

ABSTRACT
Understanding the structure of PrPSc is without doubt a sine qua non to understand not only PrPSc propagation, but also critical features of that process such as the strain phenomenon and transmission barriers. While elucidation of the PrPSc structure has been full of difficulties, we now have a large amount of structural information that allows us to begin to understand it. This commentary article summarizes a round table that took place within the Prion 2018 meeting held in Santiago de Compostela to discuss the state of the art in this matter. Two alternative models of PrPSc exist: the PIRIBS and the 4-rung β-solenoid models. Both of them have relevant features. The 4-rung β-solenoid model agrees with experimental constraints of brain derived PrPSc obtained from cryo-EM and X-ray fiber diffraction studies. Furthermore, it allows facile accommodation of the bulky glycans that decorate brain-derived PrPSc. On the other hand, the infectious PrP23-144 amyloid exhibits a PIRIBS architecture. Perhaps, both types of structure co-exist.
KEYWORDS: PrPSc structure, PrPSc syalilation, 4-rung β-solenoid, PIRIBS, PrP23-144 infectious amyloid, cryo-electron microscopy, solid state NMR, Prion2018
Introduction
‘It has not escaped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material’. With this famous understatement, Watson and Crick reflected, in their classic 1953 paper, upon the fact that contemplation of the structure of DNA, that they had just deciphered, was sufficient to understand the mechanism by which this molecule propagates [1]. PrPSc prions also propagate, although what propagates in that case is not a primary structure but rather the secondary, tertiary and quaternary structures of a specific conformation of PrP [2]. For DNA, pairing of the two helical chains within the double helix underpins the templating mechanism. For PrPSc prions, some specific elements within the PrPSc conformation must underpin the capacity of this structure to mold an incoming PrP molecule into a copy of itself. Thus, understanding the structure of PrPSc without doubt holds the key to an immediate understanding of PrPSc propagation. Furthermore, a number of critical features of the propagation process such as the strain phenomenon and transmission barriers will be immediately understood once the structure of PrPSc is known with sufficient detail.
Historically, the quest to elucidate the structure of PrPSc has been a difficult one, given that PrPSc is an analytical nightmare: difficult to isolate, insoluble in water, polymeric, and featuring variable amounts of post-translation modifications. Nevertheless, a large amount of structural information and constraints have been amassed over time by indefatigable researchers, allowing us to begin to understand the structure of this enigmatic molecule.
In this context, a round table took place within the Prion 2018 meeting held in Santiago de Compostela to discuss what we know about the structure of PrPSc. Ilia Baskakov, Byron Caughey, Witold Surewicz, and Holger Wille presented their points of view, under the moderation of Alejandro M. Sevillano and Jesús R. Requena. Ample participation by the audience in the ensuing discussion ensured a wide representation of different opinions. Here is a summary of the discussion.
Main
1. Prion diversity
In addition to the generally intractable biophysical properties of PrPSc noted above, a number of other issues complicate PrPSc structure determination. First is the multitude of prion strains as well as disease-associated, but not transmissible, aggregates of PrP that must be explained. Many lines of evidence indicate that the PrPSc of different prion strains has different underlying self-propagating structures. Even for a given prion strain, wide ranges of sizes, ultrastructures, and biochemical characteristics have been observed, and some of these characteristics can be profoundly affected by the type of host animal, e.g. whether they expressed GPI-anchored or anchorless PrPC [3]. Prion infectivity has been associated with particles ranging from small non-fibrillar PrP oligomers to amyloid fibrils hundreds of nm in length [4]. This raises the question of whether this range represents a size continuum of particles with essentially the same core structure, or more fundamentally distinct arrangements of monomers such as those indicated by PrPSc preparations with both amyloid fibrils and non-amyloid 2-D crystalline arrays (see below). Synthetic recombinant PrP prions or prion-like fibrillar assemblies have been described that can all propagate indefinitely in vitro but, when inoculated into animals, can range from being biologically inert to fully infectious, pathogenic, and transmissible in subsequent passages [e.g. 5–13]. Recent evidence suggests that the difference between these biological effects can sometimes be correlated with conformational differences [14,15]. Collectively, the diversity of prions and prion-like PrP assemblies suggest that there may be no single PrPSc structure, and that diversity in conformation, and even basic multimer architecture, should be anticipated. Indeed, a number of fungal prions have been shown to have parallel in-register intermolecular β-sheet (PIRIBS) architectures [16], while another is a β-solenoid [17]. It is notable that the latter, the [Het-s] prion of Podospora anserina, is a functional, evolutionarily selected prion, whereas most other prions, including the PrP-based mammalian prions, are the accidental consequences of the refolding of proteins that have quite distinct conformations in their normal physiological states [16]. In any case, much remains to be resolved about PrPSc structure and the diversity thereof. Careful, unbiased and dispassionate evaluation of emerging data, and empirically-based models, should help us to solve the decades-long mystery of the various ways in which PrP can misfold, aggregate, and propagate to cause devastating neurodegenerative diseases.
