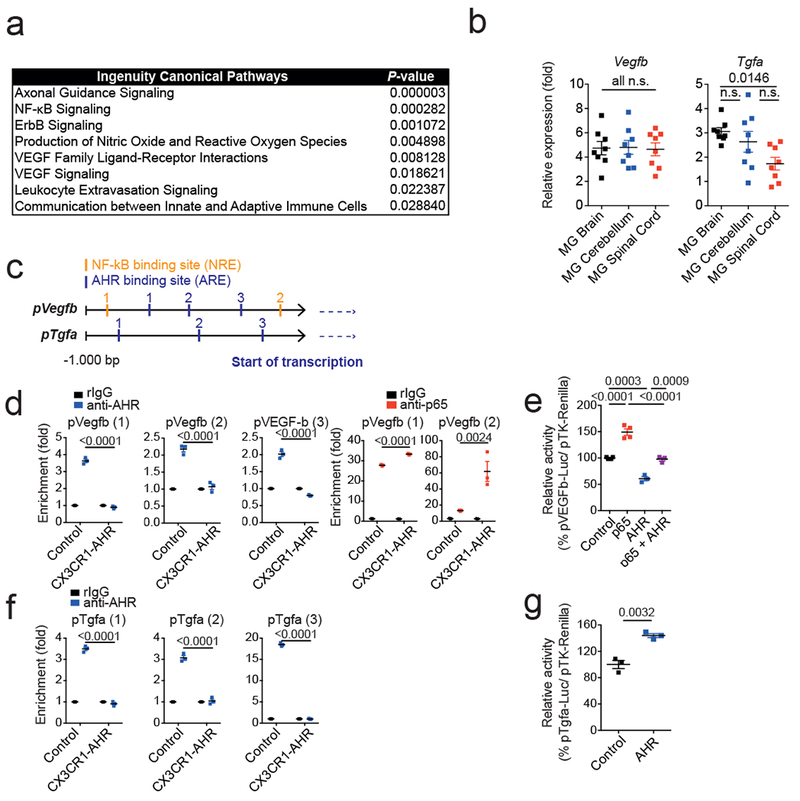
(a) Ingenuity pathway analysis of differentially regulated pathways in astrocytes from n = 3 control vs CX3CR1-AHR mice per group during EAE. (b)Tgfa and Vegfb expression determined by qPCR in microglia from brain, cerebellum and spinal cord 21 days after EAE induction (left). Data are mean ± s.e.m. and P values were determined by one-way ANOVA followed by Tukey’s post-hoc test of n = 8 mice per group. (c) Predicted NF-κB and AHR responsive sites (NREs and XREs, respectively) in Vegfb and Tgfa promoters. (d) Microglia were isolated by FACS sorting from control and CX3CR1-AHR mice during EAE. Ex vivo ChIP assay of NF-kB p65 or AHR binding to predicted binding sites in Vegfb promoter. Data are mean ± s.e.m. and P values were determined by one-way ANOVA followed by Tukey’s post-hoc test of n = 3 mice per group. Representative of 2 independent experiments. (e) Reporter assay using a construct in which the Vegfb promoter controls luciferase expression (pVegfb-Luc); luciferase activity in HEK293 cells 24 hrs after transfection with pVegfb-Luc, pTK-Renilla, and plasmids expressing AHR or NF-κB p65. Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 4 biological replicates. (f) Ex vivo ChIP assay as in (d) for AHR binding in Tgfa promoter. Data are mean ± s.e.m. and P values were determined by one-way ANOVA followed by Tukey’s post-hoc test of n = 3 mice per group. Representative of 2 independent experiments. (g) Reporter assays using a construct in which the Tgfa promoter controls luciferase expression (pTgfa-Luc); luciferase activity in HEK293 cells 24 hrs after transfection with pTgfa-Luc, pTK-Renilla, and plasmids expressing AHR or Control. Data are mean ± s.e.m. and P values were determined by two-sided Student’s t-test. Representative of 2 independent experiments with n = 3 biological replicates.
