

Abstract
Sixteen new sulfur-containing compounds targeting vesicular acetylcholine transporter (VAChT) were synthesized and assessed for in vitro binding affinities. Enantiomers (-)-8 and (-)-14a displayed high binding affinities with Ki values of 1.4 and 2.2 nM for human VAChT, moderate and high selectivity for human VAChT over σ1 (~13-fold) and σ2 receptors (> 420-fold). Radiosyntheses of (-)-[11C]8 and (-)-[18F]14a were achieved using conventional methods. Ex vivo autoradiography and biodistribution studies in Sprague Dawley rats indicated that both radiotracers had the capability to penetrate the blood-brain barrier, with high initial brain uptake at 5 min, and rapid washout. The striatal region had the highest accumulation for both radiotracers. Pretreating the rats with a VAChT ligand, (-)-vesamicol reduced brain uptake for both radiotracers. Pretreating the rats with a σ1 ligand, YUN-122 also reduced brain uptake, suggesting these two radiotracers also bind to the σ1 receptor in vivo. MicroPET study of (-)-[11C]8 in the brain of non-human primate showed high striatal accumulation that peaked quickly and washed out rapidly. Although preliminary results indicated these two sulfur-containing radiotracers had high binding affinities toward VAChT with rapid washout kinetics from the striatum, their σ1 receptor binding property limits their potential as radiotracers for quantifying VAChT in vivo.
Keywords: vesicular acetylcholine transporter, neurodegenerative diseases, radiotracers, biodistribution, PET
Graphical Abstract
Let’s image: New sulfur-containing analogues have high potency for vesicular acetylcholine transporter (VAChT). The radiosyntheses of two lead radiotracers are straightforward and effective. The rodent studies demonstrated the tracers can enter the brain and highly accumulate in the striatum. PET study suggested the lead C-11 tracer has quick washout kinetics from the macaque brain. Although moderate σ1 receptor binding affinity limited the in vivo mapping VAChT, the structure-activity relationship may provide information for the future exploring of new VAChT radioligands.
Introduction
The loss of cholinergic neurons and synapses in the brain is associated with cognitive dysfunction, a key feature of neurodegenerative diseases, such as Alzheimer disease[1] and Parkinson disease.[2] Vesicular acetylcholine transporter (VAChT) is a neurotransmitter transporter which is responsible for loading acetylcholine (ACh) into presynaptic vesicles in neurons,[3] has been widely accepted as a reliable biomarker for measuring the loss of cholinergic neurons in the central nervous system (CNS).[4] Positron emission tomography (PET) is a non-invasive molecular imaging modality that provides a sensitive three-dimensional map of tracer. Combined with a suitable VAChT radioligand, PET provides a unique methodology to quantitatively measure VAChT regional brain distribution which can advance our understanding of the pathophysiology of neurodegenerative diseases.
Currently, numerous PET tracers for VAChT have been developed using the (-)-vesamicol scaffold, but very few are approved for human use.[5] (-)-[18F]FEOBV was the first PET tracer for mapping the expression of VAChT in the human brain.[6] It provided a great advantage over [123I]IBVM, a SPECT radioligand for assessing the loss of cholinergic neuron and synapses in the brain.[7] In 2016, we reported the automated production of (-)-[18F]VAT in our current good manufacturing practice (cGMP) facility and obtained exploratory Investigational New Drug (IND) approval from the Food and Drug Administration (FDA) for the first-in-man studies.[8] Investigation of (-)-[18F]VAT in patients with different neurological diseases are ongoing. Here, we reported our exploration of a series of new sulfur-containing benzovesamicol analogues to identify VAChT PET radiotracers with novel structures related to [18F]VAT.
Sulfur is prevalent in biologically active natural products and approved drugs, like penicillin and cephalosporin β-lactam-based antibiotics. Taking advantage of the electron deficient and bivalent properties of the sulfur atom, incorporating sulfur-containing moieties into the molecules to improve pharmaceutical properties is a frequently used strategy in drug design and development. Moieties such as sulfone and sulfonamide can modulate the overall polarity or ionization state of molecules and provide convenient synthetic approaches to generate new analogues.[9] In addition, sulfur-containing compounds may engage biochemical interactions in vivo and alter the in vivo metabolic stability.[10] To test the hypothesis that incorporating sulfur into VAChT radiotracer would alter the in vivo pharmacologic properties, we interposed sulfur-containing moieties such as thioether, sulfoxide, sulfone, and sulfamide into the carbonyl-containing pharmacophore structures represented by lead VAChT radiotracers, (-)-[18F]4 and (-)-[11C]5 (Figure 1).[11, 12, 13] In current manuscript, we reported our efforts on the syntheses and in vitro biological evaluation of these new sulfur-containing analogues, the radiosyntheses and in vivo evaluation in rodent of representative (-)-[11C]8 and (-)-[18F]14a, as well as microPET study of (-)-[11C]8 in the brain of non-human primate (NHP).
Figure 1.

Chemical structures of VAChT compounds and PET radiotracers.
Results and Discussion
Chemistry
Briefly, starting with compounds (±)-6 and (±)-10, the new sulfur-containing compounds were synthesized following Scheme 1. The sulfone (±)-7 was synthesized by reacting compound (±)-6 with sodium methanesulfonate in dimethyl sulfoxide (DMSO) at 100 °C. Meanwhile, thioether (±)-8 was prepared by reacting compound (±)-6 with sodium methoxide in the presence of K2CO3 and dimethylformamide (DMF). Next, oxidation of thioether (±)-8 using one equivalent H2O2 in acetic acid afforded sulfoxide (±)-9. In addition, a novel palladium-catalyzed sulfonation of aryl halide (±)-10 in the presence of K2S2O5 and sodium formate was followed to produce the sulfinate intermediate, which directly generated sulfonamides (±)-11a-b or the sulfone (±)-12 in situ by reacting with corresponding amines or 1-bromo-3-fluoroprane, respectively. The aryl thiol (±)-13 was made using a highly efficient copper-catalyzed one-step procedure from aryl halide (±)-10 with 1,2-ethanedithiol. Compounds (±)-14a-b were prepared by reacting 1-bromo-2-fluoroethane or 1-bromo-3-fluoropropane with aryl thiol (±)-13 in the presence of tetrabutylammonium bromide (TBAB) and Cs2CO3 in DMF, respectively. All the compounds were converted to their oxalate form before performing the in vitro binding assay.
Scheme 1.

Syntheses of sulfur-containing compounds. Reagents and conditions: (a) sodium methanesulfinate, DMSO, 100 °C; (b) sodium thiomethoxide, K2CO3, DMF, 55 °C; (c) H2O2, acetic acid, 55 °C; (d) (i) K2S2O5, TBAB, sodium formate, Pd(OAc)2, PPh3, 1,10-phenanthroline, DMSO, rt; (ii) NCS, methylamine or 2-fluoroethylamine, DIPEA, THF, rt; (e) (i) K2S2O5, TBAB, sodium formate, Pd(OAc)2, PPh3, 1,10-phenanthroline, DMSO, rt; (ii)1-bromo-3-fluoropropane, THF, rt; (f) 1,2-ethanedithiol, copper(II) sulfate pentahydrate, Cs2CO3, DMSO, rt; (g) 1-bromo-2-fluoroethane or 1-bromo-3-fluoropropane, TBAB, Cs2CO3, DMF, rt.