2. The PIRIBS model
Although evidence suggests that many prions may have β-solenoid architectures (see below), there is also clear evidence that some PrP amyloids, both spontaneously nucleated [18,19] and prion-seeded [10] recombinant PrP amyloids can have PIRIBS architectures. These conclusions were discerned primarily from site-specific spin-labeling [18] and solid-state NMR experiments [10,19], which allowed determination of the basic architecture of the multimers without providing all-encompassing atomistic structures. In silico models of such structures are consistent with many empirical descriptors and constraints on PrPSc structures and appear to provide a plausible molecular basis for faithfully templated strain propagation [10]. However, as detailed in the following sections, PIRIBS models are not obviously consistent with evidence for 2D crystals, a 19.2 Å repeating unit along the fibril axis, or maximal glycosylation of all PrP monomers, as appears to be the case with some prion strains. Thus, although PIRIBS fibrils of PrP can easily be made and propagated in vitro, it remains to be determined whether such structures represent any of the diverse pathological self-propagating PrP aggregates of natural prion diseases.
3. The 4-rung β-solenoid model
The hypothesis that the structure of PrPSc is based on a β-helix or β-solenoid fold arose from studies of an alternative polymerization state for the N-terminally truncated PrP 27–30 [20]. Traditionally, PrP 27–30 polymerized into amyloid fibrils termed ‘prion rods’ [21], but here two-dimensional (2D) crystals formed by PrP 27–30 trimers were observed. By comparing the PrP 27–30 2D crystals with isomorphic 2D crystals from a redacted ‘mini-prion’ (PrPSc106) [22,23], it became apparent that traditional (i.e. elongated) β-strand architectures could not accommodate the tight packing of protein molecules within the 2D crystals. Difference mapping between 2D crystals from these two prion forms allowed the creation of the first molecular model that included a β-helix architecture [20]. A further refinement of the electron microscopy-based analyzes and the modeling approach restricted the β-helix portion to a compact, four-rung β-helix fold [24]. In subsequent years, a variety of alternate models based on the β-helix or β-solenoid hypothesis were published (reviewed in 25).
The original β-helix models still retained some α-helices (the C-terminal helices B and C) from the native PrPC structure [20,24], but a thorough re-evaluation of previously published spectroscopy data [25,26] and new H/D-exchange experiments argued against this assumption [27]. Nevertheless, the data re-evaluation and the new experiments were still compatible with the β-helix/β-solenoid hypothesis, but reduced the number of restraints as the previously α-helical part of the protein became now available to adopt β-sheet structure as well.