Extended Data 3. TGF-α and VEGF-B are regulated by AHR in highly purified astrocytes and microglia.
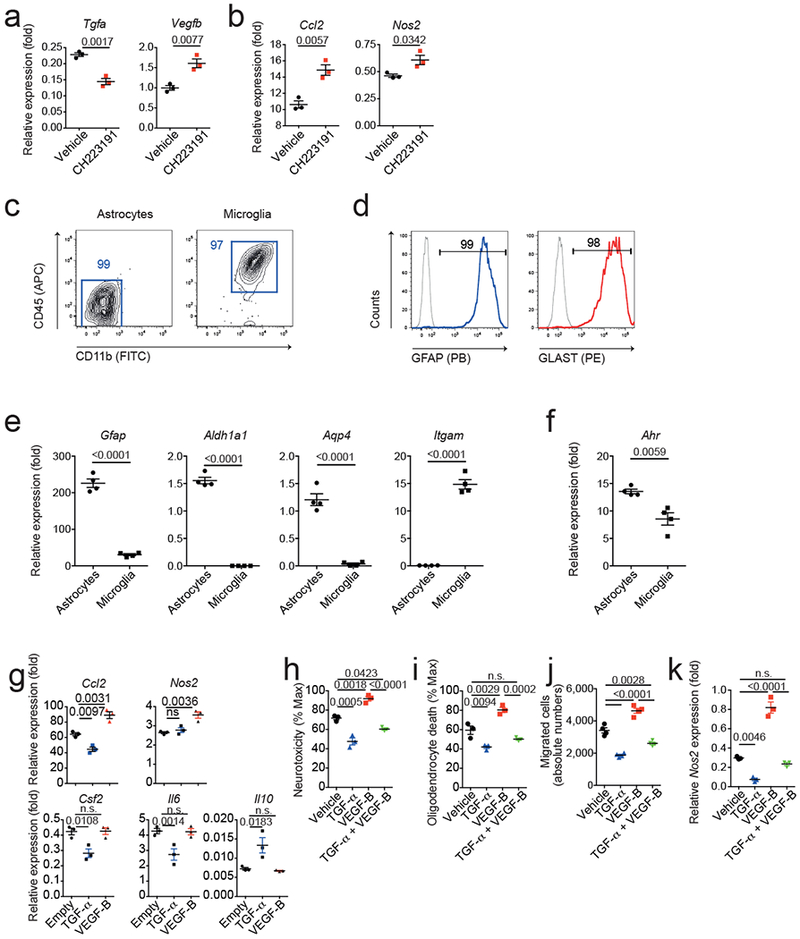
(a,b) Murine microglia were activated with LPS in the presence or absence of the AHR inhibitor CH223191. 24 hours later, activation medium was removed and substituted with fresh medium after extensive washes. 48 hours later, microglia conditioned medium (MCM) was harvested and applied to cultures of primary astrocytes. (a) Gene expression in microglia 24 hours after activation in the presence or absence of CH223191. (b) Gene expression in astrocytes after 24 hours exposure to MCM. Data are mean ± s.e.m. and P values were determined by two-sided Student’s t-test. Representative of 2 independent experiments with n = 3 biological replicates. (c) Representative FACS stainings for CD11b and CD45 in primary astrocyte and microglia cultures. Numbers indicate percentages in respective gate. Representative of n = 3 independent experiments. (d) Representative FACS stainings for GFAP and GLAST in astrocyte cultures as in (b). Representative of n = 3 independent experiments. (e,f) qPCR analysis of mRNA expression in astrocyte and microglia cultures. n=4 independent cultures, Data are mean ± s.e.m. and P values were determined by two-sided Student’s t-test. Representative of 2 independent experiments with n = 4 biological replicates. (g) Effect of TGF-α and VEGF-B on gene expression in primary astrocytes activated with TNF-α and IL-1β, determined by pPCR after 24 hrs. Representative of three independent experiments with n = 3 biological replicates. Data are mean ± s.e.m., P values determined by one-way ANOVA followed by Tukey’s post-hoc test. (h,i) Primary murine astrocytes were activated with TNF-α and IL-1β and treated with TGF-α or VEGF-B. 24 hours later, culture medium was substituted by fresh medium after extensive washes. 48 hours later, ACM was added to mouse neurons (h) and oligodendrocytes (i) in culture, and cytotoxicity was determined by quantifying lactate dehydrogenase (LDH) release after 24 hrs. n=3 biological replicates. Data are mean + s.e.m. representative of two independent experiments. P values determined by one-way ANOVA followed by Tukey’s post-hoc test. n.s. not significant. (j) CD11b+Ly6Chi monocyte migration assay performed using ACM from astrocytes activated in the presence of TGF-α or VEGF-B. n=4 biological replicates. Data are mean + s.e.m. representative of two independent experiments. P values determined by one-way ANOVA followed by Tukey’s post-hoc test. n.s. not significant. (k) qPCR analysis for Nos2 expression in microglia co-cultured with astrocytes activated in the presence of TGF-α or VEGF-B. n=3 biological replicates. Data are mean + s.e.m. representative of two independent experiments. P values determined by one-way ANOVA followed by Tukey’s post-hoc test. n.s. not significant.
Extended Data 4. Phenotypical and functional effects of knock-down of microglial TGF-α and VEGF-B.
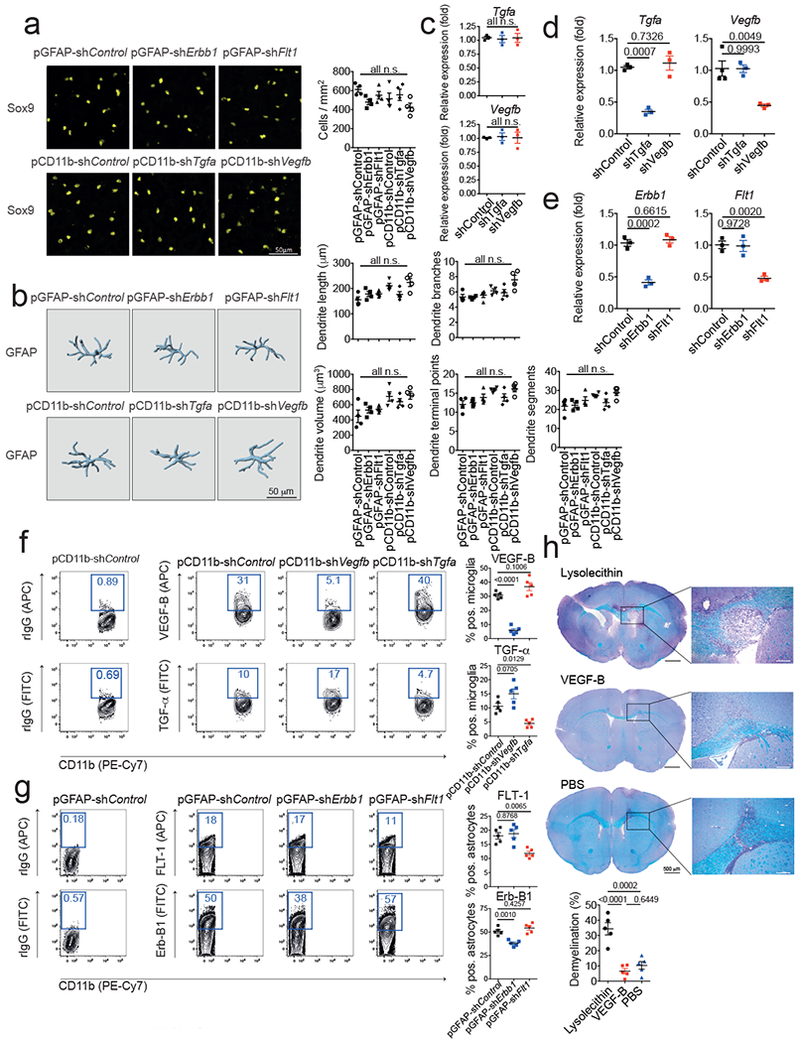
(a) Quantification of astrocyte numbers in spinal cord sections of knock-down mice. Sox9 positive astrocytes per mm2 were quantified in spinal cord sections of n=4 mice per group. n.s. (not significant) as determined by one-way ANOVA followed by Tukey’s post-hoc test. (b) IMARIS reconstruction of GFAP+ astrocytes in spinal cord sections as in (a) and quantification of dendrite length, branches, volume, terminal points, and segments of n=4 mice per group. n.s. (not significant) as determined by one-way ANOVA followed by Tukey’s post-hoc test. (c,d) qPCR analysis of Tgfa and Vegfb expression in sorted CNS-infiltrating inflammatory monocytes (c) and microglia (d) from mice injected with pCD11b-shControl, pCD11b-shTgfa, and pCD11b-shVegfb 7 days after EAE induction. Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 3 biological replicates. (e) qPCR analysis of Erbb1 and Flt1 expression in mice injected with pGFAP-shControl, pGFAP-shErbb1, and pCD11b-shFlt1 7 days after EAE induction. Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 3 biological replicates. (f) Flow cytometry analysis of VEGF-B and TGF-α expression in microglia from mice injected with pCD11b-shControl, pCD11b-shTgfa, and pCD11b-shVegfb 7 days after EAE induction (Left). Quantification of VEGF-B and TGF-α positive microglia in n=5 mice per group (Right). Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 5 biological replicates. (g) Flow cytometry analysis of FLT-1 and Erb-B1 expression in astrocytes from mice injected with pGFAP-shControl, pGFAP-shErbb1, and pCD11b-shFlt1 7 days after EAE induction (Left). Quantification of FLT-1 and Erb-B1 positive microglia in n=5 mice per group (Right). Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 5 biological replicates. (h) Naïve mice were injected with Lysolecithin, VEGF-B, or PBS into the corpus callosum by stereotaxic injection and 6 days later, brains were analyzed by myelin staining. Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 5 biological replicates.
Extended Data 5. Directionality of TGF-α and VEGF-B signaling during EAE.

(a,b) EAE development in wild type mice injected with (a) pGFAP-shControl, pGFAP-shErbb1 and pCD11b-shFlt1, or (b) pCD11b-shControl, pCD11b-shTgfa and pCD11b-shVegfb. Clinical course n=5 mice per group. Representative of 2 independent experiments with n=5 mice per group. (c) Flow cytometry analysis of TGF-α and VEGF-B expression in astrocytes as in (a) (Left). Quantification of cytokine positive astrocytes (Right). Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 4 biological replicates. (d) Flow cytometry analysis of FLT-1 and Erb-B1 expression in microglia as in (b) (Left). Quantification of surface receptor expression of microglia (Right). Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 4 biological replicates.
Extended Data 6. Regulation and transcriptional effects of TGF-α and VEGF-B during EAE.
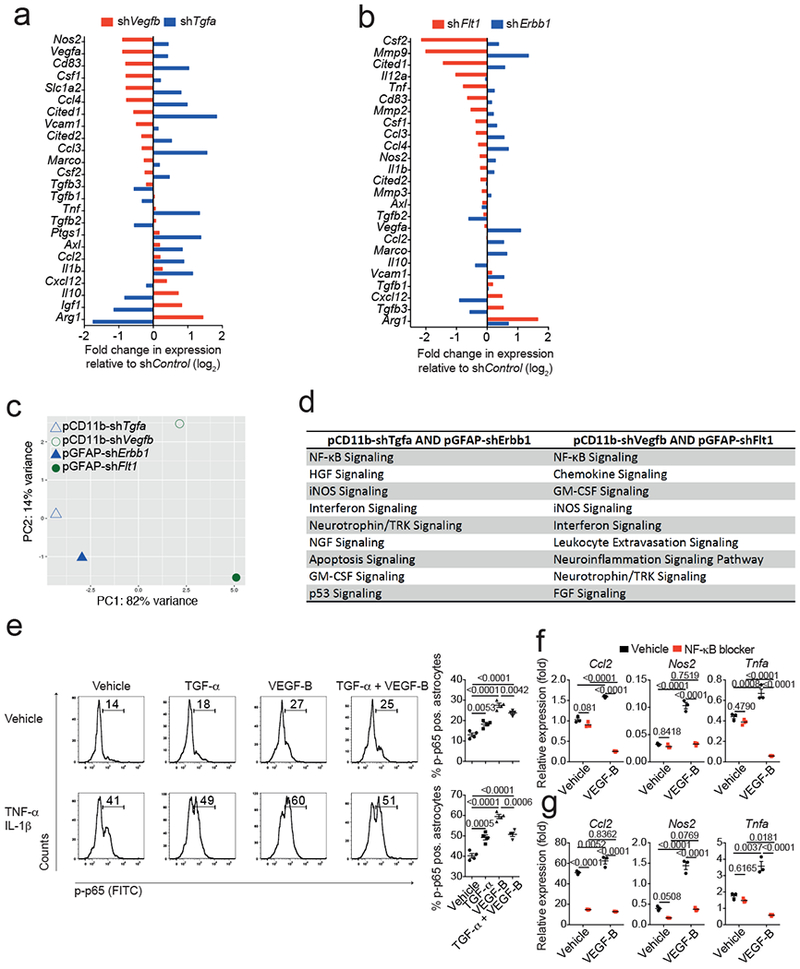
(a,b) NanoString analysis of mRNA expression in astrocytes from EAE mice injected with pCD11b-shVegfb or pCD11b-shTgfa (a) and pGFAP-shFlt1 or pGFAP-shErbb1 (b, see also Fig. 2k,l). Fold change in relative expression relative to control as determined by log2(shKD/shControl)). Representative of 2 independent experiments with pooled RNA isolated from n=3 mice per group. (c) Principal component analysis of gene expression in astrocytes isolated as in (a,b). Representative of 2 independent experiments with pooled RNA isolated from n=3 mice per group. (d) Ingenuity pathway analysis of significantly regulated pathways from astrocytes as in (a,b). Representative of 2 independent experiments with pooled RNA isolated from n=3 mice per group. (e) Left, representative flow cytometry plots depicting NF-κB p65 phosphorylation in WT astrocytes stimulated for 15 mins with Vehicle (top) or TNF-α/IL-1β (bottom) in the presence of TGF-α, VEGF-B, or their combination. Numbers indicate percentage of FITC+ cells. Bar graphs, quantification of FITC+ cells. Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 4 biological replicates. (f) Primary murine astrocytes were exposed to VEGF-B or Vehicle and pharmacological blocker of NF-κB activation. RNA was harvested after 18 hours and subjected to qPCR analyses for the indicated genes. Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 3 biological replicates. (g) Primary murine astrocytes were activated with TNF-α/IL-1β in the presence of VEGF-B or Vehicle and a pharmacological blocker of NF-κB activation. RNA was harvested after 18 hours and subjected to qPCR analyses for the indicated genes. Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 3 biological replicates.