In Vitro Binding Affinity Studies
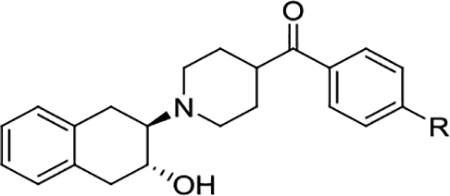
The newly synthesized compounds were first screened for in vitro VAChT binding affinities. As shown in Table 1, several of the new compounds were much more potent than (-)-vesamicol and (-)-FEOBV. Compounds (±)-7, (±)-8, (±)-14a, and (±)-14b had VAChT Ki values of 5.4, 2.4, 2.4, and 2.3 nM, respectively. The thioether compound (±)-8 (Ki = 2.4 nM) had a higher binding potency than the sulfone (±)-7 (Ki = 5.4 nM) and the sulfoxide (±)-9 (Ki = 6.5 nM). Both sulfamide ligands (±)-11a (Ki = 6.3 nM) and (±)-11b (Ki = 13.0 nM) are also very potent for VAChT. Because (-)-vesamicol structural analogues were reported to have stereo-selective binding property,[16] the racemic compounds (±)-7, (±)-8, (±)-14a, and (±)-14b were further resolved via chiral high-performance liquid chromatography (HPLC) using a semi-preparative Chiralcel OD column. The resolved enantiomers were converted to oxalates to determine their in vitro VAChT binding affinities. As shown in Table 2, all the minus enantiomers exhibited higher VAChT binding affinity than the plus counterparts.
Table 1.
Binding affinities (Ki) of sulfur-containing compounds for VAChT[a].
 | ||
|---|---|---|
| Compounds[b] | R | Ki (nM)[c] |
| (-)−vesamicol | 15.2 ± 1.1 | |
| (-)−FEOBV[14] | 19.6 ± 1.1 | |
| (±)−7 | SO2CH3 | 5.4 ± 0.6 |
| (±)−8 | SCH3 | 2.4 ± 0.3 |
| (±)−9 | SOCH3 | 6.5 ± 0.9 |
| (±)−11a | SO2NHCH3 | 6.3 ± 0.8 |
| (±)−11b | SO2NH(CH2)2F | 13.0 ± 1.0 |
| (±)−12 | SO2(CH2)3F | 12.8 ± 0.7 |
| (±)−14a | S(CH2)2F | 2.4 ± 0.5 |
| (±)−14b | S(CH2)3F | 2.3 ± 0.4 |
Ki values (mean ± SD) were determined at least three experiments.
All the compounds were tested as their oxalate forms.
Expressed human VAChT was used for the VAChT binding assay.
Table 2.
Binding affinities (Ki) of sulfur-containing compounds for the σ1 receptor, σ2 receptor, and VAChT[a].
 |
||||||
|---|---|---|---|---|---|---|
| Compounds[b] | R | Ki (nM) | Selectivity ratio | |||
| VAChT[c] | σ1[d] | σ2[e] | VAChT/ σ1 | VAChT/ σ2 | ||
| (-)−vesamicol | 15.2 ± 1.1 | 25.8 ± 8.0 | 34.5 ± 2.0 | 1.7 | 2.3 | |
| (-)−FEOBV[14] | 19.6 ± 1.1 | 209 ± 94 | - | 10.7 | - | |
| YUN-122[15] | - | 3.6 ± 1.1 | 667 ± 98 | - | - | |
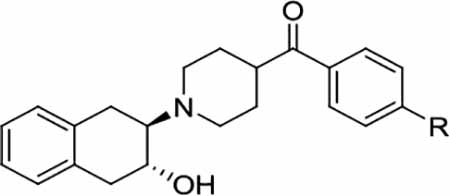
| (±)−7 | SO2CH3 | 5.4 ± 0.6 | 16.5 ± 2.1 | > 1,000 | 3.1 | > 185 |
| (+)−7 | SO2CH3 | 193 ± 17.8 | 18.5 ± 3.4 | > 1,000 | 0.1 | > 5 |
| (-)−7 | SO2CH3 | 4.1 ± 0.9 | 34.7 ± 4.9 | > 1000 | 8.5 | > 240 |
| (±)−8 | SCH3 | 2.4 ± 0.3 | 17.2 ± 4.0 | > 1,000 | 7.2 | > 420 |
| (+)−8 | SCH3 | 13.8 ± 1.1 | 23.3 ± 2.9 | > 1,000 | 1.7 | > 70 |
| (-)−8 | SCH3 | 1.4 ± 0.3 | 19.0 ± 5.6 | > 1,000 | 13.6 | > 710 |
| (±)−14a | S(CH2)2F | 2.4 ± 0.5 | 16.7 ± 2.1 | > 1,000 | 7.0 | > 420 |
| (+)−14a | S(CH2)2F | 11.8 ± 2.7 | 14.4 ± 2.0 | > 1,000 | 1.2 | > 80 |
| ()−14a | S(CH2)2F | 2.2 ± 0.4 | 30.8 ± 4.1 | > 1,000 | 13.4 | > 450 |
| (±)−14b | S(CH2)3F | 2.3 ± 0.4 | 40.8 ± 4.1 | > 1,000 | 17.7 | > 430 |
| (+)−14b | S(CH2)3F | 19.9 ± 9.2 | 38.6 ± 7.1 | > 1,000 | 1.9 | > 50 |
| (-)−14b | S(CH2)3F | 1.4 ± 0.2 | 4.1 ± 0.7 | > 1,000 | 2.9 | > 710 |
Ki values (mean ± SD) were determined at least three runs.
All the compounds were tested as their oxalate forms.
Expressed human VAChT was used for VAChT binding assay.
Homogenate of guinea pig brain was used for binding assay.
Homogenate of rat liver was used for binding assay.
To determine the selectivity of these compounds for binding VAChT relative to sigma receptors, their binding potencies for σ1 and σ2 receptors were determined (Table 2). The results showed these new compounds had no affinity for σ2 receptor (Ki > 1000 nM). Unfortunately, they had moderate to high σ1 affinity with the Ki values ranging from 4.1 to 40.8 nM, which is different from the previously reported N-substituted VAChT analogues.[11, 13] Since compounds (-)-8 and (-)-14a were the most potent VAChT ligands (Ki = 1.4 and 2.2 nM) with moderate selectivity for VAChT over σ1 receptor (~13.6-fold), and high selectivity over σ2 receptor, (-)-[11C]8 and (-)-[18F]14a were radiosynthesized and the initial evaluation as PET radiotracers for VAChT was performed via ex vivo autoradiography, distribution in Sprague-Dawley (SD) rats, and microPET study in cynomolgus macaques (for. (-)-[11C]8 only).
Radiochemistry
Precursor (-)-13 was prepared from (-)-10 which was resolved from (±)-10 using a Chiralcel OD HPLC column. The radiosynthesis of (-)-[11C]8 was accomplished via S-[11C]methylation of thiophenol (-)-13 using [11C]CH3I in the presence of Cs2CO3 and TBAB in DMF (Scheme 2) with radiochemical yield ~45%, radiochemical purity >98%, and molar activity >74 GBq/μmol (decay corrected to end of synthesis, EOS). The radiosynthesis of (-)-[18F]14a was achieved by a two-step procedure as shown in Scheme 2. First, ethylene ditosylate was reacted with [18F]KF/Kryptofix 222 in acetonitrile followed by purification on a reverse phase HPLC to afford [18F]15. Next, the precursor (-)-13 was reacted with [18F]15 in DMSO in the presence of Cs2CO3 followed by purification on HPLC afforded (-)-[18F]14a with radiochemical yield ~65%, radiochemical purity >95%, and molar activity >55 GBq/μmol (decay corrected to EOS). The radioactive (-)-[11C]8 or (-)-[18F]14a dose was authenticated by co-injection with the standard reference (-)-8 or (-)-14a on an analytical HPLC system, respectively.
Scheme 2.

Radiosyntheses of (-)-[11C]8 and (-)-[18F]14a. Reagents and conditions: (a) [11C]CH3I, Cs2CO3, TBAB, DMF, 60 °C, 5 min; (b) [18F]KF, K222, K2CO3, acetonitrile, 110 °C, 10 min; (c) [18F]15, Cs2CO3, DMSO, 100 °C, 15 min.
Ex Vivo Autoradiography Studies
Adult male SD rats were used for ex vivo autoradiography to check the radioligand’s ability to penetrate the blood-brain barrier (BBB). Rats were injected with either (-)-[11C]8 or (-)-[18F]14a. The rat was euthanized at 30 min post-injection for (-)-[11C]8 due to the short half-life of the radionuclide; for (-)-[18F]14a, the rat was euthanized at 60 min post-injection. Rat brains were immediately removed and snap-frozen then sectioned and mounted on glass slides. Radioactivity accumulation was visualized using a phosphor-imaging system as shown in Figure 2. The autoradiograms (Figure 2A and 2C) indicated that both (-)-[11C]8 and (-)-[18F]14a were able to enter the rat brain and had the highest accumulation in the VAChT-enriched striatum, consistent with previous reports.[16, 17] The uptake ratio of radioactivity in striatum versus cerebellum quantified using 30 sections was 1.75 for (-)-[11C]8 at 30 min post-injection and 1.54 for (-)-[18F]14a at 60 min post-injection (Figure 2B and 2D).
Figure 2.

Autoradiography studies of (-)-[11C]8 and (-)-[18F]14a in the male SD rats: (A) representative image of brain slice at 30 min post-injection of (-)-[11C]8; (B) the quantified ratio of striatum to cerebellum of (-)-[11C]8 from 30 slides was 1.75; (C) representative image of rat brain section at 60 min post-injection of (-)-[18F]14a; (D) the quantified ratio of striatum to cerebellum of (-)-[18F]14a from 30 brain slides was 1.54.
Biodistribution Studies in Rats
The biodistribution of (-)-[11C]8 and (-)-[18F]14a was determined using adult male SD rats. For (-)-[11C]8, rats were euthanized at 5, 30, and 60 min post-injection; for (-)-[18F]14a, an additional group were euthanized at 120 min post-injection. After euthanasia, organs and tissues of interests were collected, weighed, and counted. The uptake (%ID/g) was calculated. As shown in Table 3, the initial uptakes (%ID/g) at 5 min post-injection for blood, heart, lung, muscle, fat, pancreas, spleen, kidney, liver, and brain was 0.29, 0.45, 1.23, 0.19, 0.24, 1.03, 0.93, 1.10, 4.12, and 0.35 for (-)-[11C]8 and 0.17, 0.77, 3.79, 0.21, 0.18, 2.01, 2.19, 2.56, 3.46, 0.63, and 0.91 for (-)-[18F]14a, respectively. A rapid clearance of radioactivity was observed in blood, heart, lung, spleen, and brain for both (-)-[11C]8 and (-)-[18F]14a. Bone was collected for (-)-[18F]14a to check for in vivo defluorination. Our data indicated the bone uptake (%ID/g) was increased from 0.63 at 5 min to 2.78 at 120 min post-injection, suggesting significant in vivo defluorination for (-)-[18F]14a.
Table 3.
The uptake (%ID/g) of (-)-[11C]8 and (-)-[18F]14a in adult male SD rats[a].
| Organ | (-)-[11C]8 | (-)-[18F]14a | |||||
|---|---|---|---|---|---|---|---|
| 5 min | 30 min | 60 min | 5 min | 30 min | 60 min | 120 min | |
| Blood | 0.29 ± 0.03 | 0.12 ± 0.01 | 0.10 ± 0.02 | 0.17 ± 0.03 | 0.18 ± 0.02 | 0.20 ± 0.02 | 0.20 ± 0.02 |
| Heart | 0.45 ± 0.05 | 0.34 ± 0.07 | 0.22 ± 0.05 | 0.77 ± 0.06 | 0.46 ± 0.04 | 0.32 ± 0.04 | 0.26 ± 0.02 |
| Lung | 1.23 ± 0.06 | 0.90 ± 0.18 | 0.64 ± 0.09 | 3.79 ± 0.34 | 1.53 ± 0.25 | 0.92 ± 0.15 | 0.62 ± 0.07 |
| Muscle | 0.19 ± 0.02 | 0.17 ± 0.03 | 0.15 ± 0.03 | 0.21 ± 0.02 | 0.21 ± 0.02 | 0.19 ± 0.03 | 0.17 ± 0.01 |
| Fat | 0.24 ± 0.10 | 0.47 ± 0.06 | 0.43 ± 0.13 | 0.18 ± 0.07 | 0.34 ± 0.09 | 0.32 ± 0.11 | 0.30 ± 0.07 |
| Pancreas | 1.03 ± 0.08 | 1.26 ± 0.26 | 0.94 ± 0.14 | 2.01 ± 0.20 | 2.00 ± 0.38 | 1.37 ± 0.33 | 0.96 ± 0.16 |
| Spleen | 0.93 ± 0.06 | 0.73 ± 0.12 | 0.60 ± 0.13 | 2.19 ± 0.70 | 1.33 ± 0.18 | 0.87 ± 0.22 | 0.60 ± 0.08 |
| Kidney | 1.10 ± 0.10 | 1.29 ± 0.13 | 1.05 ± 0.18 | 2.56 ± 0.20 | 1.46 ± 0.14 | 1.18 ± 0.23 | 0.93 ± 0.09 |
| Liver | 4.12 ± 0.65 | 4.41 ± 0.43 | 3.56 ± 0.43 | 3.46 ± 0.32 | 3.24 ± 0.62 | 2.44 ± 0.18 | 2.66 ± 0.57 |
| Bone | - | - | - | 0.63 ± 0.04 | 0.96 ± 0.08 | 1.69 ± 0.24 | 2.78 ± 0.24 |
| Total brain | 0.35 ± 0.04 | 0.27 ± 0.05 | 0.15 ± 0.04 | 0.91 ± 0.05 | 0.55 ± 0.07 | 0.45 ± 0.07 | 0.32 ± 0.03 |
%ID/g values (mean ± SD) with n = 4 rats per group.
The regional brain uptake of (-)-[11C]8 and (-)-[18F]14a at 30 and 60 min post-injection was presented in Figure 3. The uptake of the radioactivity of (-)-[11C]8 at 30 min in the cerebellum, striatum, cortex, and hippocampus was 0.25, 0.30, 0.26, and 0.25, respectively; at 60 min, the uptake in abovementioned brain regions of interests was decreased gradually. For the F-18 radiotracer, (-)-[18F]14a displayed higher uptake in all brain regions at 30 and 60 min post-injection than (-)-[11C]8. Among the cerebellum, striatum, cortex, and hippocampus, the striatum displayed the highest uptake at 30 and 60 min post-injection. The regional brain uptake result suggested that both (-)-[11C]8 and (-)-[18F]14a are highly accumulated in the VAChT-enriched striatum. To check the in vivo VAChT binding specificity of (-)-[11C]8 and (-)-[18F]14a, biodistribution studies were performed in SD rats under baseline condition and after pretreatment with (-)-vesamicol, a well-known VAChT ligand (Ki- VAChT = 15.2 nM)[16, 18] and YUN-122, N-(4-benzylcyclohexyl)-2-(2-fluorophenyl)acetamide, a potent σ1 receptor selective ligand (Ki- σ1 = 3.6 nM).[15] As shown in Figure 4, reduced uptake in the striatum and total brain was observed for both (-)-[11C]8 and (-)-[18F]14a after (-)-vesamicol pretreatment, suggesting that both radiotracers bind to VAChT in the brain. The VAChT compound (-)-vesamcol reduced 55% of the striatal uptake for (-)-[11C]8 at 30 min, and 69% for (-)-[18F]14a at 60 min. Unfortunately, pretreatment using YUN-122 also reduced brain uptake for both radiotracers. YUN-122 caused 18% decrease of the striatal uptake for (-)-[11C]8 at 30 min, and 20% for (-)-[18F]14a at 60 min. These blocking study results suggested that both radiotracers bind mostly to VAChT in the striatum, but they also bind to σ1 receptor in the brain.
Figure 3.

Regional brain uptake of (-)-[11C]8 and (-)-[18F]14a in adult male SD rats at 30 and 60 min post-injection. Cerebellum (CBL), striatum (STR), cortex (CTX), and hippocampus (HIPPO).
Figure 4.

Striatum uptake of (-)-[11C]8 (30 min post-injection) and (-)-[18F]14a (60 min post-injection) in control adult male SD rats and rats pretreated with (-)-vesamicol or YUN-122.
MicroPET Imaging Studies in NHP
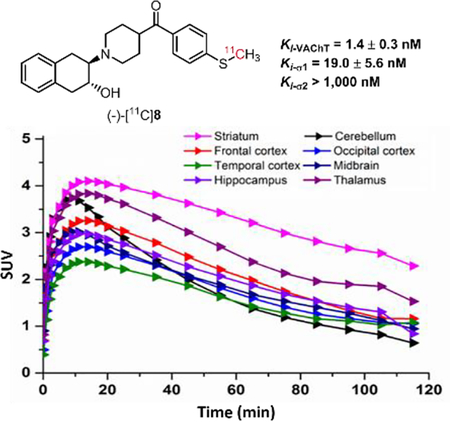
Our rodent studies showed that both (-)-[11C]8 and (-)-[18F]14a had high striatal accumulation, while (-)-[18F]14a had in vivo defluorination. To check the pharmacokinetics of the sulfur-containing radiotracers in the brain regions of interests, we performed microPET study of (-)-[11C]8 in the brain of cynomolgus macaque. As shown in Figure 5, the microPET data suggested that (-)-[11C]8 was able to enter the brain of a cynomolgus macaque and had the highest accumulation in the striatal region versus other regions. After injection, (-)-[11C]8 took ~9 min to reach the maximum striatal uptake (5157 nCi/cc) and then washed out quickly; by 120 min, the striatal uptake of (-)-[11C]8 decreased 54% from the peak value. From 20 min to 120 min, the striatum has much higher uptake than other brain regions of interests. The cerebellum has the lowest uptake after 70 min compared to the striatum, frontal, occipital and temporal cortex, midbrain, hippocampus, and thalamus. The microPET study results suggested that the sulfur-containing radiotracer (-)-[11C]8 had rapid washout in the brain of a cynomolgus macaque. Based on our in vitro and in vivo preliminary data, radiotracer (-)-[11C]8 has moderate σ1 receptor binding potency, which will interfere the quantification of VAChT in vivo, suggesting (-)-[11C]8 is not suitable PET radiotracer for iamging VAChT in vivo. No further microPET studies are ncessary to compare (-)-[11C]8 to (-)-[18F]FEOBV which is a VAChT specific radioligand.
Figure 5.

MicroPET imaging studies of (-)-[11C]8 in the NHP brain. (A) Representative PET images of (-)-[11C]8; (B) Tissue time-activity curve of (-)-[11C]8 in the NHP brain.
Conclusions
Together, 16 new sulfur-containing compounds targeting VAChT were synthesized and the enantiomers of four potent compounds were chirally resolved. In vitro binding studies suggested that compounds (-)-8 and (-)-14a exhibited high VAChT binding affinities, moderate selectivity over the σ1 receptor and high selectivity over the σ2 receptor. Both (-)-[11C]8 and (-)-[18F]14a were radiosynthesized with high radiochemical yield and purity. Ex vivo autoradiography and biodistribution studies in SD rats indicated that both (-)-[11C]8 and (-)-[18F]14a were able to enter the brain and selectively accumulated in the VAChT-enriched striatum. Blocking studies showed that both (-)-[11C]8 and (-)-[18F]14a bind mostly to VAChT but also to the σ1 receptor in vivo. The microPET studies in the brain of cynomolgus macaque showed that (-)-[11C]8 has high accumulation in the VAChT-enriched striatum and rapid washout. Although the moderate binding affinity toward σ1 receptor prevents further investigation as suitable PET radiotracers for measuring VAChT expression in vivo, the preliminary results could provide valuable information for future exploration of VAChT ligands with high specificity.
Experimental Section
General Information.
Commercially available reagents and solvents were used as purchased without further purification. Unless otherwise indicated, all reactions were conducted in oven-dried glassware. Reactions were monitored by thin-layer chromatography (TLC) and visualized under ultraviolet light (wavelength 254). Flash column chromatography was performed using Silica Flash® P60 silica gel (40–63 μm) from Silicycle Inc.1H NMR and13C NMR spectra were recorded on Varian 400 MHz and 300 MHz instruments. Chemical shifts were reported in parts per million (ppm) and calibrated using the residual undeuterated solvent as an internal reference (CDCl3: δ 7.26 ppm; CD3OD: δ 3.31 ppm; DMSO-d6: δ 2.50 ppm). A Chiralcel OD normal phase HPLC column was used to resolve enantiomers. The specific optical rotation was determined on an automatic polarimeter (Autopol 111, Rudolph Research, Flanders, NJ).
[11C]CO2 was produced via a14N(p,α)11C reaction with a 40 μA beam of 16 MeV protons on a target of 0.5% O2 in N2 for 15−30 min with an JSW BC-16/8 cyclotron. The produced [11C]CO2 was transported to a GE PETtrace MeI microlab and converted sequentially to [11C]CH3I by the previously described method.[11]
[18F]Fluoride was produced from an RDS111 cyclotron (Siemens/CTI Molecular Imaging, Knoxville, TN) by18O(p, n)18F reaction through proton irradiation of enriched18O water (95%). [18F]Fluoride was firstly passed through an ion-exchange resin and then eluted using 0.02 M potassium carbonate solution.
All animal experiments were conducted under Washington University’s Institutional Animal Care and Use Committee (IACUC)-approved protocols in accordance with the US National Research Council’s Guide for the Care and Use of Laboratory Animals.
Syntheses of Sulfur-containing Compounds.
General procedure to synthesize oxalates.
A solution of oxalic acid in ethyl acetate (0.3 M, 1 eq) was added slowly into a solution of the free base in dichloromethane (0.06 M, 1 eq). The mixture was stirred for 5 h at room temperature and filtered to afford the oxalates.
(±)-(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-(methyl-sulfonyl)phenyl)methanone ((±)-7).
To a round-bottomed flask equipped with a magnetic stir bar was added (±)-6 (0.35 g, 1.0 mmol), sodium methanesulfinate (0.11 g, 1.1 mmol), and DMSO (10 mL) under nitrogen. The reaction vessel was immersed in a 100 °C preheated oil bath for 24 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate (1/1, V/V) to afford (±)-7 (0.15 g, 37%) as a gray solid.1H NMR (400 MHz, CDCl3) δ 8.18 – 8.02 (m, 4H), 7.17 – 7.08 (m, 4H), 4.17 (s, 1H), 3.95 – 3.80 (m, 1H), 3.34 – 3.28 (m, 2H), 3.09 (s, 3H), 3.02 – 3.00 (m, 1H), 2.92 – 2.77 (m, 6H), 2.45 (t, J = 11.3 Hz, 1H), 1.98 – 1.90 (m, 3H), 1.87 – 1.80 (m, 1H).13C NMR (101 MHz, CDCl3) δ 200.84, 144.04, 140.09, 134.51, 133.88, 129.28, 129.08 (2C), 127.90, 126.21, 126.04, 66.51, 65.59, 51.69, 44.63, 44.30, 44.28, 37.76, 29.21, 28.89, 26.09. HRMS (ESI) Calculated for C23H27NO4S (M+H)+ 414.1739, found: 414.1735. (±)-7 was resolved by HPLC using a Chiralcel OD column (250 mm × 10 mm), ethanol as mobile phase, flow rate of 4.0 mL/min, and UV wavelength at 254 nM to give (+)-7 and (-)-7, respectively. The free base of (+)-7 and (-)-7 were converted to oxalate salt following the general procedure. (+)-7 oxalate, M.P.: 203 – 206 °C, = + 43° (c = 1, methanol); (-)-7 oxalate, M.P.: 207 – 211 °C, = - 43° (c = 1, methanol).
(±)-(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-(methylthio)phenyl)methanone ((±)-8).
To a round-bottomed flask equipped with a magnetic stir bar was added (±)-6 (0.18 g, 0.5 mmol), sodium thiomethoxide (0.07 g, 1.0 mmol), potassium carbonate (0.14 g, 1.0 mmol), and DMF (3 mL) under nitrogen. The reaction vessel was immersed in a 60 °C preheated oil bath for 9 h. The mixture was then cooled to room temperature and diluted with water, extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate (2/1, V/V) to afford (±)-8 (0.11 g, 58%) as a yellow solid.1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 7.13 – 7.08 (m, 4H), 4.22 (s, 1H), 3.91 – 3.81 (m, 1H), 3.36 – 3.21 (m, 2H), 3.02 – 2.93 (m, 1H), 2.92 – 2.73 (m, 6H), 2.51 (s, 3H), 2.45 – 2.36 (m, 1H), 1.95 – 1.90 (m, 3H), 1.87 – 1.75 (m, 1H).13C NMR (101 MHz, CDCl3) δ 201.34, 145.84, 134.68, 133.95, 132.16, 129.26, 129.11, 128.66, 126.15, 126.00, 125.09, 66.53, 65.60, 51.91, 44.73, 43.54, 37.82, 29.57, 29.23, 26.09, 14.77. HRMS (ESI) Calculated for C23H27NO2S (M+H)+ 382.1841, found: 382.1837. (±)-8 was resolved by HPLC using a Chiralcel OD column (250 mm × 10 mm), ethanol as mobile phase, flow rate of 4.0 mL/min, and UV wavelength at 254 nM to give (+)-8 and (-)-8, respectively. The free base of (+)-8 and (-)-8 were converted to oxalate salt following the general procedure. (+)-8 oxalate, M.P.: 199 – 205 °C, = + 66° (c = 1, methanol); (-)-8 oxalate, M.P.: 207 – 211 °C, = - 62° (c = 1, methanol).
(±)-(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-(methyl-sulfinyl)phenyl)methano-ne ((±)-9).
To a sealed tube equipped with a magnetic stir bar was added (±)-8 (0.05 g, 0.13 mmol) and acetic acid (1.0 mL), then 30% H2O2 (13 μL, 0.13 mmol) was added into the mixture. The reaction vessel was immersed in a 55 °C preheated oil bath for 2 h. The mixture was then cooled to room temperature and basified with 1 M NaOH aqueous followed by extracting with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with dichloromethane/methanol (20/1, V/V) to afford (±)-9 (30 mg, 60%) as a yellow oil.1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 8.5 Hz, 2H), 7.76 (d, J = 8.5 Hz, 2H), 7.16 – 7.06 (m, 4H), 4.17 (s, 1H), 3.94 – 3.83 (m, 1H), 3.38 – 3.29 (m, 2H), 3.07 – 2.98 (m, 1H), 2.98 – 2.79 (m, 6H), 2.77 (s, 3H), 2.51 – 2.38 (m, 1H), 2.02 – 1.92 (m, 3H), 1.90 – 1.77 (m, 1H).13C NMR (101 MHz, CDCl3) δ 201.58, 151.00, 138.26, 134.68, 134.06, 129.41, 129.22 (2C), 126.35, 126.18, 124.01, 66.71, 65.78, 51.86, 44.87, 44.17, 43.96, 37.94, 29.47, 29.15, 26.27. HRMS (ESI) Calculated for C23H27NO3S (M+H)+ 398.1790, found: 398.1786. The free base of (±)-9 was converted to oxalate salt following the general procedure. (±)-9 oxalate, M.P.: 197 – 200 °C.
(±)-4-(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperid-ine-4-carbonyl)-N-methylbenzenesulfonam-ide ((±)-11a).
To a round-bottomed flask equipped with a magnetic stir bar was added potassium metabisulfite (54.7 mg, 0.25 mmol), TBAB (43.6 mg, 0.14 mmol), sodium formate (18.4 mg, 0.27 mmol), palladium acetate (2.8 mg, 0.01 mmol), triphenylphosphine (9.7 mg, 0.04 mmol), 1,10-phenanthroline (7.3 mg, 0.04 mmol), and DMSO (2.0 mL) under nitrogen. The mixture was bubbled with nitrogen for 10 min before (±)-10 (50.8 mg, 0.12 mmol) was added. After that, the reaction vessel was immersed in a 70 °C preheated oil bath for 4 h. After cooling, diisopropylethylamine (23.8 mg, 0.19 mmol), methylamine hydrochloride (9.2 mg, 0.14 mmol), and THF (1.0 mL) were added into the above mixture. After cooling to 0 °C, a solution of N-chlorosuccinimide (32.8 mg, 0.25 mmol) in THF (1.0 mL) was added, the reaction was stirred at room temperature overnight. The mixture was then diluted with water, extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with dichloromethane/methanol (30/1, V/V) to afford (±)-11a (35 mg, 66%) as a yellow oil.1H NMR (400 MHz, CD3OD) δ 8.07 (d, J = 8.4 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H), 7.17 – 7.07 (m, 4H), 4.46 (s, 1H), 3.93 – 3.83 (m, 1H), 3.38 – 3.26 (m, 2H), 3.06 – 2.98 (m, 1H), 2.95 – 2.79 (m, 6H), 2.72 (d, J = 4.3 Hz, 3H), 2.49 – 2.38 (m, 1H), 2.04 – 1.90 (m, 3H), 1.89 – 1.77 (m, 2H).13C NMR (101 MHz, CDCl3) δ 201.24, 142.82, 139.24, 134.50, 133.89, 129.27, 129.06, 128.88, 127.58, 126.21, 126.03, 66.55, 65.62, 51.66, 44.68, 44.20, 37.77, 29.66, 29.32, 29.25, 28.94, 26.10. HRMS (ESI) Calculated for C23H28N2O4S (M+H)+ 429.1848, found: 429.1844. The free base of (±)-11a was converted to oxalate salt following the general procedure. (±)-11a oxalate, M.P.: 200 – 205 °C.
(±)-N-(2-fluoroethyl)-4-(1-(3-hydroxy-1,2,3,4-tetrahydronapht-halen-2-yl)-piperidine-4-carbonyl)benzenesulfonamide ((±)-11b).
To a round-bottomed flask equipped with a magnetic stir bar was added potassium metabisulfite (106.7 mg, 0.48 mmol), TBAB (85.1 mg, 0.26 mmol), sodium formate (35.9 mg, 0.53 mmol), palladium acetate (5.4 mg, 0.02 mmol), triphenylphosphine (18.9 mg, 0.07 mmol), 1,10-phenanthroline (14.3 mg, 0.07 mmol), and DMSO (2.0 mL) under nitrogen. The mixture was bubbled with nitrogen for 10 min before (±)-10 (100 mg, 0.24 mmol) was added. After that, the reaction vessel was immersed in a 70 °C preheated oil bath for 4 h. After cooling, diisopropylethylamine (61.9 mg, 0.48 mmol), 2-fluoroethylamine hydrochloride (29.2 mg, 0.26 mmol), and THF (1.0 mL) were added into the above mixture. After cooling to 0 °C, a solution of N-chlorosuccinimide (64.1 mg, 0.48 mmol) in THF (1.0 mL) was added, the reaction was stirred at room temperature overnight. The mixture was then diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with dichloromethane/methanol (30/1, V/V) to afford (±)-11b (20 mg, 10%) as a yellow oil.1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H), 7.18 – 7.06 (m, 4H), 4.93 (s, 1H), 4.54 (t, J = 4.7 Hz, 1H), 4.42 (t, J = 4.8 Hz, 1H), 3.94 – 3.83 (m, 1H), 3.75 (t, J = 6.3 Hz, 1H), 3.41 – 3.34 (m, 1H), 3.34 – 3.28 (m, 2H), 3.07 – 2.99 (m, 1H), 2.95 – 2.77 (m, 6H), 2.50 – 2.40 (m, 1H), 2.02 – 1.91 (m, 3H), 1.88 – 1.81 (m, 2H).13C NMR (101 MHz, CDCl3) δ 201.33, 143.93, 139.54, 134.65, 134.09, 129.46, 129.24, 129.17, 127.56, 126.42, 126.23, 82.34 (d, JC-F = 169.7 Hz), 68.13, 66.75, 65.83, 51.78, 44.91, 43.70 (d, JC-F = 21.2 Hz), 37.97, 29.86, 26.32, 25.77. HRMS (ESI) Calculated for C24H29FN2O4S (M+H)+ 461.1910, found: 461.1907. The free base of (±)-11b was converted to oxalate salt following the general procedure. (±)-11b oxalate, M.P.: 205 – 210 °C.
(±)-(4-((3-Fluoropropyl)sulfonyl)phenyl)(1-(3-hydroxy-1,2,3,4-tetrahydronaph-thalen-2-yl)piperidin-4-yl)methanone ((±)-12).
To a round-bottomed flask equipped with a magnetic stir bar was added potassium metabisulfite (88.9 mg, 0.40 mmol), TBAB (46.2 mg, 0.22 mmol), sodium formate (29.9 mg, 0.44 mmol), palladium acetate (2.2 mg, 0.01 mmol), triphenylphosphine (7.9 mg, 0.03 mmol), 1,10-phenanthroline (5.9 mg, 0.03 mmol), and DMSO (2.0 mL) under nitrogen. The mixture was bubbled with nitrogen for 10 min before (±)-10 (80 mg, 0.2 mmol) was added. After that, the reaction vessel was immersed in a 70 °C preheated oil bath for 4 h. After cooling, 1-bromo-3-fluoropropane (20.9 mg, 0.15 mmol) in DMF (1.0 mL) were added into the above mixture, the reaction was stirred at room temperature overnight. The mixture was then diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate (1/2, V/V) to afford (±)-12 (30 mg, 44%) as a gray solid.1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.4 Hz, 2H), 7.97 (d, J = 8.2 Hz, 2H), 7.09 – 7.00 (m, 4H), 4.52 (t, J = 4.8 Hz, 1H), 4.40 (t, J = 4.9 Hz, 1H), 3.88 – 3.77 (m, 1H), 3.34 – 3.15 (m, 4H), 3.01 – 2.91 (m, 1H), 2.90 – 2.69 (m, 6H), 2.38 (t, J = 10.7 Hz, 1H), 2.18 – 2.02 (m, 2H), 1.96 – 1.83 (m, 3H), 1.81 – 1.70 (m, 1H).13C NMR (101 MHz, CDCl3) δ 201.30, 142.68, 140.44, 134.62, 134.05, 129.42, 129.21, 128.71, 126.38, 126.20, 125.91, 81.51 (d, JC-F = 168.7 Hz), 66.73, 65.80, 52.57 (d, JC-F = 4.0 Hz), 51.72, 44.87, 44.40, 37.94, 29.33, 29.02, 26.29, 24.09 (d, JC-F = 20.2 Hz). HRMS (ESI) Calculated for C25H30FNO4S (M+H)+ 460.1958, found: 460.1954. The free base of (±)-12 was converted to oxalate salt following the general procedure. (±)-12 oxalate, M.P.: 201 – 205 °C.
(±)-(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-mercaptophenyl)methanone ((±)-13).
To a pressure tube containing a magnetic stir bar was added (±)-10 (124.3 mg, 0.3 mmol), copper sulfate pentahydrate (3.7 mg, 0.015 mmol), Cs2CO3 (690.5 mg, 1.5 mmol), and DMSO (2.0 mL). The mixture was bubbled with nitrogen for 10 min before 1,2-ethanedithiol (56.5 mg, 0.6 mmol) was added. The tube was sealed and immersed in a 110 °C preheated oil bath for 12 h. The mixture was then cooled to room temperature and diluted with ethyl acetate. The ethyl acetate layer was washed with water and saturated brine, and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with dichloromethane/methanol (20/1, V/V) to afford (±)-13 (53 mg, 48%) as a yellow solid.1H NMR (300 MHz, CDCl3) δ 7.90 (d, J = 8.5 Hz, 2H), 7.57 (d, J = 8.4 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.15 – 7.08 (m, 4H), 3.87 (s, 1H), 3.35 – 3.20 (m, 2H), 3.03 – 2.71 (m, 7H), 2.41 (s, 1H), 1.91 – 1.75 (m, 4H). M.P.: 171 – 175 °C.
(±)-(4-((2-Fluoroethyl)thio)phenyl)(1-(3-hydroxy-1,2,3,4-tetrahydronaph-thalen-2-yl)piperidin-4-yl)methanone ((±)-14a).
To a round-bottomed flask equipped with a magnetic stir bar was added (±)-13 (150 mg, 0.4 mmol), Cs2CO3 (133 mg, 0.4 mmol), TBAB (129 mg, 0.4 mmol), and DMF (2.0 mL) under nitrogen. After that, 1-bromo-2-fluoroethane (61 mg, 0.48 mmol) was added slowly via syringe. The reaction was stirred at room temperature for 7 h. The mixture was then cooled to room temperature and diluted with ethyl acetate. The ethyl acetate layer was washed with water, saturated brine, and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate (1/1, V/V) to afford (±)-14a (65 mg, 39%) as a white solid.1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.17 – 7.07 (m, 4H), 4.66 (t, J = 6.6 Hz, 1H), 4.55 (t, J = 6.6 Hz, 1H), 3.92 – 3.83 (m, 1H), 3.36 – 3.23 (m, 4H), 3.04 – 2.76 (m, 8H), 2.42 (t, J = 11.2 Hz, 1H), 1.99 – 1.90 (m, 3H), 1.89 – 1.75 (m, 1H).13C NMR (101 MHz, CDCl3) δ 201.33, 142.41, 134.62, 133.94, 133.36, 129.27, 129.09, 128.87, 127.39, 126.17, 126.01, 81.86 (d, JC-F = 173.7 Hz), 66.53, 65.61, 51.79, 44.74, 43.59, 37.79, 32.05 (d, JC-F = 22.2 Hz), 29.46, 29.13, 26.09. HRMS (ESI) Calculated for C24H28FNO2S (M+H)+ 414.1903, found: 414.1899. (±)-14a was resolved by HPLC using a Chiralcel OD column (250 mm × 10 mm), ethanol as mobile phase, flow rate of 4.0 mL/min, and UV wavelength at 254 nM to give (+)-14a and (-)-14a, respectively. The free base of (+)-14a and (-)-14a were converted to oxalate salt following the general procedure. (+)-14a oxalate, M.P.: 208 – 210 °C, = + 57° (c = 1, methanol); (-)-14a oxalate, M.P.: 207 – 209 °C, = - 52° (c = 1, methanol).
(±)-(4-((3-Fluoropropyl)thio)phenyl)(1-(3-hydroxy-1,2,3,4-tetrahydronaph-thalen-2-yl)piperidin-4-yl)methanone ((±)-14b).
To a round-bottomed flask equipped with a magnetic stir bar was added (±)-13 (36.7 mg, 0.1 mmol), Cs2CO3 (32.5 mg, 0.1 mmol), TBAB (32.2 mg, 0.1 mmol), and DMF (1.5 mL) under nitrogen. After that, 1-bromo-3-fluoropropane (17 mg, 0.12 mmol) was added slowly via syringe. The reaction was stirred at room temperature for 3 h. The mixture was then cooled to room temperature and diluted with ethyl acetate. The ethyl acetate layer was washed with water, saturated brine, and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under reduced pressure and the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate (1/1, V/V) to afford (±)-14b (26 mg, 61%) as a white solid.1H NMR (300 MHz, CDCl3) δ 7.87 (d, J = 8.6 Hz, 2H), 7.34 (d, J = 8.6 Hz, 2H), 7.16 – 7.07 (m, 4H), 4.66 (t, J = 5.6 Hz, 1H), 4.51 (t, J = 5.6 Hz, 1H), 3.92 – 3.83 (m, 1H), 3.35 – 3.28 (m, 2H), 3.14 (t, J = 7.2 Hz, 2H), 3.02 – 2.96 (m, 1H), 2.93 – 2.76 (m, 6H), 2.46 – 2.37 (m, 1H), 2.20 – 1.99 (m, 2H), 1.99 – 1.79 (m, 4H).13C NMR (101 MHz, CDCl3) δ 201.37, 143.63, 134.63, 133.95, 132.84, 129.27, 129.09, 128.79, 126.73, 126.16, 126.00, 82.83 (d, JC-F = 167.7 Hz), 66.52, 65.60, 51.86, 44.73, 43.57, 37.78, 29.88, (d, JC-F = 20.2 Hz), 29.50, 29.16, 27.78 (d, JC-F = 5.1 Hz), 26.08. HRMS (ESI) Calculated for C25H30FNO2S (M+H)+ 428.2060, found: 428.2056. (±)-14a was separated by HPLC using a Chiralcel OD column (250 mm × 10 mm), ethanol as mobile phase, flow rate of 4.0 mL/min, and UV wavelength at 254 nM to give (+)-14b and (-)-14b, respectively. The free base of (+)-14b and (-)-14b were converted to oxalate salt following the general procedure. (+)-14b oxalate, M.P.: 214 – 217 °C, = + 47° (c = 1, methanol); (-)-14b oxalate, M.P.: 207 – 209 °C, = - 49° (c = 1, methanol).
VAChT Binding Affinity Studies
VAChT binding affinities were determined by competition against the binding of (-)-[3H]vesamicol (~5 nM) to postnuclear supernatant prepared from PC12 cells stably expressing human VAChT according to the methods reported by Parsons and colleagues.[19]
Sigma Receptors Binding Affinity Studies
The σ1 receptor binding affinities were determined by competition against the binding of (+)-[3H]pentazocine (~5 nM) to guinea pig brain membrane homogenates. The σ2 receptor binding affinities were determined by competition against the binding of [3H]DTG (~5 nM) to rat liver membrane homogenates in the presence of (+)-pentazocine (1 μM) to block σ1 sites. The procedures for isolating the membrane homogenates and performing the σ1 and σ2 receptor binding assays were reported previously.[20]
Radiosynthesis of (-)-[11C]8
Approximately 1.0 mg of the precursor (-)-13 was placed in a reaction vessel with a saturated solution of Cs2CO3 and TBAB in DMF (0.2 mL). [11C]CH3I was bubbled into the reaction vessel and the reaction mixture was heated at 60 °C for 5 min. After quenching with 1.8 mL of HPLC mobile phase (40% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.5). The reactive mixture was loaded onto a C-18 column (Phenomenex Luna C18, 250 mm × 9.6 mm), then eluted from the column using above mentioned HPLC mobile phase at a flow rate of 4.0 mL/min. A 100 mL of glass vial that has 50 mL of sterile water was used to collect the radioactive product from 14–15 min and then passed through a C18 Sep-Pak® Plus cartridge with nitrogen gas assistance. The trapped product was eluted using 0.6 mL of ethanol and 5.4 mL of saline to formulate the injection dose. The product was authenticated using an analytical HPLC system (Agilent SB-C18 analytic column, 250 mm × 4.6 mm; mobile phase of 75% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.5; flow rate of 1.0 mL/min; UV wavelength of 254 nM; tR = 3.9 min) by co-injecting with standard reference compound (-)-8. The radiochemical yield was ~45%, the radiochemical purity was > 98%, and the specific activity was > 74 GBq/μmol (decay corrected to EOS).
Radiosynthesis of (-)-[18F]14a
Radiosynthesis of 1-[18F]fluoro-2-tosyloxyethane ([18F]15).
Following published procedure,[16] the intermediate [18F]15 was radiosynthesized.
Radiosynthesis of (-)-[18F]14a.
A solution of precursor (-)-13 (~1.0 mg) and Cs2CO3 (~2.0 mg) in DMSO (200 μL) was added to the reaction vessel containing [18F]15. The vessel was capped and heated at 100 °C for 15 min. Subsequently, the reaction mixture was diluted with 3.0 mL of HPLC mobile phase (40% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.5) and loaded onto a C-18 column (Agilent SB-C18, 250 mm × 10 mm), then eluted from the column using above mentioned HPLC mobile phase at a flow rate of 4.0 mL/min. A 100 mL of glass vial that has 50 mL of sterile water was used to collect the radioactive product from 17–18 min and then passed through a C18 Sep-Pak® Plus cartridge with nitrogen gas assistance. The trapped product was eluted using 0.6 mL of ethanol and 5.4 mL of saline to formulate the injection dose. The product was authenticated using an analytical HPLC system (Agilent SB-C18 analytic column, 250 mm × 4.6 mm; mobile phase of 70% acetonitrile in 0.1 M ammonium formate buffer, pH = 4.5; flow rate of 1.0 mL/min; UV wavelength of 254 nM; tR = 4.8 min) by co-injecting with standard reference compound (-)-14a. The radiochemical yield was ~65%, the radiochemical purity was > 95%, and the specific activity was > 55 GBq/μmol (decay corrected to EOS).
Ex Vivo Autoradiography Study
For the ex vivo autoradiography study, mature male SD rats (~250 g) were injected with ~20 MBq of (-)-[11C]8 or (-)-[18F]14a in 10% ethanol in saline via the tail vein under isoflurane/oxygen anaesthesia. The rats were euthanized after 30 min for (-)-[11C]8 or 60 min for (-)-[18F]14a, and the brains were quickly removed and frozen. The frozen brains were then sectioned into 100 μm transverse slices and mounted on glass slides. Frozen slides were directly exposed to a BAS storage phosphor screen film (BAS-IP-MS-2025) in an imaging cassette (Fuji Photo Film Co., Tokyo, Japan) for 12 h at −80 °C under the dark. The distribution of radioactivity was visualized by a Fuji Bio-Imaging Analyzer FLA-7000 (Fuji Photo Film Co., Tokyo, Japan). Photo-stimulated luminescence (PSL) from the striatum and cerebellum was quantified using Multi Gauge v3.0 software (Fuji Photo Film Co., Tokyo, Japan). Data were background-corrected and expressed as photo-stimulated luminescence signals per square millimeter (PSL/mm2), then normalized to provide the ratio of striatum to cerebellum.
Biodistribution Studies in Rats
For the biodistribution studies, mature male SD rats (180–200 g) were injected with ~ 3.7 MBq of (-)-[11C]8 or (-)-[18F]14a in 10% ethanol in saline via the tail vein under isoflurane/oxygen anaesthesia, respectively. For (-)-[11C]8, the rats were euthanized under anaesthesia at 5 min, 30 min, and 60 min post-injection (n = 4 for each time point); for (-)-[18F]14a, the rats were euthanized under anaesthesia at 5 min, 30 min, 60 min, and 120 min post-injection (n = 4 for each time point). In addition to the baseline studies, the specific uptake of (-)-[11C]8 and (-)-[18F]14a were evaluated at 30 min and 60 min post-injection using YUN-122 and (-)-vesamicol to block σ1 receptor and VAChT in the brain; the blocking agents were injected 5 min prior to the tracer injection. The whole brain was quickly harvested and dissected into regions of the cerebellum, striatum, brain stem, cortex, hippocampus, and thalamus; the rest of the brain was also collected to determine the total brain uptake. Samples of blood, heart, lung, muscle, fat, pancreas, spleen, kidney, liver, and bone (for (-)-[18F]14a only) were also collected, and all the samples were counted in an automated Beckman Gamma 8000 well counter with a standard dilution of the injectate. Tissues were weighed and the percentage injected dose per gram (%ID/g) tissue was calculated.
MicroPET Imaging Study in NHP
PET was performed on an adult male cynomolgus macaque (9.1 kg) with a microPET Focus 220 scanner (Concorde/CTI/Siemens Microsystems, Knoxville, TN). The animal was fasted for 12 h prior to PET scans. Anesthesia was induced with an intramuscular injection of ketamine (10–20 mg/kg) and glycopyrrolate (0.013–0.017 mg/kg) (to reduced secretions). The animal was intubated and anesthesia was maintained at 0.8–2.0% isoflurane/oxygen throughout the procedure. A percutaneous venous catheter was placed for radiotracer injection. During the microPET scanning session, the head was positioned supine in the adjustable head holder with the brain in the center of the field of view. A 10 min transmission scan was performed to check positioning; once confirmed, a 45 min transmission scan was obtained for attenuation correction. Subsequently, a 2 h dynamic emission scan was acquired after administration of ~400 MBq of (-)-[11C]8 via the venous catheter. PET data were collected from 0–120 min with the following time frames: 3×1 min, 4×2 min, 3×3 min and 20×5 min. PET image reconstructed resolution was < 2.0 mm full width half maximum for all 3 dimensions at the center of the field of view. Emission data were corrected for deadtime, scatter, randoms and attenuation, and then reconstructed using filtered back projection. The reconstructed PET images were co-registered with magnetization-prepared rapid gradient echo (MP-RAGE) MR images using Automated Image Registration (AIR)46, and superimposed using Analyze 10.0 (AnalyzeDirect, Overland Park, KS). For quantitative analyses, regions of interest (ROIs) were manually drawn on MRI images for striatum and cerebellum, and transformed into PET images using the co-registration transformation matrix. Time-activity curves (TACs) were then obtained from the dynamic PET images.
Acknowledgements
This work was supported by NIH grants NS061025, NS075527, NS103988, NS075321, NS103957, NS058714, and MH092797; and by the American Parkinson Disease Association (APDA), the Greater St. Louis Chapter of the APDA and the Barnes-Jewish Hospital Foundation. The authors thank William H. Margenau and Robert Dennett in the Cyclotron Facility for11C and18F radioisotope production. Optical rotation was determined in the laboratory of Dr. Douglas F. Covey in the Department of Molecular Biology and Pharmacology of Washington University. The authors thank John Hood, Emily Williams, and Darryl Craig for their assistance with the NHP MicroPET studies. The authors acknowledge NMR core facility and microPET facility for research assistance.
References:
- [1].a) Coyle J, Price D, DeLong M, Science 1983, 219, 1184–1190; [DOI] [PubMed] [Google Scholar]; b) Francis PT, Palmer AM, Snape M, Wilcock GK, J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147; P. Davies, A. J. F. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Maloney, The Lancet 1976, 308, 1403. [DOI] [PubMed] [Google Scholar]
- [2].Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, Koeppe RA, Davis JG, Mathis CA, Moore RY, Dekosky ST, Arch. Neurol 2003, 60, 1745–1748. [DOI] [PubMed] [Google Scholar]
- [3].a) Erickson JD, Varoqui H, FASEB J. 2000, 14, 2450–2458; [DOI] [PubMed] [Google Scholar]; b) Weihe E, Tao-Cheng JH, Schäfer MK, Erickson JD, Eiden LE, Proc. Natl. Acad. Sci 1996, 93, 3547–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Marien MR, Parsons SM, Altar CA, Proc. Natl. Acad. Sci 1987, 84, 876–880; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Roghani A, Feldman J, Kohan SA, Shirzadi A, Gundersen CB, Brecha N, Edwards RH, Proc. Natl. Acad. Sci 1994, 91, 10620–10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Kawamura K, Shiba K, Tsukada H, Nishiyama S, Mori H, Ishiwata K, Ann. Nucl. Med 2006, 20, 417–424; [DOI] [PubMed] [Google Scholar]; b) Kilbourn MR, Jung Y-W, Haka MS, Gildersleeve DL, Kuhl DE, Wieland DM, Life Sci. 1990, 47, 1955–1963; [DOI] [PubMed] [Google Scholar]; c) Widén L, Eriksson L, Ingvar M, Parsons SM, Rogers GA, Stone-Elander S, Neurosci. Lett 1992, 136, 1–4; [DOI] [PubMed] [Google Scholar]; d) Gage HD, Voytko ML, Ehrenkaufer RLE, Tobin JR, Efange SMN, Mach RH, J. Nucl. Med 2000, 41, 2069–2076; [PubMed] [Google Scholar]; e) Sorger D, Scheunemann M, Großmann U, Fischer S, Vercouille J, Hiller A, Wenzel B, Roghani A, Schliebs R, Brust P, Sabri O, Steinbach J, Nucl. Med. Biol 2008, 35, 185–195. [DOI] [PubMed] [Google Scholar]
- [6].a) Kuhl DE, Minoshima S, Fessler JA, Ficaro EP, Wieland DM, Koeppe RA, Frey KA, Foster NL, Ann. Neurol 1996, 40, 399–410; [DOI] [PubMed] [Google Scholar]; b) Kuhl DE, Koeppe RA, Fessler JA, Minoshima S, Ackermann RJ, Carey JE, Gildersleeve DL, Frey KA, Wieland DM, J. Nucl. Med 1994, 35, 405–410. [PubMed] [Google Scholar]
- [7].Petrou M, Frey KA, Kilbourn MR, Scott PJH, Raffel DM, Bohnen NI, Müller MLTM, Albin RL, Koeppe RA, J. Nucl. Med 2014, 55, 396–404. [DOI] [PubMed] [Google Scholar]
- [8].Yue X, Bognar C, Zhang X, Gaehle GG, Moerlein SM, Perlmutter JS, Tu Z, Appl. Radiat. Isot 2016, 107, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beno BR, Yeung K-S, Bartberger MD, Pennington LD, Meanwell NA, J. Med. Chem 2015, 58, 4383–4438. [DOI] [PubMed] [Google Scholar]
- [10].Black S, Annu. Rev. Biochem 1963, 32, 399–418. [DOI] [PubMed] [Google Scholar]
- [11].Li J, Zhang X, Zhang Z, Padakanti PK, Jin H, Cui J, Li A, Zeng D, Rath NP, Flores H, Perlmutter JS, Parsons SM, Tu Z, J. Med. Chem 2013, 56, 6216–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu H, Jin H, Li J, Zhang X, Kaneshige K, Parsons SM, Perlmutter JS, Tu Z, Eur. J. Pharmacol 2015, 752, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yue X, Jin H, Liu H, Luo Z, Zhang X, Kaneshige K, Flores HP, Perlmutter JS, Parsons SM, Tu Z, Org. Biomol. Chem 2017, 15, 5197–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kovac M, Mavel S, Deuther-Conrad W, Méheux N, Glöckner J, Wenzel B, Anderluh M, Brust P, Guilloteau D, Emond P, Bioorg. Med. Chem 2010, 18, 7659–7667. [DOI] [PubMed] [Google Scholar]
- [15].Huang Y, Hammond PS, Whirrett BR, Kuhner RJ, Wu L, Childers SR, Mach RH, J. Med. Chem 1998, 41, 2361–2370. [DOI] [PubMed] [Google Scholar]
- [16].Tu Z, Zhang X, Jin H, Yue X, Padakanti PK, Yu L, Liu H, Flores HP, Kaneshige K, Parsons SM, Perlmutter JS, Bioorg. Med. Chem 2015, 23, 4699–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gilmor ML, Nash NR, Roghani A, Edwards RH, Yi H, Hersch SM, Levey AI, Neurosci J. 1996, 16, 2179–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tu Z, Wang W, Cui J, Zhang X, Lu X, Xu J, Parsons SM, Bioorg. Med. Chem 2012, 20, 4422–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ojeda AM, Bravo DT, Hart TL, Parsons SM, in Membrane Transporters: Methods and Protocols (Ed.: Yan Q), Humana Press, Totowa, NJ U.S., 2003, pp. 155–177. [Google Scholar]
- [20].a) Tu Z, Xu J, Jones LA, Li S, Dumstorff C, Vangveravong S, Chen DL, Wheeler KT, Welch MJ, Mach RH, J. Med. Chem 2007, 50, 3194–3204; [DOI] [PubMed] [Google Scholar]; b) Xu J, Tu Z, Jones LA, Vangveravong S, Wheeler KT, Mach RH, Eur. J. Pharmacol 2005, 525, 8–17. [DOI] [PubMed] [Google Scholar]