Additional support for the β-helix hypothesis originated from X-ray fiber diffraction experiments, which revealed that the PrPSc- and PrP 27–30-fold contains a four-rung β-solenoid architecture at its core [28]. The diffraction patterns included a series of reflections at 9.6, 6.4, and 4.8 Å, which corresponded to the second, third, and fourth order signals of a 19.2 Å repeating unit (i.e. 4 times 4.8 Å). Even the ‘mini-prion’ was found to include a four-rung β-solenoid core [29], which by itself is incompatible with any presumed, residual α-helices as the ‘mini-prion’ can only adopt a four-rung β-solenoid fold if those residues are included in the β-structure.
Next, limited proteolysis followed by mass spectrometry was used to locate loops and (partially) exposed residues in PrP 27–30 that could be cleaved by proteinase K [30]. The judicious use of denaturing conditions rendered the usually compact PrP 27–30-fold more amenable to this type of analysis. The resulting proteolytic fragments were compatible with the four-rung β-solenoid architecture, and a rough threading of the PrP primary structure onto this fold was developed [31].
Most recently cryo electron microscopy was employed to analyze the structure of PrP 27–30, and the results were found to be fully compatible with a four-rung β-solenoid architecture at the core of the infectious prion [32]. Here, a GPI-anchorless variant of PrP was used [3], which reduced the complexity of the resulting amyloid fibrils. Electron micrographs of individual amyloid fibrils were used to generate three-dimensional (3D) reconstructions, revealing the presence of two protofilaments. The protein density in these protofilaments could only be accommodated with the physical dimensions and an observed average molecular height of 17–19 Å, if a four-rung β-solenoid architecture was assumed [32]. Lastly, the β-solenoid hypothesis provides constraints for the replication of prions that involves templating on the first and/or last rung of the β-solenoid structure [33]. The steric constraints on the templating process that result as a consequence of the molecular architecture will be informative for future, higher-resolution analyzes of the structure of PrPSc.
4. Constraints imposed by glycosylation
Several structural models of PrPSc that exhibit diverse PrP folding patterns including parallel in-register β-structures and two-, three- or four-rung β-solenoids have been proposed in recent years (reviewed in 33). In an effort to build a realistic molecular model of PrPSc, our knowledge about PrPSc glycosylation should be taken into consideration. The first question to ask, is which of the proposed models can accommodate N-linked glycans? To address this question, a tri-antennary glycan with a size average of those found in PrP sialoglycoforms was used for modeling PrPSc [34]. In-register parallel β-sheet structure, the glycans of neighboring PrP molecules have to be spaced at a distance of 4.7 Å bringing them into substantial spatial overlap that precludes such arrangements [34]. Considerable spatial overlap between glycans still exists in two-rung solenoid that separates glycans at a distance of 2 × 4.7 Å. However, three- or four-rung solenoids permit recruitment of diglycosylated PrP molecules [34]. The result of modeling supports the hypothesis that glycans limit the diversity of folding patterns accessible to glycosylated PrPC.
To better understand how glycans might be enrolled in defining strain-specific PrPSc structures, two alternative views can be considered. According to one view, prion strains can partially overcome constraints imposed by glycans by selectively recruiting mono- and un-glycosylated PrPC sialoglycoforms at the expense of diglycosylated sialoglycoforms. Only those PrPC glycoforms are recruited that fit into strain-specific PrPSc structures. Alternative view proposes that recruitment is not selective, i.e. PrPC sialoglycoforms are incorporated into PrPSc proportionally to their relative presentations in a pool of PrPC molecules expressed in a cell. If this is the case, the spectrum of PrPSc structures is limited to those that can accommodate all sialoglycoforms. To answer the question on selectivity of recruitment, composition of PrPSc sialoglycoform was analyzed using 2D gels [35,36]. Because N-linked glycans carry negatively charged sialic acid residues, the sialoglycoforms can be separated in horizontal dimension of 2D according to their charge [37]. The 2D analysis revealed that PrPSc strains exhibits broad range of strain-specific selectivity with respect to PrPC sialoglycoforms [34,35]. Consistent with the first mechanism, a group of strains shows strong preferences, as they excluded highly sialylated molecules as well as diglycosylated molecules [34,35]. At the same time, in support of the second mechanism, a group of strains did not display any preferences with respect to glycosylation or sialylation status [34,35]. Analysis across all examined strains revealed a great correlation between glycosylation and sialylation status of PrP sialoglycoforms within PrPSc [34,35]. This analysis also demonstrated a broad range of selectivity displayed by prion strains in recruiting PrPC sialoglycoforms, ranging from non-selective to highly selective. Notably, for the group of non-selective strains, the composition of sialoglycoforms within PrPSc was very similar to that of PrPC.
A broad range of selectivities displayed by prion strains could be attributed to strain-specific variations in quaternary structures and does not require significant variations in strain-specific folding patterns (Figure 1). In particular, quaternary assembly of non-selective strains could involve considerable twist or rotation between neighboring PrP molecules. Owing to such rotations, glycans of neighboring PrP molecules extend into different directions avoiding spatial interference and minimizing electrostatic repulsion between sialic acid residues (Figure 1). In strains that select against diglycosylated and highly sialylated PrPC, the rotation between neighboring PrP molecules is proposed to be very small or absent. Such modes of assembly can create spatial and electrostatic interference between glycans and limit recruitment of diglycosylated and highly sialylated PrPC (Figure 1). The three- or four-rung solenoid models offer the best opportunity for accommodating both selective and non-selective strains.
Figure 1.
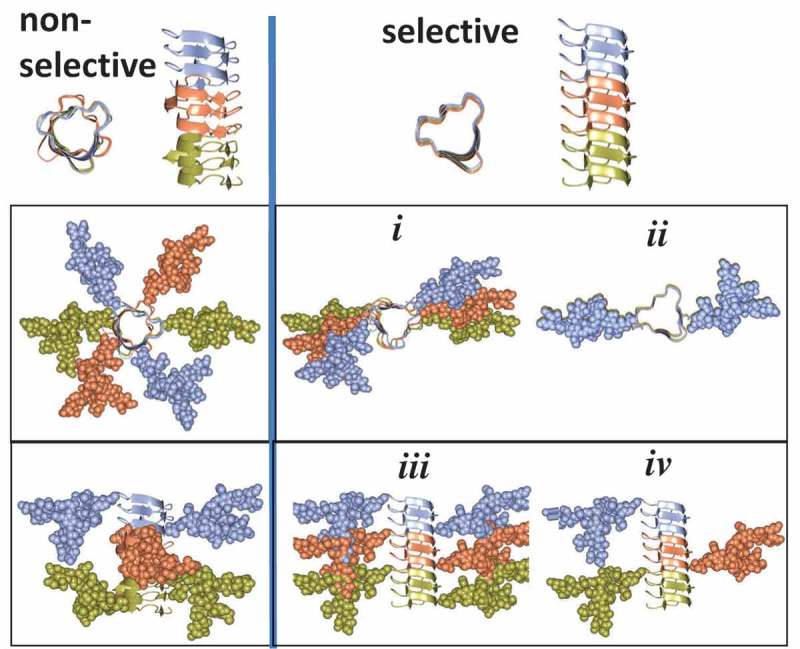
Schematic diagram illustrating differences in quaternary assembly between non-selective (left panels) and selective (right panels) strains. Non-selective strains can accommodate diglycosylated sialoglycoforms due to rotation between neighboring PrP molecules that allows spatial separation of glycans. In selective strains, the rotation between neighboring PrP molecules is very small (a) or absent (b). Recruitment of diglycosylated molecules by selective strains would lead to spatial interference between glycans (c). Negative selection of diglycosylated molecules helps to minimize spatial and electrostatic interference between glycans (d).
To summarize our view on constraints imposed by glycosylation, prion strains display a broad range of selectivity toward PrPC sialoglycoforms. Some strains recruit sialoglycoforms proportionally to their presentation in PrPC, whereas others avoid diglycosylated and highly sialylated PrPC isoforms. Strain-specific ratio of the glycoforms within PrPSc is a result of negative selection of heavily sialylated PrP molecules with bulky glycans. The extent to which heavily sialylated glycoforms are excluded is believed to be controlled by a strain-specific structure. Glycan volume and electrostatic repulsion due to sialylation have to be taken into consideration for modeling PrPSc structures.
5. Lessons from studies with PrP23–144 amyloid fibrils
An important aspect of human prion diseases is the presence of a large number of mutations in the human PrP gene (PRNP) that segregate with familial CJD, Gerstmann-Sträussler-Scheinker (GSS) disease or fatal familial insomnia. All these disorders are autosomal dominant. One of these disease-related mutations, associated with GSS-like subtypes, is the tyrosine to stop codon mutation at position 145, resulting in a C-terminally truncated PrP fragment corresponding to residues 23–144 (PrP23-144) (38; 39). Bacterially expressed recombinant PrP23-144 from different species readily form amyloid fibrils under physiologically relevant conditions, and studies in vitro with this truncated PrP variant provided important insights into mechanistic principles of the conformational basis of species- and strain-dependent seeding specificity of prion protein amyloids [40,41].
The potential value of PrP23-144 amyloid as a model for exploring molecular aspects of mammalian prion propagation is further indicated by recent studies showing that mouse PrP23-144 fibrils are infectious, causing clinical prion disease in mice [42]. An intriguing and highly unusual feature of the latter disease is the accumulation in mouse brain of two types of self-propagating PK-resistant PrP fragments: one of them about 6–7 kDa in size (with N- and C-termini mapping to residues ~80–89 and 150–159, respectively) and the second one with molecular mass upon deglycosylation of ~17–18 kDa. While the shorter fragments are reminiscent of human PrPres in GSS subtypes, the longer fragments are similar to those observed in classical mouse-adapted scrapie strains.
The finding that PrP23-144 fibrils are infectious is of particular importance given that, in contrast to fibrils formed from full-length PrP, the former fibrils give rise to high quality solid-state NMR (ssNMR) spectra and, thus, are amenable to high-resolution structural characterization. Different types of ssNMR studies with human PrP23-144 amyloid fibrils published over the past ten years revealed that (i) The rigid β-core of the amyloid spans an approximately 30-amino acid segment that maps to residues ~112–140, with the reminder of the protein dynamically disordered; (ii) This rigid core region consists of three β-strands encompassing residues ~112–113 (strand 1), ~120–123 (strand 2), and ~130–140 (strand 3); and (iii) The core region displays a parallel in-register organization of β-strands [43–45]. Furthermore, a recently published structural model [46] reveals that these fibrils consist of two protofilaments with β-sheet regions running parallel to the long fibril axis. The compact hydrophobic core of constituent monomers consists of Ala, Gly and Val-rich segment between residues ~115–122, and this structure is stabilized by a highly specific interaction between the side chains of Ala117 in this hydrophobic core and Ile139 in the longest β-strand. Interestingly, a GSS-related substitution of Ala117 with Val disrupts this stabilizing interaction, resulting in a different amyloid fold. Apart from this high-resolution insight into the structure of human PrP23-144 amyloid, ssNMR studies also revealed the nature of structural differences between PrP23-144 amyloid fibrils from different species, providing a structural basis for understanding species-dependent seeding barriers [45].
Even though mouse PrP23-144 amyloid fibrils can seed in vivo the conversion of full-length PrP to an infectious, self-propagating structure that displays PK-resistance similar to that of classical scrapie strains, it is at present unknown whether the product of this seeding reaction retains the parallel in-register structural motif of the seed. In any case, structural and biological data for PrP23-144 amyloid clearly indicate that prion protein fibrils with parallel in-register organization can be infectious. Combined with recent evidence for a β-solenoid structure of anchorless prions, this raises an intriguing possibility that entirely different structural motifs may be present in distinct prion strains.
