

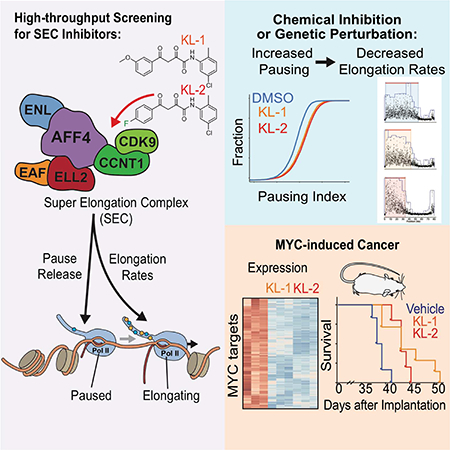
SUMMARY
The Super Elongation Complex (SEC) is required for robust and productive transcription through release of RNA Polymerase II (Pol II) with its P-TEFb module and promoting transcriptional processivity with its ELL2 subunit. Malfunction of SEC contributes to multiple human diseases including cancer. Here, we identify peptidomimetic lead compounds, KL-1 and its structural homolog KL-2, which disrupt the interaction between the SEC scaffolding protein AFF4 and PTEFb, resulting in impaired release of Pol II from promoter-proximal pause sites and a reduced average rate of processive transcription elongation. SEC is required for induction of heat shock genes and treating cells with KL-1 and KL-2 attenuates the heat shock response from Drosophila to human. SEC inhibition downregulates MYC and MYC-dependent transcriptional programs in mammalian cells and delays tumor progression in a mouse xenograft model of MYC-driven cancer, indicating that small molecule disruptors of SEC could be used for targeted therapy of MYC-induced cancer.
Keywords: Super Elongation Complex (SEC), transcription elongation, promoter-proximal pausing, processive elongation, MYC, transcriptional addiction in cancer.
ETOC
Targeting transcriptional elongation with a small molecule inhibitor of the Super Elongation Complex blocks transcriptional programs driven by the oncogene MYC
Graphical Abstract
INTRODUCTION
In most metazoans, the majority of Pol II-transcribed genes are regulated at a step called promoter-proximal pausing (Chen et al., 2018; Jonkers and Lis, 2015). Release from pausing requires phosphorylation of the Pol II C-terminal domain (CTD) by cyclin-dependent kinase 9 (CDK9)-containing positive transcription elongation factor b (P-TEFb) (Peterlin and Price, 2006; Zhou et al., 2012). Proper regulation of this transcriptional checkpoint is vital for physiological responses in metazoan development and misregulation of this checkpoint has been found to contribute to human diseases, including cancer (Lin et al., 2010; Bradner et al., 2017; Shilatifard et al., 1996; Smith et al., 2011; Takahashi et al., 2011).
The majority of P-TEFb, comprised of CDK9 and its cyclin partner Cyclin T1 (CCNT1), is sequestered in an inactive form by RNA binding proteins HEXIM1 or HEXIM2 associating with 7SK snRNP (Luo et al., 2012a; Zhou et al., 2012). The inactive complex occupies promoter-proximal regions on chromatin (Ji et al., 2013; McNamara et al., 2016). P-TEFb can be released from the 7SK/HEXIM complex into active complexes such as BRD4/P-TEFb (Yang et al., 2005) and the Super Elongation Complex (SEC) (Lin et al., 2010; Luo et al., 2012b; Zhou et al., 2012). Inhibition of all P-TEFb activity can be achieved with inhibitors such as flavopiridol (Chao and Price, 2001). The activity of BRD4/P-TEFb can be inhibited with small molecules blocking its recruitment to chromatin (Dawson et al., 2011; Filippakopoulos et al., 2010) and phthalimide-conjugated analogs can induce rapid degradation of BRD4 (Winter et al., 2015; Winter et al., 2017). Blocking BRD4/P-TEFb with these compounds inhibits release of paused Pol II into productive elongation with profound effects on MYC targets (Bradner et al., 2017; Delmore et al., 2011). However, studies of SEC in cells are more limited due to a lack of small molecular inhibitors specifically targeting this form of P-TEFb.
In addition to P-TEFb, SEC contains AF4/FMR2 (AFF) family proteins (AFF1–4), the YEATS domain-containing proteins ENL or AF9 (encoded by the MLLT1 and MLLT3 genes), the Pol II elongation factors eleven-nineteen lysine-rich leukemia (ELL) proteins, and ELL-associated factor 1 (EAF1) or EAF2 (Lin et al., 2010; Chen et al., 2018; Luo et al., 2012b). Within SEC, the AFF proteins function as scaffolds for binding the other subunits. P-TEFb is required for phosphorylation of Pol II CTD on serine 2 (Ser2P) and transcription elongation factor SPT5 on its C-terminal region to promote release from the promoter-proximal pausing, while ELL proteins have been demonstrated to enhance processivity of elongation by RNA Pol II using in vitro transcription assays (Shilatifard et al., 1997; Shilatifard et al., 1996) and in cells (Hu et al., 2013).
SEC is required for rapid induction of transcription in response to cellular signals (Galbraith et al., 2013; Lin et al., 2010; Lin et al., 2011; Takahashi et al., 2011) and is hijacked by HIV Tat to activate proviral genome transcription (He et al., 2010; Sobhian et al., 2010). Mistargeting of SEC is essential for leukemogenesis driven by rearrangements of the mixed lineage leukemia (MLL) gene (Liang et al., 2017; Mohan et al., 2010; Yokoyama et al., 2010). Mutations that stabilize SEC lead to the developmental syndrome CHOPS (Izumi et al., 2015). SEC is a regulatory factor of MYC (Erb et al., 2017; Luo et al., 2012a; Takahashi et al., 2011; Wan et al., 2017), which is a master regulator for cancer cell proliferation and contributes to the pathogenesis of a majority of human cancers by coordinated amplification of transcription (Lin et al., 2012; Nie et al., 2012; Sabo et al., 2014; Walz et al., 2014), and is particularly required for expression of cell division and pre-mRNA splicing factors (Hsu et al., 2015; Koh et al., 2015). Given the importance of transcription elongation control in cancer pathogenesis and the paradigm of BET domain inhibitors in targeting MYC expression and the transcriptional elongation misregulation in cancers (Bradner et al., 2017), developing inhibitors of SEC can serve as a tool for both mechanistic studies of SEC and for cancer therapeutics.
RESULTS
Peptidomimetic identification of disruptors of the Super Elongation Complex
To identify small molecule disruptors of the Super Elongation Complex (SEC), we first examined the crystal structure (PDB: 4IMY) showing AFF4 bound to the CCNT1 subunit of PTEFb (Gu et al., 2014; Schulze-Gahmen et al., 2013). Five residues of the N-terminus of AFF4 (L34, F35, A36, E37 and P38) interacted with the binding groove of CCNT1, comprised of the residues W221, Y224, L163, V164, R165, Y175, F176, D169, W207, W210 and E211 (Figure 1A). Mutation data of CCNT1 residues Y175, E211, D169, F176, R165, W210 and W207 demonstrated the importance of this pocket for interactions with AFF4 (Schulze-Gahmen et al., 2013). A three-tiered grid-based ligand docking glide algorithm was employed to screen for potential compounds binding to the CCNT1 binding pocket (Figure 1A).
Figure 1. Peptidomimetic identification of disruptors of the AFF4-CCNT1 interaction within the Super Elongation Complex.

(A) Schematic for identifying small molecule disruptors of the AFF4-CCNT1 interaction using in silico screening. Compounds from the ZINC database were docked with the AFF4-CCNT1 structure (4IMY) using a three-tier glide-docking algorithm, which led to 40 candidates. Residues in the binding pocket of CCNT1 are labeled orange and residues of the AFF4 peptide are labeled red.
(B) Validation screening identified KL-1 as a potential SEC disruptor using AlphaLISA screening. GST-CCNT1 (AA 1–300) and biotin-labeled AFF4 peptide (AA 32–67) were used to measure the AFF4-CCNT1 interaction with the AlphaLISA assay.
(C) Similarity search for KL-1-like molecules identified KL-2. The peptidomimetic potential of KL-1 and KL-2 can be seen by overlaying them with the AFF4 peptide LFAEP structure.
(D) Dose-dependent inhibition of KL-1 and KL-2 on AFF4-CCNT1 interaction. The Ki constants of both compounds were measured with the AlphaLISA assays.
(E) KL-1 and KL-2 treatment results in reduced protein levels of SEC components AFF1 and AFF4 but not CDK9 or CCNT1. HEK293T cells were treated with 20 μM of SEC inhibitors KL1 or KL-2 for 6 hr. 5 μl, 10 μl and 20 μl cell lysates were loaded and the protein levels of AFF1, AFF4, CCNT1, CDK9 and Tubulin (load control) were determined by western blotting.
(F-H) ChIP-seq analysis demonstrates that KL-1 and KL-2 treatment results in decreased occupancy of AFF1 and AFF4 on chromatin as seen at the HSPA8 gene (F) or by metaplot analysis at AFF1 (G) and AFF4 peak regions (H). See also Figure S1.
To test if candidate compounds can disrupt the AFF4-CCNT1 interaction in vitro, we used a bead-based AlphaLISA assay with recombinant CCNT1 (1–300) protein and synthesized BiotinAFF4 peptides (AA 32–67) (Figure S1A, S1B and S1C). We tested the 40 compounds identified from the in silico screening at 20 μM with the AlphaLISA assay (Figure 1B) and found that the compound (2Z)-N-(5-Chloro-2-methylphenyl)-2-hydroxy-4-(3-methoxyphenyl)-4-oxo-2butenamide, referred to as “KL-1” (Figure 1C), could inhibit the interaction of AFF4-CCNT1, while neither the pan-CDK9 kinase inhibitor flavopiridol nor the BET domain inhibitors disrupted the AFF4-CCNT1 interaction (Figure 1B).
Based on the structure of KL-1 (Figure 1C), we performed a KL-1 similarity search with ChemDiv compounds and tested the top 32 most similar compounds. The compound (2Z)-N-(5Chloro-2-methylphenyl)-4-(4-fluorophenyl)-2-hydroxy-4-oxo-2-butenamide (referred to as “KL2”) shares the same scaffold as KL-1 and modeling of their structures reveals similar peptidomimetic potential for the AFF4 pentapeptide LFAEP (Figure 1C). Both compounds exhibited dose-dependent inhibitory effects on the AFF4-CCNT1 interaction with observed Ki of 3.48 μM and 1.50 μM, respectively, for KL-1 and KL-2 (Figure 1D). In order to study the functions of KL-1 and KL-2 in depth, we synthesized these compounds in house as shown in Figure S1D.
KL-1 and KL-2 treatments led to depletion of SEC components AFF1 and AFF4 in both HEK293T and HCT-116 cells within 6 hr (Figure 1E, Figure S1E and S1F), but no major effect on the protein levels of P-TEFb components CCNT1 and CDK9, or on Pol II, BRD2 and BRD4. We confirmed that treating cells with these two compounds results in decreased AFF1 and AFF4 protein using doxycycline-inducible Flag-AFF1 and Flag-AFF4 HEK293T cells (Figure S1G and S1H). KL-1 and KL-2 treatments do not result in decreased mRNA levels of SEC components (Figure S1I). Together, these data demonstrate that KL-1 and KL-2 disrupt the interaction of CCNT1 and the SEC scaffolding AFF proteins, which results in reduced levels of cellular SEC. To investigate the consequences of AFF1 and AFF4 protein reduction on their association with chromatin, we performed ChIP-seq of AFF1 and AFF4 in HEK293T cells treated with KL-1 and KL-2 for 6 hr. Genome browser views of the HSPA8 gene demonstrate that both inhibitors lead to decreased occupancy of AFF1 and AFF4 (Figure 1F) and reduced AFF1 and AFF4 chromatin occupancy is observed by genome-wide analysis (Figure 1G and 1H).
Small molecule disruption of SEC increases promoter-proximal pausing
Treatment of cells with KL-1 and KL-2 resulted in increased Pol II occupancy at the promoter-proximal regions of HSPA8 and SRSF4 loci (Figure 2A). Analysis of the 6,119 Pol II occupied genes in HEK293T cells that are also occupied by AFF1 or AFF4, showed that these genes have increased Pol II occupancy at the promoter-proximal regions after treatment of KL-1 and KL-2 (Figure 2B). We also tested the SEC inhibitors in HCT-116 and Jurkat (J-Lat 6.3 clone) (Jordan et al., 2003) cells and observed similar increases of Pol II occupancy in promoter-proximal regions (Figure S2A, S2B and S2C).
Figure 2. Small molecule disruption of SEC increases promoter-proximal pausing.

(A) SEC inhibitors increase Pol II occupancy near transcription start sites (TSS) of the HSPA8 and SRSF4 genes. Pol II ChIP-seq was performed in HEK293T cells with 20 μM SEC inhibitors KL-1 and KL-2 for 6 hr. Coverage is in reads per million (rpm).
(B) Heatmap analysis of Pol II occupancy at SEC-occupied genes in cells treated with KL-1 and KL-2 shows increased Pol II occupancy at promoter-proximal regions. Rows are sorted by Pol II occupancy in the DMSO condition and metaplots of Pol II occupancy are shown at the bottom. Corresponding AFF1 and AFF4 occupancy (rpm) in the vehicle-treated condition is shown.
(C-D) Scatter plots of log2 fold changes of Pol II occupancy versus log2 fold changes of AFF4 (left panels) or AFF1 in (right panels) in KL-1 (C) or KL-2 (D) treated cells. SEC inhibitor treatments result in increased Pol II occupancy and decreased AFF4 and AFF1 occupancy at most of the 6,119 expressed and SEC-occupied genes.
(E) Illustration of the pausing index calculation based on the ratio of Pol II occupancy around the TSS to Pol II occupancy in the gene body.
(F) Empirical cumulative density function (ECDF) plots of Pol II pausing index in vehicle and SEC inhibitors-treated cells at the 6,840 expressed genes in 293T cells.
(G-H) Knockdown of AFF1 and AFF4 by shRNA-mediated RNAi shows similar pausing index changes as SEC inhibitors. (G) Genome browser tracks of Pol II occupancy at the SRSF4 gene. (H) ECDF plot of Pol II pausing index in non-targeting (shGFP), AFF1 knockdown (shAFF1) and AFF4 knockdown (shAFF4). See also Figure S2.
To further investigate the relationship between Pol II and SEC occupancy changes due to SEC inhibitor treatments, we used k-means clustering with the 6,119 Pol II and SEC-occupied genes into three groups: Group I, II and III (Figure S2D). Group I genes exhibited the highest foldchange increase in Pol II occupancy and have the highest AFF1 (Figure S2E) and AFF4 (Figure S2F) occupancy in the control condition, suggesting that the increased Pol II occupancy by KL-1 and KL-2 can be attributed to changes in SEC occupancy. Scatter plot analysis shows that KL-1 (Figure 2C) and KL-2 (Figure 2D) globally reduce AFF1 and AFF4 occupancy (leftward shift) and increases Pol II occupancy (upward shift) at co-occupied genes, suggesting that disruption of SEC by small molecules leads to increases in promoter-proximal Pol II occupancy.
We calculated levels of Pol II pausing based on the ratio of Pol II reads in the gene body and promoter (Figure 2E). Analysis of pausing index revealed that SEC disruptors KL-1 and KL-2 could increase promoter-proximal pausing as shown by the Empirical Cumulative Distribution Function (ECDF) plot (Figure 2F). ECDF analysis of pausing indexes in HCT-116 and Jurkat cells showed similar effects as seen in HEK293T (Figure S2G and S2H), demonstrating that SEC inhibition leads to increased promoter-proximal pausing. To further demonstrate that SEC inhibition through KL-1 and KL-2 increases pausing at the early stage of transcription elongation, we depleted AFF1 and AFF4 proteins in HEK293T cells with short hairpin RNAs (shRNA) and found that co-knockdown of AFF1 and AFF4 leads to increased Pol II occupancy in promoter-proximal regions, as can be seen at the SRSF4 gene (Figure 2G) and by ECDF analysis (Figure 2H) similar to the use of KL-1 and KL-2 (figure 2F).
Disruption of SEC phenocopies slow Pol II mutants
Examination of Pol II changes at the 3’-end of genes, such as SRSF4 and HSPA8, revealed a 5’end shift of Pol II from the transcription termination sites (Figure S2I and S2J), which is reminiscent of the recently published phenotype of slow Pol II mutants (Figure 3A) (Fong et al., 2015). Therefore, we tested the effect of KL-1 and KL-2 treatments with the Pol II speed-mutant cells generously provided by the Bentley laboratory (University of Colorado). KL-1 and KL-2 treatments shift the Pol II profile downstream of the TES more 5’ in the fast, wild-type (WT) and slow Pol II mutant cells at the ACTB and PIM3 gene (Figure 3B and3C). Performing genome-wide analysis of genes with Pol II termination signals around the TES sites (Figure S3A), we observed similar Pol II profiles observed by Bentley and colleagues in the slow and fast Pol II cells (Figure 3D) (Fong et al., 2015). Treatment of fast Pol II mutant cells with SEC inhibitors leads to a similar Pol II pattern as the slow Pol II mutant, with a 5’ shift in read coverage at the 3’-end of genes (Figure 3D, 3E and S3B), indicating that disruption of SEC phenocopies slow Pol II mutants. To further verify these phenotypes, we performed time and dose-dependent treatment of KL-2 in the 293T cells and observed dose- and time-dependent 5’ shifts of Pol II signal around TES sites (Figure S3C and S3D).
Figure 3. Disruption of SEC phenocopies slow Pol II mutants and reduces Pol II processivity.

(A) Schematic of alpha-amanitin resistant (N792D) versions of Pol II that are otherwise wildtype (WT) or have a mutation in the trigger loop (E1126G) that results in faster Pol II or the funnel domain (R749H) that results in slower Pol II (Fong et al., 2014). Alpha-amanitin resistant Pol II is induced with doxycycline while alpha-amanitin is used to inhibit and cause the degradation of endogenous Pol II.
(B-C) Genome browser tracks of Pol II ChIP-seq coverage at the ACTB (B) and PIM3 (C) genes in the fast, WT and slow Pol II mutant cells after both SEC inhibitors. Dotted line indicates the position of the annotated transcription end site (TES) site.
(D) Metaplots of Pol II ChIP-seq coverage at the 3’-end of genes in the fast, WT and slow Pol II mutant cells. The region from TES to 7.5 kb downstream of the TES for the 1,057 genes with typical Pol II termination signals in HEK293T cells is plotted (Figure S3A). Pol II appears to terminate earlier in the R749H slow Pol II mutant relative to WT Pol II cells, while the E1126G fast Pol II mutant appears to have a delayed termination with a less prominent peak of Pol II signal 3’ of the TES.
(E) Treatment of the fast Pol II mutant expressing cells with SEC inhibitors leads to a Pol II phenotype similar to slower Pol II mutant expressing cells as viewed at the 3’-end of genes.
(F-H) PRO-seq analysis in the presence or absence of SEC inhibitors. (F) HEK293T cells were treated for 6 hr with the indicated compounds before nuclear isolation and precision nuclear run-on sequencing (PRO-seq). (G) Genome browser tracks of PRO-seq signal at the SRSF1 gene in vehicle and SEC inhibitor-treated cells. (H) Metagene plot of PRO-seq signal from pausing sites to TES. (H) Heatmap of Pol II occupancy (rpm) and log2 fold changes in vehicle or the indicated SEC inhibitors at single-nucleotide resolution. Rows represent genes and are sorted by gene length from shortest to longest and are shown from the predicted pausing site determined by PRO-seq to 50 kb downstream.
(I-J) SEC inhibitors reduce the Pol II processivity at gene body, especially, the 3’-end of the gene body. Metagene plotting (I) and Metaplot analysis of PRO-seq signal in the 10 kb region surrounding the annotated TES (J) were performed with all of the expressing genes (N=6,840). See also Figure S3.
Since slow Pol II mutants exhibited hyperphosphorylation of the CTD on Ser2 at the 5’-end of genes due to higher “dwell-time” (Fong et al., 2017), we asked if SEC inhibitor treatments mimicked this phenotype as well. We performed ChIP-seq for the Ser2P form of Pol II in HEK293T cells after 6 hr of KL-1 and KL-2 treatments and observed that KL-1 or KL-2 elevates Pol II Ser2P levels around transcription start sites and their downstream regions, as can be seen at the SRSF1 gene (Figure S3E) and by metagene analysis of Pol II Ser2P changes (Figure S3F and S3G), further suggesting KL-1 and KL-2 treatments could slow down Pol II transcription elongation.
Since both ELL and the related ELL2 were originally biochemically and mechanistically identified to function as transcription elongation factors to regulate the processive rate (Vmax) of transcription elongation in vitro (Shilatifard et al., 1997; Shilatifard et al., 1996) and KL-1 and KL-2 treated cells exhibited phenotypes indicative of less processive Pol II, we therefore measured ELL2 protein levels after KL-1 and KL-2 treatments. We found that KL-1 and KL-2 led to reduced ELL2 protein levels in cells (Figure S3H), suggesting that ELL2 reduction may account for the observed slow-down of Pol II elongation rate (Figure 3E and S3B). To test this, we depleted ELL2 in HEK293T cells (Figure S3I) and found a 5’ shift of Pol II signal at TES sites (Figure S3J, S3K and S3L) similar to the KL-1 and KL-2 treatments or the slow Pol II mutant, albeit to a lesser extent. Together, these data suggest that ELL2 reduction contributes to the slow Pol II phenotypes resulting from KL-1 and KL-2 treatments.
To better delineate changes in Pol II occupancy in response to KL-1 and KL-2, we performed precision nuclear run-on and sequencing (PRO-seq) (Kwak et al., 2013), which allows single-nucleotide resolution of polymerase position (Figure 3F and 3G). PRO-seq analysis confirmed that KL-1 and KL-2 treatments result in increased promoter-proximal pausing (Figure S3M and S3N). Increased occupancy of engaged Pol II in the gene body, particularly at the 3’-end of genes as can be seen by metagene analysis (Figure 3H and 3I). Heatmap analysis of expressed genes ranked by gene length is consistent with KL-1 and KL-2 treatments reducing Pol II processivity, which in turn leads to premature termination due to slower transcription elongation of Pol II (Figure 3H). Furthermore, PRO-seq analysis confirms a 3’-end transcriptional defect (Figure 3H and 3J), which is consistent with the Pol II profiles observed in the slow Pol II mutant cells.
Disruption of SEC by KL1/KL2 slows Pol II elongation rates
To measure the consequence of SEC inhibition on the rate and processivity of transcription elongation, we used a 4sU-FP-seq strategy employing flavopiridol (FP)-induced pausing followed by release in the presence of 4sU (Fuchs et al., 2014) (Figure 4A). When cells are pretreated with KL-1 and KL-2, the distance Pol II traveled after release from flavopiridol was markedly reduced, as seen at individual genes (Figure 4B) and by heatmap (Figure 4C) and metaplot analysis (Figure 4D).
Figure 4. Small molecule disruption of SEC slows Pol II elongation rates.

(A) Workflow of 4sU-FP-seq based measurement of transcription elongation rates. HEK293T cells were pretreated with vehicle or 20 μM SEC inhibitors for 5 hr before addition of the CDK9 inhibitor flavopiridol for 1 hr to arrest Pol II at the promoter-proximal pause site. Inhibitors are washed out with PBS before allowing transcription to proceed in the presence of fresh medium containing 500 μM 4-Thiouridine (4sU) for 15 min. The 4sU-labeled RNA was extracted and fragmented before purification, and followed with RNA-seq.
(B) Genome browser tracks of 4sU-FP-seq for vehicle and SEC inhibitor-treated cells at the ACTN2 and MTR loci.
(C) Heatmap analysis of 4sU-FP-seq in vehicle and the indicated SEC inhibitors in HEK293T cells. All genes longer than 50 kb (N= 5,568) were plotted and ordered using the total 4sU-FPseq signals in the vehicle-treated cells.
(D) Metaplot of strand-specific 4sU-FP-seq signals in vehicle and SEC inhibitors-treated cells.
(E) Hidden Markov Model (HMM) for elongation rate analysis. Raw changes in 4sU-FP-seq read counts in non-overlapping 50 bp windows were used to infer elongation rates for the MTR gene. Boxes show the span of advancing wave inferred by a 3-state HMM analysis.
(F-G) Histograms (F) and boxplots (G) comparing transcription elongation rates for 982 genes in HEK293T cells for which high confidence elongation rates could be determined. Statistical analysis was performed with the Wilcoxon test. See also Figure S4.
We used two different approaches to measure elongation rates in the 4sU-FP-seq experiments as the result of KL1 and/or KL2 treatment. First, we employed a Hidden Markov Model (HMM) method for calculating elongation rates (Danko et al., 2013). We were able to measure elongation rates across all samples for 982 genes (the number of genes for which the model could detect a wave front of Pol II elongation). This analysis demonstrated that both KL-1 and KL-2 treatments decreased the elongation rate of Pol II at the ACTN2 and MTR genes (Figure 4E and S4A), and globally as can be seen by boxplot and histogram analyses of these 982 genes (Figure 4F and 4G). Second, we used the island-based peak caller SICER to use the width of peaks overlapping TSS regions to measure distance traveled (Figure S4B, S4C, S4D and S4E). Together, these data suggest that KL1/KL2-dependnet disruption of SEC results in slower elongating Pol II.
SEC inhibitors block paused Pol II release in SEC-dependent rapid response models
SEC has been shown to mediate transcription elongation in rapid response models such as heat shock induced gene expression (Lin et al., 2010; Takahashi et al., 2011) and Tat-induced HIV proviral transcription (He et al., 2010; Sobhian et al., 2010). Therefore, we tested if KL-1 and KL-2 could inhibit the function of SEC in these rapid-response models. For heat shock induction, we pretreated HCT-116 cells with KL-1 or KL-2 for 5 hr to block SEC function and heat shocked cells at 42 °C for 1 hr. Pol II ChIP-seq demonstrates that SEC inhibitors KL-1 and KL-2 block induction of known heat shock inducible genes such as FOS, HSPD1, HSPE1 and EGR1 (Figure 5A, 5B and S5A). Analysis of Pol II occupancy at 136 genes induced by heat shock under vehicle conditions (Figure 5C and S5B) demonstrates that KL-1 and KL-2 treatments led to an impaired heat shock response (Figure 5D and 5E).
Figure 5. SEC inhibitors block transcription elongation in SEC-dependent rapid response models.

(A-B) Genome browser views of Pol II ChIP-seq at FOS, HSPD1 and HSPE1 after 1 hr heat shock of HCT-116 cells treated with vehicle (DMSO) or the indicated SEC inhibitors. HCT-116 cells were pretreated with inhibitors for 5 hr at 37 °C before exchanging medium with conditioned 42 °C medium. Dark and light colors indicate the 37 and 42 °C conditions, respectively.
(C) Genome-wide identification of heat shock-induced genes with fold change of reads per million (rpm) in gene bodies and pausing index. Red dots are the 136 heat shock-induced genes according to Pol II signals (fold change rpm in gene bodies > 1.5 and pausing index decreased > 1.5-fold in the DMSO treatment).
(D-E) Metagene plot of the 136 heat-shock induced genes shows attenuated induction with both inhibitors (D). Box plot analysis depicts the log2 fold changes (rpm) in gene bodies (E) (y axis) after heat shock with pretreatment of vehicle or SEC inhibitors.
(F) Sequence alignment of the CCNT1 interacting region in human AFF family proteins and the Drosophila homolog Lilliputian.
(G) Genome-wide analysis of SEC inhibitors effect on the heat shock response in Drosophila S2 cells (N=215). S2 cells treated with and without 20 μM SEC inhibitors were heat shocked 10 min at 37 °C before performing Pol II ChIP-seq. Statistical analysis was performed with the Wilcoxon test. See also Figure S5.
KL-1 and KL-2 structures mimic the AFF4 pentapeptide LFAEP (Figure 1C) that is conserved across the AFF family in humans and in the sole Drosophila member, Lilliputian (Figure 5F). Therefore, we tested the effect of KL-1 and KL-2 in the heat shock response in Drosophila S2 cells. As shown in Figure 5G, treatment of S2 cells with KL-1 and KL-2 attenuated heat shock induction of the 215 heat shock induced genes in these cells (Figure 5G).
Since SEC interacts with, and is an essential coactivator for the HIV transactivator Tat (He et al., 2010; Sobhian et al., 2010), we examined the effect of KL-1 and KL-2 in this process using the J-Lat 6.3 clone, a derivative of Jurkat cells that has an integrated HIV genome in which GFP replaces the HIV nef gene (Jordan et al., 2003). In this system, the activation of the HIV genome can be achieved by treatment with 10 nM Phorbol 12-myristate 13-acetate (PMA) and the expression of the HIV genome can be monitored with GFP fluorescence (Figure S5C). Treating cells with 20 μM KL-1 or KL-2 resulted in a strong inhibition of GFP expression in J-Lat 6.3 cells after PMA induction as revealed by FACS analysis (Figure S5D), and this inhibition is dose-dependent (Figure S5E). ChIP-seq of Pol II in the J-Lat 6.3 cells confirmed that SEC inhibitors block transcription elongation of the Tat-dependent integrated HIV genome (Figure S5F). Together, these studies demonstrate that KL-1 and KL-2 can inhibit SEC-mediated transcription elongation in both heat-shock and Tat-mediated rapid-transcriptional induction models.
Chemical inhibition of SEC by KL1/KL2 downregulates MYC and MYC-dependent transcriptional programs
We performed mRNA-seq of 293T cells after KL-1 and KL-2 treatments for 24 hr and found a large overlap in gene expression changes, with 1,911 genes being downregulated and 1,242 genes being upregulated by both treatments (Figure 6A, 6B and 6C). Gene ontology analysis revealed that RNA splicing-related factors, MYC target gene sets, and cell proliferation-related terms are among the top enriched terms for downregulated genes in response to SEC inhibitors (Figures 6D and S6A). DNA repair, apoptosis, and cellular response to unfolded protein were modestly enriched terms for upregulated genes, suggesting a stress response of these cells after SEC inhibition (Figure S6B).
Figure 6. SEC disruption targets MYC and MYC target genes.

(A-B) Venn diagram of deregulated genes in 293T cells by KL-1 and KL-2. 1,911 genes were downregulated and 1,242 genes were upregulated by both inhibitors.
(C) Heatmap showing expression changes in response to SEC inhibitors, with differentially expressed canonical MYC targets, RNA splicing factors, and core SNRP assembly genes highlighted. Z score-normalized values are displayed (n=3).
(D) Network enrichment analysis with Metascape (Tripathi et al., 2015) of the 1,911 genes downregulated by both KL-1 and KL-2. Each cluster is represented by different colors and a circle node denotes an enriched term.
(E) MA plots of RNA splicing and Hallmark MYC target genes in KL-2 treated, AFF1 and AFF4 co-depletion, and ELL2-depleted cells. Circles mark downregulated splicing and Hallmark MYC target genes. The MYC and PRMT5 genes, and genes encoding SEC components are denoted as red squares.
(F) Western analysis of MYC levels in the MYC lowly expressed small cell lung cancer cell line SW2171 and MYC-amplified small cell lung cancer cell line H2171.
(G) Increased SEC at MYC binding sites in MYC highly expressed H2171 cells. Genome browser views of MYC and SEC occupancy around the PRMT5 gene in SW1271 and H2171 cells are shown.
(H) ChIP-seq analysis of MYC binding peaks in SW1271 and H2171 cells. Venn diagram of MYC peaks showing that H2171 cells gain more MYC binding sites.
(I) MYC and SEC overlap in SW1271 and H2171 cells. The SEC and MYC peaks were determined by MACS with a p-value cutoff at 1E-5.
(J) Heatmap of SEC occupancy at the 50,819 gained MYC binding sites in H2171 cells. The heatmap is separated based on whether gained SEC peaks could be called by MACS with a p-value cutoff of 1E-5.
(K) MYC-amplified H2171 cells are more sensitive to KL-1 and KL-2 inhibition than MYC lowly expressed SW2171 cells. Both cell lines were treated with increasing concentrations of KL-1 and KL-2 for 3 days and cell proliferation was measured with CellTiter-Glo (Promega) (n=3–6). See also Figure S6.
KL-1 and KL-2 treatments led to decreased expression of MYC, canonical MYC targets (Zeller et al., 2003), and RNA splicing-related genes (Figure 6C), which were recently identified as direct MYC targets important for MYC-driven cancers (Hsu et al., 2015; Koh et al., 2015). Interestingly, the PRMT5 gene, which encodes an enzyme responsible for methylation of splicing machinery proteins, is among the downregulated genes, suggesting that SEC inhibitors perturb the MYC–PRMT5 axis (Koh et al., 2015), which could potentially be used to target splicing vulnerabilities in these cancers. We compared mRNA-seq after SEC depletion with either AFF1 and AFF4 co-knockdown or ELL2 knockdown with gene expression changes after SEC chemical inhibition. We found that 1,221 genes out of the 1,911 (63.8%) SEC inhibitors-downregulated genes were significantly downregulated by SEC subunit depletion, and accordingly, gene ontology analysis demonstrated enrichment for RNA splicing and MYC target gene terms (Figure S6C and 6E), which were also significantly down-regulated after acute-degradation of SEC subunit ENL (Erb et al., 2017).
These findings led us to investigate the potential of using SEC inhibitors in cancer cells exhibiting transcriptional addiction (Bradner et al., 2017), such as those with high expression of MYC, which leads to increased transcription of downstream genes necessary for cancer cell proliferation (Lin et al., 2012; Sabo et al., 2014). We used the previously characterized MYC-amplified small cell lung carcinoma H2171 cells (Lin et al., 2012) and a corresponding low-MYC expressing small cell lung cell line, SW1271 (Figure 6F). ChIP-seq of MYC protein in both H2171 and SW1271 cells confirms that H2171 has more MYC-occupied sites, consistent with a previous study (Lin et al., 2012) (Figure 6G and 6H).
To elucidate the role of SEC in MYC-mediated transcriptional regulation, we performed ChIPseq of SEC subunits in both H2171 and SW1271 cells and found SEC co-localized with MYC on chromatin (Figure 6G). Genome-wide analysis of MYC and SEC occupancy shows that H2171 cells have more MYC and SEC co-bound regions and increased occupancy of SEC (Figure 6H, 6I and 6J), suggesting that SEC is involved in MYC-mediated transcriptional regulation in these cancer cells. We found that H2171 cells (Lin et al., 2012) are more sensitive to KL-1 and KL-2 treatments than SW1271 cells (Figure 6K), and knockdown of MYC in H2171 cells resulted in decreased sensitivity to SEC inhibition (Figure S6E), indicating that SEC inhibitors could be useful for abrogating the growth advantage of high MYC expressing cancer cells.
Since MYC has been shown to recruit the P-TEFb complex (Rahl et al., 2010) and co-localizes with the P-TEFb containing SEC complex (Figure 6I), we examined the role of MYC in SEC recruitment and productive transcription elongation. We depleted MYC in 293T cells for 2 days and found that MYC depletion led to reduced chromatin occupancy of SEC subunits AFF1 and AFF4 (Figure S6F and S6G). We also performed 4sU-FP-seq in MYC-knockdown cells and found that MYC depletion for 2 days results in reduced processivity at the MTR loci (Figure S6H) and observed a global defect in distance traveled after release from flavopiridol (Figure S6I). HMM analysis demonstrated that MYC knockdown led to decreased elongation rates (Figure S6J). These data suggest that SEC acts as a MYC cofactor by enhancing transcription processivity.
KL1/KL2 disruption of SEC delays tumor progression and improve survival of mice bearing MDA231-LM2 tumor
To determine if the SEC disruptors KL-1 and KL-2 could be used in vivo as possible cancer therapeutics through affecting the rate of transcription elongation, we employed the murine MDA231-LM2 tumor model (Figure 7A), which has been extensively characterized as a MYC-dependent tumor model (Hsu et al., 2015) and has been recently established in our lab (Wang et al., 2017). Both KL-1 and KL-2 could inhibit colony formation of MDA231-LM2 cells in vitro (Figure S7A) and both SEC disruptors increase apoptosis as shown by Annexin V staining (Figure S7B). We assessed the toxicity of KL-1 and KL-2 in mice with increasing doses and found that injection of 5 doses of 50 mg/kg KL-1 or 10 mg/kg KL-2 for 5 days does not result in significant weight loss in mice after monitoring for 35 days, and no obvious sign of sickness was observed during this period (Figure S7C).
Figure 7. SEC disruptors delay tumor progression and improve survival of MDA231-LM2 tumor mice.

(A) Schematic of MDA231-LM2 tumor development in athymic nude mice (Wang et al., 2017). 4×106 MDA231-LM2 cells were inoculated into fat pads of nude mice. 17 days after injection, when the tumor reached 100 mm3, mice were divided randomly into three groups. Drug treatments were performed once daily with 50 mg/kg KL-1, 10 mg/kg KL-2 or PBS (vehicle) for a total of 15 intraperitoneal injections.
(B-C) KL-1 and KL-2 delay tumor growth in the MDA231-LM2 tumor mouse model. Average tumor sizes of the vehicle (n=6), KL-1 (n=7), and KL-2 (n=5) treated groups were plotted from day 5 to day 36 after inoculation (B). Representative tumors sizes are shown (C).
(D) Dot plots of tumor sizes at day 36 after inoculation and vehicle, KL-1, and KL-2 treatment, indicating that SEC disruptors KL-1 and KL-2 delay tumor progression in vivo. A 2-way unpaired student’s t-test was used for statistical analysis.
(E) Kaplan-Meier survival curves of vehicle, KL-1, and KL-2 treated nude mice transplanted with 4×106 MDA231-LM2 cells. 17 days after inoculation, vehicle (n=6), KL-1 (n=7), and KL-2 (n=5) were administered daily for 15 intraperitoneal injections. Mice were euthanized when the tumor size reached 1000 mm3. The p values were calculated using the log-rank test. See also Figure S7.
To measure the potential of SEC inhibition in the MDA231-LM2 tumor model, we initiated injection of mice with SEC inhibitors on day 17 after inoculation, when the average tumor size reached 100 mm3 (Figure 7A). After once daily administration for 15 days, we further monitored tumor weights and mice were euthanized when the tumor size reached 1000 mm3. Both KL-1 and KL-2 delayed tumor progression as monitored by tumor sizes (Figure 7B-D). Our study demonstrated that both SEC inhibitors significantly extended survival of recipient mice (Figure 7E). Together, these data suggest that SEC disruptors could potentially be used to delay progression and improve the survival of MYC-dependent cancer.
DISCUSSION
The Super Elongation Complex functions as a transcriptional cofactor that is required for driving high rates of transcription for immediate-early genes, heat shock genes under stress, for production of the HIV provirus, and contributes to oncogenesis by driving high rates of coordinated transcriptional programs such as occurs in MYC amplified cancers (Lin et al., 2010; Smith et al., 2011; Luo et al., 2012b). Treatment of mammalian cells with KL-1 and KL-2 result in reduced levels of subunits of SEC AFF1, AFF4 and ELL2. Germline mutations that stabilize AFF4 cause the human developmental disorder CHOPS syndrome (Izumi et al., 2015). Both AFF4 and ELL2 are targeted by the SIAH1 E3 ligase (Liu et al., 2012). The SEC destabilizing property of KL-1 and KL-2 likely enhances the efficacy of our lead compounds in the transcription elongation assays and in vivo animal tumor model. Proteolysis-Targeting Chimera (PROTAC) methods that allow targeted degradation of proteins with small molecules have been shown to be much more efficacious than the small molecule inhibitor alone (Neklesa et al., 2017; Winter et al., 2015; Winter et al., 2017). For example, JQ1 and related compound IBET-151 bind to the bromodomains of BRD4 and block its interactions with acetylated histones on chromatin (Dawson et al., 2011; Filippakopoulos et al., 2010). When JQ1-like molecules are fused to phthalimides to target BRD4 degradation by the endogenous cellular ubiquitin ligase cereblon, the loss of BRD4 protein obviates the need for constant interaction of JQ1 with BRD4 (Lu et al., 2015; Winter et al., 2015; Winter et al., 2017).
In this study, we found that SEC disruptors KL1 and KL2 could block transcription elongation in multiple SEC-dependent transcriptional models, demonstrating these compounds can be used a convenient chemical perturbation tool for the mechanistic and functional studies of SEC in other SEC-related cellular and developmental processes. Interestingly, we found that the MYC-dependent cancer cells are sensitive to the SEC inhibitors, providing the mechanistic finding that SEC is co-localized with MYC and exhibit increased occupancy in the MYC highly expressing cells, suggesting a dependency of transcription elongation for MYC-dependent cancers. Indeed, we also showed that SEC complex is involved in MYC-dependent transcription through promotion of transcription elongation rates and these SEC disruptors can be potentially used in vivo.
MYC hyperactivation induces transcriptional amplification and increases messenger RNA synthesis, which leads to an increased burden on the core spliceosome to properly process mRNA, suggesting that RNA splicing is a therapeutic vulnerability in MYC-driven cancer (Hsu et al., 2015; Lee and Abdel-Wahab, 2016). RNA expression profiling analysis shows that KL-1 and KL-2 treatment leads to a significantly decreased output of the MYC transcriptional program, including RNA splicing-related genes including the PRMT5 gene, which is a key regulator among the MYC-upregulated genes (Bezzi et al., 2013; Koh et al., 2015), suggesting that KL-1 and KL-2 could directly target MYC to lead to an impaired downstream MYC-PRMT5 axis.
KL-1 and KL-2 share the same scaffold and have similar activities toward SEC disruption and Pol II processivity, suggesting that this scaffold could function as a lead for future optimization. These leads already exhibit efficacy in impairing SEC function in rapid response models and delaying the progression of a MYC-dependent tumor. Therefore, we anticipate that development of small molecule inhibitors targeting SEC or otherwise slowing RNA Pol II processivity will be useful both for understanding the regulation of transcription elongation in cells and as therapeutic tools for the treatment of human disease including cancer.
STAR*Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Ali Shilatifard (ASH@Northwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293T (female cell line, ATCC CRL-3216), HCT-116 (male cell line, ATCC CCL-247), MDA231-LM2 (female cell line), Flag-AFF1 and Flag-AFF4 HEK293T stable cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, catalog No. F6178, Sigma). NCI-H2171 [H2171] (male cell line, ATCC CRL-5929) and SW1271 (male cell line, ATCC CRL-2177) small cell lung cancer cells were maintained in RPMI-1640 and DMEM/F12 medium supplemented with 20% FBS. The Jurkat J-Lat full-length cells (6.3) (male cell line, NIH-ARP Cat# 9846–446) were provided by the NIH AIDS Reagent Program and cultured in RPMI-1640 medium with 10% FBS. Drosophila melanogaster S2 cells, a male cell line, were maintained in Schneider’s medium. The wild-type Pol II, slow Pol II (R749H) and fast Pol II (E1126G) mutant HEK293T cell lines (Fong et al., 2014) were provided by Dr. David Bentley (University of Colorado School of Medicine) and cultured in DMEM with 10% FBS. After induction with doxycycline (2.0 μg/mL) for 16 hr, speed-mutant cells were treated with α-amanitin (2.5 μg/mL, Santa Cruz) for 42 hr prior to ChIP-seq analysis.
Plasmids, Peptides and Chemicals
pGEX-2TK cyclin T1 (1–300) (Addgene P#432) was purchased from Addgene and used to express recombinant GST-CCNT1(1–300) in Rosetta cells. GST-CCNT1 (AA1–300) recombinant protein was purified with glutathione superflow agarose (Thermo, Cat# 25236). shRNAs for human AFF1 (TRCN0000021975 and TRCN0000330908), and AFF4 (TRCN0000426769 and TRCN0000015825) were obtained from Sigma. ELL2 was also depleted with shRNAs targeting the sequences AAC GCC AGA ATT ATA AGG ATG and AAA TGA TCC CCT CAA TGA AGT.
Biotin labeled AFF4 peptide (AA32–67) and mutant AFF4 peptide abolishing the binding with CCNT1 were synthesized and purified (purity > 96%) by VCPBIO with further Trifluoroacetic acid removal. The sequence for wild-type AFF4 peptide is Biotin-GABA-SPL FAE PYK VTS KED KLS SRI QSM LGN YDE MKD FIG-amide and the mutant AFF4 peptide sequence is Biotin-GABA-SAA AAE PYK VTS KAA KLSS RIQ SAA GNY DEM KDF IG-amide where Biotin indicates N-terminal biotin labeling and GABA indicates a γ-amino-butyric acid spacer. The candidate chemicals from the in silico screening were purchased from the vendors ChemDiv, ChemBridge and Enamine.
MDA231-LM2 Tumor Model
MDA231-LM2 tumor model was established as previously reported (Wang et al., 2017). Briefly, Six-week-old female athymic mice (nu/nu genotype, BALB/c background) were purchased from Envigo (Indianapolis, IN) and housed under aseptic conditions. All protocols, described below, were approved by the Northwestern University Institutional Animal Care and Use Committee. 4×106 MDA231-LM2 cells, in 0.4 ml of cell culture media with matrigel (BD Bioscience) were injected in the right mammary pad of mice under anesthetization by isoflurane. For the in vivo therapy-response study, mice were randomly assigned to vehicle (DMSO,) KL-1, and KL-2 treatment groups when the size of tumor reached 100 mm3. Mice were treated with drug administration by intraperitoneal injection at 50 mg/kg of KL1 and 10 mg/kg of KL2, with once daily administration for 15 days for 3 weeks. The tumor sizes were measured twice a week and the mice were euthanized when the tumor size reached 1000 mm3.
METHOD DETAILS
Chemical Synthesis
All chemical reagents were obtained from commercial suppliers and used without further purification unless otherwise stated. Anhydrous solvents were purchased from Sigma-Aldrich and dried over 3 Å molecular sieves when necessary. Normal phase flash column chromatography was performed using Biotage KP-Sil 50 μm silica gel columns and ACS grade solvents on a Biotage Isolera flash purification system. Analytical thin layer chromatography (TLC) was performed on EM Reagent 0.25 mm silica gel 60 F254 plates and visualized by UV light. Proton (1H), and carbon (13C) NMR spectra were recorded on a 500 MHz Bruker Avance III with direct cryoprobe spectrometer. Chemical shifts were reported in ppm (δ) and were referenced using residual non-deuterated solvent as an internal standard. The chemical shifts for 1H NMR and 13C NMR are reported to the second decimal place. Proton coupling constants are expressed in hertz (Hz). The following abbreviations were used to denote spin multiplicity for proton NMR: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, brs = broad singlet, dd = doublet of doublets, dt = doublet of triplets, quin = quintet, tt = triplet of triplets. Low resolution liquid chromatography/mass spectrometry (LCMS) was performed on a Waters Acquity-H UPLC/MS system with a 2.1 mm × 50 mm, 1.7 μm, reversed phase BEH C18 column and LCMS grade solvents. A gradient elution from 95% water +0.1% formic acid/5% acetonitrile +0.1% formic acid to 95% acetonitrile +0.1% formic acid/5% water +0.1% formic acid over 2 min plus a further minute continuing this mixture at a flow rate of 0.85 mL/min was used as the eluent. Total ion current traces were obtained for electrospray positive and negative ionization (ESI+/ESI-). High-resolution mass spectra were obtained using an Agilent 6210 LC-TOF spectrometer in the positive ion mode using electrospray ionization with an Agilent G1312A HPLC pump and an Agilent G1367B autoinjector at the Integrated Molecular Structure Education and Research Center (IMSERC), Northwestern University.
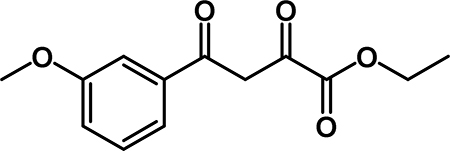
Ethyl 4-(3-methoxyphenyl)-2,4-dioxobutanoate (2a): To a solution of diisopropylamine (1.4 mL, 10 mmol) in THF (33 mL) at −78 °C was added n-BuLi (4.0 mL, 10 mmol). 3’-methoxyacetophenone (0.91 mL, 6.7 mmol) was added slowly, and the reaction was stirred at 78 °C for 15 min. Diethyl oxalate (1.4 mL, 10 mmol) was added slowly, and the reaction stirred at −78 °C for 1.5 hr. The reaction was slowly warmed to room temperature, then was quenched by the addition of 1M HCl (10 mL). The aqueous layer was extracted with EtOAc (3 × 75 mL) and then combined organic layers were washed with 1M HCl (10 mL), saturated aqueous NaHCO3 (10 mL), water (10 mL), and brine (10 mL). The organic phase was dried over Na2SO4, decanted into a round bottom flask and concentrated by rotary evaporation. The crude material was recrystallized from EtOH to obtain 2a (0.764 g, 46% yield) as an off-white solid. 1H NMR (500 MHz, CDCl3) δ 15.47 (s, 1H), 7.78 (dt, J = 7.7, 1.3 Hz, 1H), 7.73 (t, J = 2.1 Hz, 1H), 7.62 (t, J = 7.9 Hz, 1H), 7.47 (s, 1H), 7.36 (dd, J = 8.3, 2.6 Hz, 1H), 4.61 (q, J = 7.2 Hz, 2H), 4.09 (s, 3H), 1.63 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 190.98, 169.47, 162.36, 160.17, 136.53, 130.03, 120.64, 120.38, 112.38, 98.38, 62.79, 55.67, 14.26.
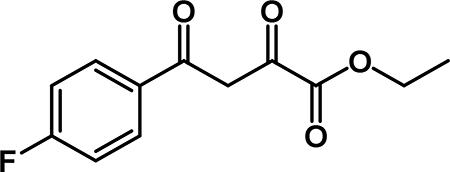
Ethyl 4-(4-fluorophenyl)-2,4-dioxobutanoate (2b): To a solution of diisopropyl amine (6.2 mL, 44 mmol) in THF (44 mL) at 0 °C was added n-BuLi (16.2 mL, 40.5 mmol). The cloudy yellow solution was stirred at 0 °C for 30 min., then cooled to −78 °C. 4’-fluoroacetophenone (3.2 mL, 26 mmol) was added slowly along the sides of the flask and was stirred for 15 min. Diethyl oxalate (7.9 mL, 58 mmol) was added and the reaction stirred at −78 °C for 1 hour. The mixture was warmed to room temperature and stirred for 20 min and the reaction was quenched by the addition of 1M HCl. The organic solvent was removed by rotary evaporation. The aqueous phase was extracted with EtOAc (3 × 75 mL) and the combined organic layers were washed with 1M HCl (25 mL), saturated aqueuos NaHCO3 (25 mL), and brine (25 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash column chromatography and recrystallized from EtOH to obtain 2b (3.38 g, 54 % yield) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 15.83 – 15.03 (m, 1H), 8.42 – 8.09 (m, 2H), 7.50 (s, 1H), 7.42 (t, J = 8.5 Hz, 2H), 4.64 (q, J = 7.1 Hz, 2H), 1.65 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 189.86, 169.33, 166.36 (d, J = 256.5 Hz), 162.27, 131.50 (d, J = 2.5 Hz), 130.72 (d, J = 9.5 Hz)*, 116.3 (d, J = 22.0 Hz)*, 97.96, 62.83, 14.25.
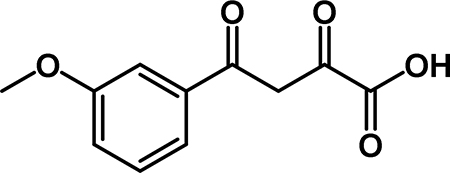
4-(3-methoxyphenyl)-2,4-dioxobutanoic acid (3a): To a solution of 2a (0.764 g, 3.05 mmol) in THF (15 mL) was added a solution of NaOH (1.22 g, 30.5 mmol) in 15 mL of water. The reaction stirred at room temperature for 15 min. The organic solvent was removed by rotary evaporation. The aqueous phase was extracted with Et2O (3 × 30 mL), then acidified with 1M HCl. The aqueous layer was extracted wtih EtOAc (3 × 50 mL), and the combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated by rotary evaporation to obtain 3a (0.450 g, 66% yield) as an off-white solid. 1H NMR (500 MHz, CDCl3) δ 15.32 (s, 1H), 7.74 – 7.66 (m, 1H), 7.66 – 7.59 (m, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.37 (s, 1H), 7.29 (dd, J = 8.7, 3.0 Hz, 2H), 4.00 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 186.77, 174.75, 161.49, 160.07, 134.35, 130.09, 120.69, 120.49, 112.27, 95.27, 55.56.
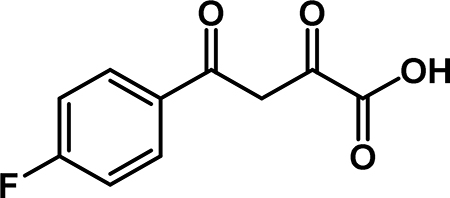
4-(4-fluorophenyl)-2,4-dioxobutanoic acid (3b): To a solution of 2b (3.38 g, 14.2 mmol) in THF (47 mL) was added a solution of NaOH (5.68 g, 142 mmol) in 45 mL of water. The reaction stirred at room temperature for 15 min., then the organic solvent was removed by rotary evaporation. The aqueous phase was extracted with Et2O (3 × 50 mL), then acidified with conc. HCl. The aqueous layer was extracted with EtOAc (3 × 100 mL), and the combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated to obtain 3b (1.92 g, 64% yield) as an off-white solid. 1H NMR (500 MHz, CDCl3) δ 15.21 (s, 1H), 8.25 – 8.01 (m, 2H), 7.27 (s, 1H), 7.22 (t, J = 8.6 Hz, 2H), 7.14 (s, 1H). 1H NMR (500 MHz, DMSO-D6) δ 8.30 – 8.02 (m, 2H), 7.41 (t, J = 8.8 Hz, 2H), 7.10 (s, 1H), (Carboxylic acid -OH and enol-OH not observed). 13C NMR (125 MHz, CDCl3) δ 185.90, 174.19, 166.49 (d, J = 257.7 Hz), 161.63, 130.62 (d, J = 9.5 Hz)*, 129.42 (d, J = 3.1 Hz), 116.44 (d, J = 22.1 Hz)*, 95.08. * Indicates two equivalent carbons with the same chemical shift that couple with 19F.
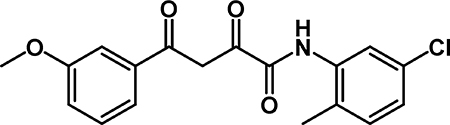
N-(5-chloro-2-methylphenyl)-4-(3-methoxyphenyl)-2,4-dioxobutanamide (4a): Acid 3a (0.400 g, 1.80 mmol) was dissolved in THF (9.00 mL) and 5-chloro-2-methylaniline (0.33 mL, 2.7 mmol) was added, followed by EEDQ (0.467 g, 1.89 mmol). The reaction stirred at room temperature for 18 hr then was diluted with EtOAc. The organic phase was washed with 1M HCl (2 × 20 mL), saturated aqueous NaHCO3 (2 × 20 mL), water (20 mL) and brine (20 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The crude material was recrystallized from MeOH to obtain 4a (0.412 g, 66% yield) as a yellow powder. 1H NMR (500 MHz, CDCl3) δ 15.65 (s, 1H), 9.02 (s, 1H), 8.28 (d, J = 2.2 Hz, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.59 – 7.49 (m, 1H), 7.43 (t, J = 8.0 Hz, 1H), 7.31 (s, 1H), 7.25 – 7.15 (m, 2H), 7.10 (dd, J = 8.1, 2.2 Hz, 1H), 3.90 (s, 3H), 2.35 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 185.77, 179.73, 160.15, 159.08, 135.81, 134.96, 132.59, 131.55, 130.12, 126.23, 125.43, 121.28, 120.48, 120.38, 112.19, 94.18, 55.68, 17.20.
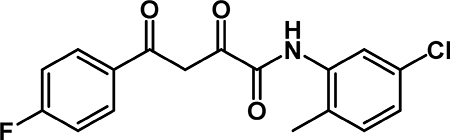
N-(5-chloro-2-methylphenyl)-4-(4-fluorophenyl)-2,4-dioxobutanamide (4b): To a solution of 3b (0.208 g, 0.990 mmol) in THF (5 mL) was added 5-chloro-2-methylaniline (0.18 mL, 1.5 mmol), followed by EEDQ (0.257 g, 1.04 mmol). The reaction stirred at room temperature for 18 hr, then was diluted with EtOAc. The organic phase was washed wtih 1M HCl (3 × 10 mL), saturated aqueous NaHCO3 (3 × 10 mL), water (10 mL), and brine (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated by rotary evaporation. The crude material was recrystallized from MeOH to obtain 4b (0.061 g, 59% yield) as a yellow powder. 1H NMR (500 MHz, CDCl3): δ 15.65 (s, 1H), 9.00 (s, 1H), 8.27 (d, J = 2.3 Hz, 1H), 8.07 (dd, J = 8.5, 5.3 Hz, 2H), 7.21 (t, J = 8.4 Hz, 2H), 7.16 (d, J = 8.1 Hz, 1H), 7.10 (dd, J = 8.3, 2.2 Hz, 1H), 2.35 (s, 3H). 1H NMR (500 MHz, DMSO-D6) δ 10.24 (s, 1H), 8.17 (dd, J = 8.5, 5.3 Hz, 2H), 7.58 (d, J = 2.5 Hz, 1H), 7.46 – 7.35 (m, 3H), 7.31 (d, J = 8.3 Hz, 1H), 7.24 (dd, J = 8.3, 2.3 Hz, 1H), 7.17 (s, 1H), 2.22 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 184.81, 179.44, 166.34 (d, J = 256.6 Hz), 158.98, 135.76, 132.60, 131.57, 130.50 (d, J = 9.4 Hz)*, 129.91 (d, J = 2.85 Hz), 126.26, 125.48, 121.30, 116.42 (d, J = 22.2 Hz)*, 93.80, 17.19. * Indicates two equivalent carbons with the same chemical shift that couple with 19F.
In Silico High-throughput Screening
In Silico Filtering of The Small Molecule Database for Ligand Preparation
The ZINC database (Irwin et al., 2012), which contains approximately 41 million commercially available compounds, was used for virtual high-throughput screening (vHTS). All compounds in the ZINC library were subjected to a panel of PAINS substructures filters with Smiles ARbitrary Target Specifications (SMARTS) strings (Baell and Holloway, 2010) to eliminate promiscuous and non-drug-like molecules that interfere with functionality of the target proteins. Filtering generated a list of approximately 10 million commercially available compounds for further screening. The 10 million compound dataset was then subjected to the LigPrep module of Schrodinger (Small-Molecule Drug Discovery Suite 2017–2, Schrödinger, LLC, New York) in OPLS2005 force field at pH 7.4 ± 1 (physiological pH) retaining the specific chirality. A low energetic 3D structure for each molecule was generated in this ligand preparation panel.
Protein Preparation for Small Molecule Screening and Grid Generation
The protein preparation (prot-prep) engine implemented in the Schrödinger software suite was utilized to prepare the protein for small molecule docking simulations. Analysis of the tripartite complex crystal structure (4IMY.pdb) having a resolution 2.94 Å reveals the binding of the AFF4 protein to CCNT1, a subunit of P-TEFb. We observed that five terminal residues of AFF4 (L34, F35, A36, E37 and P38) are having good interactions with the binding groove of CCNT1containing the residues W221, Y224, L163, V164, R165, Y175, F176, D169, W207, W210 and E211. Furthermore, the mutation data of Y175, E211, D169, F176, R165, W210 and W207 of CCNT1 reported in the literature (Schulze-Gahmen et al., 2013) guided us to select the small molecule ligand-binding site. A 12 Å3 grid was generated considering the centroid of the above mention critical residues in the CCNT1 groove.
Virtual Screening Workflow
For vHTS, we began with the curated library of approximately 10 million drug-like compounds described above and the OPLS 2005 force field was set. The ligand van der Waals radii was scaled to 0.80 Å with partial atomic charges <0.15 esu. A three-tier Glide docking algorithm (Small-Molecule Drug Discovery Suite 2017–2, Schrödinger, LLC) was employed that incorporates vHTS followed by Standard Precision (SP) and Extra Precision (XP) docking protocols. The output of this three-tier docking engine was analyzed using the XP-visualization tools by considering the interactions of the compounds with the critical residues reported above. Based on the docking scores, a list of 122 compounds was selected for cross validation using a 5point structure focus pharmacophore generated by the pharmacophore module implemented in BIOVIA software considering the interactions of residues of AFF4 and Cyclin T1. Using the 5point pharmacophore as the query, the glide hits were scored. Based on fitting scores and low energetics conformers, 67 hits were selected. We selected 40 available compounds having a Glide docking score < −6.0. The Glide score is a function of the binding energy (Small-Molecule Drug Discovery Suite 2017–2, Schrödinger, LLC)
AlphaLISA Assay
The interaction of CCNT1-AFF4 was measured by Perkin Elmer’s bead-based AlphaLISA assays. Recombinant GST-CCNT1 (1–300), which was purified by Glutathione Superflow Agarose from Rosetta cells, and AFF4 peptides were diluted in incubation buffer (25 mM HEPES, PH 7.4, 100 mM NaCl, 0.1% NP-40). GST-CCNT1 (AA1–300) and AFF4 peptides at indicated concentrations were mixed together with 0.5 μg of AlphaScreen Streptavidin Donor beads, and 0.5 μg of Glutathione AlphaLISA Acceptor Beads. For inhibition assays, inhibitors were added right after the mixture of CCNT1 and AFF4 peptide. Reactions were subsequently incubated for 2 hr with agitation in the dark. Plates were read with a Tecan INFINITE M1000 PRO. The dissociation constant Kd of CCNT1-AFF4 interaction was calculated based on the hyperbolic binding equation in Prism 7 (Graphpad). The IC50 values of the KL-1 and KL-2 were calculated with a four-parameter sigmoid fitting equation in Prism 7 and converted to the inhibitory Constants (Ki) with the Cheng and Prusoff equation.
Heat Shock Induction
Heat shock of mammalian cells was performed using ~70–80% confluent HCT-116 cells by adding pre-heated (42 °C) conditioned media collected from identically growing cells (Mahat et al., 2016b). The heat shock cells were incubated at 42 °C for 1 hour. After washing with PBS, the heat shock and non-heat shock HCT-116 cells were fixed with 1% formaldehyde in PBS for downstream ChIP-seq analysis. For heat shock induction of Drosophila S2 cells, the S2 cells were mixed with pre-heated medium to instantly increase the medium temperature from 24 °C to 37 °C and maintained in a water bath at 37 °C for 10 min before fixation for ChIP-seq.
Induction of J-Lat 6.3 Cells
The J-Lat 6.3 cell line was derived from human Jurkat cells with the integration of a full-length green fluorescent protein (GFP)-encoding HIV-1 vector (HIV-R7/E−/GFP) under the control of the viral 5′-LTR (Jordan et al., 2003). To measure the effects on Tat-mediated HIV inducibility with flow cytometry, J-Lat 6.3 cells were incubated with 10 nM PMA (Phorbol 12-myristate 13acetate) in the presence of vehicle or SEC inhibitors at the indicated concentrations for 17 hr. Cells were washed in PBS and GFP fluorescence was measured with a FACSVantage instrument (Becton Dickinson, San Jose, CA). Analysis was gated on live cells according to forward and side scatter. A two-parameter analysis to distinguish GFP-derived fluorescence from background fluorescence was used: GFP was measured in FL1 and cellular autofluorescence was monitored in FL2. The percentage of GFP-positive cells was calculated based on live cells (Jordan et al., 2003). The J-Lat 6.3 cells were also induced with 10 nM PMA for 11 hr and then treated with Vehicle or SEC inhibitors for 6 hr prior to ChIP-seq analysis.
Chromatin Immunoprecipitation Sequencing
Chromatin Immunoprecipitation Sequencing (ChIP-seq) was performed according to a previously published protocol (Liang et al., 2015). Briefly, cells were crosslinked with 1% paraformaldehyde for 10 min and were quenched with glycine for 5 min at room temperature. Fixed chromatin was sonicated with a Covaris Focused-ultrasonicator for 6 min and immunoprecipitated with the indicated antibody and Dynabeads Protein G. Libraries were prepared with the HTP Library Preparation Kit for Illumina (KAPA Biosystems) and sequenced on a NextSeq 500. ChIP-seq reads were aligned to the Drosophila genome (UCSC dm3) or human genome (UCSC hg19). Alignments were processed with Bowtie version 1.1.2, allowing only uniquely mapping reads with up to two mismatches within the 50 bp read. The resulting reads were extended to 150 bp toward the interior of the sequenced fragment and normalized to total reads aligned (reads per million, r.p.m.). Peaks were called using MACS (model based analysis of ChIP-Seq) (Zhang et al., 2008) version 1.4.2 using default parameters. Ensembl version 75 transcripts were chosen with the highest total coverage from the annotated TSS to 200 nt downstream for protein coding genes that also have a RefSeq identifier, were at least 2 kb long, and 2 kb away from the nearest gene. The genes with SEC and Pol II occupancy were defined by the overlapping of peaks with Pol II and SEC peaks by MACS 1.4.2 using default parameters. For pausing indexes, the promoter region was defined as −200 bp upstream to 400 bp downstream, and the body region was the remainder of the entire gene body. The ratio of the average coverage (r.p.m.) of the promoter over the average coverage of the gene body was then taken to be the pausing index. ECDF plots were made in R version 3.3.3 using the ecdf function. P-values were calculated with a two-sided Kolmogorov-Smirnov test. Heatmap tables were made for the indicated windows around the TSS or TES using the average coverage (r.p.m.) in 25 bp & 50 bp bins (50 bp bins for 50 kb downstream of the TSS). Metagene tables were made by approximating the coverage across all genes to the same length. All ChIP-seq heatmaps were sorted by the decreasing coverage in indicated windows by the control samples and visualized using JavaTreeView version 1.6.4 (Saldanha, 2004). Average profile plots were made by averaging the coverage for all genes using colMeans in R.
Precision Nuclear Run-on and Sequencing
Precision Nuclear Run-on and Sequencing (PRO-seq) was performed according to the previously published protocol (Mahat et al., 2016a) with minor modifications. All 4 biotinylated nucleotides were used at 25 μM each final concentration for the run-on reaction. RPPH (NEB) was used to remove the 5’ RNA cap. Libraries were size selected using a 2% agarose gel on a Pippin HT programmed to elute 140–350 bp. After sequencing, adaptors were removed with cutadapt version 1.14 (Martin, 2011). Reads were trimmed from the 3’-end to 36 bp with removing low quality bases using Trimmomatic version 0.33 (Bolger et al., 2014) requiring a minimal read length of 16nt. Reads were then mapped to the human genome (UCSC hg19) using Bowtie version 1.1.2 (Langmead et al., 2009). Only uniquely mapped reads with up to 2 mismatches in the entire read were used for further analysis. Read where then converted to single nucleotide 3’ BigWig strand specific tracks by taking 5’ positions of the read (using bedtools genomecov version 2.17 (Quinlan and Hall, 2010) with options -strand -bg −5. Strands were then swapped to give the correct orientation with the 5’-end now becoming the 3’-end of the read (Mahat et al., 2016a). PRO-seq genome browser track examples show coverage of the entire length of the read for easier visualization. The single nucleotide 3’ BigWig strand specific tracks were used to generate all other figures. For the Pol II-selected genes described above, we found the site of maximum coverage, in the region from the annotated TSS to 500bp downstream, to which we assign the pausing site. Heatmap tables were made as described above but instead centering at this calculated pausing site.
4sU-FP-seq
Cell Labeling with 4sU, RNA Extraction and Fragmentation
20–50 million cells were treated with flavopiridol for 1–2 hr to pause the Pol II at the TSS sites. For the release of Pol II and measurement of elongation rates, the cells were labeled with 4thiouridine (4sU, Sigma-Aldrich, St. Louis, MO, USA) either in water bath or in plates. For water bath labeling, the cells were harvested through centrifugation for 3 min at 350 g and washed with PBS twice. Then, the cells were released with prewarmed medium containing 500 μM 4-thiouridine for 15 min, and harvested by centrifuge at 1800 g for 3 min. For labeling in plates, the cells were washed with PBS twice after flavopiridol treatment and released with prewarmed medium containing 500 μM 4-thiouridine in CO2 incubator at 37 °C for 15 min. RNA was extracted with 4 mL Trizol (Invitrogen) and 5 μL 20 mg/mL glycogen. The extracted RNA was further fragmented by base hydrolysis in 0.2 M NaOH on ice for 18 min, neutralized by adding 1x volume of 1 M Tris-HCl pH 6.8 and precipitated with isopropanol.
Biotinylation of RNA
Biotinylation of 4sU-labeled RNA was performed using EZ-Link Biotin-HPDP (Pierce) dissolved in dimethylformamide (DMF, Sigma) at a concentration of 1 mg/mL and stored at 4 °C. Biotinylation was carried out in 10 mM Tris (pH 7.4), 1 mM EDTA, and 0.2 mg/mL BiotinHPDP at a final RNA concentration of 200 ng/μL for 1.5 hrs. at room temperature. After biotinylation, unbound Biotin-HPDP was removed by extracting twice with chloroform and phase lock gel. Afterward, a 1/10 volume of 5 M NaCl and an equal volume of isopropanol was added to precipitate RNA. RNA was collected by centrifugation at 20,000g for 20 min and the pellet was washed with an equal volume of 80% ethanol. The pellet was resuspended in 200 μL RNAse-free water.
Purification of 4sU-Labeled RNA, Library Preparation and Alignment
After denaturation of RNA samples at 65 °C for 5 min followed by rapid cooling on ice for 5 min, biotinylated RNA was captured using streptavidin beads. Up to 200 μg of biotinylated RNA were incubated with 50 μL of Dynabeads® MyOne™ Streptavidin C1 with rotation for 15 min at room temperature. Beads were washed two times with 65 °C wash buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 M NaCl, 0.1% Tween20) followed by four washes with room temperature wash buffer. Labeled RNA was eluted twice for 5 min with 100 μL of freshly prepared 100 mM dithiothreitol (DTT). RNA was recovered from the elute fractions and purified using the RNeasy MinElute Spin columns (Qiagen). Libraries were made with the TruSeq RNA Sample Prep kit (Illumina) and subjected to Illumina sequencing. 4sU-FP-seq reads were aligned to the human genome (UCSC hg19). Alignments were processed with Bowtie version 1.1.2, allowing only uniquely mapping reads with up to two mismatches within the 50 bp read. The resulting reads were extended to 150 bp toward the interior of the sequenced fragment and normalized to total reads aligned (reads per million, rpm) for each strand.
Elongation Rate Analysis
Genes used for calculating elongation rates were required to have an observed transcription start site with 4sU RNA signals, a minimum gene length of 50 kb and must be at least 2 kb away from the transcription start site of another gene. Genes were further filtered for activity/coverage by filtering on the reads-per-million count within each gene body (+400 to TES) in our untreated, wild-type data. Thus, the read coverage must be present at levels above background (r.p.m. > 1). We first used the Hidden Markov Model (HMM) to calculate elongation rates. Advancing waves were identified using a three state Hidden Markov Model (HMM) that was previously developed and implemented on GRO-seq data from a human cell line (Danko et al., 2013). We also used SICER peak calling to determine elongation rates. Peaks were called with SICER version 1.1 (Zang et al., 2009) with the following options --windowSize 150 --fragSize 150 --gapSize 3 with strand separated reads over input. These strand-specific peaks were filtered for an FDR < 0.01. Peaks were merged if there was a gap less than 2 kb. Peaks were then overlapped with TSS’s of genes on the same strand that were greater than 50 kb. The distance traveled was calculated from the TSS to the 3’-end of the merged peak and the elongation rate in kb/min was calculated using the time after release.
RNA-seq Analysis
Total RNA-seq reads were trimmed from the 3′ end until the final base had a quality score > 30, using Trimmomatic version 0.33 (Bolger et al., 2014) and then aligned to the human genome (UCSC hg19, using Tophat version 2.1.0 (Kim et al., 2013)) with the following options –no-novel-juncs --read-mismatches 2 --read-edit-dist 2 --num-threads 10 --max-multihits 20 then post filtering for uniquely mapped reads using the NH flag. Protein coding genes from Ensembl version 75 that also had a RefSeq identifier were only considered for analysis. Raw read counts were normalized to r.p.m. per sample and then displayed in the UCSC genome browser as bigWig-formatted coverage tracks. Exonic reads were assigned to specific genes from Ensembl release 75 using Bioconductor package GenomicRanges countOverlaps. The R Bioconductor package edgeR (Robinson et al., 2010), version 3.12.0 was used to fit the data to a negative binomial generalized log–linear model and estimate a dispersion parameter. To filter out lowly expressed genes, genes had to have at least 1 count per million (c.p.m) in at least 2 samples in each comparison. The total number of uniquely mapped reads was provided to edgeR for the calcNormFactors normalization rather than the default column sums. An adjusted-p value threshold of 0.01 and a log2 r.p.m cut off of 3 was used to identify genes significantly differentially expressed in one experimental condition relative to another. GO term analysis was done using Metascape (Tripathi et al., 2015).
QUANTIFICATION AND STATISTICAL ANALYSES
Data are presented as Mean ± SD. The sample sizes (n) in the figure legends indicate the number of replicates in each experiment and is provided in the corresponding figure legends. The peak or gene size (N) in the heatmaps indicates the number of peaks or genes included. For Figures S1I and S5E, One-Way ANOVA tests were performed with Prism 7 (GraphPad Software, La Jolla, CA) to determine the statistical significance. P value < 0.005 (∗∗) was considered as highly significantly different, p value < 0.05 was considered as significantly different, n.s, not significantly different, p ≥ 0.05. For Figures 2F, 2H, S2G and S2H, the two-sided Kolmogorov-Smirnov test was performed for the ECDF curves and the p values were provided in each figure. For Figures S2E, S2F, 4G, S4E, 5E and 5G, the statistical significance was determined by a two-sided Wilcoxon signed-rank test using R 3.3.3 package with the p values provided in each figure. For Figures 7D, a 2-tailed unpaired t-test was used for comparison the tumor size between each treatment group. For Figure 7E, the Kaplan-Meier survival curves were plotted with GraphPad Prism 7 and the p values were calculated using the log-rank test.
DATA AND SOFTWARE AVAILABILITY
The accession number for the raw and processed ChIP-seq, RNA-seq, PRO-seq and 4sU-FP-seq data reported in this paper is GEO: GSE112608.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-AFF1 Antibody, Affinity Purified | Bethyl Laboratories | Bethyl Cat# A302-344A, RRID:AB_1850255 |
| Rabbit anti-ELL2 Antibody, Affinity Purified | Bethyl Laboratories | Bethyl Cat# A302-505A, RRID:AB_1966087 |
| Pol II Rpb1 NTD (D8L4Y) Rabbit mAb | Cell Signaling Technology | Cell Signaling Technology Cat# 14958, RRID:AB_2687876 |
| BRD2 (D89B4) Rabbit mAb | Cell Signaling Technology | Cell Signaling Technology Cat# 5848, RRID: AB_10835146 |
| BRD4 (E2A7X) Rabbit mAb | Cell Signaling Technology | Cell Signaling Technology Cat# 13440, RRID: AB_2687578 |
| MED26 (D4B1X) Rabbit mAb | Cell Cell Signaling Technology | Cell Signaling Technology Cat# 14950 |
| c-Myc (D3N8F) Rabbit mAb | Cell Signaling Technology | Cell Signaling Technology Cat# 13987, RRID:AB_2631168 |
| Tubulin beta antibody | DSHB | DSHB Cat# E7, RRID:AB_528499 |
| AFF4 Antibody | Proteintech | Proteintech Group Cat# 14662–1-AP, RRID:AB_2242609 |
| Cyclin T1 Antibody (H-245) | Santa Cruz | Santa Cruz Biotechnology Cat# sc-10750, RRID:AB_2073888 |
| HSP 90α/β Antibody | Santa Cruz | Santa Cruz Biotechnology Cat# sc-7947, RRID:AB_2121235 |
| CDK9 Antibody (C-20) | Santa Cruz | Santa Cruz Biotechnology Cat# sc-484, RRID:AB_2275986 |
| Anti-RNA polymerase II subunit B1 (phospho CTD Ser2) Antibody, clone 3E10 | Active Motif | Active Motif Cat# 61083, RRID:AB_2687450 |
| FLAG-synthetic antibody | Sigma-Aldrich | Sigma-Aldrich Cat# F3165, RRID:AB_259529 |
| ANTI-FLAG M2 Affinity Gel | Sigma-Aldrich | Sigma-Aldrich Cat# A2220, RRID:AB_10063035 |
| Anti-SPT5 Antibody, clone 6F1 | Millipore | Millipore Cat# MABE1803 |
| Rabbit anti-AFF1 serum | (Lin et al., 2010) | N/A |
| Rabbit anti-ELL2 serum | (Lin et al., 2010) | N/A |
| Rabbit anti-AFF4 serum | (Lin et al., 2010) | N/A |
| Rabbit anti-Drosophila Rpb1 antibody | (Lin et al., 2010) | N/A |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Flavopiridol | Cayman | Cat# 10009197 |
| 4-Thiouridine | Sigma-Aldrich | Cat# T4509 |
| Screening compounds | ChemDiv, ChemBridge and Enamine | N/A |
| EZ-Link™ HPDP-Biotin | Thermo | Cat# 21341 |
| KL-1 | This study | N/A |
| KL-2 | This study | N/A |
| Biotin-AFF4 (32–67) peptides | VCPBIO | N/A |
| Biotin-AFF4 mutant peptides | VCPBIO | N/A |
| Glycogen | Sigma-Aldrich | Cat# 10901393001 |
| GST-CCNT1 (1–300) | This study | N/A |
| α-Amanitin | Santa Cruz | CAS 23109–05-9 |
| Biotin-11-ATP | Perkin Elmer | Cat# NEL544001EA |
| Biotin-11-CTP | Perkin Elmer | Cat# NEL542001EA |
| Biotin-11-GTP | Perkin Elmer | Cat# NEL545001EA |
| Biotin-11-UTP | Perkin Elmer | Cat# NEL543001EA |
| RNA 5’ Pyrophosphohydrolase (RppH) | NEB | Cat# M0356S |
| 2% Agarose, PippinHT, 100–600 bp.10/pkg. | Sage science | Cat# HTC2010 |
| BD Matrigel Matrix | BD Biosciences | Cat# 354234 |
| Critical Commercial Assays | ||
| TruSeq® Stranded Total RNA LT - (with Ribo-ZeroTM Human/Mouse/ Rat) - Set A | Illumina | RS-123–2201 |
| TruSeq® Stranded Total RNA LT - (with Ribo-ZeroTM TM Human/ Mouse/Rat) - Set B | Illumina | RS-123–2202 |
| High-Throughput Library Preparation Kit Standard PCR Amp Module - 96 rxn | KAPA Biosystems | KK8234 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Vi-CELL Reagent Quad Pak | Beckman Coulter | Cat# 383198 |
| AlphaScreen GST Detection Kit, 500 assay points | Perkin Elmer | Cat# 6760603C |
| AlphaScreen Streptavidin Donor beads | Perkin Elmer | Cat# 6760002S |
| RNeasy MinElute Cleanup Kit | Qiagen | Cat# 74204 |
| Glutathione Superflow Agarose | Thermo | Cat# 25236 |
| Dynabeads® MyOne™ Streptavidin C1 | Thermo | Cat# 65001 |
| Streptavidin M280 beads | Invitrogen | Cat# 11206D |
| Dynabeads Protein G | Invitrogen | Cat# 10003D |
| Phase Lock Gel Heavy | VWR | Cat# 10847–802 |
| Trizol Reagent | Invitrogen | Cat# 15596–018 |
| Crystal violet solution | Sigma-Aldrich | Cat# HT90132–1L |
| Dead Cell Apoptosis Kit with Annexin V Alexa Fluor® 488 & Propidium Iodide (PI) | Thermo | Cat# V13245 |
| CellTiter-Glo® 2.0 Assay | Promega | Cat# G9242 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE112608 |
| Human reference genome GRCh37/hg19 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/ |
| Drosophila reference genome BDGP Release 5/dm3 | Genome Reference Consortium | http://www.fruitfly.org/sequence/release3genomic.shtml |
| Experimental Models: Cell Lines | ||
| HCT-116, human, male | ATCC | ATCC Cat# CCL-247, RRID:CVCL_0291 |
| J-Lat 6.3 clone (Jurkat cells), human, male | NIH AIDS Program | NIH-ARP Cat# 9846-446, RRID:CVCL_8280 |
| HEK-293T, human, male | ATCC | ATCC Cat# CRL-3216, RRID:CVCL_0063 |
| HEK293T Flag-AFF1 | (Lin et al., 2010) | N/A |
| HEK293T Flag-AFF4 | (Lin et al., 2010) | N/A |
| NCI-H2171 [H2171] (ATCC® CRL-5929™), human, male | ATCC | ATCC Cat# CRL-5929, RRID:CVCL_1536 |
| SW 1271 [SW1271] (ATCC® CRL-2177™), human, male | ATCC | ATCC Cat# CRL-2177, RRID:CVCL_1716 |
| FAST Pol II Mutant E1126G HEK293 cells, human, female | (Fong et al., 2014) | N/A |
| WT Pol II Mutant HEK293 cells, human, female | (Fong et al., 2014) | N/A |
| Slow Pol II Mutant R749H HEK293 cells, human, female | (Fong et al., 2014) | N/A |
| MDA231-LM2, human, female | (Wang et al., 2017) | N/A |
| S2 cells, Drosophila, male | Invitrogen | Cat# R690–07 |
| Experimental Models: Organisms/Strains | ||
| athymic nude mice, nu/nu | Envigo | Hsd:Athymic NudeFoxn1nu |
| Oligonucleotides | ||
| Recombinant DNA | ||
| shRNA targeting sequence: AFF1 #1 GCCTCAAGTGAAGTTTGACAA | Sigma-Aldrich | Clone ID: TRCN0000021975 |
| shRNA targeting sequence: AFF1 #2 TAGGTTGGGAAAGCCGAAATA | Sigma-Aldrich | Clone ID: TRCN0000330908 |
| shRNA targeting sequence: AFF4 #1 GCACGACCGTGAGTCATATAA | Sigma-Aldrich | Clone ID: TRCN0000426769 |
| shRNA targeting sequence: AFF4 #2 GCACCAGTCTAAATCTATGTT | Sigma-Aldrich | Clone ID: TRCN0000015825 |
| shRNA targeting sequence: ELL2 #1 AACGCCAGAATTATAAGGATG | This paper | N/A |
| shRNA targeting sequence: ELL2 #2 AAATGATCCCCTCAATGAAGT | This paper | N/A |
| Lenti-sh1368 knockdown c-myc shRNA targeting sequence: GACGAGAACAGTTGAAACA | Addgene | Addgene plasmid # 29435 |
| pGEX-2TK cyclin T1 (1–300) | Addgene | Addgene plasmid # P432 |
| Software and Algorithms | ||
| TopHat 2.1.0 | (Kim et al., 2013) | https://ccb.jhu.edu/software/tophat/index.shtml |
| MACS 1.4.2 | (Zhang et al., 2008) | http://liulab.dfci.harvard.edu/MACS/ |
| EdgeR 3.12.0 | (Robinson et al., 2010) | http://bioconductor.statistik.tu-dortmund.de/packages/2.11/bioc/html/edgeR.html |
| Bowtie version 1.1.2 | (Langmead et al., 2009) | http://bowtiebio.sourceforge.net/index.shtml |
| Trimmomatic 0.33 | (Bolger et al., 2014) | http://www.usadellab.org/cms/index.php?page=trimmomatic |
| Cutadapt 1.14 | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/guide.html |
| Bedtools 2.17 | (Quinlan and Hall, 2010) | https://launchpad.net/ubuntu/+source/bedtools/2.17.0-1 |
| Metascape | (Tripathi et al., 2015) | http://metascape.org/gp/index.html |
| R 3.2.1 | https://www.rproject.org/ | |
| ZINC database | (Irwin et al., 2012) | http://zinc.docking.org |
| Small-Molecule Drug Discovery Suite 2017–2 | Schrödinger | https://www.schrodinger.com/suites/small-molecule-drugdiscovery-suite |
| Prism 7 | GraphPad Software | https://www.graphpad.com |
| FlowJo | FlowJo, LLC | https://www.flowjo.com |
| Dassault Systèmes BIOVIA | Dassault Systèmes | https://www.3ds.com/productsservices/biovia/ |
| Other | ||
Supplementary Material
(A) Schematic of AlphaLISA assay of the AFF4-CCNT1 interaction. Anti-GST AlphaLISA acceptor beads and AlphaScreen streptavidin donor beads (Perkin Elmer) were used to detect the interaction of GST-CCNT1 (AA1–300) and biotin-AFF4 (AA32–67).
(B-C) Optimization of the AlphaLISA assay with various biotin-AFF4 peptide and GST-CCNT1 concentrations. No CCNT1 (B) and mutant AFF4 (C) were used as negative controls.
(D) Scheme for chemical synthesis of SEC inhibitors KL-1 and KL-2. Reagents and conditions: a. LDA, THF, −78 °C, 15 min., then diethyl oxalate, −78 °C to room temperature. b. NaOH, THF/H2O, room temperature, 15 min. c. EEDQ, 5-chloro-2-methylaniline, THF, room temperature, 24 hr.
(E) 6 hr of treatment with KL-1 or KL-2 does not significantly reduce the protein levels of BRD2, BRD4, Pol II (RPB1) and MYC in 293T cells. 293T cells were treated with 20 μM of KL-1 or KL-2 for 6 hr, and the protein levels of BRD2, BRD4, Pol II (RPB1), SPT5, MED26, MYC, HSP90 (load control) and Tubulin (load control) were measured by western blotting.
(F) Treatment of HCT-116 cells with KL-1 and KL-2 reduced protein levels of SEC components AFF1 and AFF4 but not CDK9 or CCNT1. HCT-116 cells were treated with 20 μM of KL-1 or KL-2 for 6 hr, and the protein levels of AFF1, AFF4, CCNT1, CDK9 and Tubulin (load control) were determined by western blotting.
(G-H) Treating cells with KL-1 and KL-2 results in reduced levels of AFF1 and AFF4. FLAGAFF1 (G) and FLAG-AFF4 (H) were expressed under the Tet-inducible promoter in HEK293T cells. Cells were treated with 20 μM of the indicated inhibitors for 6 hr for western blotting.
(I) KL1 and KL-2 do not lead to reduced mRNA expression of SEC subunits AFF1, AFF4, ELL2, CCNT1 and CDK9 in HEK293T cells. RNA-seq was performed in HEK293T cells treated for 12 hr with 20 μM of the indicated inhibitors. The log2 counts per million (CPM) SEC genes were calculated with HTseq. KL-1 and KL-2 had no significant effects on CCNT1, CDK9 and AFF1 expression, but modest increases in the mRNA levels of AFF4 and ELL2 were observed (n=3). Unpaired two-way ANOVA was used for the statistical testes. **, p < 0.05, ***, p < 0.001.
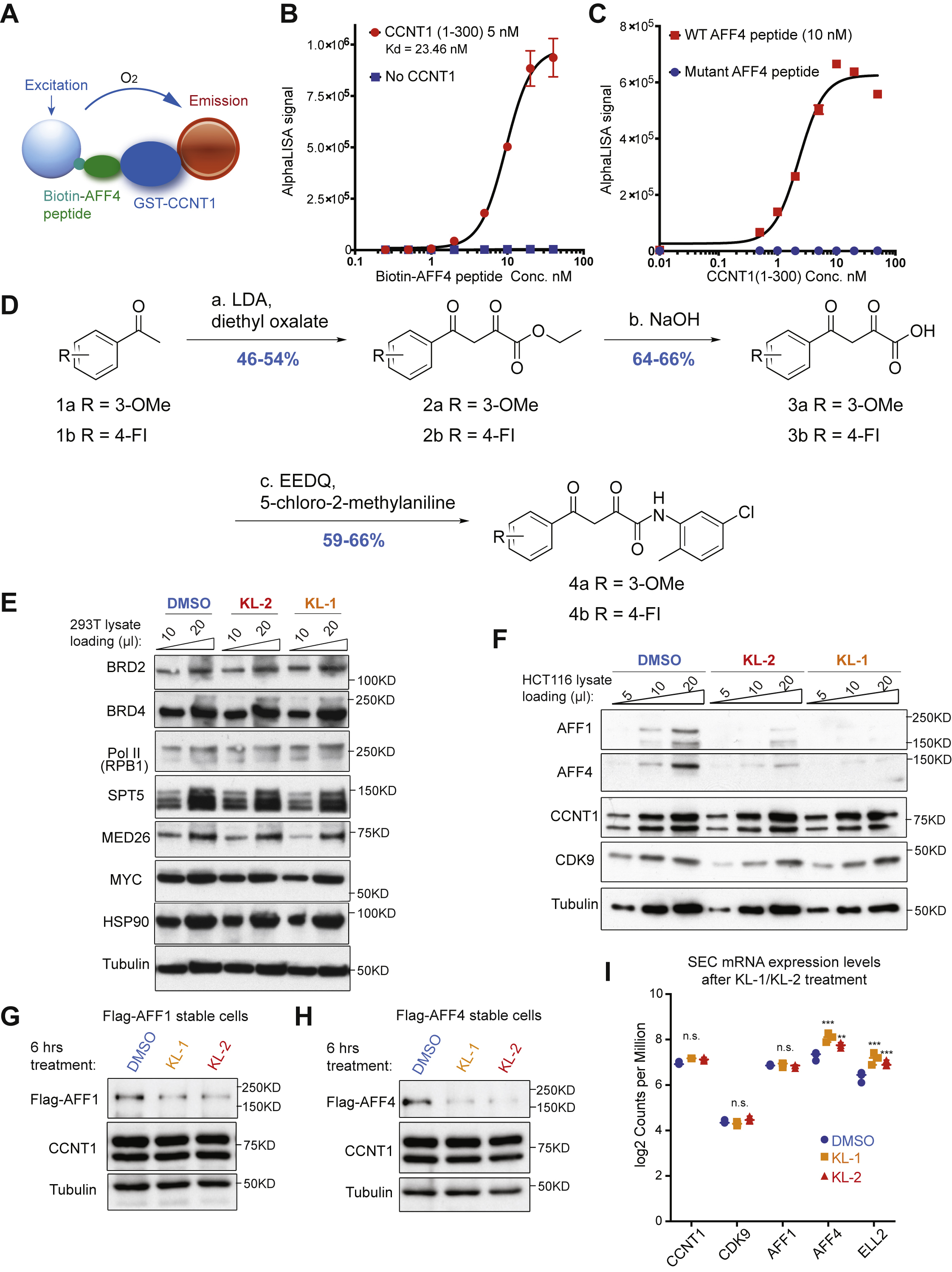{kind=link}
(A) Genome browser tracks of Pol II occupancy at the SRSF4 gene in vehicle or SEC inhibitortreated cells gene in HCT-116 (top panel) and Jurkat cells (bottom panel).
(B-C) Metaplots of Pol II occupancy for all expressing genes with Pol II occupancy in HCT-116 (B) and Jurkat cells (C). SEC inhibitor treatments result in increased Pol II occupancy around the TSS. Pol II density is plotted in a −2 kb and +2 kb window around the TSS.
(D) K-means 3 clustered heatmap of Pol II fold change of the 6,840 genes after SEC inhibitor treatments. The region around the TSS is shown. Group I genes are preferentially affected by the SEC inhibitors.
(E-F) Boxplot analysis of AFF1 (E) and AFF4 (F) occupancy at the three clusters from (D).
(G-H) ECDF plots of Pol II pausing indexes for all expressing genes in HCT-116 (G) and Jurkat cells (H) in the presence of vehicle or SEC inhibitors.
(I-J) SEC inhibition results in a 5’ shift of Pol II density near the Transcription End Site (TES) at the HSPA8 and SRSF4 genes. Pol II coverage in HEK293T cells is displayed in the UCSC genome browser.
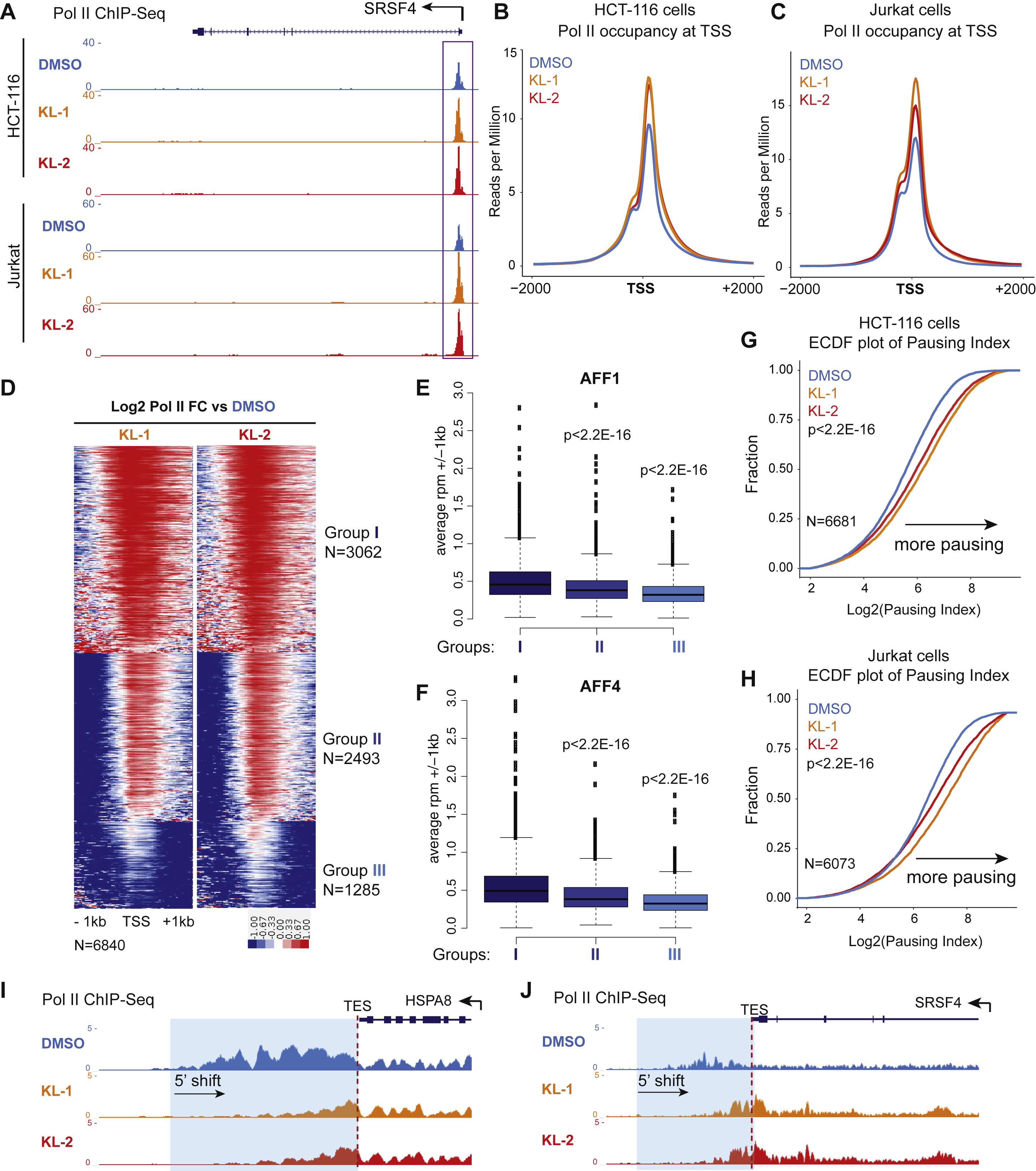{kind=link}
(A) Identification of the 1,057 genes with typical Pol II termination signals around the TES. Pol II signals from WT Pol II mutant expressing cells were K-means clustered into 5 groups with a window of 15 kb around the TES. The expressing genes from clusters with a typical Pol II termination signals were selected and plotted.
(B) SEC inhibitor treatments of the WT Pol II mutant expressing cells lead to a Pol II phenotype similar to slower Pol II mutant expressing cells as viewed at the 3’-end of genes.
(C-D) Time-dependent and dose-dependent shift of Pol II signals around the TES sites by KL-2 treatment (N=1,057).
(E) Genome browser tracks of Serine 2 phosphorylated (Ser2P) Pol II at the SRSF1 gene in HEK293T cells treated with vehicle or SEC inhibitors. SEC inhibitor treatments result in increased Pol II occupancy at the promoter-proximal region and increased occupancy of Ser2P Pol II in the gene body.
(F-G) Metagene plot of Pol II Ser2P occupancy (F) and log2 Ser2P/total Pol II ratio (G) for 6,840 well-expressed genes after SEC inhibitor treatments, indicating that disruption of SEC leads to altered Pol II dynamics, with increased CTD Ser2 phosphorylation ratio near TSS sites and decreased Ser2P occupancy after the annotated TES sites.
(H) SEC inhibition results in decreased protein levels of the SEC subunit ELL2. 293T cells were treated with 20 μM of the indicated inhibitors for 6 hr before harvesting cells for western blotting.
(I-K) Depletion of ELL2 in HEK293T cells (I) results in apparent early termination of Pol II with reduced Pol II occupancy downstream of the PIM3 (J) and ACTB (K) annotated TES.
(L) Metaplots of Pol II occupancy at TES regions for all of the 1,057 genes in Figure S3A. ELL2 knockdown results in a 5’ shift of Pol II in these regions.
(M-N) Metaplot and heatmap analysis of PRO-seq signal in HEK293T cells treated with vehicle or SEC inhibitors for the region 500 bp upstream and 500 bp downstream of the empirical promoter-proximal pausing site identified in the control condition. Rows represent genes and are sorted according to total PRO-seq signal in the control condition (N). The right two panels display log2 fold changes in PRO-seq signal with the indicated inhibitors. Increased Pol II occupancy in promoter-proximal regions is observed along with decreased occupancy in the region downstream.
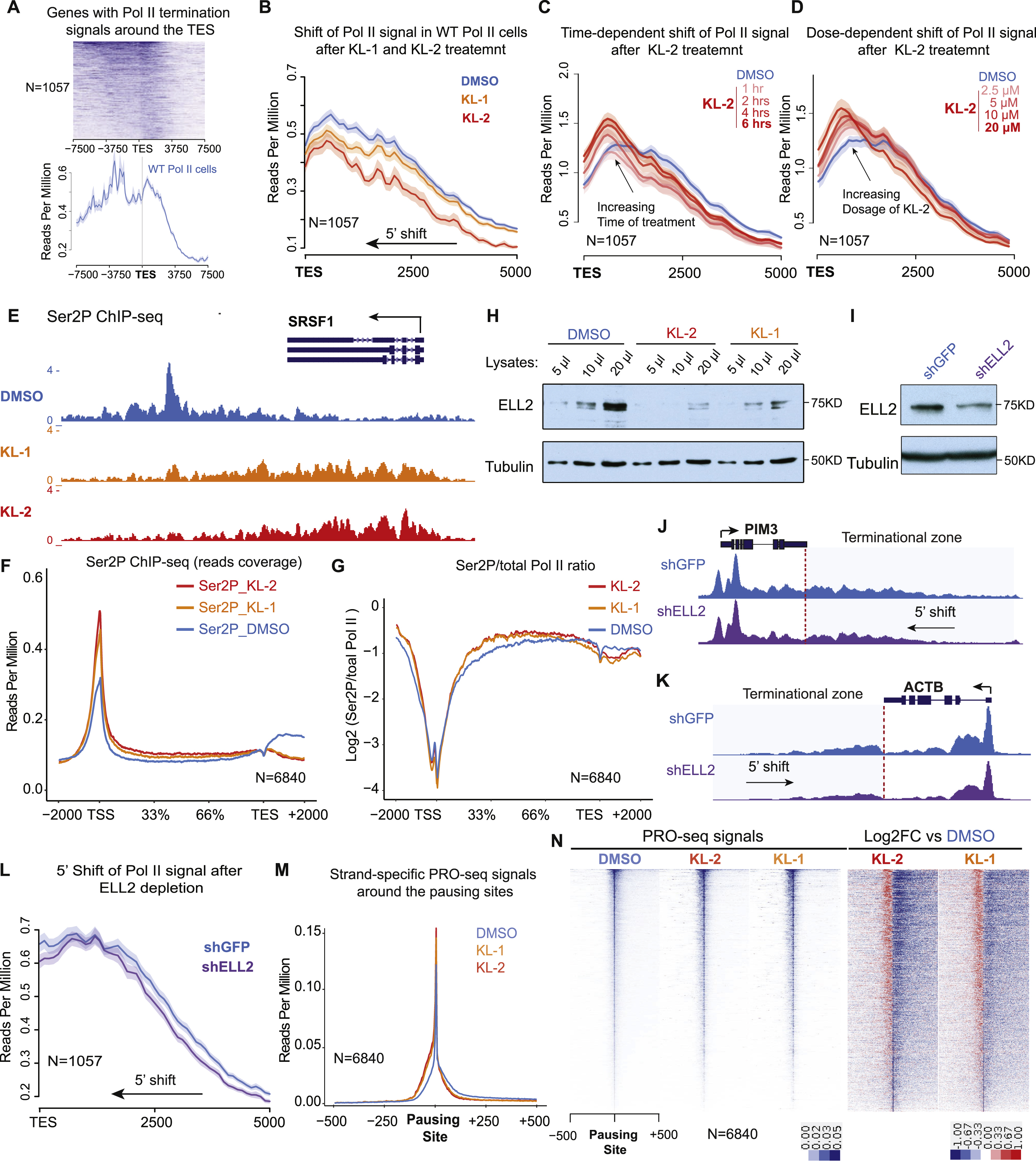{kind=link}
(A) Hidden Markov Model (HMM) for elongation rates analysis. Raw changes in 4sU-FP-seq read counts in non-overlapping 50 bp windows used to infer elongation rates for the ACTN2 gene. The boxes show the span of advancing wave inferred by a 3-state HMM analysis, demonstrating that SEC inhibitors decrease the elongation rates of Pol II elongation at the ACTN2 gene.
(B) Calculation of Pol II elongation rate using the length of SICER-called peaks of 4sU-FP-seq data to measure distance traveled. Genome browser tracks of 4sU-FP-seq signals at the MTR gene in vehicle and SEC inhibitor-treated HEK293T cells. Black bars beneath the tracks indicate SICER-called peaks. Note that in the DMSO condition, Pol II has traveled further during the 15 minutes of release from flavopiridol resulting in a longer distance traveled.
(C-D) Histograms of elongation rates (N=1,484) calculated form SICER data for DMSO (C-D) and SEC inhibitors KL-1 (C) and KL-2 (D).
(E) Boxplot analysis of SICER-based elongation rates (N=1,484, left panel) and log2 fold change of elongation rates (right panel) for the indicated treatments Wilcoxon test was used for the statistical analysis.
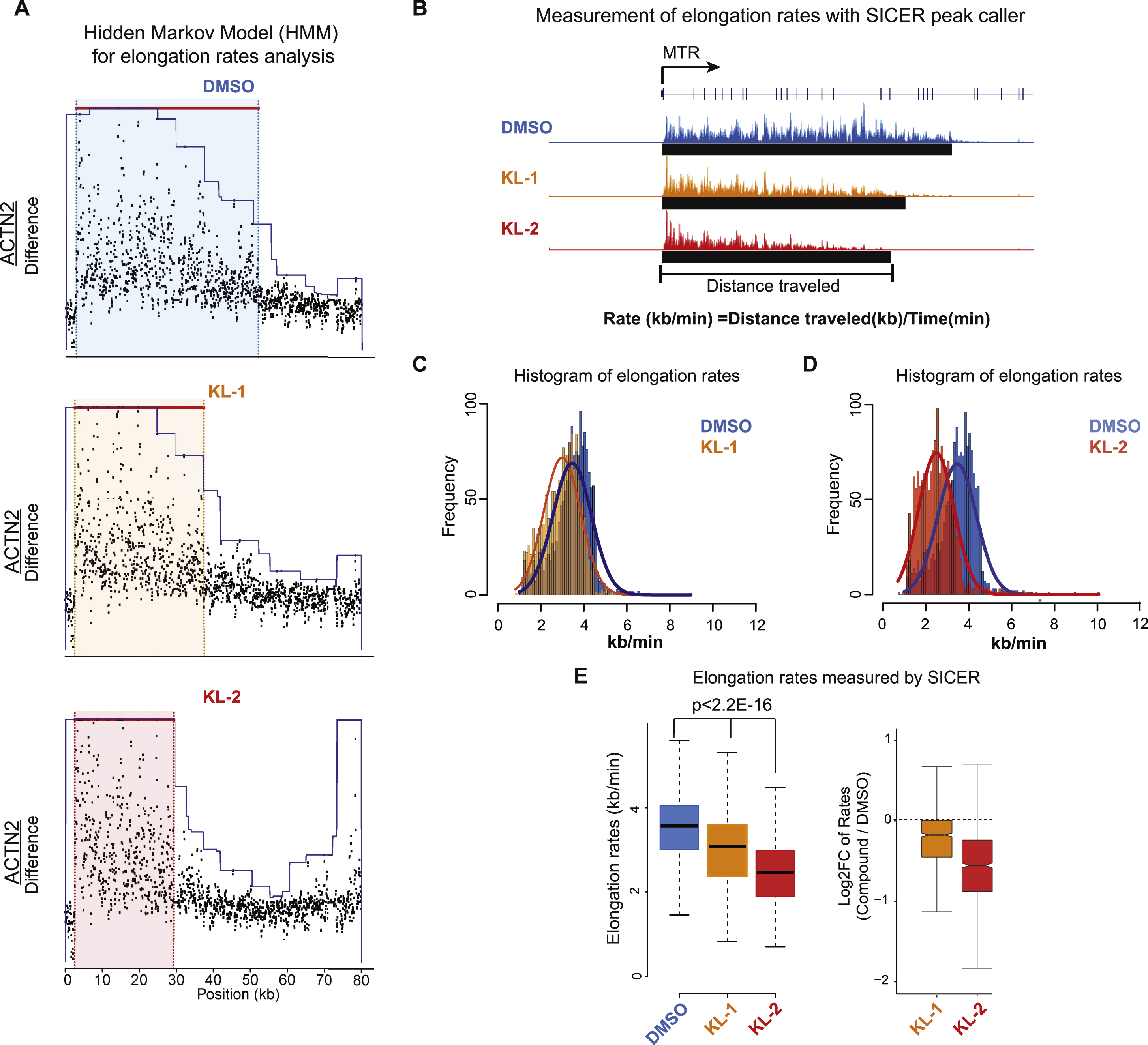{kind=link}
(A) Genome browser tracks of Pol II occupancy demonstrating that SEC inhibitors attenuate the induction of heat shock-induced gene EGR1.
(B) Gene Ontology analysis of the 136 identified heat shock-induced genes in HCT-116 cells.
(C) Workflow of Tat-induced HIV genome activation in J-Lat 6.3 cells. J-Lat 6.3 cells contain an integrated copy of the HIV genome that has GFP replacing HIV nef and could be activated by Phorbol-12-Myristate-13-Acetate (PMA). J-Lat 6.3 cells were treated with vehicle or 20 μM SEC inhibitor along with PMA to activate the HIV genome transcription, which could be monitored by flow cytometry analysis.
(D-E) SEC inhibitors block Tat-induced HIV genome activation in J-Lat 6.3 cells. (D) FACS analysis of J-Lat 6.3 cells treated with 10 nM PMA and SEC inhibitors for 17 hr. Treatment with SEC inhibitors attenuates HIV genome activation in a dose-dependent manner (E) as revealed by flow cytometry analysis with GFP (n=3–5). Unpaired two-way ANOVA was used to compare the groups between DMSO and SEC inhibitors. ***, p < 0.001.
(F) Genome browser views demonstrating that SEC inhibitors inhibit the Tat-dependent induction of the integrated HIV genome. J-Lat 6.3 cells were first induced with 10 nM PMA for 11 hr to induce the Tat expression and then treated with 20 μM SEC inhibitors for 6 hr prior to Pol II ChIP-seq.
{kind=link}
(A-B) Gene ontology analysis of the deregulated genes performed with Metascape (Tripathi et al., 2015), showing that MYC target genes and RNA splicing terms are highly enriched, while the upregulated genes are related to stress response and apoptosis. The enriched terms are shown with FDR-adjusted q-values.
(C) Gene ontology analysis of the 1,226 common genes downregulated by SEC inhibitors and SEC depletion. 1,226 out of the 1,911 genes downregulated by SEC inhibitor treatments are also downregulated by SEC depletion with either AFF1 and AFF4 co-knockdown or ELL2 knockdown. Analysis of these 1,226 genes shows that RNA splicing and MYC target genes are highly enriched terms with FDR-adjusted q-values indicated.
(D) Heatmap showing the SEC occupancy at the shared MYC peaks (upper panel) and SW1271unique MYC peaks (bottom panel). The heatmap is separated based on whether the SEC peaks could be called by MACS with a p-value cutoff of 1E-5.
(E) SEC inhibition reduces the proliferative capacity of MYC highly expressed small cell lung cancer cells. H2171 cells with MYC amplification were subjected to either shGFP or MYC shRNA for 3 days, followed by seeding cells at 0.3 million cells/mL in the presence of the indicated doses of SEC inhibitors for 3 days. Viable cells were counted with a Vi-CELL XR (Beckman Coulter) (n=3).
(F-G) Depletion of MYC reduces the SEC recruitment at the MYC binding sites in 293T cells. 293T cells were depleted with either shGFP (control) or shMYC shRNAs for 2 days, and then used for ChIP-seq and 4sU-FP-seq for measurement of elongation rates (F). Heatmap of MYC, AFF1 and AFF4 occupancy showing that MYC knockdown decreases both MYC occupancy and the SEC occupancy at the overlapped MYC and SEC peaks (G).
(H) Genome browser tracks of 4sU-FP-seq after shGFP and MYC knockdown in 293T cells at the MTR loci. Cells were paused with flavopiridol and release with fresh medium in plates. Knockdown of MYC reduces the distance Pol II travels following Pol II release, suggesting decreased elongation rates after MYC depletion.
(I) Metaplot of strand-specific 4sU-FP-seq signals in shGFP and MYC depleted cells.
(J) Boxplots showing the log2 fold change of elongation rates as determined by HMM after MYC depletion (N=1,021). Three replicates of MYC knockdown were plotted and showed that MYC depletion decreases the elongation rates in 293T cells.
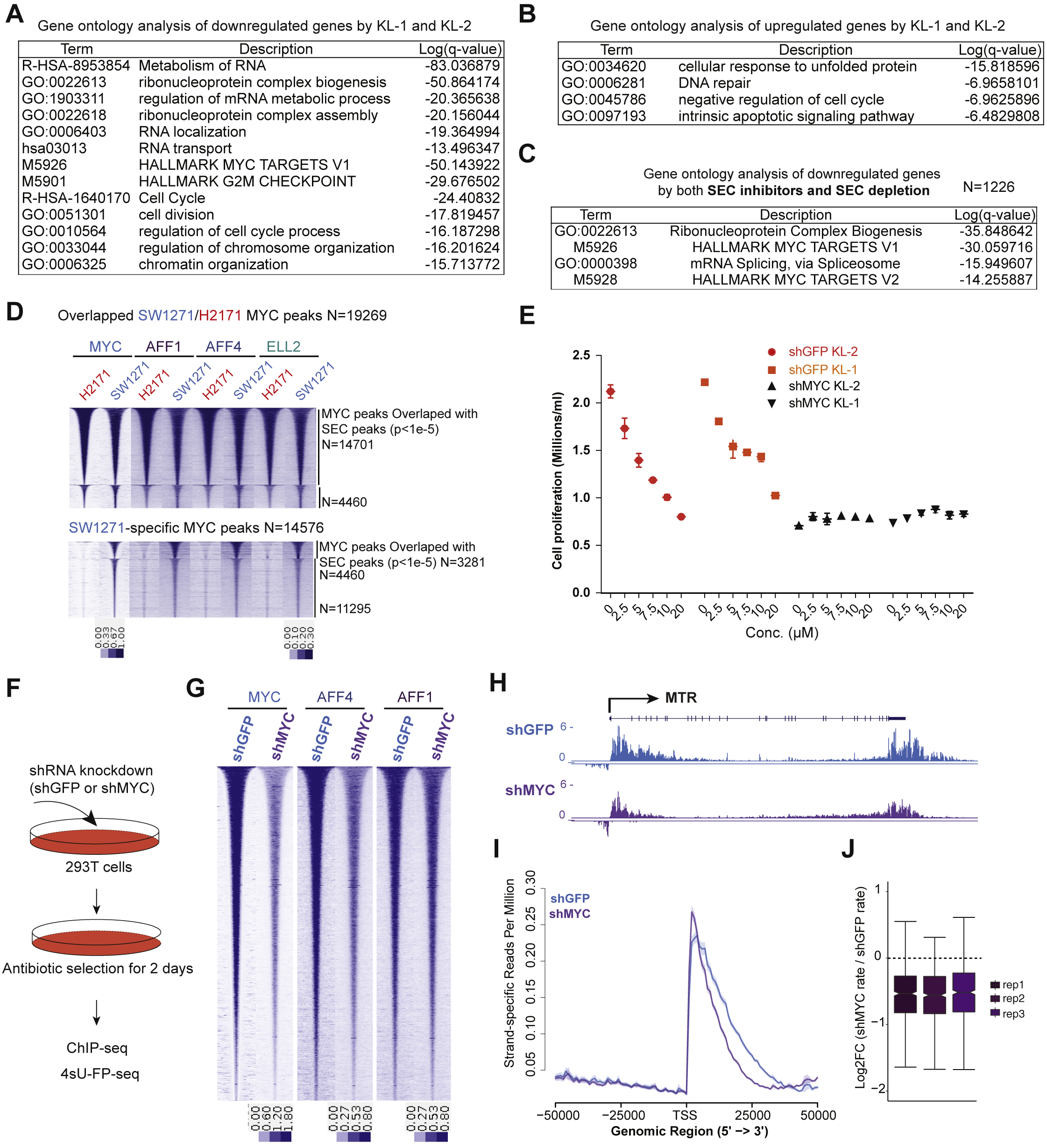{kind=link}
(A) KL-1 and KL-2 inhibit the colony formation of MDA231-LM2 cells in vitro. 5×103 MDA231-LM2 cells were seeded in 6-well plates and treated with KL-1 or KL-2 at the indicated doses for 5 days. Crystal violet staining was performed to stain the colonies.
(B) KL-1 and KL-2 induce apoptosis in MDA231-LM2 cells. Bar graph depiction of Annexin V positive MDA231-LM2 cells after 24 hr of KL-1 and KL-2 treatments at the indicated concentrations.
(C) Five intraperitoneal injections of 50 mg/kg of KL-1 or 10 mg/kg of KL-2 do not cause significant weight loss in mice after monitoring for 35 days. Each individual line indicates the weights of each mouse during toxicity test.
{kind=link}
HIGHLIGHTS.
Discovery of small molecule inhibitors of SEC and transcription elongation by Pol II
KL-1 and KL-2 disrupt the Cyclin T1-AFF4 interaction within SEC
SEC inhibitors attenuate SEC-dependent rapid transcriptional responses
MYC transcriptional programs are inhibited by SEC chemical disruptors KL-1/KL-2
ACKNOWLEDGMENTS
We thank Dr. David Bentley for the slow and fast Pol II mutant cells and Dr. Yibing Kang for the MDA231-LM2 cells. We thank Dr. Lihua Zou for help with the Hidden Markov analysis. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: J-Lat Full Length Clone (6.3) from Dr. Eric Verdin. A part of this work was performed by the Northwestern University ChemCore which is funded by Cancer Center Support Grant P30CA060553 from the National Cancer Institute awarded to the Robert H. Lurie Comprehensive Cancer Center, and the Chicago Biomedical Consortium with support from The Searle Funds at The Chicago Community Trust. L.W. was supported by the Training Program in Signal Transduction and Cancer (T32CA070085). Y.A. was supported by a JSPS Research Fellowship for Young Scientists. E.R.S. was supported by NCI R50CA211428. Studies on transcription elongation control in the Shilatifard laboratory were supported by the National Cancer Institute’s NCI R01 CA214035–16 to A.S.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Baell JB, and Holloway GA (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Bonday ZQ, and Guccione E (2013). Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev 27, 1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SH, and Price DH (2001). Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 276, 31793–31799. [DOI] [PubMed] [Google Scholar]
- Chen FX, Smith ER, and Shilatifard A (2018). Born to run: control of transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 19, 464–478. [DOI] [PubMed] [Google Scholar]
- Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, and Kraus WL (2013). Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol Cell 50, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. (2011). Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Brannan K, Erickson B, Kim H, Cortazar MA, Sheridan RM, Nguyen T, Karp S, and Bentley DL (2015). Effects of Transcription Elongation Rate and Xrn2 Exonuclease Activity on RNA Polymerase II Termination Suggest Widespread Kinetic Competition. Mol Cell 60, 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, and Bentley DL (2014). Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev 28, 2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Saldi T, Sheridan RM, Cortazar MA, and Bentley DL (2017). RNA Pol II Dynamics Modulate Co-transcriptional Chromatin Modification, CTD Phosphorylation, and Transcriptional Direction. Mol Cell 66, 546–557 e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs G, Voichek Y, Benjamin S, Gilad S, Amit I, and Oren M (2014). 4sUDRB-seq: measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome Biol 15, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, Long HW, Daniels DL, Hahn WC, Dowell RD, et al. (2013). HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 153, 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Babayeva ND, Suwa Y, Baranovskiy AG, Price DH, and Tahirov TH (2014). Crystal structure of HIV-1 Tat complexed with human P-TEFb and AFF4. Cell Cycle 13, 1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, and Zhou Q (2010). HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell 38, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. (2015). The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Smith ER, Garruss AS, Mohaghegh N, Varberg JM, Lin C, Jackson J, Gao X, Saraf A, Florens L, et al. (2013). The little elongation complex functions at initiation and elongation phases of snRNA gene transcription. Mol Cell 51, 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, and Coleman RG (2012). ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, Rajagopalan R, Venditti CP, Gripp K, Samanich J, et al. (2015). Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet 47, 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, and Fu XD (2013). SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 153, 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, and Lis JT (2015). Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 16, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, and Verdin E (2003). HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22, 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, Al-Haddawi M, Tan SY, Osato M, Sabo A, et al. (2015). MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100. [DOI] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, and Abdel-Wahab O (2016). Therapeutic targeting of splicing in cancer. Nat Med 22, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Volk AG, Haug JS, Marshall SA, Woodfin AR, Bartom ET, Gilmore JM, Florens L, Washburn MP, Sullivan KD, et al. (2017). Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell 168, 59–72 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Woodfin AR, Slaughter BD, Unruh JR, Box AC, Rickels RA, Gao X, Haug JS, Jaspersen SL, and Shilatifard A (2015). Mitotic Transcriptional Activation: Clearance of Actively Engaged Pol II via Transcriptional Elongation Control in Mitosis. Mol Cell 60, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Garrett AS, De Kumar B, Smith ER, Gogol M, Seidel C, Krumlauf R, and Shilatifard A (2011). Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev 25, 1486–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, and Shilatifard A (2010). AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. (2015). Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Lin C, Guest E, Garrett AS, Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP, and Shilatifard A (2012a). The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol 32, 2608–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Lin C, and Shilatifard A (2012b). The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol 13, 543–547. [DOI] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016a). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Salamanca HH, Duarte FM, Danko CG, and Lis JT (2016b). Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol Cell 62, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnetjournal 17, 10–12. [Google Scholar]
- McNamara RP, Bacon CW, and D’Orso I (2016). Transcription elongation control by the 7SK snRNP complex: Releasing the pause. Cell Cycle 15, 2115–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan M, Lin C, Guest E, and Shilatifard A (2010). Licensed to elongate: a molecular mechanism for MLL-based leukaemogenesis. Nat Rev Cancer 10, 721–728. [DOI] [PubMed] [Google Scholar]
- Neklesa TK, Winkler JD, and Crews CM (2017). Targeted protein degradation by PROTACs. Pharmacol Ther 174, 138–144. [DOI] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlin BM, and Price DH (2006). Controlling the elongation phase of transcription with PTEFb. Mol Cell 23, 297–305. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al. (2014). Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Schulze-Gahmen U, Upton H, Birnberg A, Bao K, Chou S, Krogan NJ, Zhou Q, and Alber T (2013). The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. Elife 2, e00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A, Duan DR, Haque D, Florence C, Schubach WH, Conaway JW, and Conaway RC (1997). ELL2, a new member of an ELL family of RNA polymerase II elongation factors. Proc Natl Acad Sci U S A 94, 3639–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A, Lane WS, Jackson KW, Conaway RC, and Conaway JW (1996). An RNA polymerase II elongation factor encoded by the human ELL gene. Science 271, 18731876. [DOI] [PubMed] [Google Scholar]
- Smith E, Lin C, and Shilatifard A (2011). The super elongation complex (SEC) and MLL in development and disease. Genes Dev 25, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, and Benkirane M (2010). HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 38, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Parmely TJ, Sato S, Tomomori-Sato C, Banks CA, Kong SE, Szutorisz H, Swanson SK, Martin-Brown S, Washburn MP, et al. (2011). Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 146, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, DumayOdelot H, Karim S, Bartkuhn M, et al. (2014). Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YE, Erb MA, et al. (2017). ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Collings CK, Zhao Z, Cozzolino KA, Ma Q, Liang K, Marshall SA, Sze CC, Hashizume R, Savas JN, et al. (2017). A cytoplasmic COMPASS is necessary for cell survival and triple-negative breast cancer pathogenesis by regulating metabolism. Genes Dev 31, 2056–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell 67, 5–18 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, and Zhou Q (2005). Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 19, 535–545. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Lin M, Naresh A, Kitabayashi I, and Cleary ML (2010). A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 17, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, O’Donnell KA, and Dang CV (2003). An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol 4, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li T, and Price DH (2012). RNA polymerase II elongation control. Annu Rev Biochem 81, 119–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic of AlphaLISA assay of the AFF4-CCNT1 interaction. Anti-GST AlphaLISA acceptor beads and AlphaScreen streptavidin donor beads (Perkin Elmer) were used to detect the interaction of GST-CCNT1 (AA1–300) and biotin-AFF4 (AA32–67).
(B-C) Optimization of the AlphaLISA assay with various biotin-AFF4 peptide and GST-CCNT1 concentrations. No CCNT1 (B) and mutant AFF4 (C) were used as negative controls.
(D) Scheme for chemical synthesis of SEC inhibitors KL-1 and KL-2. Reagents and conditions: a. LDA, THF, −78 °C, 15 min., then diethyl oxalate, −78 °C to room temperature. b. NaOH, THF/H2O, room temperature, 15 min. c. EEDQ, 5-chloro-2-methylaniline, THF, room temperature, 24 hr.
(E) 6 hr of treatment with KL-1 or KL-2 does not significantly reduce the protein levels of BRD2, BRD4, Pol II (RPB1) and MYC in 293T cells. 293T cells were treated with 20 μM of KL-1 or KL-2 for 6 hr, and the protein levels of BRD2, BRD4, Pol II (RPB1), SPT5, MED26, MYC, HSP90 (load control) and Tubulin (load control) were measured by western blotting.
(F) Treatment of HCT-116 cells with KL-1 and KL-2 reduced protein levels of SEC components AFF1 and AFF4 but not CDK9 or CCNT1. HCT-116 cells were treated with 20 μM of KL-1 or KL-2 for 6 hr, and the protein levels of AFF1, AFF4, CCNT1, CDK9 and Tubulin (load control) were determined by western blotting.
(G-H) Treating cells with KL-1 and KL-2 results in reduced levels of AFF1 and AFF4. FLAGAFF1 (G) and FLAG-AFF4 (H) were expressed under the Tet-inducible promoter in HEK293T cells. Cells were treated with 20 μM of the indicated inhibitors for 6 hr for western blotting.
(I) KL1 and KL-2 do not lead to reduced mRNA expression of SEC subunits AFF1, AFF4, ELL2, CCNT1 and CDK9 in HEK293T cells. RNA-seq was performed in HEK293T cells treated for 12 hr with 20 μM of the indicated inhibitors. The log2 counts per million (CPM) SEC genes were calculated with HTseq. KL-1 and KL-2 had no significant effects on CCNT1, CDK9 and AFF1 expression, but modest increases in the mRNA levels of AFF4 and ELL2 were observed (n=3). Unpaired two-way ANOVA was used for the statistical testes. **, p < 0.05, ***, p < 0.001.
(A) Genome browser tracks of Pol II occupancy at the SRSF4 gene in vehicle or SEC inhibitortreated cells gene in HCT-116 (top panel) and Jurkat cells (bottom panel).
(B-C) Metaplots of Pol II occupancy for all expressing genes with Pol II occupancy in HCT-116 (B) and Jurkat cells (C). SEC inhibitor treatments result in increased Pol II occupancy around the TSS. Pol II density is plotted in a −2 kb and +2 kb window around the TSS.
(D) K-means 3 clustered heatmap of Pol II fold change of the 6,840 genes after SEC inhibitor treatments. The region around the TSS is shown. Group I genes are preferentially affected by the SEC inhibitors.
(E-F) Boxplot analysis of AFF1 (E) and AFF4 (F) occupancy at the three clusters from (D).
(G-H) ECDF plots of Pol II pausing indexes for all expressing genes in HCT-116 (G) and Jurkat cells (H) in the presence of vehicle or SEC inhibitors.
(I-J) SEC inhibition results in a 5’ shift of Pol II density near the Transcription End Site (TES) at the HSPA8 and SRSF4 genes. Pol II coverage in HEK293T cells is displayed in the UCSC genome browser.
(A) Identification of the 1,057 genes with typical Pol II termination signals around the TES. Pol II signals from WT Pol II mutant expressing cells were K-means clustered into 5 groups with a window of 15 kb around the TES. The expressing genes from clusters with a typical Pol II termination signals were selected and plotted.
(B) SEC inhibitor treatments of the WT Pol II mutant expressing cells lead to a Pol II phenotype similar to slower Pol II mutant expressing cells as viewed at the 3’-end of genes.
(C-D) Time-dependent and dose-dependent shift of Pol II signals around the TES sites by KL-2 treatment (N=1,057).
(E) Genome browser tracks of Serine 2 phosphorylated (Ser2P) Pol II at the SRSF1 gene in HEK293T cells treated with vehicle or SEC inhibitors. SEC inhibitor treatments result in increased Pol II occupancy at the promoter-proximal region and increased occupancy of Ser2P Pol II in the gene body.
(F-G) Metagene plot of Pol II Ser2P occupancy (F) and log2 Ser2P/total Pol II ratio (G) for 6,840 well-expressed genes after SEC inhibitor treatments, indicating that disruption of SEC leads to altered Pol II dynamics, with increased CTD Ser2 phosphorylation ratio near TSS sites and decreased Ser2P occupancy after the annotated TES sites.
(H) SEC inhibition results in decreased protein levels of the SEC subunit ELL2. 293T cells were treated with 20 μM of the indicated inhibitors for 6 hr before harvesting cells for western blotting.
(I-K) Depletion of ELL2 in HEK293T cells (I) results in apparent early termination of Pol II with reduced Pol II occupancy downstream of the PIM3 (J) and ACTB (K) annotated TES.
(L) Metaplots of Pol II occupancy at TES regions for all of the 1,057 genes in Figure S3A. ELL2 knockdown results in a 5’ shift of Pol II in these regions.
(M-N) Metaplot and heatmap analysis of PRO-seq signal in HEK293T cells treated with vehicle or SEC inhibitors for the region 500 bp upstream and 500 bp downstream of the empirical promoter-proximal pausing site identified in the control condition. Rows represent genes and are sorted according to total PRO-seq signal in the control condition (N). The right two panels display log2 fold changes in PRO-seq signal with the indicated inhibitors. Increased Pol II occupancy in promoter-proximal regions is observed along with decreased occupancy in the region downstream.
(A) Hidden Markov Model (HMM) for elongation rates analysis. Raw changes in 4sU-FP-seq read counts in non-overlapping 50 bp windows used to infer elongation rates for the ACTN2 gene. The boxes show the span of advancing wave inferred by a 3-state HMM analysis, demonstrating that SEC inhibitors decrease the elongation rates of Pol II elongation at the ACTN2 gene.
(B) Calculation of Pol II elongation rate using the length of SICER-called peaks of 4sU-FP-seq data to measure distance traveled. Genome browser tracks of 4sU-FP-seq signals at the MTR gene in vehicle and SEC inhibitor-treated HEK293T cells. Black bars beneath the tracks indicate SICER-called peaks. Note that in the DMSO condition, Pol II has traveled further during the 15 minutes of release from flavopiridol resulting in a longer distance traveled.
(C-D) Histograms of elongation rates (N=1,484) calculated form SICER data for DMSO (C-D) and SEC inhibitors KL-1 (C) and KL-2 (D).
(E) Boxplot analysis of SICER-based elongation rates (N=1,484, left panel) and log2 fold change of elongation rates (right panel) for the indicated treatments Wilcoxon test was used for the statistical analysis.
(A) Genome browser tracks of Pol II occupancy demonstrating that SEC inhibitors attenuate the induction of heat shock-induced gene EGR1.
(B) Gene Ontology analysis of the 136 identified heat shock-induced genes in HCT-116 cells.
(C) Workflow of Tat-induced HIV genome activation in J-Lat 6.3 cells. J-Lat 6.3 cells contain an integrated copy of the HIV genome that has GFP replacing HIV nef and could be activated by Phorbol-12-Myristate-13-Acetate (PMA). J-Lat 6.3 cells were treated with vehicle or 20 μM SEC inhibitor along with PMA to activate the HIV genome transcription, which could be monitored by flow cytometry analysis.
(D-E) SEC inhibitors block Tat-induced HIV genome activation in J-Lat 6.3 cells. (D) FACS analysis of J-Lat 6.3 cells treated with 10 nM PMA and SEC inhibitors for 17 hr. Treatment with SEC inhibitors attenuates HIV genome activation in a dose-dependent manner (E) as revealed by flow cytometry analysis with GFP (n=3–5). Unpaired two-way ANOVA was used to compare the groups between DMSO and SEC inhibitors. ***, p < 0.001.
(F) Genome browser views demonstrating that SEC inhibitors inhibit the Tat-dependent induction of the integrated HIV genome. J-Lat 6.3 cells were first induced with 10 nM PMA for 11 hr to induce the Tat expression and then treated with 20 μM SEC inhibitors for 6 hr prior to Pol II ChIP-seq.
(A-B) Gene ontology analysis of the deregulated genes performed with Metascape (Tripathi et al., 2015), showing that MYC target genes and RNA splicing terms are highly enriched, while the upregulated genes are related to stress response and apoptosis. The enriched terms are shown with FDR-adjusted q-values.
(C) Gene ontology analysis of the 1,226 common genes downregulated by SEC inhibitors and SEC depletion. 1,226 out of the 1,911 genes downregulated by SEC inhibitor treatments are also downregulated by SEC depletion with either AFF1 and AFF4 co-knockdown or ELL2 knockdown. Analysis of these 1,226 genes shows that RNA splicing and MYC target genes are highly enriched terms with FDR-adjusted q-values indicated.
(D) Heatmap showing the SEC occupancy at the shared MYC peaks (upper panel) and SW1271unique MYC peaks (bottom panel). The heatmap is separated based on whether the SEC peaks could be called by MACS with a p-value cutoff of 1E-5.
(E) SEC inhibition reduces the proliferative capacity of MYC highly expressed small cell lung cancer cells. H2171 cells with MYC amplification were subjected to either shGFP or MYC shRNA for 3 days, followed by seeding cells at 0.3 million cells/mL in the presence of the indicated doses of SEC inhibitors for 3 days. Viable cells were counted with a Vi-CELL XR (Beckman Coulter) (n=3).
(F-G) Depletion of MYC reduces the SEC recruitment at the MYC binding sites in 293T cells. 293T cells were depleted with either shGFP (control) or shMYC shRNAs for 2 days, and then used for ChIP-seq and 4sU-FP-seq for measurement of elongation rates (F). Heatmap of MYC, AFF1 and AFF4 occupancy showing that MYC knockdown decreases both MYC occupancy and the SEC occupancy at the overlapped MYC and SEC peaks (G).
(H) Genome browser tracks of 4sU-FP-seq after shGFP and MYC knockdown in 293T cells at the MTR loci. Cells were paused with flavopiridol and release with fresh medium in plates. Knockdown of MYC reduces the distance Pol II travels following Pol II release, suggesting decreased elongation rates after MYC depletion.
(I) Metaplot of strand-specific 4sU-FP-seq signals in shGFP and MYC depleted cells.
(J) Boxplots showing the log2 fold change of elongation rates as determined by HMM after MYC depletion (N=1,021). Three replicates of MYC knockdown were plotted and showed that MYC depletion decreases the elongation rates in 293T cells.
(A) KL-1 and KL-2 inhibit the colony formation of MDA231-LM2 cells in vitro. 5×103 MDA231-LM2 cells were seeded in 6-well plates and treated with KL-1 or KL-2 at the indicated doses for 5 days. Crystal violet staining was performed to stain the colonies.
(B) KL-1 and KL-2 induce apoptosis in MDA231-LM2 cells. Bar graph depiction of Annexin V positive MDA231-LM2 cells after 24 hr of KL-1 and KL-2 treatments at the indicated concentrations.
(C) Five intraperitoneal injections of 50 mg/kg of KL-1 or 10 mg/kg of KL-2 do not cause significant weight loss in mice after monitoring for 35 days. Each individual line indicates the weights of each mouse during toxicity test.
Data Availability Statement
The accession number for the raw and processed ChIP-seq, RNA-seq, PRO-seq and 4sU-FP-seq data reported in this paper is GEO: GSE112608.