

ABSTRACT
Immune escape is a hallmark of cancer. In human lung cancer, we have identified a unique microRNA (miR)-based pathway employed by tumor cells to repress detection by immune cells via the NKG2D-MICA/B receptor-ligand system. MICA/B is readily induced by cell transformation and serves as a danger signal and ligand to alert NK and activated CD8+ T cells. However, immunohistochemical analysis indicated that human lung adenocarcinoma and squamous cell carcinoma specimens express little MICA/B while high levels of miR-183 were detected in both tumor types in a TCGA database. Human lung tumor cell lines confirmed the reverse relationship in expression of MICA/B and miR-183. Importantly, a miR-183 binding site was identified on the 3ʹuntranslated region (UTR) of both MICA and MICB, suggesting its role in MICA/B regulation. Luciferase reporter constructs bearing the 3ʹUTR of MICA or MICB in 293 cells supported the function of miR-183 in repressing MICA/B expression. Additionally, anti-sense miR-183 transfection into H1355 or H1299 tumor cells caused the upregulation of MICA/B. Abundant miR-183 expression in tumor cells was traced to transforming growth factor-beta (TGFβ), as evidenced by antisense TGFβ transfection into H1355 or H1299 tumor cells which subsequently lost miR-183 expression accompanied by MICA/B upregulation. Most significantly, anti-sense miR-183 transfected tumor cells became more sensitive to lysis by activated CD8+ T cells that express high levels of NKG2D. Thus, high miR-183 triggered by TGFβ expressed in lung tumor cells can target MICA/B expression to circumvent detection by NKG2D on immune cells.
KEYWORDS: Immune evasion, NKG2D-MICA/B, non-small cell lung cancer, NK cells, T cells, mirR-183
Introduction
Lung cancer remains a deadly disease worldwide, due in part to lack of reliable means for early diagnosis as well as lack of detailed understanding of immune escape mechanisms developed by the advancing tumor cells.1,2 Emerging evidence indicates that microRNAs (miRs) may play a critical role in cancer and could serve as biomarkers, depending on the tumor type.3 These non-coding small RNAs function via RNA interference-mediated post-transcriptional gene regulation, and their dysregulation is of particular importance in cancer development and progression due to their potency to control genes involved in tumorigenesis, cell cycle control, metabolism, apoptosis and tumor progression.4 Recently, miR-183 has garnered considerable attention because of its overexpression in numerous human cancers, including lung cancer.5-7 It is part of the highly-conserved miR-183-96-182 cluster, located on human chromosome 7. In addition to lung cancer, upregulation of miR-183 has been associated with poor prognosis in carcinomas of the breast,8 colon,9,10 liver,11 esophagus,12 prostate,13 and pancreas,14 and is driven by the presence of a number of promoter elements specific for β-catenin/TCF/LEF-1 in its 5ʹ UTR15 and thus is associated with cancer development16. Moreover, tumor-associated factors such as Transforming Growth Factor-beta (TGFβ)17 and AKT18 have been identified as additional upstream regulators of miR-183 transcription. Additional effects of mir-183 include the induction of HIF-1α, which has been reported to protect against hypoxia and starvation.19 Also, miR-183 inhibits apoptosis and promotes proliferation and invasion by downregulation of Programmed Cell Death 4 (PDCD4) in tumor cells,11,12 and is reported to target protein phosphatase 2A,20 EGR1,21 PTEN21 and FoxO1,22 all of which are involved in tumor cell survival and proliferation. Although a clear role of miR-183 is emerging as a tumor promoter, it is not known whether it plays a role in immune escape by the tumor.
In order for a tumor to flourish, it must dampen the immune system and avoid detection by immune cells, including natural killer (NK) cells. NK cells are poised to kill aberrant cells, including tumor cells, by virtue of high expression of activating receptors, such as NKG2D.23,24 NKG2D is a C-type, lectin-like, type II transmembrane glycoprotein expressed on activated NK, CD8 T and γδT cells that can recognize ligands on target cells induced by stress, DNA damage, or cell transformation.25 It utilizes a specific adaptor protein, DAP10, to signal downstream for mobilization of lytic granules towards target cells.26 NKG2D recognizes a number of ligands, which include two members of the major histocompatibility complex class I chain-related (MIC) proteins, MICA and MICB, as well as 6 members of UL16-binding proteins, ULBPs 1-6.27-29 MICA/B constitutes a separate family of highly-glycosylated membrane-anchored MHC class I-like molecules that share structural homology to the MHC-I heavy chain but does not bind β-2 microglobulin or transporter-associated with antigen processing (TAP).27 However, this phenomenon is relevant for human NK cells only, as MICA/B are not conserved in the mouse, unlike Rae1 and ULBP1,30,31 so it has not been investigated in depth in this context. Unlike classical MHC-I molecules, MICA/B proteins are rarely displayed on normal cells and are only induced upon viral infection, DNA damage or transformation to serve as danger signals for clearance by NK cells. In fact, MICA/B has been reported on numerous human cancer types, including lung cancer, and its level of expression can be prognostic.32-34 Thus, a potent means for transformed tumor cells to escape immune detection is to downregulate the surface expression of NKG2D ligands. Despite the ability of NK cells to penetrate the tumor bed, the absence of NKG2D ligands could blind the NK effector cells to the surrounding tumor cells. To test this concept, we analyzed whether miR-183 could target NKG2D ligands on tumor cells, and uncovered a seminal pathway for miR-183 specific suppression of MICA/B ligands on human lung tumor cells. Therefore, in addition to promoting tumor cell survival and proliferation, miR-183 has an immunoregulatory property that disarms NK cells by depleting MICA/B from the tumor cell surface.
Results
miR-183 is overexpressed and MICA/B is underexpressed in human lung cancer
Expression of MICA/B can be heterogeneous among tumor types and tumor cell lines.27,32 In analysis of MICA/B expression in human non-small cell lung carcinoma (NSCLC) cell lines, we found low or absent levels of expression on H1155, H1355, H2170, H1299, A549 and Wi-38. Figure 1(a). This was in sharp contrast to human bronchiolar epithelial cells, HBE4, virally transformed by Epstein-Barr E6/7, as well as K562 chronic myelogenous leukemia cells, both of which express high levels of MICA/B. This data initially suggested to us that this may be simply a solid tumor phenomenon, however we additionally analyzed Panc-1 and MiaPaCa and found high levels of MICA/B, similar to that seen on HBE4, K562, and HeLa (Fig S1A, S1B). This indicated to us that this is perhaps a novel, lung-exclusive phenomenon. Reports have shown that tumor cells can downregulate NKG2D ligands, including MICA/B, to evade NK and effector CD8+ T cell responses.35-38 Although MICA/B reportedly can be shed from tumor cell surfaces, downregulation of these genes is another important mechanism to disrupt MICA/B expression. Emerging data indicates that miRs are aberrantly expressed by tumors and can control critical genes associated with proliferation and metastasis. To understand if miRs might also control immune escape in tumor cells, we focused our efforts on miR-183 which is known to be overexpressed by tumors of diverse origin, but may be of particular interest in the solid tumor realm, particularly that of lung cancer, as shown in our previous work.17 Bioinformatics analysis of the MICA and MICB 3ʹ UTRs by miRANDA and TargetScan revealed high affinity miR-183 binding sites in both genes (Figure 1(b)), suggesting that both MICA and MICB could be repressed by miR-183. Of particular interest, no predicted miR-183 binding sites were present in the 3ʹUTR of the six ULBPs that are also ligands for the common receptor, NKG2D, indicating that they are under a separate regulatory machinery than that which controls MICA/B. We then sought to address whether there was an inverse correlation between miR-183 and MICA/B expression in the human lung cell lines. Analysis of miR-183 expression among the cell lines revealed that, in contrast to virally-transformed HBE4 cells, and leukemic K562 cells, which expressed little miR-183, all of the lung lines expressed heightened endogenous levels of miR-183 and the level of miR183 expression occurred in reverse direction of that of MICA/B. (Figure 1(c)). Further, analysis of The Cancer Genome Atlas (TCGA) human lung dataset revealed a striking increase in miR-183 expression in tumors as compared to normal tissue (Figure 1(d)). In contrast, let-7c, a microRNA known to suppress expression of oncogenes thus leading to cellular proliferation,39 was sharply downregulated from normal levels in the same lung tissues, as reported by others.40 The inverse relationship between miR-183 and MICA/B, in combination with the miR-183 binding sites on MICA/B and elevated miR183 expression in the TCGA lung tumor database, suggested potential MICA/B dysregulation by miR-183 in lung cancer.
Figure 1.
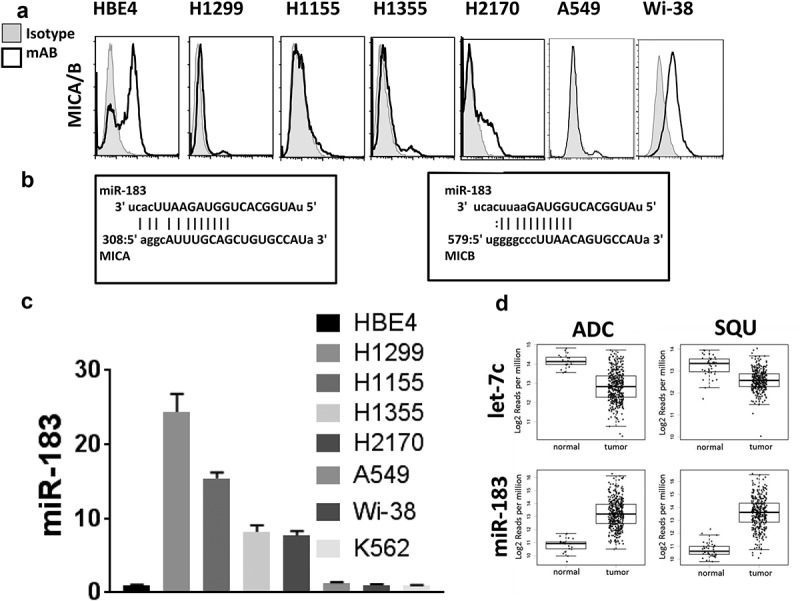
Human lung tumor cells express high levels of miR-183 but low levels of MICA/B. (a) Flow cytometric analysis of MICA/B in human lung tumor cell lines, H1299, H1155 H1355, H2170, A549, and Wi-38, in comparison to Epstein-Barr virus E6/7-transformed HBE4 lung epithelial cells. (b) Alignment of human MICA and MICB 3ʹUTR mRNA and miR-183 as depicted by the miRanda algorithm (microRNA.org).The number labeling the 5ʹ end of the MICA/B mRNA represents the nucleotide position downstream of the translation termination codon. Capital letters constitute the miR seed sequences and vertical lines depict miR binding to mRNA. (c) qPCR analysis of miR-183 in the same lung cell lines as (A) showed a reverse correlation with MICA/B expression. (d) Using the TCGA database, meta-analysis was conducted for miR-183 expression in 422 human lung adenocarcinoma (ADC) samples as compared to 46 normal adjacent tissues, and 332 squamous (SQU) cell carcinoma samples as compared to 45 normal adjacent tissues. Unlike let-7 miR, which is known to be downregulated in lung cancer, miR-183 was elevated.
To confirm clinically that elevated miR-183 in the TCGA database correlated with low to absent expression of MICA/B in lung cancer, we next evaluated surface expression of MICA/B in paraffin-embedded lung tumor tissues containing adenocarcinoma and squamous NSCLC. The Allred scoring system was used and a representative figure of Scores 0–3 is shown in Figure 2(a), as well as non-neoplastic lung parenchyma in Figure 2(b).41 Using this representative data, we see that immunohistochemical analysis demonstrates that the intensity of MICA/B staining in Grade 1, Grade 2 and Grade 3 squamous cell carcinoma specimens is no different from that seen in normal adjacent tissue. All tumor grades have approximately the same staining intensity for both squamous cell and adenocarcinomas. (Figure 2(c)). In addition, the percentage of MICA-positive tumor cells was not higher than that of adjacent normal tissues, using the Allred scoring system of 1 = 1/100, 2 = 1/10, 3 = 1/3, 4 = 2/3, 5 = >2/3 (Figure 2(d)). In adenocarcinoma specimens, again the intensity of MICA/B staining in all the grades was minor (Score 2) similar to that of adjacent normal tissues. Despite this, there exists a higher percentage of tumor cells expressing MICA/B (Figure 2(e,f)), which correlates with earlier Grades 1–2, but not Grade 3. Thus, MICA/B expression is minor, and is not overexpressed beyond normal levels in lung cancer tissues. We previously conducted meta-analysis of MICA and MICB mRNA gene expression in patient lung adenocarcinoma and normal tissues taken from data in a published report42 and also found little MICA/B gene expression in the tumors, equivalent to the levels seen in normal tissues.17 Thus, similar to the low levels of MICA/B seen in lung cell lines, primary patient lung cancer tissues do not express MICA/B transcripts or proteins beyond the levels seen in adjacent normal tissues.
Figure 2.
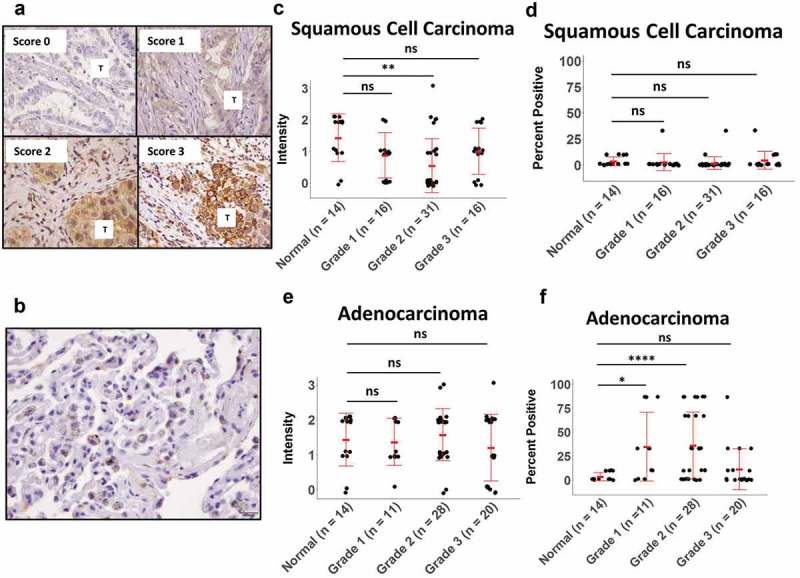
Immunohistochemical analysis of MICA/B expression in human lung tumor tissues. (a, b) Paraffin-embedded adenocarcinoma and squamous cell carcinoma tissues were stained with anti-MICA/B and scored 0–3 using the Allred Intensity scoring system. In figure 2A, tumor with different IHC scores are represented. In figure 2B, the adjacent non neoplastic lung parenchyma is represented with macrophages containing carbon deposits. (c, d) Staining Intensity and percentage of MICA/B+ positive tumor cells in squamous cell carcinoma, using the Allred Percentage scoring system of 1 = 1/100, 2 = 1/10, 3 = 1/3, 4 = 2/3, 5 = >2/3. (e, f) Staining Intensity and percentage of MICA/B+ positive tumor cells in adenocarcinoma. ns: not significant, *p < 0.05, **p < 0.001, ****p < 0.0001.
miR-183 targets MICA in lung tumor cells
To validate the putative miR-183 binding sites on wildtype (WT) MICA/B, we generated luciferase reporter constructs containing either the MICA 3ʹUTR or the MICB 3ʹUTR. We also mutated the 6–8 nucleotide seed sequence in the 3ʹUTR of MICA complementary to miR183, which is required for effective transcriptional repression (Figure 3(a)). Co-transduction of 293 cells with MICA-WT and miR-183 markedly reduced luciferase expression, as compared to MICA-WT together with scrambled control transduction, while mutant MICA-MUT, when co-expressed with miR183, resisted luciferase repression, suggesting that miR183 binding is critical for MICA gene regulation (Figure 3(b)). Similar experiments using MICB were conducted and, while miR-183 could repress luciferase activity in MICB-WT transfected cells, it could not interfere with luciferase activity in MICB-mut transfected cells. Thus, both MICA and MICB are subject to the same control by miR183.
Figure 3.
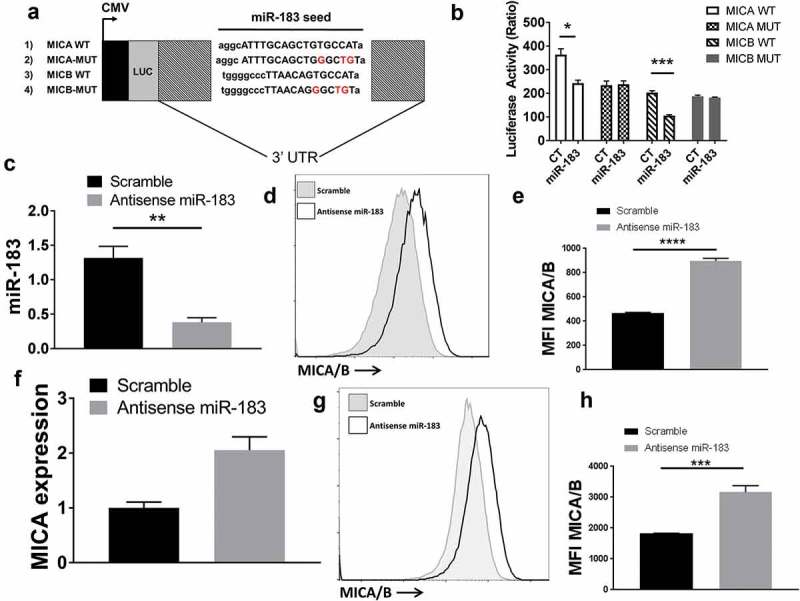
miR-183 targets the 3ʹUTR of MICA/B mRNA. (a) Schematic of luciferase reporter constructs carrying wild-type (WT) MICA or MICB 3ʹUTR as well as mutated (MUT) MICA or MICB 3ʹUTR. The miR seed sequences are depicted as capital letters and the mutated nucleotides are in red. CMV, cytomegalovirus promoter; LUC, luciferase. (b) 293T cells were transfected with luciferase constructs containing MICA-WT, MICA-MUT, MICB-WT or MICB-MUT (500ng), renilla luciferase (5ng) together with miR-183 (25nM). After 24 h, the cells were lysed and quantified for firefly luciferase activity. (c) Lentiviral constructs containing scramble control or anti-sense miR-183 were transfected into H1355 lung tumor cells. qPCR analysis was conducted to confirm the downregulation of miR-183 expression by antisense miR-183 (d, e) Flow cytometric analysis using a monoclonal antibody that detects a common sequence in MICA and MICB indicated that antisense miR-183 transfected H1355 tumor cells upregulated MICA/B expression. D is a representative staining of MICA/B. (f, g, h) H1299 lung tumor cells also transfected with antisense miR-183 showed upregulation of both MICA mRNA by qPCR and protein by flow cytometry analysis. G, H is a representative staining of MICA/B. Each of the experiments is representative of at least 3 experiments performed.
We next verified that miR-183 directly controls MICA/B in lung tumor cells. To accomplish this, we transfected antisense miR183 into H1355 lung tumor cells to analyze if loss of miR183 expression could increase MICA/B levels on the transfected tumor cells. We first confirmed that antisense miR-183 transfection was successful in suppressing miR-183 expression by qPCR analysis (Figure 3(c)). Next, we examined the level of MICA and MICB expression in these transfected cells by flow cytometry using a monoclonal antibody that recognizes both MICA/B. MICA/B were significantly upregulated in antisense miR-183-transfected tumor cells (mean fluorescence intensity (MFI) of 4660) as compared to scramble control (MFI of 1958) (Figure 3(d,e)). To ensure that miR-183 is a common regulator of MICA/B, we also tested another lung cell line, H1299, in a similar manner. Expression of antisense miR-183 in H1299 cells markedly increased MICA/B levels (MFI of 8263) in comparison to scramble (MFI of 3997) and upregulated MICA mRNA (Figure 3(f–h)). Thus, blockade of miR-183 expression was sufficient to enhance MICA/B expression in lung tumor cells. Together, the results of the luciferase reporter constructs in HeLa cells together with the anti-sense miR-183 overexpression in lung tumor cells indicate that it is a direct effect of miR183 binding to the MICA/B 3ʹUTR that regulates its expression.
TGFβ drives miR183 expression to downregulate MICA/B
We had earlier documented that TGFβ was highly elevated in lung tumor cell lines as well as primary lung tumor tissues.17 In fact, we reported the novel observation that TGFβ can induce miR-183 from NK cells leading to the loss of a critical adaptor protein required in mediating tumor lysis. This is highly relevant to all aspects of cancer immunology and immunotherapy, as NK cells are critical to antitumor immunity. Since we showed that the exposure of TGFβ reduces the effectiveness of NK cells, which induces miR-183 and thus suppresses NK cells, we felt that this topic deserved more attention. Relevant to our past work and thus current study, TGFβ has additionally been reported by others to downregulate NKG2D ligands.38 Based on the conclusions of the above cited works, and the knowledge that TGFβ is elevated in lung cell lines, we thus endeavored to explore whether TGFβ was involved in inducing miR-183 expression in the tumor cell themselves, consequently leading to suppression of MICA/B. The novelty of this concept lies in the ability of the lung cancer cells to contribute to tumor induced immune suppression. Thus, to pursue this exciting concept, we transfected H1355 lung tumor cells with shRNA TGFβ or scramble control shRNA prior to analysis of miR-183 and MICA/B expression. We first confirmed that TGFβ mRNA was effectively knocked down by shRNA TGFβ transfection, as compared to the scramble control (Figure 4(a)). Loss of TGFβ resulted in a corresponding reduction in miR-183 expression (Figure 4(b)). In contrast, TGFβ depletion led to increased MICA/B expression, with MFI rising from 659 in scramble control-shRNA transfected cells to 1077 in the shRNA TGFβ-transfected cells (Figure (4c,d)). We reproduced similar results in H1299 lung tumor cells. Depletion of TGFβ with specific shRNA (Figure 4(e)) also caused significant depletion of miR-183 in H1299 cells (Figure 4(f)), accompanied by an upregulation of MICA/B (Figure 4(g,h)). Since lentiviral transfection can induce a type I interferon response, we utilized a pan-Type I Interferon neutralizing antibody43 to ensure that our observed biologic effects were due to the silencing of TGFβ, and not to secondary viral transfection effects or type I interferon itself. The results were consistent with that of the original experiment, where the loss of TGFB led to increased MICA/B expression by flow cytometry the H1299 cell line(Fig. S2B, S2C). Thus, TGFβ appears to induce miR-183 which can bind the 3ʹUTR of MICA/B to disrupt its expression, and this phenomenon is not Type I interferon dependent.
Figure 4.
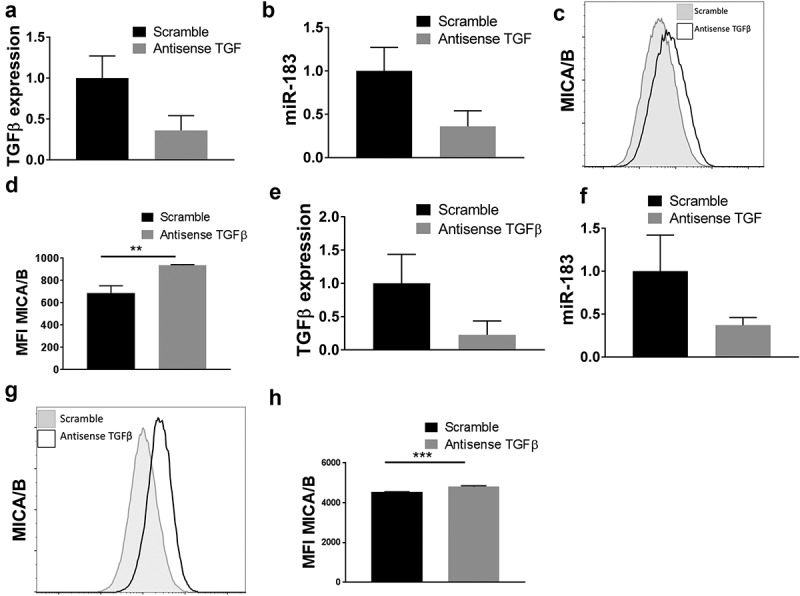
TGFβ suppresses MICA/B expression via miR-183. (a, b) H1355 tumor cells were transfected with antisense TGFβ or scramble control prior to analysis of TGFβ mRNA or miR-183 by qPCR. Reduced TGFβ expression correlated with reduced miR-183 expression in these cells. (c, d) In these same cells, flow cytometric analysis and the mean fluorescence intensity indicated that antisense TGFβ-transfected cells expressed higher levels of MICA/B than scramble control. C, D is a representative staining of MICA/B. (e, f, g h) The experiments in H1355 tumor cells were repeated in H1299 tumor cells and confirmed that antisense TGFβ transfection reduces TGFβ mRNA with a corresponding downregulation of miR-183, accompanied by heightened expression of MICA/B in tumor cells. G, H is a representative staining of MICA/B. Each of the experiments is representative of at least 3 experiments performed.
miR-183 expression blocks NKG2D-mediated lysis of tumor cells
To analyze the biological consequence of miR-183 expression in tumor cells, we next assessed the sensitivity of lung tumor cells to NKG2D-mediated lysis by cytotoxic immune cells. First, we assessed the MHC class I expression of tumor cell lines, as class I presence is important for NK response. We examined healthy peripheral blood mononuclear cells (PBMC) as a positive control, K562 immortalized myologenous leukemic cells as a negative control, and H1299 and H1355 lung tumor cells. We found moderate expression in H1355, and higher expression in H1299 cells (Fig. S3). These cell lines are thus class I competent, a factor that is prominent because lung tumor cells often lose class I expression as a mechanism of tumor immune escape.44 NK cells sense and detect MHC Class I, and through the missing self hypothesis, recognize and eliminate cells missing Class I.45 NK cells, however, cannot be used as effector cells to study NKG2D-specific lytic events, because IL-2 used to culture human NK cells induces not only high levels of NKG2D but also upregulated other activating NK receptors such as activatory Killer Immune Receptors (aKIRs),46 NKp44,47 NKG2C,48 and NKG2E,49 which recognize tumor-associated ligands unrelated to MICA/B. To overcome this obstacle, we generated cytotoxic CD8+ T cells by first purifying CD8+ T cells via negative selection from normal peripheral blood mononuclear cells and then placing them in culture for 7 days with anti-CD3/CD28 containing medium plus IFNγ. We demonstrated that such short term stimulation induces primarily NKG2D but no other activating receptors, including NKp44, NKp46, KIR2DS4, or NKG2C (Figure 5(a)). Indeed, these CD8+ T cells lysed H1355 lung tumor cells which could be effectively blocked by anti-NKG2D (Figure 5(b)). A CD8+ T cell response is restricted through MHC Class I. These tumor cells are Class I competent, however the T cell receptor on CD8+ T cells is not involved because H1355 tumor cells are allogeneic to the T effector cells and have no matching MHC-Class I.50 We next transfected antisense miR-183 into H1355 tumor cells and tested their susceptibility to lysis by CD8+ T cells. In 2 separate normal donors, we found that CD8+ T cells had markedly increased tumoricidal activity against H1355 mir-183 transfected tumor cells, as compared to scramble control (Figure 5(d,e)). To prove that this relationship between mir-183 and MICA/B sensitizes effector cells, we transfected H1299 and H1355 tumor cells with a vector to overexpress MICA/B. As expected, the tumor lysis phenotype was similar (Figure 5(c). The CD8+ T cells had a similar trend in tumoricidal acitivity as did the antisense miR-183 transfected cells. This confirms that this relationship does indeed work both ways. Taken together, these results indicate that miR-183 suppression, either by means of silencing mir-183 or overexpressing MICA/B in tumor cells sensitizes them to be better recognized by T cells via the NKG2D-MICA/B pathway.
Figure 5.
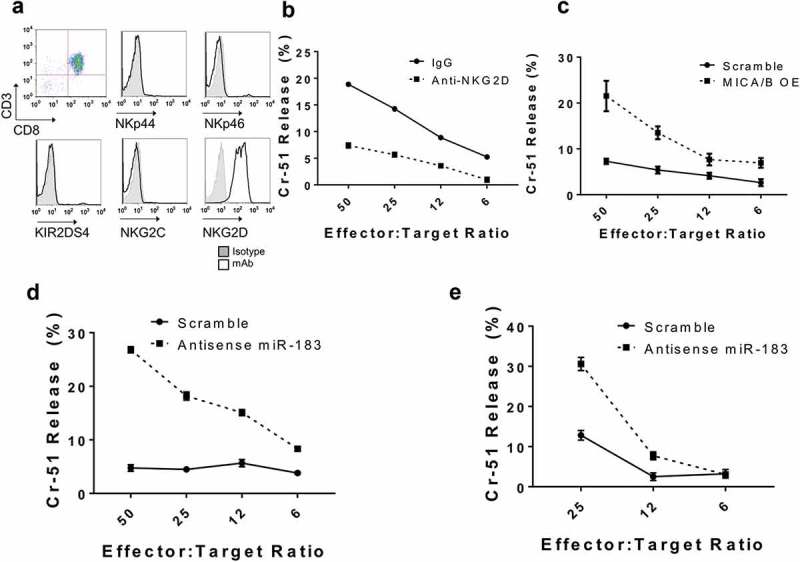
Depletion of miR-183 in tumor cells enhances sensitivity to lysis by activated T cells. (a) CD8+ T cells, purified by negative selection from normal peripheral blood mononuclear cells, were activated with IFNγ and anti-CD3/CD28 and cultured in IL2 for 7 days. Flow cytometric analysis indicated that such short term culture induced only NKG2D but not the other NK receptors on the T cells. (b) Short term-activated CD8+ T cells kill H1355 tumor cells in a 5 h Cr release assay, which can be blocked by anti-NKG2D. (c) Short term-activated CD8+ T cells were transfected with MICA/B overexpressing vector or scramble control, and then tested for cytotoxicity against H1299 tumor cells. MICA/B transfected tumor cells were more susceptible to lysis than scramble control, indicating overexpression of MICA/B in tumor cells increases sensitivity to lysis by activated T cells. (d, e) Short term-activated CD8+ T cells from 2 separate donors were transfected with antisense miR-183 or scramble control, and then tested for cytotoxicity against H1355 tumor cells. Antisense miR-183 transfected tumor cells were more susceptible to T cell lysis than scramble control. Each of the experiments is representative of at least 3 experiments performed.
Discussion
In this study, we provide firm evidence of a novel molecular process that promotes immune escape in human lung tumor cells. It identifies a new link between TGFβ and miR-183 that drives repression of a critical NK ligand, MICA/B, causing the loss of detection by immune cells expressing its receptor, NKG2D. Immunohistochemical assessment of tissues from lung cancer patients with either adenocarcinoma or squamous cell carcinoma indicated a lack of MICA/B expression while meta-analysis of the TGCA database for human lung adenocarcinoma and squamous carcinoma revealed that both lung tumor types display a high expression of miR-183. In light of the reverse correlation of MICA/B expression with miR-183, we conducted a search for miR binding sites in the 3ʹUTR of both MICA and MICB and identified a 7-mer binding sequence that tightly complements the mature 22 nucleotide miR-183 in MICA and an even longer 9-mer sequence that completely mirrors miR-183 in MICB. Using luciferase reporter constructs carrying the MICA or MICB 3ʹUTR, we were able to demonstrate that miR-183 effectively repressed MICA and MICB. Sequence-specific mutation of the binding site on MICA and MICB further corroborated the requirement of miR-183 in regulating MICA/B in these reporter constructs. Besides such molecular elucidation of miR-183 binding to the MICA/B 3ʹUTR, analysis of the biological process in H1355 lung tumor cells indicated that antisense miR-183 transduction can upregulate MICA/B expression and sensitize these cells to lysis by activated allogeneic T cells expressing NKG2D. This process was reproduced in another lung tumor cell line, H1299. Use of short term activated allogeneic T cells allowed for the recognition of tumor cells by NKG2D without TCR which is self-MHC Class I specific. It also circumvents the issue posed by activated NK cells which express other activating receptors such as aKIRs, NKp30,NKp44, NKG2C that could mask specific MICA/B-targeted killing.23,24 We also provide evidence of the role of TGFβ in initiating the process, by demonstrating that anti-sense TGFβ transfection into human lung tumor cells can downregulate miR-183 expression, with corresponding increase in MICA/B expression.
Earlier studies have reported that MICA can be controlled by several other miRs, namely miR-10, miR-20, miR-93, miR-106, miR-373, miR-520.51,52 However, these miRs were uncovered through the TargetSCAN algorithm to search for miR consensus sites on the 3ʹUTR of the MICA/B gene and are not linked to the tumor microenvironment. It is not known if any of these miRs are inducible by TGFβ, as we have shown for miR-183 expression in our lung tumor system. However, miR-96 has been shown to be regulated by TGFβ, which targets a negative regulator of mTOR, AKT1S153 and promotes prostate-to-bone metastases. As miR-96 is a family member of the miR183-96-182 cluster, it is likely that the miR-183-TGFB axis may affect mTOR signaling in some way, which is important for NK cell metabolism and functionality.54 An additional link between mTOR signaling has been linked to TGFβ and is implicated in NK dysfunction.55 Additionally, the miR-183-mTOR pathway has been explored in spinal muscular atrophy (SMA), but not in the context of cancer and NK cells. This represents a unique pathway that will be relevant for further studies.56 Another interesting observation pertaining to MICA regulation is the report that IFNγ can repress MICA via miR-520.57 Thus, other miRs may respond to signals related or unrelated to TGFβ, and impact NK functionality through multiple pathways, including mTOR.
Shedding of MICA/B is another process utilized by tumor cells but it is still not settled whether shed MICA/B blocks or enhances NKG2D-mediated NK lysis.35-37 This shedding phenomenon could explain our consistent observation of better detection of cytoplasmic MICA/B by western blot analysis than by flow cytometric analysis of surface MICA/B.
One of the hallmarks of cancer is immune escape.2 Our work constitutes the first report of a tumor-promoting miR, namely miR-183, which can also drive immune escape of tumor cells. In addition to numerous reports of the ability of miR-183 to enhance tumor progression via direct interference with key regulators of cell survival and proliferation, we show here that miR-183 expressed within lung tumor cells can repress MICA/B expression to avoid detection by NKG2D+ CD8+ T cells. This dual effect of miR-183 on tumor cells can be a powerful driver of tumor cell progression. Remarkably, there is a third property of miR-183 that we have recently uncovered. Rather than a direct effect on tumor cells, we reported that TGFβ-inducible miR-183 can repress NK cells within the tumor microenvironment. Here, miR-183 targets a separate NK lytic mechanism unrelated to the MICA/B/NKG2D ligand/receptor system. In this case, the ligation of TGFβ transcriptionally induces miR-183 in NK cells, and it is this NK-associated miR-183 that specifically represses DAP12, a vital adaptor protein, required to anchor numerous NK activating receptors on the cell surface, including activatory Killer Immune Receptors (KIRs),46 NKp44,47 NKG2C,48 and NKG2E.49 It is to be noted that the ligands for these receptors are entirely different from MICA/B and ULBPs1-6. Activatory KIRs specifically target classical MHC-I molecules, thus facilitating the recognition of allogeneic tumor cells, useful in bone marrow transplantation in the allogeneic setting.58 NKG2C and NKG2E primarily recognize HLA-E, which belongs to the MHC-I heavy chain family, but bind a restricted subset of peptides derived from the leader peptides of other MHC-I molecules.59 HLA-E is often overexpressed in malignant cells, rendering them sensitive to NK lysis.60 The ligand for NKp44 is unclear, although viral hemagglutinin and heparin derivatives have been reported to bind NKp44.61,62 However, anti-NKp44 can dampen NK lysis of various tumor types, suggesting a putative common ligand expressed on tumor cells. MiR-183 repression of DAP12 can thus have a potent effect on NK cells, suppressing a variety of activating receptors to essentially blind them to the tumor cells.
The lesson learnt from our studies of miR-183 on immune modulation, together with other reports on tumor cell proliferation and metastasis, is profound. The tumor cell, working through TGFβ, is endowed with a powerful transcriptional regulator in miR-183 with three essential subversive properties. First, miR-183 can target endogenous genes to favor aggressive growth. Second, it can dial down NK ligands, such as MICA/B, to avoid immune detection by a highly potent NK receptor, NKG2D, that is expressed on both activated NK and CD8+ T cells. NKG2D is also present on γδT cells which infiltrate a number of human solid cancers.63 Third, miR-183 can not only modulate the tumor itself to hide from the immune system, it can also disarm the surrounding NK cells by targeting yet another NK recognition system through repression of DAP12 that anchors multiple NK receptors, including KIRs, NKp44, NKG2C and NKG2E. It is notable that tumor cells have evolved to regulate only DAP12 in NK cells instead of its associated NK receptors, leaving them intact but cloistered in the cytoplasm, representing an economic means to silence NK cells. In the context of the NKG2D receptor, tumor cells opted to utilize miR-183 to repress its ligands, MICA/B. Tumor-associated miR-183 thus represents a global strategy by the tumor to disable immune recognition by innate immune receptors and simultaneously promote tumor growth. Whether this TGFβ/miR-183 network is operative in other tumor types has not yet been explored but it seems possible that it is a common strategy of most tumor cells, given the abundance of reports on TGFβ and miR-183 co-expression in breast, colorectal, hepatic, pancreatic and prostate cancer.
Materials and methods
Cell culture and reagents
H1299, H1355, H1155, A549, and H2170 human lung tumor cells were maintained in 10% (vol/vol) FBS, RPMI (Thermo Fisher Scientific) supplemented with 1% Pen/Strep, 1% L-glutamine (Thermo Fisher Scientific), and 1% MEM Non-Essential Amino Acids Solution (NEAA) (Corning). HBE4 human lung epithelial cells transformed with E6/E7 of the Epstein-Barr virus (American Type Culture Collection) were maintained in 10% (vol/vol) FBS Keratinocyte SFM supplemented with cholera toxin (CTX 10 ng/mL), bovine pituitary extract (50 ng/ml), and epidermal growth factor (5 ng/ml). MiaPaCa and PANC-1 human pancreatic ductal adenocarcinoma cell lines (American Type Culture Collection), Wi-38 human lung cells, HeLa human cervical adenocarcinoma cells, and K562 human chronic myelogenous leukemia cells were maintained in were maintained in 10% (vol/vol) FBS, Dulbecco’s Modified Eagle’s Medium (DMEM) (Thermo Fisher Scientific) supplemented with 1% Pen/Strep, 1% L-glutamine (Thermo Fisher Scientific), and 1% MEM Non-Essential Amino Acids Solution (NEAA) (Corning).
Primary cytotoxic T lymphocytes (CTLs) were obtained from peripheral blood of healthy donors from OneBlood. CD8+ T cells were purified from peripheral blood mononuclear cells by negative selection (CD8+ T cell Isolation Kit; Miltenyi Biotech). Purifications yielded >95% CD8+CD3+ cells by FACS analysis. Cells were then incubated overnight at 37ºC in culture media supplemented with 10% FBS RPMI (Thermo Fisher Scientific), 1% NEAA (Corning), 1% Sodium Pyruvate (NaPyr) (Corning), 1% Pen/Strep, 1% L-glutamine (Thermo Fisher Scientific), MycoZap-PR reagent (Lonza), and IFNγ (1000 units/ml), followed by activation with anti-CD3/anti-CD28 and expansion in recombinant human IL-2 (100 U/mL) for 6 days.
Flow cytometry analysis
For analysis of surface receptors, 1 × 106 cells of each cell line were stained with 5 µl anti-human PE-conjugated antibody to MICA/B (eBioscience; clone 6D4) or 2.5 uL PE-conjugated antibody to HLA-ABC (BD: Clone W6/32)Following incubation with anti-MICA/B antibody or anti-HLA-ABC, cells were washed and resuspended in FACs buffer (1X PBS containing 2% BSA and 0.1% sodium azide) and acquired on a LSR II Flow Cytometer (BD Biosciences). Primary CTLs were stained with the following antibodies: CD8-FITC (BD Pharmingen; clone SK1), CD3-PE (Biolegend; clone SK7), NKp44-APC (R&D Systems; clone 253415), NKp46-APC (BD Pharmingen; clone 9-E2), KIR2DS4-APC (R&D Systems; clone 179315), NKG2C-APC (R&D Systems; clone 134591), and NKG2D-APC (BD Pharmingen; clone 1D11). Data were analyzed using FlowJo version 10 and figures were prepared in GraphPad Prism 7.
Type I interferon neutralization
For neutralizing of Type I interferon, cells were treated with Human Type 1 Interferon Neutralizing Antibody (PBL Assay Science) for 30 minutes at a concentration of 1:50. After incubation at 37ºC, cells were harvested and washed, and used for further downstream assays. Data were analyzed, and figures were prepared in GraphPad Prism 7.
Chromium release assay
Chromium release assay was performed as described.17 Anti-sense miR-183 lentiviral transduced H1299 or H1355 cells were used at targets. Targets were labeled with 200μCi of Na [51Cr] chromate (Amersham) for 1 hr at 37°C. Target cells were then washed and plated at 5 × 103 cells/well with CD8+ T cells to give an effector/target ratio of 50/1 to 6/1 in a round bottom microtiter plate. After incubation at 37ºC for 5 hr, supernatants were harvested and counted in a γ-counter. The percent specific 51Cr release was then calculated as: [(experimental cpm – spontaneous cpm)/total cpm incorporated] x 100. All determinations were performed in triplicate, and the SEM of all assays was calculated typically ~5% of the mean or less. Figures were prepared in GraphPad Prism 7.
Quantitative RT-PCR
To detect mature microRNA (miR) transcripts, PCR was performed with Taqman reagents (Thermo Fisher Scientific). Purified RNA was reverse transcribed with primer for has-miR-183 (miR-183) and RNU6B followed by real-time PCR amplification with 6-carboxyfluorescein-conjugated primers for miR-183 and RNU6B in TaqMan Universal PCR Master Mix, no AmpErase UNG (Thermo Fisher Scientific). Experimental miRs were normalized to RNU6B. To detect MICA, MICB and TGFβ transcripts, total RNA was extracted using Trizol Reagent (Thermo Fisher Scientific). Total RNA was then reverse transcribed using the qScript cDNA Supermix (Quanta). Real-time PCR amplification was performed with primers specific for the respective mRNA, using the Power SYBR Green Master Mix (Thermo Fisher Scientific). Relative fold changes in expression were determined by using the comparative cycle threshold method (2 −∆∆CT). Figures were prepared in GraphPad Prism 7.
Immunohistochemical staining of lung tmas
Tissue cores (0.6 mm) from 59 adenocarcinoma (ADC), 67 squamous cell carcinoma and 14 normal adjacent human lung tissues purchased from US Biomax were fixed in 10% (vol/vol) neutral buffered formalin for construction of a tissue microarray (TMA). Sections of the TMA (4 μm) were stained for MICA/B expression as described using anti-MICA/B (R&D Systems)17 at a dilution of 1:100, for 2 h at room temperature with the Ventana automated immunostainer Discovery XT (Ventana Medical Systems, Tucson, AZ). As a negative control, non-immune mouse sera were used, omitting the MICA/B antibody during the primary antibody incubation step. The slides were read by a certified pathologist and co-author (DC) in a blinded fashion and the MICA/B protein expression levels were measured using the Allred semiquantitative scoring system.41
Bioinformatics analysis of miR binding
Analysis of the MICA (NM_000247) and MICB (NM_005931) 3ʹUTR was performed using the publically available algorithm miRANDA (microRNA.org) or TargetScan.
Cloning, transfection and luciferase assay
The oligonucleotide containing the MICA or MICB 3ʹ UTR (synthesized by Integrated DNA Technologies) was amplified by RT-PCR (HotSTar Mastermix, Qiagen). Amplified products were run on a 1% agarose gel, and extracted using QIAquick Gel Extraction Kit, Qiagen) and subcloned into the pMIR-Report luciferase vector (Promega) to generate either MICA or MICB-luciferase reporter vector. The scrambled miR-183 seed-site sequence was generated by Mutagenex. The constructs were verified for correct insert sequence and orientation by DNA sequencing. For luciferase reporter assays, 293 cells were grown (70% confluent) in 12-well plates and transfected (Lipofectamine 2000; Thermo Fisher Scientific) with reporter constructs (500 ng per well), renilla luciferase (5 ng per well), and premiR-precursors (25 nm) (Applied Biosystems). At 24 h later, cells were lysed, and luciferase activity was quantified (Dual-Luciferase Reporter Assay; Promega) on a single automatic injection luminometer (Turner Biosystems). Ratios of renilla to firefly luciferase were quantified, and quadruplicates were averaged; experimental miRs were normalized to control scramble miRs. Figures were prepared in GraphPad Prism 7.
Lentiviral transfection of cell lines
HIV-based lentiviral expression constructs (purchased from SBI) containing miR-183 (pCDH-CMV-MCS-EF1), antisense miR-183 (pGreenPuro shRNA), or scramble control were packaged with third-generation packaging plasmids (PMD-g, PMD-Lg, Rev; a kind gift of Todd Fehniger, Washington University St. Louis) in 293T cells. For TGFβ shRNA, plasmids (psi-LVRH1GP) were purchased from GeneCopoeia and packaged as mentioned above. Viral supernatants were harvested, filtered, and concentrated (Lenti-X concentrator; Clontech). H1355 or H1299 cells (5 × 105 cells per well in a 6-well plate) were spin-transduced with lentiviral particles at a multiplicity of infection (MOI) of 20 in RPMI. After two infections, cells were maintained in 10% (vol/vol) FBS RPMI supplemented with 1% Pen/Strep, 1% L-glutamine (Thermo Fisher Scientific), and puromycin to select for transduced cells.
Analysis of published lung cancer datasets
Expression of miR-183 and let7 in human ADC and SQU lung cancer in comparison to normal tissues were analyzed using the publically available Cancer Genome Atlas (TCGA) directories:
LUAD:https://tcgadata.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/luad/cgcc/bcgsc.ca/illuminahiseq_mirnaseq/mirnaseq/bcgsc.ca_LUAD.IlluminaHiSeq_miRNASeq.Level_3.1.12.0/
LUSC:https://tcgadata.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/lusc/cgcc/bcgsc.ca/illuminahiseq_mirnaseq/mirnaseq/bcgsc.ca_LUSC.IlluminaHiSeq_miRNASeq.Level_3.1.7.0/.
TCGA IlluminaHiSeq miRNASeq level 3 data were downloaded from the open http directory of the TCGA data portal. The level 3 (normalized) miRNA quantification data analyses were utilized: LUAD = 1.12.0, LUSC = 1.7.0. Sample counts were as follows: LUAD (tumor = 422, normal = 46); LUSC (tumor = 332, normal = 45). Figures were prepared in R.
Funding Statement
This work was funded by the Manuel and Adeline Garcia Endowed Chair (to J.Y.D.), DOD LC140277 (to J.Y.D.), Florida Biomedical 6JK01 (to J.Y.D.), DOD LC140270 (to T.Le), the T32 Training Grant CA115308 (N.T.) and K01CA187020 (E.A.E).
Acknowledgments
This work has been supported by the Flow cytometry Core Facility, Molecular Genomics, Tissue Core Facilities and the Microscopy Core Facility of the H. Lee Moffitt Cancer Center, a comprehensive cancer center designated by the NCI (Cancer Center support grant P30CA076292).
Disclosure of potential conflicts of interest
The authors declare no conflict of interest
Author contributions
T.L., W.K., S.S.D, J.Y.D., E.A.E, and S.W. designed research; T.L., W.K., S.S.D., N.T., M.M.T., D.L.G., A.B., and W.A. conducted experiments; J.T. conducted bioinformatics; D.C. performed histopathology; T.L., W.K., S.S.D, N.T., J.Y.D., J.L., S.W. analyzed data; T.L., W.K., J.Y.D., and S.W. wrote the paper.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Torre LA, Siegel RL, Jemal A.. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. doi: 10.1007/978-3-319-24223-1_1. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA.. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. EMBO Mol Med. 2012;4:143–159. doi: 10.1002/emmm.201100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 5.Zhu W, Liu X, He J, Chen D, Hunag Y, Zhang YK. Overexpression of members of the microRNA-183 family is a risk factor for lung cancer: a case control study. BMC Cancer. 2011;11:393. doi: 10.1186/1471-2407-11-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu W, Zhou K, Zha Y, Chen D, He J, Ma H, Liu X, Le H, Zhang Y, Zheng SG. Diagnostic value of serum miR-182, miR-183, miR-210, and miR-126 levels in patients with early-stage non-small cell lung cancer. PLoS One. 2016;11:e0153046. doi: 10.1371/journal.pone.0153046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Y, Liang AJ, Fan YP, Huang YR, Zhao XM, Sun Y, Chen X-F. Dysregulation and functional roles of miR-183-96-182 cluster in cancer cell proliferation, invasion and metastasis. Oncotarget. 2016;7:42805–42825. doi: 10.18632/oncotarget.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li P, Sheng C, Huang L, Zhang H, Huang L, Cheng Z, Andrulis IL, Spurdle AB, Schmidt MK, Schmutzler RK, et al. MiR-183/-96/-182 cluster is up-regulated in most breast cancers and increases cell proliferation and migration. Breast Cancer Res. 2014;16:473. doi: 10.1186/s13058-014-0492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarver AL, French AJ, Borralho PM, Thayanithy V, Oberg AL, Silverstein KA, Morlan BW, Riska SM, Boardman LA, Cunningham JM, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer. 2009;9:401. doi: 10.1186/1471-2407-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou T, Zhang GJ, Zhou H, Xiao HX, Li Y. Overexpression of microRNA-183 in human colorectal cancer and its clinical significance. Eur J Gastroenterol Hepatol. 2014;26:229–233. doi: 10.1097/MEG.0000000000000002. [DOI] [PubMed] [Google Scholar]
- 11.Li J, Fu H, Xu C, Tie Y, Xing R, Zhu J, Qin Y, Sun Z, Zheng X. miR-183 inhibits TGF-beta1-induced apoptosis by downregulation of PDCD4 expression in human hepatocellular carcinoma cells. BMC Cancer. 2010;10:354. doi: 10.1186/1471-2407-10-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren LH, Chen WX, Li S, He XY, Zhang ZM, Li M, Cao R-S, Hao B, Zhang H-J, Qiu H-Q, et al. MicroRNA-183 promotes proliferation and invasion in oesophageal squamous cell carcinoma by targeting programmed cell death 4. Br J Cancer. 2014;111:2003–2013. doi: 10.1038/bjc.2014.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mihelich BL, Khramtsova EA, Arva N, Vaishnav A, Johnson DN, Giangreco AA, Martens-Uzunova E, