

Abstract
Acute exposure to high concentrations of H2S causes severe brain injury and long-term neurological disorders, but the mechanisms involved are not known. To better understand the cellular and molecular mechanisms involved in acute H2S-induced neurodegeneration we used a broad-spectrum proteomic analysis approach to identify key molecules and molecular pathways involved in the pathogenesis of acute H2S-induced neurotoxicity and neurodegeneration. Mice were subjected to acute inhalation exposure of up to 750 ppm of H2S. H2S induced behavioral deficits and severe lesions including hemorrhage in the inferior colliculus (IC). The IC was microdissected for proteomic analysis. Tandem mass tags (TMT) liquid chromatography mass spectrometry (LC-MS/MS)-based quantitative proteomics was applied for protein identification and quantitation. LC-MS/MS identified 598, 562, and 546 altered proteomic changes at 2 h, and on days 2 and 4 post-H2S exposure, respectively. Of these, 77 proteomic changes were statistically significant at any of the 3 time points. Mass spectrometry data were subjected to Perseus 1.5.5.3 statistical analysis, and gene ontology heat map clustering. Expressions of several key molecules were verified to confirm H2S-dependent proteomics changes. Webgestalt pathway overrepresentation enrichment analysis with Panther engine revealed H2S exposure disrupted several biological processes including metabotropic glutamate receptor group 1 and inflammation mediated by chemokine and cytokine signaling pathways among others. Further analysis showed that energy metabolism, integrity of blood-brain barrier, hypoxic, and oxidative stress signaling pathways were also implicated. Collectively, this broad-spectrum proteomics data has provided important clues to follow up in future studies to further elucidate mechanisms of H2S-induced neurotoxicity.
Keywords: Hydrogen sulfide, Proteomic profiling, Proteomic analysis, TMT labeled LC-MS/MS, Neurotoxicity, Neurodegeneration
1. Introduction
Hydrogen sulfide (H2S) is a highly neurotoxic colorless gas with a “rotten egg” odor (Chou et al., 2016). It is as an environmental pollutant and an occupational hazard in a variety of industries including the oil and gas industry, intensive animal farming operations, sewer and waste water treatment plants, pulp and paper plants, and gas storage facilities, among several others (Chou et al., 2016). It is the second leading cause of fatal gas exposure in the workplace after carbon monoxide (Greenberg and Hamilton, 1998). It is estimated that there are > 1000 reports of human exposures to H2S each year in the United States (Chou et al., 2016). Besides exposures under environmental and industrial settings, intentional H2Spoisoning for suicide has recently increased in Western and Asian societies (Morii et al., 2010; Reedy et al., 2011). This is possible because raw chemical ingredients used to generate H2S for suicide are readily accessible in local stores (Morii et al., 2010). Also law enforcement and first responders called to help, as well as innocent bystanders are at risk of acute exposure to H2S (Sams et al., 2013). H2S was previously developed as a chemical weapon (Foulkes, 2009). There are concerns of potential nefarious use of this gas by terrorists, particularly in confined spaces, such as underground transit systems. For this reason, H2S has been identified as a priority chemical for research by the US Department of Homeland security (Security, 2017). The central nervous system (CNS) is the primary target organ of acute H2S-intoxication and neurotoxicity is the primary cause of death (Tvedt et al., 1991a; Tvedt et al., 1991b; Snyder et al., 1995; Guidotti, 2015). Short-term effects of acute H2S poisoning by inhalation include knockdown, seizures, dyspnea, comma, and death. Currently, there is consensus that survivors of acute H2S poisoning develop neurological sequelae, which can be incapacitating and last many years often leading to disability (Tvedt et al., 1991a; Snyder et al., 1995; Guidotti, 2015). A number of neurological sequelae have been reported. These include dizziness, vertigo, ataxia with tendency to fall, insomnia, fatigue, anxiety, learning and cognition deficits, hearing impairment, recurrent seizures, lack of libido, and increased sensitivity to H2S, among others (Tvedt et al., 1991a; Tvedt et al., 1991b; Snyder et al., 1995). Extreme cases progress to permanent vegetative states. Neurotoxicity, is manifested in individuals acutely exposed to H2S at concentrations > 500 ppm (Snyder et al., 1995; Guidotti, 2015; Rumbeiha et al., 2016).
Currently, there are no FDA approved drugs for treatment of either short-term or long-term effects of acute H2S-induced neurotoxicity. The development of effective therapeutics requires a good understanding of the molecular mechanisms and pathways of acute H2S-inducedneurotoxicity. These mechanisms remain largely unknown. There is an acute need for countermeasures for treatment of mass civilian casualties of acute H2S poisoning in the field, such as following catastrophic industrial meltdowns or intentional terrorist activities. Elucidating molecular mechanisms underlying H2S-induced neurotoxicity is essential in identifying suitable therapeutic targets to counter both immediate and delayed neurotoxic effects of acute H2S poisoning in humans.
We recently developed an inhalation mouse model of acute H2S intoxication that exhibits key phenotypes of short- and long-term effects of acute H2S poisoning in victims and survivors (Anantharam et al., 2017). Briefly, this model is unique because normal walking mice without anesthesia are exposed to H2S by inhalation, recapitulating the typical exposure scenario, following accidents, or during suicide. Clinically, mice suddenly collapse, have seizures, and are dyspneic during exposure. Surviving mice manifest motor deficits on rotarod and open field tests, and developed neurodegeneration, faithfully recapitulating the human condition. The objective of this study was to use this mouse model to investigate proteomic changes following acute H2S-exposure to identify novel toxic mechanisms that could potentially be targeted for therapeutic intervention. These studies focused on the central inferior colliculus (IC), part of the brainstem, because our previous studies revealed this to be the most sensitive brain region to H2S-induced neurodegeneration (Anantharam et al., 2017). However, we also previously observed histopathological changes in other parts of the brain, especially the thalamus and cortex (Anantharam et al., 2017). This ground breaking H2S-study demonstrated, for the first time, that H2S exposure induces significant proteomic changes in the IC, including albumin (Alb) leakage, neuroinflammation, and oxidative stress, which collectively play an important role in the execution of H2S-induced neurotoxicity and neurodegeneration.
2. Materials and methods
2.1. Materials
H2S gas was purchased from Airgas (La Porte, TX). RNeasy mini kit was purchased from Qiagen (Germantown, MD). High Capacity cDNA RT kit were purchased from ThermoFisher Scientific (Waltham, MA). RT2 SYBR Green ROX qPCR Mastermix and primers for Gapdh were purchased from Qiagen (Valencia, CA). Primary antibodies against Alb, hypoxia inducing factor 1 alpha (Hif-1α), and Vimentin (Vim) were purchased from Cell signaling (Danvers, MA). Primary antibodies against nuclear factor-like 2 (Nrf2) and 3-Oxoacid CoA Transferase 1 (Oxctl) were purchased from Abeam (Cambridge, MA). Primary antibody against Fas was purchased from SantaCruz Biotechnology (SantaCruz, CA). Primary antibody against NeuN was purchased from Millipore (Billerica, MA). U-PLEX combo kit against TNF-α was purchased from Meso Scale Diagnostics (Rockville, MD).
2.2. Animals and treatment
This study was approved by Iowa State University Animal Care and Use Committee. Seven- to eight-week-old male C57 BL/6 J mice were used because previous studies from our lab showed that males were more sensitive than females. The mice were housed at room temperature of 20–22 °C under a 12-h light cycle, and a relative humidity of 35–50%. Protein rodent maintenance diet (Teklad HSD Inc., WI, US) and water were provided ad libitum. Prior to H2S exposure on day 1, all mice were acclimated to breathing air for 40 min for two consecutive days. Freely moving unanesthetized mice were exposed either to normal breathing air (Control) or to 650–750 ppm H2S (H2S-treated) either once or to 2–7 short-term exposures. Both breathing air and H2S were supplied from compressed gas cylinders. The inhalation dosage of H2S was selected basing on published literature indicating concentrations of H2S > 500 ppm are associated with knockdown and neurotoxicity in human (Snyder et al., 1995; Guidotti, 2015; Rumbeiha et al., 2016). Typically, humans are exposed once, but repeated exposures have been reported (Ahlborg, 1951). For purposes of studying the progression of neurotoxicity, separate groups of mice in this study were euthanized after receiving either only one or 2–7 acute exposures. For proteomic studies, the first batch of mice was terminated on the first day, day 1, only 2 h post exposure. Others were terminated on days 2 and 4. Negative controls were exposed to normal breathing air daily and euthanized on day 4. The gas exposure paradigm is summarized in Fig. 1. Animals were cared for in accordance with the Institutional Animal Care and Use committee guidelines.
Fig. 1.
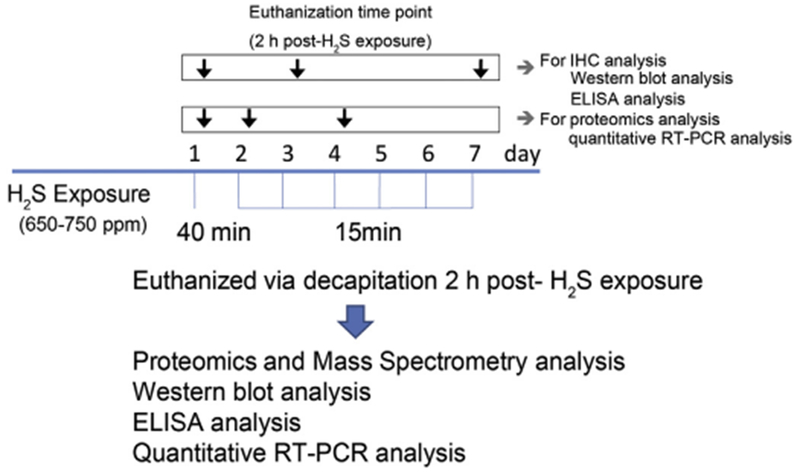
Acute exposure paradigm of hydrogen sulfide in C57 black mice.
Mice were exposed to 765 ppm of H2S in a chamber for 40 min either once only (day 1) and for 15 min on the subsequent days up to day 7. Mice were sacrificed 2h post-H2S exposure on specified days of the study. Negative control mice were exposed to breathing air from a cylinder daily up to day 7. Separate groups of mice were sacrificed on days 1 (2 h post-exposure), 3, and 7 for immunohistochemistry, Western blot assay, and ELISA analysis. Groups of mice for proteomics studies and quantitative RT-PCR analysis were sacrificed on days 1 (2 h post-exposure), 2 and 4.
2.3. Behavioral assessment
The VersaMax open field test was used to assess motor deficits induced by H2S. We used this test because previous studies in the lab had indicated it to be sensitive to acute H2S intoxication in this mouse model (Anantharam et al., 2017). Ataxia and other movement disorders are frequently reported neurological sequelae in survivors of acute H2S poisoning (Ahlborg, 1951; Tvedt et al., 1991a; Tvedt et al., 1991b). Spontaneous activity was measured using an automated computer device (Model RXYZCM-16; Accuscan, Columbus, OH, USA). The activity chamber’s dimensions are 40 × 40 × 30.5 cm, and it is made of Plexiglas with a Plexiglas lid. The lid has holes for ventilation. Data was analyzed using VersaMax Analyzer (Model CDA-8; Accuscan). Mice were placed in the activity chamber for acclimation 2 min prior to recording for 10 min. Vertical activity, horizontal activity, and distance traveled were measured.
2.4. Histopathology and immunohistochemistry
Mice were euthanized 2 h after the last H2S exposure. They were deeply anesthetized with a cocktail of 100 mg/kg BW ketamine and 10 mg/kg BW xylazine via intraperitoneal (IP) injection. The thoracic cavity was opened to expose the heart and fresh 4% paraformaldehyde (PFA, pH 7.4) was injected through the left ventricle. After perfusion, the skull was opened and brains were post-fixed in 4% PFA for 24–48 h before removal from the skull. Brains were processed in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E), and examined microscopically. Immunohistochemistry was used to visualize live and degenerating neurons. Neurons were stained with NeuN antibody (abl77487, Abeam, Cambridge, MA) using an indirect immunostaining protocol. Diaminobenzidine was used as chromogen. Stained sections were examined using a Nikon Eclipse Ci-L microscope equipped with a DS-Fi2 camera. For image analysis to count neurons, NeuN-positive cells (sites of DAB chromogen deposition) were enumerated in each of five 400× photomicrographs of the IC from mice exposed to H2S or breathing air, and the mean number of NeuN-positive cells were compared between groups. Degenerating and dying neurons were identified by Fluoro-Jade C staining. Briefly, deparaffinized, hydrated sections were incubated in 0.06% potassium permanganate solution for 10 min, rinsed for 2 min in distilled water and incubated in a 0.0001% solution of Fluoro-Jade C (Histo-Chem, Inc., Jefferson, AR) for 10 min at room temperature. Dark conditions were maintained after application of Fluoro-Jade C to prevent photobleaching. Sections were rinsed three times in distilled water, incubated at 4 °C in 6-diamidino-2-phenylindole (DAPI, Sigma, St. Louis, MO), rinsed, air-dried and cover slipped using DPX non-fluorescent mounting medium (Sigma, St. Louis, MO). Sections were examined using an EVOS FL Cell Imaging System (ThermoFisher Scientific).
2.5. Mass spectrometry proteomic analysis
Mice were euthanized by decapitation 2 h after the last exposure to H2S on days 1, 2 and 4 for proteomics analysis, quantitative gene expression analysis by real-time PCR, and by Western blot. Following decapitation, brains were immediately removed from the skull. The IC was microdissected on ice and immediately flash-frozen using liquid nitrogen, and stored at −80 °C until further use. Samples were processed according to a previously published method with some modifications (Meade et al., 2015). Briefly, IC tissues were placed in 100 μL of urea lysis buffer and homogenized with a handheld pestle homogenizer. Protein samples were further reduced and alkylated. Small aliquots of each sample were taken to measure protein concentration using the Bradford assay from Bio-Rad (Hercules, CA). The remaining samples were diluted and trypsin-digested overnight, followed by desalting using a C18 peptide trap from Michrom (Auburn, CA). The desalted samples were vacufuged prior to individual sample labeling using TMT-6plex labels according to the manufacturers’ instructions (Thermo Fisher Scientific, Waltham, MA). Labeled samples of each exposure group were combined with the control for sample comparison (Table 1). Peptides were separated on a Waters BEH C18 capillary column prior to online analysis using a 240-min linear increasing gradient of acetonitrile with 0.1% formic acid. Following elution from the column, ions were generated using 2.6 kV on a taper tip in a New Objective Nanosource and entered into an LTQ-OrbitrapVelos mass spectrometer (Thermo Fisher, San Jose, CA). A full scan was taken in the LTQ, followed by data-dependent MS/MS analysis of the top 6 peaks. MS/MS analysis included collision-induced dissociation (CID) in the LTQ for structural information and higher-energy collisional dissociation (HCD) in the Orbitrap for quantitation. MS/MS data were aligned and quantitated using MaxQuantl.5.4.1 (Cox and Mann, 2008) Analytics Platform with PTXQC (Bielow et al., 2016) quality control data management. Peptide alignment was executed with the mouse Uniprot protein database UP000000589_10090.fasta; enzyme: trypsin; carboxymethyl (C), and oxidation (M), FDR < 1% based on peptide q-value under standard settings (see supplemental data). The secondary computational analysis was executed using Perseus 1.5.5.3 (Tyanova et al., 2016) for statistical rendering, and web-based software Morpheus for heatmap rendering. Prior to analysis of experimental samples, small aliquots of individually labeled control samples were analyzed to determine individual variations in controls. Due to low variability in the controls, a pooled control setting was used. Sample analysis included modification of the MaxQuant protein output list by normalization of individual TMT reporter ion intensities by division through the median intensity followed by determination of fold TMT expression ratios by division of individual experimental TMT values (126,127,128,129,and 130) through the TMT value (131) of the pooled control.
Table 1.
A summary of mass spectrometry and distribution of TMT sample labeling. GA = group A; GB = group B; GC = group C; GD = group D.
| Group | Total mice/animal ID | TMT label | Treatment | Exposure/dose | Euthanasia |
|---|---|---|---|---|---|
| GA control | (n = 5) | 40 min day 1 | Inhalation/breathing air | Day 4 | |
| 1,2,3,4,5, pooled | 131 | 15 min day 2 | |||
| 15 min day 3 | |||||
| 15 min day 4 | |||||
| + 2 h recovery | |||||
| GB 1 exposure | (n = 5) | 40 min day 1 | Inhalation/650–700 ppm H2S | Day 1 | |
| 1 | 126 | + 2 h recovery | |||
| 2 | 127 | ||||
| 3 | 128 | ||||
| 4 | 129 | ||||
| 5 | 130 | ||||
| GC 2 exposures | (n = 5) | 40 min day 1 | Inhalation/650–700 ppm H2S | Day 2 | |
| 1 | 126 | 15 min day 2 | |||
| 2 | 127 | + 2 h recovery | |||
| 3 | 128 | ||||
| 4 | 129 | ||||
| 5 | 130 | ||||
| GD 4 exposures | (n = 4) | 40 min day 1 | Inhalation/650–700 ppm H2S | Day 4 | |
| 1 | 126 | 15 min day 2 | |||
| 2 | 127 | 15 min day 3 | |||
| 3 | 128 | 15 min day 4 | |||
| 4 | 130 | + 2 h recovery |
2.6. Perseus statistical analysis
The previously modified Maxquant protein list was entered into the Perseus 1.5.5.3 software program followed by annotating Maxquant defined Swissprot protein accession numbers with mouse gene identifiers. Statistical analysis included one-sample t-tests of fold expression values by considering the deviation of samples from 1 fold expression (no change in TMT reporter ion intensities vs. control intensity values) using p < .05 as a criterion for significance. Scatter Plots were established by plotting one-sample t-test difference fold protein expression vs. –log one-sample t-test p value fold protein expression according to the associated Perseus tool set.
2.7. Morpheus Heatmap rendering
The Perseus processed protein list containing mouse gene identifiers, protein names and gene ontology (GO) biological process, one-sample t-test, levels of significantly and non-significantly modulated proteins was entered into the web-based Morpheus heatmap-rendering tool (https://software.broadinstitute.org/morpheus/). A gradient coloring scheme was applied to display upregulated (above 1.2 fold expression (in log2 display above 0.263), red color) vs. downregulated (below 0.83 fold expression (in log2 display below −0.263) blue color) protein nodes. White color nodes represent changes of non-modulated nodes to control levels (between 0.83 and 1.2 fold expression). Normalization of individual TMT reporter ion intensities was done by division through the median intensity followed by determination of fold TMT expression ratios by division of individual experimental TMT values (126,127,128,129,and 130) through the TMT value (131) of the pooled control. The cut-off limit of 1.2 fold was chosen because these individual values are already highly normalized expression values and the maximum fold values achieved were below 2.5. A 1.2 fold cut-off value was used as in a previous publication (Meade et al., 2015). The heatmap was hierarchically clustered by Euclidean distance using row average linkage and grouping of rows by GO biological process. Functional enrichment analysis was performed using Webgestalt pathway overrepresentation enrichment analysis with Panther engine (http://www.webgestalt.org/option.php). Up to ten highest probable pathways for each time point of H2S treatment data were presented.
2.8. Western blot assay
Microdissected tissue samples were lysed in modified RIPA lysis buffer (1% Triton X-100, ImM EDTA, 100 mM NaCl, ImM EGTA, 1 mM NaF, 20 mM Na4P207, 2mM Na3V04, 10% glycerol, 0.1% SDS, 0.5% deoxycholate, 50 mM Tris-HCl, and pH 7.4) via sonication, followed by centrifugation as described previously (Kim et al., 2016). After protein concentration of samples were measured using the Bradford assay, equal amounts of protein samples were loaded to a 10–12% SDS-PAGE gel and transferred onto a nitrocellulose membrane. The membranes were blocked with 5% bovine serum albumin in TBS supplemented with 0.1% Tween-20. Specific primary antibodies were incubated with the membrane overnight at 4 °C. After rinsing thoroughly in PBS supplemented with 0.1% Tween-20, the membrane was incubated with secondary antibodies. β-Actin antibody was used for loading control. Immunoblot imaging was performed with an Odyssey Infrared Imaging system (LI-COR, Lincoln, NE). ImageJ software (National Institutes of Health, Bethesda, MD) was used to quantify western blot bands.
2.9. Enzyme-linked immunosorbent assay
Microdissected tissue samples were immediately frozen. Samples were lysed in modified RIPA lysis buffer (150 mM NaCl, 20 mM Tris pH 7.5, 1 mM EGTA, 1 mM EDTA, 1% NP-40, 0.1% SDS) with protease inhibitors via sonication (Dalgard et al., 2012; Homji et al., 2012). Lysed samples were centrifuged at 10,000 × g for 10 min at 4 °C. Expression of TNFα was measured using Meso Scale Discovery system following manufacturer’s protocol.
2.10. Quantitative real-time RT-PCR
Following exposure to H2S, tissues were dissected and immediately stored at −80 °C till analysis. Total RNA was extracted from frozen tissues using the RNeasy® Plus Mini kit with treatment of DNase I according to the manufacturer’s protocol. Validated primers for Gapdh (Qiagen, #PPR57734E) were used as the housekeeping gene controls. The threshold cycle (Ct) was calculated from the instrument software, and fold change in gene expression was calculated using the ΔΔt method as described earlier (Kim et al., 2016). The following primers were used to check the quantitative transcriptional level of Prkab1, Vim, and Ahsal; 5′-TCCGATGTGTCTGAGCTGTC-3′ and 5′-CCCGTGTCCTTGTTCAAGAT-3′ for Prkab1 (Bandow et al., 2015), 5′-TCCACACGCACCTACAGTCT-3′ and 5′-CCGAGGACCGGGTCACATA-3′ for Vim (Ulmasov et al., 2013), 5′-CAGAGGGGCACTTTGCCACCA-3′ and 5′-CACGGCCTTCCATGCACAGCT-3′ for Ahsal.
2.11. Statistical analysis
Data were analyzed using Prism 4.0 (GraphPad Software, San Diego, CA). Non-paired Student’s t-test was used when two groups were being compared. Differences were considered statistically significant for p-values < 0.05. Data are represented as the mean ± S.E.M. of at least two separate experiments performed at least in triplicate.
3. Results
3.1. Acute exposure to H2S induces motor behavioral deficits and seizures in C57 black mice
Locomotor activities of mice were assessed on days 2, 4, and 6. Results indicated that the horizontal and vertical activities were decreased by > 50% and were statistically different in mice exposed to H2S compared to those that received breathing air (Fig. 2A and B). Total distance traveled was also decreased by > 50% and was also statistically different in H2S-exposed mice compared to controls (Fig. 2C). Most importantly, mice exposed to H2S exhibited severe seizure activity. On day 1, during the first exposure, seizure activity was observed, on average, starting at 15 min of H2S exposure. More than 50% of mice had seizures at 40 min of exposure. We also observed that mice that were exposed to H2S on subsequent days were more sensitive to H2S than naive mice. For example, mice exposed to H2S more than once had seizures after only 5 min of H2S exposure compared to 15 min for naive mice. Also, consistent with increased sensitivity, 40% of the mice exhibited seizures at 10 min of H2S exposure on day 6 compared to about 40 min in naive mice on day 1. Collectively, these results indicate increased susceptibility of mice to H2S after each successive H2S exposures, suggesting that the toxic effects of H2S exposure are cumulative (Fig. 2D).
Fig. 2.
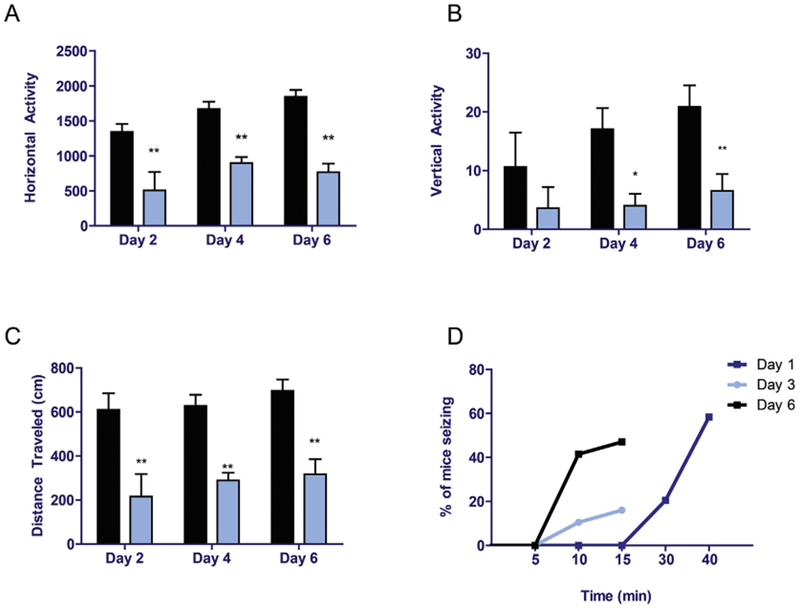
Acute exposure to hydrogen sulfide induced motor deficits and seizures.
C57 black mice were exposed to H2S as shown in Fig. 1. Locomotor activity was measured using an automated VersaMax locomotor activity monitor for 10 min on days 2, 4, and 6. Horizontal activity (A), vertical activity (B), and total distance traveled (C) were analyzed between groups. For seizures, time to seizure and number of mice seizing were monitored (D). Asterisks (*, p < 0.05; **, p < 0.01) indicate statistically significant differences between H2S and breathing air negative control groups.
3.2. Acute hydrogen sulfide exposure induced brain damage
Motor behavioral deficits induced by exposure to H2S may be a sign of injury to the CNS. Therefore, histopathology was performed to identify the effects of H2S exposure in the IC. Mice were exposed to H2S as designated in Fig. 1. For this portion of the study, in order to assess the progression of neuropathology, mice were sacrificed at multiple time points (2 h, days 3 or 7). Microscopic examination of PFA-perfused brains revealed H2S-induced neurodegeneration and loss of neurons in the IC (Fig. 3A). By day 7, mice exposed to H2S exhibited severe lesions in the IC, with necrosis, vacuolar change, and infiltration by neuroglial cells. H2S exposure frequently induced hemorrhage in IC (Fig. 3B). In order to further characterize and quantify the loss of neurons, IC tissues were analyzed by immunohistochemical staining using the neuron specific marker, NeuN. On day 7, there was a marked loss of neurons in the IC of H2S-exposed mice, compared with the breathing air group. Enumeration of neurons in the IC revealed approximately a 70% loss of IC neurons by day 7 of H2S exposure (Fig. 3C and D) with infiltration by glial cells. However, in H2S exposed mice, neurons in the midbrain region adjacent to the IC appeared unaffected morphologically and glial cell numbers and activation state were not altered. Neurodegeneration was further confirmed using Fluoro-Jade C staining in the IC of H2S-exposed mice (Fig. 3E). There was an increase in Fluoro-Jade C positive degenerating neurons in the IC. A reduction in numbers of NeuN positive cells and an increase in population of Fluoro-Jade C stained cells demonstrate H2S-induced selective loss of neurons and neurodegeneration in the IC.
Fig. 3.
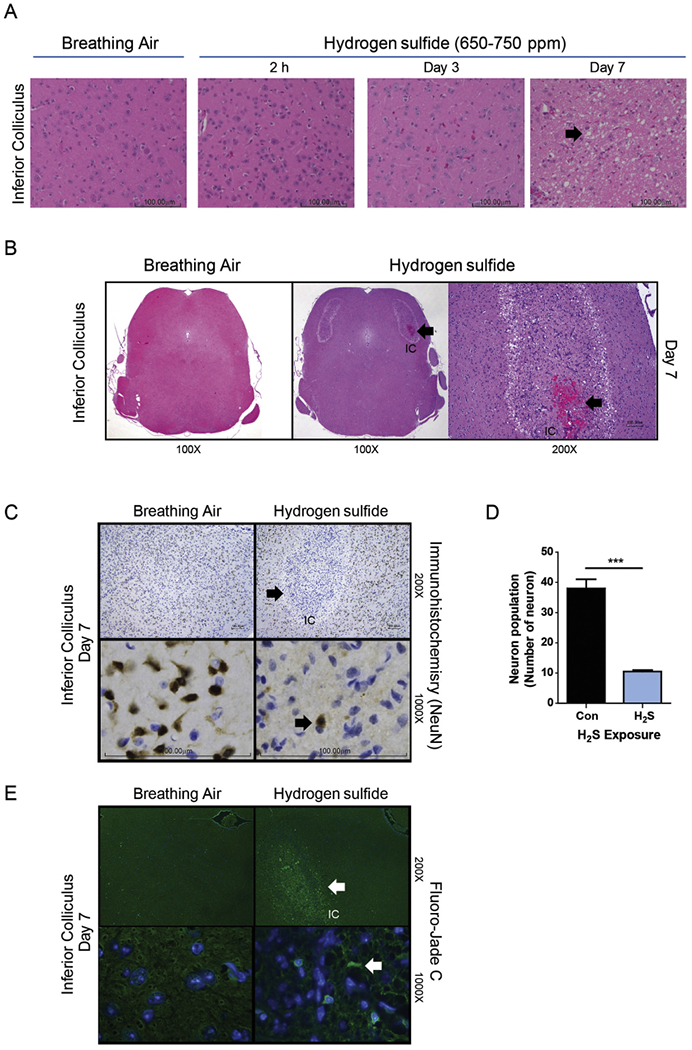
Neurodegeneration and necrosis in the IC of mice exposed to 650–750 ppm H2S in acute short-term repeated inhalation exposures over 7 days. Note the loss of neurons and development of clear vacuoles in the neuropil (arrow) on day 7 in mice exposed to H2S and the minimal morphologic effects to earlier H2S exposures or breathing air (A). H2S exposure induced hemorrhage (thick arrow) in the IC. An insert at higher magnification is included to show the hemorrhage. Hemorrhage was not seen in control mice (B). NeuN staining in brown color (arrow) of neurons (C). H2S caused marked and selective loss of neurons in the IC (1000× magnification images, C), with retention of neurons in the regions surrounding the IC (200× magnification image) (C). Computer aided image analysis of NeuN immunostained sections reveals marked loss of neurons (D) in the IC of mice exposed to H2S (p < 0.001, t-test). Representative photomicrographs of mice exposed to breathing air or H2S, hematoxylin and eosin (A) and NeuN immunohistochemistry (C). Neurons in the IC region of the H2S exposed mice on day 7 were enumerated and compared to breathing air control (D). Degenerating neurons were visualized with Flouro-Jade C staining (E). Arrow indicates degenerative neurons. Neurodegeneration was not seen in the control group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. Acute H2S exposure caused changes in the broad spectrum proteome
Proteomic profiles of the IC were determined using TMT peptide labeling coupled with LC/MS/MS analysis by comparison of the breathing air negative control group and the H2S exposed group is described in the methods section. Mass spectrophotometry analysis was able to identify 598, 562, and 546 altered proteins for 2 h, days, 2 and 4 of H2S exposure, respectively. Subsequent analyses showed alterations of protein expressions (Fig. 4, heatmap) with 36, 12 and 14 proteins being significantly downregulated (below 0.83 fold) and 9, 7, and 13 proteins being significantly upregulated (above 1.2 fold) compared to the control for 2 h and days 2 and 4 of H2S exposure, respectively. The cut-off limit of 1.2 fold was chosen because individual experimental TMT values for each sample were highly normalized as described above and has been used in previous publication (Meade et al., 2015). A single H2S exposure, in which mice were euthanized only 2 h post exposure on day 1, demonstrated the highest range of fold protein expression changes compared to the control with the majority of proteins being in the downregulation cluster. This was followed by day 2, in which mice received only 2 acute exposures, with equal distribution of upregulated and downregulated proteins, while mice euthanized on day 4 manifested the lowest range of fold changes with the least changes in the proteomic profile (Fig. 5A, B, C, D, scatter plots). Several proteins including cytoplasmic FMR1-interacting protein 2 (Cyfip2), Alb, and hemoglobin subunit proteins were increased on all three time-points, while some proteins including activator of HSP90 ATPase activity 1 (Ahsal) were consistently downregulated at the three time-points (Fig. 4 and Table 2). Scatter plots to show overall distribution of proteomic profile changes in the IC following H2S exposure are shown in Fig. 5. Fig. 5 display is a common method to display proteomics data.
Fig. 4.

Heatmap of changes in proteomic profile in the IC following acute H2S exposure.
Morpheus rendered Heatmap of determined changes in protein expression at 2 h and on days 2 and 4. Hierarchical clustering by Euclidean distance, row average linkage and grouping of rows by gene ontology for biological process (GOBP). Heatmap displays fold expression values of five (2 h and day 2) or four (day 4) IC tissue samples, and average fold expression values, mouse gene identifiers (gene names), protein names, and GOBP. Red color indicates upregulation of protein expression (above 1.2 fold expression vs. control), whereas blue color indicates downregulation of protein expression (below 0.83 fold expression vs. control) Grey color indicates proteins not identified. Protein expression change were converted into a log2 data display. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 5.
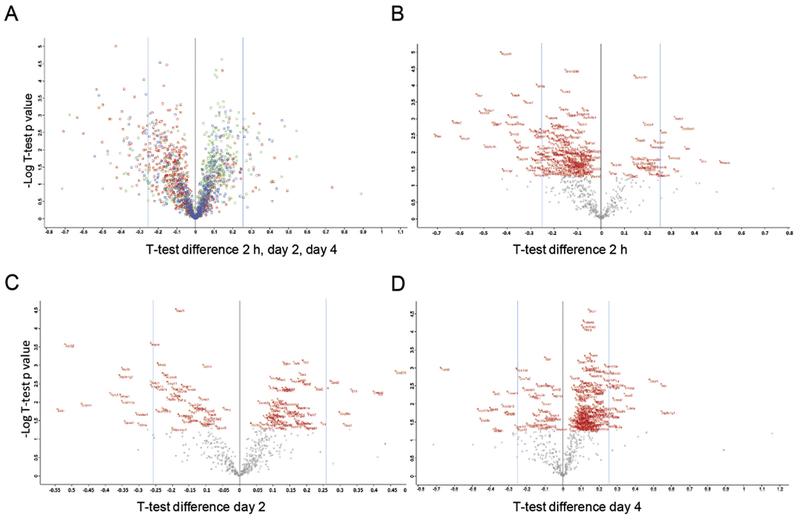
Overall distribution of proteomic profile changes in the IC following H2S exposure.
A) Scatter Plot to display overall distribution of one-sample t-test differences in protein expression profiles vs. –Log one-sample t-test p values 2 h (red), day 2 (blue), and day 4 (green) following H2S exposure. Scatter Plots of one-sample t-test fold protein expression differences vs. –Log one-sample t-test p values from B) 2 h, C) day 2, and D) day 4 of H2S exposure. One-sample t-test of significantly modulated proteins in the H2S exposure group vs. the control according to fold expression values are displayed with official mouse gene symbols in red. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
Table of changes in proteomic profile in IC following acute H2S exposure.
| Gene names | Protein names | GOBP name | 2h |
Day 2 |
Day 4 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fold change | Avg | sthev | t-Test p | Fold change | Avg | sthev | t-Test p | Fold change | Avg | sthev | t-Test p | |||
| Arpc4 | Actin-related protein 2/3 complex subunit 4 | Actin cytoskeleton organization | Up | 0.32 | 0.08 | 0 | Down | −0.26 | 0.05 | 0 | 0 | 0.06 | 0 | |
| Dstn | Destrin | Actin cytoskeleton organization | 0 | 0.11 | 0 | Down | −0.52 | 0.3 | 0.0013 | Up | 0.28 | 0.12 | 0.0004 | |
| Iqsecl | IQ motif and SEC7 domain-containing protein 1 | Actin cytoskeleton organization | Down | −0.33 | 0.24 | 0.0006 | ||||||||
| Kif2a | Kinesin-like protein | Actin cytoskeleton organization | Down | −0.28 | 0.1 | 0 | ||||||||
| Trf | Serotransferrin | Actin cytoskeleton organization | 0.26 | 0.18 | 0.0003 | 0.25 | 0.18 | 0.0002 | Up | 0.29 | 0.06 | 0 | ||
| Lmna | Prelamin-A/C | Adhesion | Up | 0.36 | 0.13 | 0.0004 | ||||||||
| Vim | Vimentin | Adhesion | Up | 0.44 | 0.24 | 0.0009 | ||||||||
| Eprs | Bifunctional glutamate/proline-tRNA ligase | Amine metabolic process | Down | −0.28 | 0.16 | 0.0001 | ||||||||
| Vdac1 | Voltage-dependent anion-selective channel protein 1 | Anion transport | −0.26 | 0.08 | 0 | −0.08 | 0.08 | 0 | Down | −0.27 | 0.19 | 0.0015 | ||
| Gdap1l1 | Ganglioside-induced differentiation-associated protein 1-like 1 | Axon guidance | Down | −0.27 | 0.1 | 0 | 0 | 0.11 | 0 | −0.05 | 0.06 | 0 | ||
| Dpysl4 | Dihydropyrimidinase-related protein 4 | Axon guidance | Down | −0.35 | 0.19 | 0.0003 | ||||||||
| Nef1 | Neurofilament light polypeptide | Axon guidance | Down | −0.36 | 0.14 | 0.0001 | −0.19 | 0.18 | 0.0002 | 0.03 | 0.09 | 0.0001 | ||
| Plp1 | Myelin proteolipid protein | Axon guidance | Down | −0.38 | 0.37 | 0.0031 | −0.04 | 0.38 | 0.0042 | Down | −0.36 | 0.23 | 0.0025 | |
| Alb | Serum albumin | Blood | Up | 0.37 | 0.16 | 0.0002 | Up | 0.42 | 0.17 | 0.0002 | Up | 0.55 | 0.12 | 0.0003 |
| Hbbt1 | Hemoglobin subunit beta-2 | Blood | Up | 0.45 | 0.4 | 0.0069 | Up | 0.33 | 0.23 | 0.0007 | Up | 0.49 | 0.32 | 0.0066 |
| Slc2a1 | Solute carrier family 2, facilitated glucose transporter member 1 | Carbohydrate transport | Down | −0.28 | 0.34 | 0.0024 | 0.01 | 0.36 | 0.0034 | Down | −0.37 | 0.24 | 0.0029 | |
| Cend1 | Cell cycle exit and neuronal differentiation protein 1 | Cell cycle | −0.24 | 0.16 | 0.0002 | −0.13 | 0.28 | 0.0012 | Up | 0.28 | 0.08 | 0.0001 | ||
| Dnah6 | Dynein Axonemal Heavy Chain 6 | Cell motility | Up | 0.41 | 0.16 | 0.0002 | ||||||||
| Ehd3 | EH domain-containing protein 3 | Cellular transport | 0.01 | 0.14 | 0.0001 | −0.24 | 0.06 | 0 | Down | −0.21 | 0.28 | 0.0045 | ||
| Exoc1 | Exocyst complex component 1 | Cellular transport | Down | −0.26 | 0.12 | 0 | 0.01 | 0.17 | 0.0002 | 0.08 | 0.07 | 0.0001 | ||
| Lgi3 | Leucine-rich repeat LGI family member 3 | Cellular transport | −0.11 | 0.17 | 0.0002 | 0.07 | 0.07 | 0 | Up | 0.28 | 0.06 | 0.0001 | ||
| H3f3a | Histone H3.3 | DNA metabolism | Down | −0.34 | 0.29 | 0.0014 | Down | −0.29 | 0.22 | 0.0005 | −0.24 | 0.21 | 0.0018 | |
| Hist1h3a | Histone H3.1 | DNA metabolism | −0.09 | 0.1 | 0 | Down | −0.34 | 0.17 | 0.0002 | −0.21 | 0.4 | 0.0139 | ||
| Set | Protein SET | DNA metabolism | Up | 0.33 | 0.22 | 0.0006 | −0.07 | 0.09 | 0 | −0.11 | 0.09 | 0.0002 | ||
| Atp5j2 | ATP synthase subunit f, mitochondrial | Energy metabolism | −0.09 | 0.08 | 0 | Down | −0.52 | 0.1 | 0 | 0.04 | 0.07 | 0.0001 | ||
| Ndufab1 | NADH:Ubiquinone Oxidoreductase Subunit AB1 | Energy metabolism | −0.2 | 0.11 | 0 | Down | −0.3 | 0.18 | 0.0002 | −0.04 | 0.02 | 0 | ||
| Cox6c | Cytochrome c oxidase subunit 6C | Energy metabolism | −0.09 | 0.15 | 0.0001 | −0.06 | 0.09 | 0 | Down | −0.44 | 0.22 | 0.0021 | ||
| Atp6v1g2 | V-type proton ATPase subunit G 2 | Energy metabolism | Down | −0.36 | 0.11 | 0 | Down | −0.31 | 0.31 | 0.0061 | ||||
| Ndufa13 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 13 | Energy metabolism | −0.1 | 0.07 | 0 | −0.26 | 0.09 | 0 | Down | −0.33 | 0.12 | 0.0003 | ||
| Atp6v1h | V-type proton ATPase subunit H | Energy metabolism | Down | −0.49 | 0.21 | 0.0004 | −0.04 | 0.07 | 0 | −0.01 | 0.02 | 0 | ||
| Idh3a | Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial | Energy metabolism | Down | −0.28 | 0.04 | 0 | 0.04 | 0.08 | 0 | 0.07 | 0.05 | 0 | ||
| Mtco2 | Cytochrome c oxidase subunit 2 | Energy metabolism | Down | −0.34 | 0.15 | 0.0001 | −0.04 | 0.1 | 0 | −0.14 | 0.12 | 0.0003 | ||
| Atp6v1g1 | V-type proton ATPase subunit G 1 | Energy metabolism | Up | 0.56 | 0.22 | 0.0022 | ||||||||
| Me1 | Malic enzyme | Energy metabolism | Down | −0.53 | 0.09 | 0 | ||||||||
| Ogdhl | Oxoglutarate Dehydrogenase-Like | Energy metabolism | 0.09 | 0.23 | 0.0006 | Up | 0.3 | 0.11 | 0.0003 | |||||
| Oxct1 | Succinyl-CoA:3-ketoacid coenzyme A transferase 1, mitochondrial | Energy metabolism | −0.01 | 0.07 | 0 | 0.06 | 0.07 | 0 | Up | 0.48 | 0.1 | 0.0002 | ||
| Hsd17b10 | 3-hydroxyacyl-CoA dehydrogenase type-2 | Fatty acid and steroid metabolism | −0.24 | 0.14 | 0.0001 | Down | −0.46 | 0.18 | 0.0012 | |||||
| Gpd2 | Glycerol-3-phosphate dehydrogenase | Glucose metabolism | −0.01 | 0.15 | 0.0001 | Up | 0.27 | 0.09 | 0 | 0.2 | 0.19 | 0.0013 | ||
| Gpi | Glucose-6-phosphate isomerase | Glucose metabolism | Down | −0.46 | 0.13 | 0.0001 | −0.23 | 0.07 | 0 | −0.1 | 0.01 | 0 | ||
| Pfkp | ATP-dependent 6-phosphofructokinase | Glucose metabolism | Down | −0.35 | 0.1 | 0 | 0.12 | 0.1 | 0 | 0.05 | 0.05 | 0 | ||
| Pgam2 | Phosphoglycerate mutase 2 | Glucose metabolism | Down | −0.39 | 0.1 | 0 | ||||||||
| C1qbp | Complement component 1 Q subcomponent-binding protein, mitochondrial | Immune response | 0.03 | 0.21 | 0.0004 | 0.17 | 0.1 | 0 | Up | 0.34 | 0.08 | 0.0001 | ||
| Mif | Macrophage migration inhibitory factor | Immune response | Down | −0.35 | 0.16 | 0.0001 | −0.07 | 0.07 | 0 | 0 | 0.16 | 0.0008 | ||
| Acat2 | Acetyl-CoA acetyltransferase, cytosolic | Lipid metabolism | 0.03 | 0.08 | 0 | −0.02 | 0.24 | 0.0007 | Up | 0.34 | 0.09 | 0.0002 | ||
| Tuba1b | Tubulin alpha-1B chain | Microtubule organization | Down | −0.28 | 0.09 | 0 | −0.25 | 0.09 | 0 | Down | −0.31 | 0.08 | 0.0001 | |
| Tubb6 | Tubulin beta-6 chain | Microtubule organization | Down | −0.67 | 0.1 | 0.0002 | ||||||||
| Mp68 | 6.8 kDa mitochondrial proteolipid | Mitochondrial proteolipid | Down | −0.35 | 0.09 | 0 | Down | −0.4 | 0.15 | 0.0007 | ||||
| Slc25a51 | Solute carrier family 25 member 51 | Mitochondrial transport | Up | 0.35 | 0.1 | 0 | ||||||||
| Sccpdh | Saccharopine dehydrogenase-like oxidoreductase | oxidoreductase | down | −0.4 | 0.11 | 0 | ||||||||
| Pcyt2 | Ethanolamine-phosphate cytidylyltransferase | Phosphatidylethanolamine biosynthesis | Down | −0.3 | 0.13 | 0.0004 | ||||||||
| Psmc4 | 26S protease regulatory subunit 6B | Protein degradation | Down | −0.51 | 0.12 | 0 | ||||||||
| Ahsa1 | Activator of 90 kDa heat shock protein ATPase homolog 1 | Protein folding | Down | −0.59 | 0.22 | 0.0004 | Down | −0.35 | 0.15 | 0.0001 | −0.06 | 0.08 | 0.0001 | |
| Hsbp1 | Heat shock factor-binding protein 1 | Protein folding | Down | −0.3 | 0.1 | 0 | ||||||||
| Hspa4l | Heat shock 70 kDa protein 4 L | Protein folding | Down | −0.43 | 0.03 | 0 | −0.2 | 0.21 | 0.0004 | |||||
| Gm8797 | Ubiquitin-60S ribosomal protein L40 | RNA metabolism | Up | 0.46 | 0.41 | 0.0078 | Up | 0.47 | 0.13 | 0.0001 | 0.16 | 0.13 | 0.0005 | |
| Hnrnpd | Heterogeneous nuclear ribonucleoprotein D0 | RNA metabolism | Up | 0.3 | 0.15 | 0.0001 | −0.1 | 0.12 | 0.0004 | |||||
| Pcbp1 | Poly(rC)-binding protein 1 | RNA metabolism | Down | −0.49 | 0.11 | 0 | −0.16 | 0.1 | 0 | −0.2 | 0.14 | 0.0006 | ||
| Pabpc2 | Polyadenylate-binding protein | RNA metabolism | Down | −0.29 | 0.16 | 0.0001 | ||||||||
| Hnrnpl | Heterogeneous nuclear ribonucleoprotein L | RNA metabolism | Down | −0.4 | 0.29 | 0.0013 | ||||||||
| Rps20 | 40S ribosomal protein S20 | RNA metabolism | Down | −0.31 | 0.13 | 0.0005 | ||||||||
| Rp17 | 60S ribosomal protein L7 | RNA metabolism | Down | −0.45 | 0.13 | 0.0001 | −0.08 | 0.08 | 0 | −0.16 | 0.07 | 0.0001 | ||
| Pcbp2 | Poly(rC)-binding protein 2 | RNA metabolism | Down | −0.63 | 0.17 | 0.0002 | 0.02 | 0.03 | 0 | 0.17 | 0.04 | 0 | ||
| Git1 | ARF GTPase-activating protein GIT1 | Signaling | 0.04 | 0.08 | 0 | Up | 0.34 | 0.13 | 0.0001 | |||||
| Arf3 | ADP-ribosylation factor 3 | Signaling | −0.03 | 0.05 | 0 | −0.09 | 0.08 | 0 | Down | −0.38 | 0.1 | 0.0002 | ||
| Arhgap35 | Rho GTPase-activating protein 35 | Signaling | Down | −0.27 | 0.22 | 0.0005 | ||||||||
| Gnaq | Guanine nucleotide-binding protein G(q) subunit alpha | Signaling | Down | −0.3 | 0.22 | 0.0004 | −0.17 | 0.12 | 0 | −0.07 | 0.06 | 0 | ||
| Rhot1 | Mitochondrial Rho GTPase 1 | Signaling | Down | −0.33 | 0.06 | 0 | ||||||||
| Rab1 | Ras-related protein Rab-1A | Signaling | Down | −0.27 | 0.14 | 0.0001 | 0 | 0.09 | 0 | 0.14 | 0.07 | 0.0001 | ||
| Plaa | Phospholipase A-2-activating protein | Signaling | Down | −0.69 | 0.25 | 0.0006 | ||||||||
| Prkacb | cAMP-dependent protein kinase catalytic subunit beta | Signaling | Up | 0.53 | 0.29 | 0.0021 | −0.05 | 0.08 | 0 | −0.12 | 0.09 | 0.0001 | ||
| Synj1 | Synaptojanin-1 | Synaptic transmission | Down | −0.38 | 0.16 | 0.0001 | 0.16 | 0.09 | 0.0001 | |||||
| Cldn11 | Claudin-11 | Tight junction component | −0.21 | 0.19 | 0.0003 | Down | −0.46 | 0.23 | 0.0006 | −0.11 | 0.13 | 0.0005 | ||
| Eef1b2 | Elongation factor 1-beta | Translation | Down | −0.29 | 0.19 | 0.0003 | 0.21 | 0.11 | 0.0002 | |||||
| Napb | Beta-soluble NSF attachment protein | Vesicle transport | Down | −0.38 | 0.06 | 0 | −0.09 | 0.06 | 0 | −0.02 | 0.04 | 0 | ||
| Dnm3 | Dynamin-3 | Vesicle transport | Down | −0.38 | 0.13 | 0.0001 | 0.08 | 0.05 | 0 | |||||
| Vps26b | Vacuolar protein sorting-associated protein 26B | Vesicle transport | Up | 0.31 | 0.17 | 0.000223 | ||||||||
| Sv2a | Synaptic vesicle glycoprotein 2A | Vesicle transport | −0.21 | 0.12 | 0.0001 | 0.08 | 0.1 | 0 | Up | 0.29 | 0.16 | 0.0008 | ||
Table displays the Perseus processed protein list containing gene names, protein names, GO biological process names, one-sample t-test, and change of significantly modulated proteins on 2 h and days 2 and 4. This table is complimentary to Morpheus rendered Heatmap of determined changes in protein expression on 2 h and days 2 and 4 (Fig. 4).
3.4. Validation of proteomic changes of genes after H2S exposure
To confirm the observed changes in the IC proteomic profile following acute exposure to H2S, several genes were analyzed by Western blot or quantitative RT-PCR analysis. Mice were sacrificed on each designated time points to measure cellular response. The rationale for selection of proteomic changes for verification was based on a; proteomic changes which were consistently up or down regulated; and b) proteomic changes which fit the overall hypothesis of H2S-induced neurotoxicity.
H2S exposure consistently induced protein expression of Alb in proteomic analysis at 2 h, days 2 and 4. Alb, a major blood protein, is mainly produced in liver (Dodson et al., 2001). The Western blot analysis showed increased protein expression of Alb in the IC in H2S-exposed mice (Fig. 6A). Oxctl is a homodimeric mitochondrial matrix enzyme in the 3-oxoacid CoA-transferase gene family. Oxctl protein was increased in IC of H2S exposed mice (Fig. 6B). Vim encodes intermediate filaments and is part of cytoskeleton (Challa and Stefanovic, 2011). It signals neuroinflammation (Liu et al., 2014). To confirm potential H2S exposure dependent pro-inflammatory and ischemic effects, mRNA expression of Vim was measured. Vim mRNA expression demonstrated a steady increase at 2 h through day 4 (Fig. 7B). This supports the increased protein expression of Vim at 2h. The increased mRNA expression of Vim was consistent with increased Vim protein expression on days 3 and 7 (Fig. 6C).
Fig. 6.
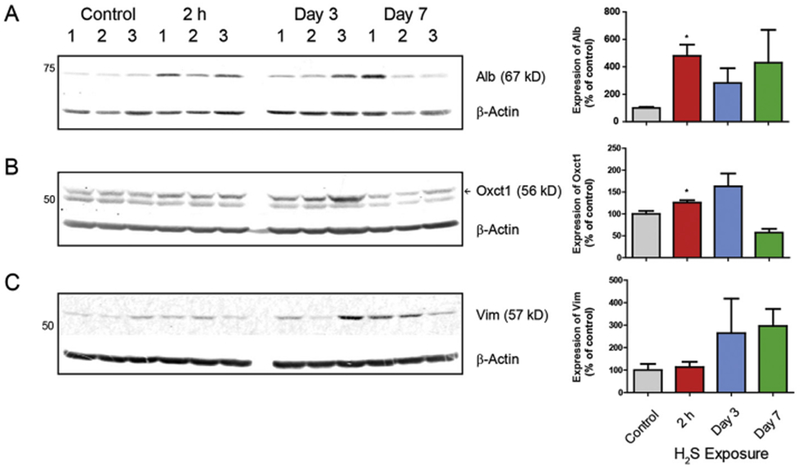
Validation of selected protein expression changes in IC following H2S exposure. Protein expression was measured by Western blot analysis. Quantitative results are shown in graphs next to Western blot images. Samples were normalized to reference gene, β-Actin. Note the significant increase in Alb and Oxctl and a trend of increased Vim. Data are presented as the mean ± S.E.M. Asterisk (*p < 0.05) indicates a significant difference between H2S group and Control group.
Fig. 7.
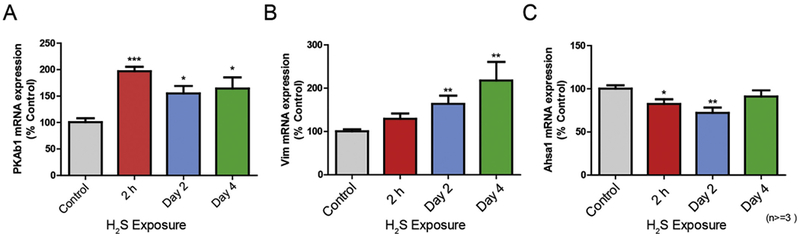
Validation of expression changes of three genes at the mRNA level in IC following H2S exposure. The transcriptional level of three genes (A; Prkab1, B; Vim, C; Ahsal) was measured by quantitative PCR. Samples were normalized with the reference gene Gapdh. Data are presented as the mean ± S.E.M. Note that only a single acute exposure to H2S (2 h) was needed to cause upregulation of gene expression of PkAbl and Vim or downregulation of Ahsal gene expression. Asterisks (***p < 0.001, **p < 0.01, *p < 0.05) indicate a significant difference between H2S group and Control group.
Protein kinase AMP-activated non-catalytic subunit beta 1 (Prkab1) is known to correlate with calcium fluctuations as a measure of cellular calcium response (Yong et al., 2010). Due to unavailability of reliable antibody for the specific Prkab1, Prkab1 mRNA expression was analyzed as a means to measure the calcium-dependent cellular response following H2S exposure. Prkab1 mRNA expression was upregulated following H2S exposure on 2 h and days 2 and 4 which is in line with the mass spectrophotometry analysis observed upregulation of protein Kinase cAMP-activated catalytic subunit alpha (Prkca) and calcium activated protein kinase C beta (Prkacb) on exposure 2 h.
We observed downregulation of Ahsal at 2 h and on day 2 in proteomic profile analysis. Expression of Ahsal was examined by RT-PCR (Fig. 7C). Similar to the protein expression of Ahsal in proteomic profile analysis, mRNA expression of Ahsal demonstrated a significant 30–40% downregulation on 2 h and day 2, compared to the breathing air control group.
3.5. H2S exposure induced signaling pathways in IC
We further analyzed H2S-dependent alterations in the proteome using Webgestalt pathway overrepresentation enrichment analysis with Panther engine and displayed the 10 highest probable pathways for each day of treatment, when applicable, by integrating gene cards information (www.genecards.org). Using this approach, we found that several molecular pathways were involved in H2S-induced neurotoxicity (Table 3). Acute exposure to H2S resulted in alteration of metabotropic glutamate receptor group I pathway, endothelin signaling pathway, inflammation mediated by chemokine and cytokine signaling pathway, and glycolysis at 2 h and on day 2. Fas signaling pathway, gonadotropin-releasing hormone receptor pathway, and cytoskeletal regulation by Rho GTPase pathway were altered on day 4. Remarkably, biological pathways involved at 2h and on day 2 were similar, but patterns of altered biological pathways on day 4 were different (Table 3). The metabotropic glutamate receptor group 1 pathway, which was shown to be involved during early response of H2S exposure at 2h and on day 2, is consistent with our previous findings incriminating dysregulation of glutamate in H2S-induced neurotoxicity (Anantharam et al., 2017). These results also underscore the role of neuroinflammation and are consistent with our previously published data incriminating inflammation in the pathogenesis of H2S-induced neurotoxicity (Anantharam et al., 2017). In addition, expression of Fas was measured by Western blot to check activation of Fas signaling in H2S-induced neurotoxicity (Fig. 8A). The expression of Fas was significantly increased on day 3.
Table 3.
Functional annotation of significantly modulated protein expression in the IC following H2S exposure at 2h, days 2, and 4. Table displays dysregulated molecular pathways and total number of proteins associated with GO-term definition. Note the similarity in pathways at 2 h and on day 2.
| ID | Name | #Gene | FDR |
|---|---|---|---|
| 2h | |||
| P00041 | Metabotropic glutamate receptor group I pathway | 2 | 5.31E-01 |
| P02772 | Pyruvate metabolism | 1 | 1.00E + 00 |
| P00019 | Endothelin signaling pathway | 2 | 1.00E + 00 |
| P02746 | Heme biosynthesis | 1 | 1.00E + 00 |
| P00031 | Inflammation mediated by chemokine and cytokine signaling pathway | 3 | 1.00E + 00 |
| P06664 | Gonadotropin-releasing hormone receptor pathway | 3 | 1.00E + 00 |
| P00024 | Glycolysis | 1 | 1.00E + 00 |
| P04386 | Histamine H2 receptor mediated signaling pathway | 1 | 1.00E + 00 |
| P05913 | Enkephalin release | 1 | 1.00E + 00 |
| P00017 | DNA replication | 1 | 1.00E + 00 |
| Day 2 | |||
| P00041 | Metabotropic glutamate receptor group I pathway | 2 | 5.79E-01 |
| P00017 | DNA replication | 2 | 5.79E-01 |
| P02772 | Pyruvate metabolism | 1 | 1.00E + 00 |
| P02746 | Heme biosynthesis | 1 | 1.00E + 00 |
| P00019 | Endothelin signaling pathway | 2 | 1.00E + 00 |
| P00031 | Inflammation mediated by chemokine and cytokine signaling pathway | 3 | 1.00E + 00 |
| P06664 | Gonadotropin-releasing hormone receptor pathway | 3 | 1.00E + 00 |
| P00024 | Glycolysis | 1 | 1.00E + 00 |
| P04386 | Histamine H2 receptor mediated signaling pathway | 1 | 1.00E + 00 |
| P05913 | Enkephalin release | 1 | 1.00E + 00 |
| Day 4 | |||
| P00029 | Huntington disease | 2 | 1.00E + 00 |
| P00020 | FAS signaling pathway | 1 | 1.00E + 00 |
| P06664 | Gonadotropin-releasing hormone receptor pathway | 2 | 1.00E + 00 |
| P00016 | Cytoskeletal regulation by Rho GTPase | 1 | 1.00E + 00 |
Fig. 8.
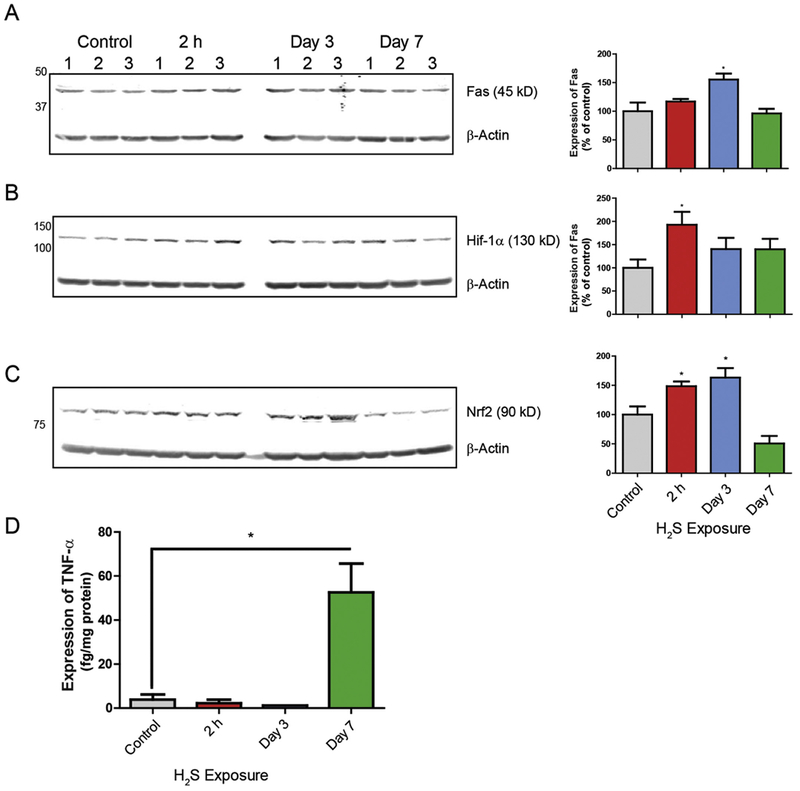
Exposure to H2S induced hypoxic signaling, Fas signaling and inflammatory response in IC. Expression of Fas, Hif-1α, and Nrf2 in IC were analyzed by Western blot assay (A, B and C). Target protein expression was normalized to β-Actin. Quantification of Fas, Hif-1α, and Nrf2 expressions are shown graphically on the right. Expression of TNF-α was measured by ELISA (D). Note that TNF-α was significantly increased in the IC on day 7. Data are presented as the mean ± S.E.M. Asterisk (*p < 0.05) indicates a significant difference between H2S group and Control group.
3.6. H2S exposure induced hypoxia, oxidative stress, and inflammatory signaling pathways in the IC
Hypoxia is implicated in H2S-induced neurotoxicity, but little or no evidence was presented before (Li et al., 2000; Shen et al., 2011). To check if H2S exposure activates hypoxic signaling, expression of Hif-1α was measured by Western blot analysis. Expression of Hif-1α was increased 2 fold after a one time exposure and 1.5 fold on days 3 and 7 (Fig. 8B). To examine oxidative stress signaling in H2S-induced neurotoxicity, expression of Nrf2 was measured by Western blot analysis (Fig. 8C). Protein expression of Nrf2 was significantly increased at 2 h and on day 3. Functional enrichment analysis showed inflammation response was involved in H2S-induced toxicity (Table 3). TNF-α is a key pro-inflammatory cytokine. The expression of TNF-α was measured by ELISA and was found significantly increased on day 7 (Fig. 8D).
4. Discussion
Early events involved in acute H2S exposure or those leading to persistent neurological sequelae are not well known. Neurological sequelae among human survivors of acute H2S poisoning are widely reported (Matsuo et al., 1979; Tvedt et al., 1991a; Tvedt et al., 1991b; Kilburn, 1993; Snyder et al., 1995; Woodall et al., 2005). However, the exact cellular and molecular mechanisms underlying the pathogenesis of delayed neurological sequelae after acute H2S exposure are poorly understood. We have previously identified and herein confirmed that the IC in the brainstem region is highly sensitive to H2S-induced neurodegeneration (Anantharam et al., 2017).
The relative sensitivity of the IC to H2S-induced injury reflects the unique elements and cell populations in this region of the brain. We observed histologic features of neurodegeneration including neuronal death, and reactive gliosis in the IC starting on day 3. Massive cell necrosis and glial scarring were noted in the IC on day 7. Immunohistochemistry with neuron specific antibody (NeuN) revealed severe neuronal loss in the IC following H2S exposure. Whereas neuronal loss was minimal in mice from the breathing air control group, there was > 70% loss in neurons in IC of mice exposed to H2S. In contrast, brain stem regions adjacent to the IC were shown to be relatively unaffected, indicating unique susceptibility of the IC region to H2S toxicity. The function of the IC is to integrate auditory and other sensory signals. IC is reportedly a region with high metabolic rate requirements (Ridgway et al., 2006; Houser et al., 2010) while it is also the most highly vascularized brain region (Gonzalez-Lima et al., 1997). H2S is a systemic metabolic toxicant that reportedly interferes with ATP synthesis. It is reasonable to infer that brain regions with high blood supply and metabolic rates such as the IC are more vulnerable to H2S-induced toxicity. Hearing loss is a frequently reported sequelae of acute H2S poisoning and these results are consistent with that observation.
In this study, we used proteomic analysis to define cellular and molecular mechanisms of H2S induced neurotoxicity and neurodegeneration during early stages of injury. Exposure to H2S induced significant proteomic changes in IC. Proteins commonly and constantly upregulated during H2S exposure were Cyfip2, hemoglobin subunit alpha (Hba), hemoglobin subunit beta-2 (Hbb-b2), and Alb. In contrast, Ahsal was consistently downregulated on 2h and days 2 and 4. The increased expression of Alb was confirmed by Western blot. Alb is a major blood protein and its presence in the IC suggests a breach of the blood-brain barrier (BBB), leading to protein leakage. Indeed, hemorrhage was observed in IC of mice following H2S exposure. Disruption of BBB triggered by a variety of factors has been reported to cause neuroinflammation (Erickson et al., 2012). This is a preliminary observation and the role of breach of BBB in H2S-induced neurotoxicity should be evaluated further in the future. The cause of the BBB breach is beyond the scope of this current study, but we also observed changes in other proteins triggered by hypoxic conditions including adenosine Kinase, Tropomodulin 2, vimentin, Neurogranin, and Poly(RC) binding protein 2. It is possible hypoxia induced the breach in the BBB.
Other key altered molecular biological pathways include metabotropic glutamate receptor group I pathway, inflammation mediated by chemokine and cytokine signaling pathway, and altered glycolysis. We have previously reported that H2S increases the glutamate:GABA ratio (Anantharam et al., 2017), an indication that H2S may activate glutamate receptors. These results suggesting a role for metabotropic glutamate I pathway in the pathogenesis of H2S-induced neurotoxicity is yet more evidence for glutamate-induced excitotoxicity. Glutamate excitotoxicity causes seizures (Jett, 2012).
H2S-induced neurotoxicity has been suggested to resemble brain injury caused by ischemic hypoxic conditions (Doujaiji and Al-Tawfiq, 2010; Rumbeiha et al., 2016). We observed increased Hif-1α protein expression in IC of H2S exposed mice. These results further support the role of hypoxia in H2S-induced neurotoxicity. The trigger for hypoxia is likely inhibition of cytochrome c oxidase in mitochondria, but this remains to be elucidated. Also, characterizing the downstream cascade of Hif-1α signaling pathway remains to be studied. Regardless, this is the first study to directly tie hypoxia to Hif-1α signaling in H2S-induced neurotoxicity. Previous studies have suggested neurofilament Nefl, and collapsing Response Mediator Protein 3 (Dpysl4) as biomarkers of hypoxia and cerebral ischemia (Hou et al., 2006; Lian et al., 2015). Dpysl4 is cleaved by activated calpain reaction in response to cerebral ischemia (Hou et al., 2006). Both Nefl and Dpysl4 were downregulated during H2S exposure. In addition, mass spectrophotometry analysis detected downregulation of phosphoglycerate mutase 2 (Pgam2), an ischemia biomarker, on day 1. This biomarker is suggested to correlate with the ischemic conditions (Li et al., 2012). Vimentin (Vim) has been shown as another biomarker for ischemia (Li et al., 2008). Mass spectrophotometry also detected a steady increase in protein and mRNA expression of the inflammatory and ischemia biomarker Vim for the entire duration of the study. These data collectively support H2S-dependent ischemia-like conditions during acute H2S exposures, further supporting the role of hypoxia as a mechanism of H2S-induced neurotoxicity.
It is well known that H2S inhibits cytochrome c oxidase which plays a crucial role in mitochondrial ATP synthesis (Nicholls and Kim, 1982; Dorman et al., 2002). Using variations of this animal model we have consistently shown inhibition of cytochrome c oxidase (Anantharam et al., 2017). In this proteomic study, expression levels of cytochrome c oxidase-related proteins were not changed, but mitochondrial cytochrome c oxidase subunit 2 (mt-Co2) and cytochrome c oxidase subunit 6C were decreased at 2 h and on day 4, respectively, supporting the notion of potential impairment of energy production following H2S exposure. Several other proteins related to energy production were also altered. For instance, ATP synthase subunits Atp5j2 and ATP51 together with cytochrome C oxidases 6C were downregulated on days 2, and 4, respectively. In addition, citric acid cycle proteins malic enzyme (Mel) and phosphoglycerate mutase (Pgam2) were downregulated on 2 h post a single dose. Oxctl was upregulated on day 2 and 4. Oxctl was upregulated and induced ATP production from ketone bodies when hepatocellular carcinoma were deprived of serum (Galluzzi and Kroemer, 2016). Increase of Oxctl during H2S exposure may indicate a switch from glucose metabolism to alternative sources of ATP production in IC of mice exposed acutely to H2S. Further studies are warranted to check whether Coxy6c, mt-Co2, and Oxctl are the bona fide targets of H2S toxicity. Collectively, these results suggest hypoxia, resulting in low oxygen tissue delivery, and dysregulation of cytochrome C related proteins, likely working in concert leading to energy deprivation and eventual neuronal death in the IC.
We have previously implicated neuroinflammation in this mouse model of acute H2S intoxication (Anantharam et al., 2017). Inflammation mediated by chemokine and cytokine signaling pathway was a key early finding in this study, and strongly upregulated starting 2h following a single acute exposure (Table 3). We have previously reported activated astrocytes and microglia following acute H2S exposure (Anantharam et al., 2017). Changes in immunogenic biomarkers represent one of earliest responses to cytotoxicity, and inflammation (Anantharam et al., 2017). Expression of TNF-α was shown to be significantly activated and increased in the IC on day 7 of H2S exposure. Moreover, anti-inflammatory protein macrophage migration inhibition factor (Mif) was downregulated on 2 h, and complement binding protein Clqbp was increased on day 4. Other pro-inflammatory related proteomic changes observed in this study include upregulations of Cyfip2 and nuclear hormone receptor and transcriptional repressor family (Nr1d2) which were observed throughout the course of the study. Also, increased expression of Cyfip2 was previously shown by others to have a p53-response element in its promoter region and to be one of the direct targets of p53 (Jackson et al., 2007). In contrast, the anti-apoptotic and immunogenic protein Bcl2 associated athanogene (Bag5) was downregulated at 2 h. Endoplasmic reticulum stress-induced downregulation of Bag5 has previously reported (Bruchmann et al., 2013; Gupta et al., 2016). Furthermore, expression of Fas was increased on day 3. Fas was previously responsible for initiating apoptotic signaling pathways and inflammation response (Cullen and Martin, 2015). The exact roles of these molecules in H2S-induced neurotoxicity remain to be studied further. However, collectively, these results are consistent with our previous observations that inflammation may play an important role in H2S-induced neurotoxicity (Anantharam et al., 2017).
In summary, this seminal proteomic study of the IC from mice acutely H2S-intoxicated mice has yielded both novel and supporting results of our previous observations. H2S-induced neurotoxicity is complex. A summary of our current overarching hypothesis of acute H2S-induced neurotoxicity is summarized in Fig. 9. We have explained how proteomic changes observed in this study fit key nodes of this summary scheme. We have also, to the extent possible, validated select proteomic changes using quantitative RT-PCR and Western blot analyses. Sufficient concordance was observed for the validated proteomic changes using transcript and/or protein expression. A very interesting observation was the close similarity in the top 10 proteomic pathways early in the course of H2S-intoxication (2 h and day 2). The study also uncovered novel pathways with a potential role in H2S-induced toxicity which we would not have predicted. These include the endothelin signaling pathway, the gonadotropin releasing hormone pathway, and the histamine H2 receptor mediated signaling pathway, among others summarized in Table 3. These likely play a role in any of the pathogenesis of multiple sequelae reported following acute H2S-intoxication, some of which are listed in Fig. 9. More research is certainly needed to study the role of these novel pathways in acute H2S-induced neurotoxicity.
Fig. 9.
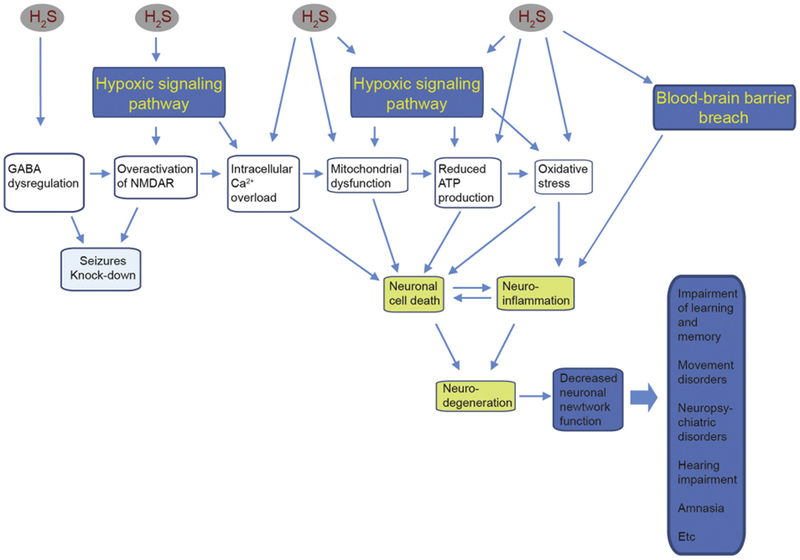
Overarching scheme of H2S induced neurotoxicity in the IC.
This figure is a summary of the overarching hypothesis of acute H2S-induced neurotoxicity. H2S activates glutamate excitotoxicity, mitochondrial injury, hypoxia, and a breach of the blood brain barrier, and mitochondrial injury trigger neuronal cell death, neuro-inflammation and oxidative stress which ultimately leads to neurodegeneration. Key pathways uncovered in the present study fit in various nodes in this overarching scheme.
5. Conclusion
In this study we examined the early effects of acute H2S-induced neurotoxicity in mice. The study focused on the IC, the most sensitive brain region to acute H2S poisoning. Results show that H2S modulated several proteins and biological pathways in the IC. Specifically, results suggest glutamate-induced excitotoxicity, immune mediated inflammatory response, pro-apoptosis mechanisms, and hypoxia/ischemia-like signaling to be involved in H2S-induced neurotoxicity, among others. These results are in concert with our previously published observations, including dysregulated glutamate and GABA, mitochondrial injury, neuroinflammation and oxidative stress in the pathogenesis of acute H2S-induced neurotoxicity. An overall scheme of key events we hypothesize to be involved in the pathogenesis of acute H2S induced neurotoxicity is shown in Fig. 9. However, this research has also uncovered novel pathways which potentially may not fit the above scheme. This suggests that acute H2S-induced neurotoxicity is a complex process. Further research is recommended to better understand the singular and/or collective role of these potential mechanisms in H2S-induced mortality, neurotoxicity, and evolution of neurological sequelae.
Acknowledgment
Funding
This work was partially supported by the Iowa State University College of Veterinary Medicine Seed grant, Startup funds and incentive account funds for Rumbeiha.
Footnotes
The authors do not have any conflict of interest.
References
- Ahlborg G, 1951. Hydrogen sulfide poisoning in shale oil industry. A.M.A. Arch. Ind. Hygiene Occup. Med 3, 247–266. [PubMed] [Google Scholar]
- Anantharam P,, Whitley EM, Mahama B, Kim DS, Imerman PM, Shao D, Langley MR, Kanthasamy A, Rumbeiha WK, 2017. Characterizing a mouse model for evaluation of countermeasures against hydrogen sulfide-induced neurotoxicity and neurological sequelae. Ann. N. Y. Acad. Sci 1400, 46–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandow K, Kusuyama J, Kakimoto K, Ohnishi T, Matsuguchi T, 2015. AMP-activated protein kinase (AMPK) activity negatively regulates chondrogenic differentiation. Bone 74, 125–133. [DOI] [PubMed] [Google Scholar]
- Bielow C, Mastrobuoni G, Kempa S, 2016. Proteomics quality control: quality control software for MaxQuant results. J. Proteome Res 15, 777–787. [DOI] [PubMed] [Google Scholar]
- Bruchmann A, Roller C, Walther TV, Schafer G, Lehmusvaara S, Visakorpi T, Klocker H, Cato AC, Maddalo D, 2013. Bcl-2 associated athanogene 5 (Bag5) is overexpressed in prostate cancer and inhibits ER-stress induced apoptosis. BMC Cancer 13, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challa AA, Stefanovic B, 2011. A novel role of vimentin filaments: binding and stabilization of collagen mRNAs. Mol. Cell. Biol 31, 3773–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S, Ogden JM, Pohl HR, Scinicariello F, Ingerman L, Barber L, Citra M, 2016. In: Sciences, D.o.T.a.H.H (Ed.), Toxicological Profile for Hydrogen Sulfide and Carbonyl Sulfide. Environmental Toxicology Branch. [Google Scholar]
- Cox J, Mann M, 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cullen SP, Martin SJ, 2015. Fas and TRAIL ‘death receptors’ as initiators of inflammation: implications for cancer. Semin. Cell Dev. Biol 39, 26–34. [DOI] [PubMed] [Google Scholar]
- Dalgard CL, Cole JT, Kean WS, Lucky JJ, Sukumar G, McMullen DC, Pollard HB, Watson WD, 2012. The cytokine temporal profile in rat cortex after controlled cortical impact. Front. Mol. Neurosci 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson CS, Rengarajan K,, Gewant HD, Stodulkova E, Nguyen HT, Boatright JH, Nickerson JM, 2001. Extra-hepatic expression of serum albumin mRNA in mouse retina. Curr. Eye Res 22, 182–189. [DOI] [PubMed] [Google Scholar]
- Dorman DC, Moulin FJ, McManus BE, Mahle KC, James RA, Struve MF, 2002. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol. Sci 65, 18–25. [DOI] [PubMed] [Google Scholar]
- Doujaiji B, Al-Tawfiq JA, 2010. Hydrogen sulfide exposure in an adult male. Ann. Saudi Med 30, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MA, Dohi K, Banks WA, 2012. Neuroinflammation: a common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation 19, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes CH, 2009. “Gas!” The Story of the Special Brigade. Naval and Military. [Google Scholar]
- Galluzzi L, Kroemer G, 2016. Aberrant ketolysis fuels hepatocellular cancer progression. Cell Res 26, 1077–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lima F, Valla J, Matos-Collazo S, 1997. Quantitative cytochemistry of cytochrome oxidase and cellular morphometry of the human inferior colliculus in control and Alzheimer’s patients. Brain Res 752, 117–126. [DOI] [PubMed] [Google Scholar]
- Greenberg M, Hamilton R, 1998. The epidemiology of deaths related to toxic exposures in the US workplace, 1992–1996. J. Toxicol 5, 430. [Google Scholar]
- Guidotti TL, 2015. Hydrogen sulfide intoxication. Handb. Clin. Neurol 131, 111–133. [DOI] [PubMed] [Google Scholar]
- Gupta MK, Tahrir FG, Knezevic T, White MK, Gordon J, Cheung JY, Khalili K, Feldman AM, 2016. GRP78 interacting partner Bag5 responds to ER stress and protects cardiomyocytes from ER stress-induced apoptosis. J. Cell. Biochem 117, 1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homji NF, Mao X, Langsdorf EF, Chang SL, 2012. Endotoxin-induced cytokine and chemokine expression in the HIV-1 transgenic rat. J. Neuroinflammation 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou ST, Jiang SX, Desbois A, Huang D, Kelly J, Tessier L, Karchewski L, Kappler J, 2006. Calpain-cleaved collapsin response mediator protein-3 induces neuronal death after glutamate toxicity and cerebral ischemia. J. Neurosci 26, 2241–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser DS, Moore PW, Johnson S, Lutmerding B, Branstetter B, Ridgway SH, Trickey J, Finneran JJ, Jensen E, Hoh C, 2010. Relationship of blood flow and metabolism to acoustic processing centers of the dolphin brain. J. Acoust. Soc. Am 128, 1460–1466. [DOI] [PubMed] [Google Scholar]
- Jackson RS 2nd, Cho YJ, Stein S, Liang P, 2007. CYFIP2, a direct p53 target, is leptomycin-B sensitive. Cell Cycle 6, 95–103. [DOI] [PubMed] [Google Scholar]
- Jett DA, 2012. Chemical toxins that cause seizures. Neurotoxicology 33, 1473–1475. [DOI] [PubMed] [Google Scholar]
- Kilburn KH, 1993. Case report: profound neurobehavioral deficits in an oil field worker overcome by hydrogen sulfide. Am J Med Sci 306, 301–305. [DOI] [PubMed] [Google Scholar]
- Kim DS, Jin H, Anantharam V, Gordon R, Kanthasamy A, Kanthasamy AG, 2016. p73 gene in dopaminergic neurons is highly susceptible to manganese neurotoxicity. Neurotoxicology 59, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GL, Farooque M, Lewen A, Lennmyr F, Holtz A, Olsson Y, 2000. MAP2 and neurogranin as markers for dendritic lesions in CNS injury. An immunohistochemical study in the rat. APMIS 108, 98–106. [DOI] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Stahlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, Nilsson M, Pekny M, 2008. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab 28, 468–481. [DOI] [PubMed] [Google Scholar]
- Li H, Li J, Wang Y, Yang T, 2012. Proteomic analysis of effluents from perfused human heart for transplantation: identification of potential biomarkers for ischemic heart damage. Proteome Sci 10, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian T, Qu D, Zhao X, Yu L, Gao B, 2015. Identification of site-specific stroke biomarker candidates by laser capture microdissection and labeled reference peptide. Int. J. Mol. Sci 16, 13427–13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li Y, Cui Y, Roberts C, Lu M, Wilhelmsson U, Pekny M, Chopp M, 2014. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia 62, 2022–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo F, Cummins JW, Anderson RE, 1979. Neurological sequelae of massive hydrogen sulfide inhalation. Arch. Neurol 36, 451–452. [DOI] [PubMed] [Google Scholar]
- Meade ML, Hoffmann A, Makley MK, Snider TH, Schlager JJ, Gearhart JM, 2015. Quantitative proteomic analysis of the brainstem following lethal sarin exposure. Brain Res 1611, 101–113. [DOI] [PubMed] [Google Scholar]
- Morii D, Miyagatani Y, Nakamae N, Murao M, Taniyama K, 2010. Japanese experience of hydrogen sulfide: the suicide craze in 2008. J. Occup. Med. Toxicol 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls P, Kim JK, 1982. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can. J. Biochem 60, 613–623. [DOI] [PubMed] [Google Scholar]
- Reedy SJ, Schwartz MD, Morgan BW, 2011. Suicide fads: frequency and characteristics of hydrogen sulfide suicides in the United States. West J Emerg Med 12, 300–304. [PMC free article] [PubMed] [Google Scholar]
- Ridgway S, Houser D, Finneran J, Carder D, Keogh M, Van Bonn W, Smith C, Scadeng M,, Dubowitz D, Mattrey R, Hoh C, 2006. Functional imaging of dolphin brain metabolism and blood flow. J. Exp. Biol 209, 2902–2910. [DOI] [PubMed] [Google Scholar]
- Rumbeiha W, Whitley E, Anantharam P, Kim DS, Kanthasamy A, 2016. Acute hydrogen sulfide-induced neuropathology and neurological sequelae: challenges for translational neuroprotective research. Ann. N. Y. Acad. Sci 1378, 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sams RN, Carver HW 2nd, Catanese C, Gilson T, 2013. Suicide with hydrogen sulfide. Am J Forensic Med Pathol 34, 81–82. [DOI] [PubMed] [Google Scholar]
- Security D.o.H, 2017. Appendix A to Part 27. – DHS Chemicals of Interest. Federal Regulation. [Google Scholar]
- Shen HY, Lusardi TA, Williams-Karnesky RL, Lan JQ, Poulsen DJ, Boison D, 2011. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J. Cereb. Blood Flow Metab 31, 1648–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JW, Safir EF, Summerville GP, Middleberg RA, 1995. Occupational fatality and persistent neurological sequelae after mass exposure to hydrogen sulfide. Am. J. Emerg. Med 13, 199–203. [DOI] [PubMed] [Google Scholar]
- Tvedt B, Edland A, Skyberg K, Forberg O, 1991a. Delayed neuropsychiatric sequelae after acute hydrogen sulfide poisoning: affection of motor function, memory, vision and hearing. Acta Neurol. Scand 84, 348–351. [DOI] [PubMed] [Google Scholar]
- Tvedt B, Skyberg K, Aaserud O, Hobbesland A, Mathiesen T, 1991b. Brain damage caused by hydrogen sulfide: a follow-up study of six patients. Am. J. Ind. Med 20, 91–101. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J, 2016. The Perseus computational platform for comprehensive analysis of (prote) omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Ulmasov B, Oshima K, Rodriguez MG, Cox RD, Neuschwander-Tetri BA, 2013. Differences in the degree of cerulein-induced chronic pancreatitis in C57BL/6 mouse substrains lead to new insights in identification of potential risk factors in the development of chronic pancreatitis. Am. J. Pathol 183, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodall GM, Smith RL, Granville GC, 2005. Proceedings of the hydrogen sulfide health research and risk assessment symposium October 31-November 2, 2000. Inhal. Toxicol 17, 593–639. [DOI] [PubMed] [Google Scholar]
- Yong QC, Choo CH, Tan BH, Low CM, Bian JS, 2010. Effect of hydrogen sulfide on intracellular calcium homeostasis in neuronal cells. Neurochem. Int 56, 508–515. [DOI] [PubMed] [Google Scholar]