Extended Data 7. Role of AHR in astrocytes and microglia during EAE.

(a-c) EAE was induced in Control (WT), GFAP-Cr AHRflox/flox (GFAP-AHR), or CX3CR1-AHR EAE mice. Starting from day 7, mice were injected daily intraperitoneally with Indoxyl-3-sulfate (I3S), given a tryptophan depleted diet (TDD), or kept on a control diet (Control). Clinical course of EAE mice under treatment conditions as indicated. Representative of 2 independent experiments with n = 4 mice per group. Data are mean ± s.e.m. and P values were determined by two-way ANOVA. (d-f) EAE was induced in WT mice, which were treated with lentiviruses to knock-down AHR in astrocytes (pGFAP-shAhr) or microglia (pCD11b-shAhr). A noncoding RNA was used as control. Flow cytometry quantification of AHR expression in astrocytes and microglia by FACS. (d) Representative histograms of n=4 mice per group. Numbers indicate percentage of AHR positive cells, thin lines isotype control, thick lines AHR staining. (e) Quantification of AHR positive astrocytes and microglia as in (d). Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 4 biological replicates. (f) EAE mice with knock down of AHR in astrocytes, microglia, or both as in (d) were subjected to daily I3S injections, TDD, or Control diet conditions starting on day 14 after disease induction. Clinical course of n=4 mice per group. Representative of 2 independent experiments with n = 4 mice per group. Data are mean ± s.e.m. and P values were determined by two-way ANOVA. (g) Quantification of CNS infiltrating pro-inflammatory monocytes as determined by FACS at day 28 of EAE. Data are mean ± s.e.m. and P values were determined by one way ANOVA followed by Tukey’s post-hoc test. Representative of 2 independent experiments with n = 3 biological replicates.
Extended Data 8. Dietary factors influence mouse and human TGF-α and VEGF-B expression.

(a) Ingenuity pathway analysis of NF-κB signaling comparing TDD to TDD+Trp diet in control animals. Colors code for up- and down-regulation of individual members in red (up) and blue (down). Normalized reads of n = 2 independent samples per group. (b) mRNA expression determined by qPCR in from EAE mice as in Fig. 3a. Data are mean + s.e.m. and representative of two independent experiments with n = 3 replicates. P values determined by one-way ANOVA followed by Tukey’s post-hoc test. (c) Quantification of co-expression of AHR and CD14, VEGF-B and CD14, TGF-α and CD14 in immunofluorescence stainings of human white matter brain tissue of NAWM, active, or chronic MS lesions for AHR (left), VEGF-B (middle), or TGF-α (right), CD14 (green), and DAPI (blue). Data shown are representative of n = 12 fields from three distinct MS brains. (d) Ratio of VEGF-B to TGF-α intensities. Ratio of means from (g) + s.e.m of n = 25 fields. P values derived by one-way ANOVA followed by Tukey’s post-hoc test.
Acknowledgements:
This work was supported by grants NS087867, ES02530, AI126880 and AI093903 from the National Institutes of Health, RSG-14-198-01-LIB from the American Cancer Society and RG4111A1 and JF2161-A-5 from the National Multiple Sclerosis Society to FJQ. FJQ and JA received support from International Progressive Multiple Sclerosis Alliance grant PA-1604-08459. V.R. received support from an educational grant from Mallinkrodt Pharmaceuticals (A219074) and by a fellowship from the German Research Foundation (DFG RO4866 1/1). M.P. is supported by the BMBF-funded competence network of multiple sclerosis (KKNMS), the Sobek-Stiftung and the DFG (SFB 992, SFB1140, SFB/TRR167, Reinhart-Koselleck-Grant) and the Ministry of Science, Research and the Arts, Baden-Wuerttemberg (Sonderlinie “Neuroinflammation”). Human fetal tissue came from the Human Fetal Tissue Repository (Albert Einstein College of Medicine) and from the University of Washington Birth Defects Research Laboratory (BDRL, Project Number 5R24HD000836-51).
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Ben Haim L & Rowitch DH Functional diversity of astrocytes in neural circuit regulation. Nature reviews. Neuroscience 18, 31–41, doi: 10.1038/nrn.2016.159 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Khakh BS & Sofroniew MV Diversity of astrocyte functions and phenotypes in neural circuits. Nature neuroscience 18, 942–952, doi: 10.1038/nn.4043 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sofroniew MV Astrocyte barriers to neurotoxic inflammation. Nature reviews. Neuroscience 16, 249–263, doi: 10.1038/nrn3898 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liddelow SA & Barres BA Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46, 957–967, doi: 10.1016/j.immuni.2017.06.006 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Prinz M & Priller J Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nature reviews. Neuroscience 15, 300–312, doi: 10.1038/nrn3722 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Buttgereit A et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nature immunology 17, 1397–1406, doi: 10.1038/ni.3585 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Lee YH et al. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia 63, 1138–1154, doi: 10.1002/glia.22805 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Goldmann T et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nature neuroscience 16, 1618–1626, doi: 10.1038/nn.3531 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Croxford AL et al. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 43, 502–514, doi: 10.1016/j.immuni.2015.08.010 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Rothhammer V et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med, doi: 10.1038/nm.4106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeste A et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Science signaling 9, ra61, doi: 10.1126/scisignal.aad0612 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Matcovitch-Natan O et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science (New York, N.Y.) 353, aad8670, doi: 10.1126/science.aad8670 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487, doi: 10.1038/nature21029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quintana FJ & Sherr DH Aryl hydrocarbon receptor control of adaptive immunity. Pharmacological reviews 65, 1148–1161, doi: 10.1124/pr.113.007823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stockinger B, Di Meglio P, Gialitakis M & Duarte JH The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol 32, 403–432, doi: 10.1146/annurev-immunol-032713-120245 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Mayo L et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med 20, 1147–1156, doi: 10.1038/nm.3681 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothhammer V et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proceedings of the National Academy of Sciences of the United States of America 114, 2012–2017, doi: 10.1073/pnas.1615413114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wheeler MA & Quintana FJ Regulation of Astrocyte Functions in Multiple Sclerosis. Cold Spring Harbor perspectives in medicine, doi: 10.1101/cshperspect.a029009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutierrez-Vazquez C & Quintana FJ Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 48, 19–33, doi: 10.1016/j.immuni.2017.12.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wikoff WR et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proceedings of the National Academy of Sciences of the United States of America 106, 3698–3703, doi: 10.1073/pnas.0812874106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Girolamo F, Coppola C, Ribatti D & Trojano M Angiogenesis in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta neuropathologica communications 2, 84, doi: 10.1186/s40478-014-0084-z (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosher KI et al. Neural progenitor cells regulate microglia functions and activity. Nature neuroscience 15, 1485–1487, doi: 10.1038/nn.3233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mor F, Quintana FJ & Cohen IR Angiogenesis-inflammation cross-talk: vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. Journal of immunology (Baltimore, Md. : 1950) 172, 4618–4623 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Gaal EI et al. Comparison of vascular growth factors in the murine brain reveals placenta growth factor as prime candidate for CNS revascularization. Blood 122, 658–665, doi: 10.1182/blood-2012-07-441527 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Li X, Kumar A, Zhang F, Lee C & Tang Z Complicated life, complicated VEGF-B. Trends in molecular medicine 18, 119–127, doi: 10.1016/j.molmed.2011.11.006 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Nag S, Eskandarian MR, Davis J & Eubanks JH Differential expression of vascular endothelial growth factor-A (VEGF-A) and VEGF-B after brain injury. Journal of neuropathology and experimental neurology 61, 778–788 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Junier MP What role(s) for TGFalpha in the central nervous system? Progress in neurobiology 62, 443–473 (2000). [DOI] [PubMed] [Google Scholar]
- 28.White RE, Yin FQ & Jakeman LB TGF-alpha increases astrocyte invasion and promotes axonal growth into the lesion following spinal cord injury in mice. Exp Neurol 214, 10–24, doi: 10.1016/j.expneurol.2008.06.012 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson MA et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200, doi: 10.1038/nature17623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kigerl KA et al. Gut dysbiosis impairs recovery after spinal cord injury. The Journal of experimental medicine 213, 2603–2620, doi: 10.1084/jem.20151345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional References:
- 31.Mildner A et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 11159–11171, doi: 10.1523/jneurosci.6209-10.2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mildner A et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nature neuroscience 10, 1544–1553, doi: 10.1038/nn2015 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Trapnell C et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578, doi: 10.1038/nprot.2012.016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan Y et al. CNS-specific therapy for ongoing EAE by silencing IL-17 pathway in astrocytes. Mol Ther 20, 1338–1348, doi: 10.1038/mt.2012.12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jack CS et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. Journal of immunology (Baltimore, Md. : 1950) 175, 4320–4330 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA sequencing data have been uploaded and is accessible under the access code https://figshare.com/s/c109f251b149b7a843b3.