Conclusions
The earlier large trove of structural models of PrPSc [47] has now shrunk to only two remaining models: the PIRIBS and the 4-rung β-solenoid models. Both of them have important features that make them relevant. The 4-rung β-solenoid model agrees with experimental constraints of brain derived PrPSc obtained through cryo-EM and X-ray fiber diffraction studies [28,32]. Furthermore, it allows facile accommodation of the bulky glycans present in brain-derived PrPSc [34]. However, the PrP23-144 amyloid, which exhibits a PIRIBS architecture [46] has also been demonstrated to be infectious [42]. How can these facts be reconciled? The most parsimonious explanation is that both types of structure co-exist. The first recombinant PrP prions were generated in 2004 using a technique that is known to yield amyloid fibers [5]. In retrospect, it is likely that these prions were structurally similar to amyloids produced at the later time, whose architecture is known to conform to the PIRIBS model, as assessed by site-directed spin labeling, solid-state NMR [10,18,19], and X-ray fiber diffraction analyzes [28]. In some cases, such PrP amyloids can propagate in the brain of recipient experimental animals without causing a clinical disease (v.g. 11,13). Atypical PrPres is one of the example of self-replicating and transmissible, yet clinically silent state [48–50]. However, upon second or third passage they evolve to classical PrPSc prions that lead to TSE disease [13,17,49–51]. This conformational transformation can be easily tracked biochemically, as the pattern of PK-resistant fragments evolves throughout passages. Therefore, it is conceivable that different types of PrP amyloids with a propagative, infectious properties can co-exist in the brain. Even though some of them exhibit a PIRIBS architecture, it is likely that the most infectious ones might exhibit an alternative 4-rung β-solenoid structure. Furthermore, PIRIBS structures might be able to template 4-rung β-solenoids and 4-rung β-solenoids might template PIRIBS amyloids, as was seen in the amyloid seeding assay [52]. The deformed templating model might provide a useful framework for explaining mutual templating of self-propagating structures with alternative folding patterns [53]. To add further layers of complexity, different subtypes of PIRIBS and 4-rung β-solenoid conformations/architectures might have different degrees of infectivity, and some of them may be even innocuous while maintaining the capacity to self-propagate [11,13,14,49]. However, it should be remembered that infectivity is an operational, not an absolute property of a potentially infectious agent. In this respect, thousands of British citizens are believed to harbor PrPSc in their bodies, but it will hopefully not cause any clinical neurodegenerative disease in their lifetime [54]. In this context, it might be necessary to acknowledge that PrPSc is not the only infectious PrP amyloid, that in fact some PrPSc strains and/or subtypes can propagate within brains without causing disease, just as some other propagative PrP amyloids that are not PrPSc, and that interconversions among these could happen. In other words, that the terms ‘PrPSc’ and ‘PrP prion’ might perhaps not be synonyms.
Funding Statement
Supported by grants BFU2013-48436-C2-1-P and BFU2017-86692-P from the Spanish Ministries of Economy and Competitiveness and Science, Innovation and Universities, respectively, to JRR and grant 201600029 from the Alberta Prion Research Institute to HW. This work was also supported in part by the Intramural Research Program of the NIAID (BC) and by the National Institute of Health grants R01 NS045585 (IVB), P01 AI106705 (WKS), R01 NS083687 (WKS) and R01 NS103848 (WKS).
Acknowledgments
We thank the NeuroPrion Association for support in the organization of the Prion 2018 conference (May 22-25, 2018, www.prion2018.org), at which the round table on the structure of PrPSc took place.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Watson J, Crick F.. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. [DOI] [PubMed] [Google Scholar]
- [2].Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chesebro B, Trifilo M, Race R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. [DOI] [PubMed] [Google Scholar]
- [4].Silveira JR, Raymond GJ, Hughson AG, et al. The most infectious prion protein particles. Nature. 2005;437:257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Legname G, Baskakov IV, Nguyen HO, et al. Synthetic mammalian prions. Science. 2004;305:673–676. [DOI] [PubMed] [Google Scholar]
- [6].Deleault NR, Harris BT, Rees JR, et al. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang F, Wang X, Yuan CG, et al. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim JI, Cali I, Surewicz K, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285:14083–14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Makarava N, Savtchenko R, Baskakov IV. Two alternative pathways for generating transmissible prion disease de novo. Acta Neuropathol Comm. 2015;3:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Groveman BR, Dolan MA, Taubner LM, et al. Parallel in-register intermolecular β-sheet architectures for prion-seeded prion protein (PrP) amyloids. J Biol Chem. 2014;289:24129–24142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Groveman BR, Raymond GJ, Campbell KJ, et al. Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Pathog. 2017;13(9):e1006623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sevillano AM, Fernández-Borges N, Younas N, et al. Recombinant PrPSc shares structural features with brain-derived PrPSc: insights from limited proteolysis. PLoS Pathog. 2018;14(1):e1006797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kraus A, Raymond GJ, Race B, et al. PrP P102L and nearby lysine mutations promote spontaneous in vitro formation of transmissible prions. J Virol. 2017;91:e01276–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang F, Wang X, Orrú CD, et al. Self-propagating, protease-resistant, recombinant prion protein conformers with or without in vivo pathogenicity. PLoS Pathog. 2017;13(7):e1006491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Q, Wang F, Xiao X, et al. Structural attributes of mammalian prion infectivity: insights from studies with synthetic prions. J Biol Chem. 2018. October 1:pii: jbc.RA118.005622. Epub ahead of print DOI: 10.1074/jbc.RA118.005622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wickner RB, Edskes HK, Son M, et al. Yeast prions compared to functional prions and amyloids. J Mol Biol. 2018;430:3707–3719. [DOI] [PubMed] [Google Scholar]
- [17].Wasmer C, Lange A, Van Melckebeke H, et al. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. [DOI] [PubMed] [Google Scholar]
- [18].Cobb NJ, Sonnichsen FD, Mchaourab H, et al. Molecular architecture of human prion protein amyloid. Proc Natl Acad Sci USA. 2007;104:18946–18951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tycko R, Savtchenko R, Ostapchenko VG, et al. The α-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel β-sheet structure in PrP fibrils: evidence from solid state nuclear magnetic resonance. Biochemistry. 2010;49:9488–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wille H, Michelitsch MD, Guenebaut V, et al. Structural studies of the scrapie prion protein by electron crystallography. Proc Natl Acad Sci USA. 2002;99:3563–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Prusiner SB, McKinley MP, Bowman KA, et al. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–358. [DOI] [PubMed] [Google Scholar]
- [22].Muramoto T, Scott M, Cohen FE, et al. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc Natl Acad Sci USA. 1996;93:15457–15462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Supattapone S, Bosque P, Muramoto T, et al. Prion protein of 106 residues creates an artifical transmission barrier for prion replication in transgenic mice. Cell. 1999;96:869–878. [DOI] [PubMed] [Google Scholar]
- [24].Govaerts C, Wille H, Prusiner SB, et al. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci USA. 2004;101:8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Caughey BW, Dong A, Bhat KS, et al. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. [DOI] [PubMed] [Google Scholar]
- [26].Pan KM, Baldwin M, Nguyen J, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Smirnovas V, Baron GS, Offerdahl DK, et al. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol. 2011;18:504–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wille H, Bian W, McDonald M, et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc Natl Acad Sci USA. 2009;106:16990–16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wan W, Wille H, Stöhr J, et al. Structural studies of truncated forms of the prion protein PrP. Biophys J. 2015;108:548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vázquez-Fernández E, Alonso A, Pastrana MA, et al. Structural organization of mammalian prions as probed by limited proteolysis. PLoS ONE. 2012;7:e50111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Silva CJ, Vázquez-Fernández E, Onisko B, et al. Proteinase K and the structure of PrPSc: the good, the bad and the ugly. Virus Res. 2015;207:120–126. [DOI] [PubMed] [Google Scholar]
- [32].Vázquez-Fernández E, Vos MR, Afanasyev P, et al. The structural architecture of an infectious mammalian prion using electron cryomicroscopy. PLoS Pathog. 2016;12(9):e1005835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wille H, Requena JR. The structure of PrPSc prions. Pathogens. 2018;7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baskakov IV, Katorcha E. Multifaceted role of sialylation in prion diseases. Front Neurosci. 2016;10:e358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Katorcha E, Makarava N, Savtchenko R, et al. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci Rep. 2015;5:e16912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Srivastava S, Makarava N, Katorcha E, et al. Post-conversion sialylation of prions in lymphoid tissues. Proc Acad Natl Sci USA. 2015;112:6654–6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Katorcha E, Makarava N, Savtchenko R, et al. Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLoS Pathog. 2014;10:e1004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kitamoto T, Iizuka R, Tateishi J. An amber mutation of prion protein in GSS syndrome with mutant PrP plaques. Biochem Biophys Res Commun. 1993;192:525–531. [DOI] [PubMed] [Google Scholar]
- [39].Ghetti B, Piccardo P, Spillantini MG, et al. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codón 145 mutation in PRNP. Proc Natl Acad Sci USA. 1996;93:744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vanik DL, Surewicz KA, Surewicz WK. Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell. 2004;14:139–145. [DOI] [PubMed] [Google Scholar]
- [41].Jones EJ, Surewicz WK. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell. 2005;121:63–72. [DOI] [PubMed] [Google Scholar]
- [42].Choi JK, Cali I, Surewicz K, et al. Amyloid fibrils from the N-terminal prion protein fragment are infectious. Proc Natl Acad Sci USA. 2016;113:13851–13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Helmus JJ, Surewicz K, Nadaud PS, et al. Molecular conformation and dynamics of the Y145 Stop variant of human prion protein in amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:6284–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Helmus JJ, Surewicz K, Surewicz WK, et al. Conformational flexibility of Y145Stop human prion protein amyloid fibrils probed by solid-state nuclear magnetic resonance spectroscopy. J Am Chem Soc. 2010;132:2393–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Theint T, Xia Y, Nadaud PS, et al. Structural studies of amyloid fibrils by paramagnetic solid-state nuclear magnetic resonance spectroscopy. J Am Chem Soc. 2018;140: 13161–13166. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Theint T, Nadaud PS, Aucoin D, et al. Species-dependent structural polymorphism of Y145Stop prion protein amyloid revealed by solid-state NMR spectroscopy. Nature Commun. 2017;8(753). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Requena JR, Wille H. The structure of the infectious prion protein: experimental data and molecular models. Prion. 2014;8:60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kovacs GG, Makarava N, Savtchenko R, et al. Atypical and classical forms of the disease-associated state of the prion protein exhibit distinct neuronal tropism, deposition patterns, and lesion profiles. Am J Pathol. 2013;183:1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Makarava N, Kovacs GG, Savtchenko R, et al. A new mechanism for transmissible prion diseases. J Neurosci. 2012;32:7345–7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Makarava N, Kovacs GG, Savtchenko R, et al. Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 2011;7(12):e1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Makarava N, Kovacs GG, Bocharova O, et al. Recombinant prion protein induces a new transmissible prion disease in wild type animals. Acta Neuropathol. 2010;119:177–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Colby DW, Zhang Q, Wang S, et al. Prion detection by an amyloid seeding assay. Proc Natl Acad Sci U S A. 2007;104:20914–20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Makarava N, Baskakov IV, True HL. The evolution of transmissible prions: the role of deformed templating. PLOS Pathog. 2013;9:e1003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gill ON, Spencer Y, Richard-Loendt A, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ. 2013;347:f5675. [DOI] [PMC free article] [PubMed] [Google Scholar]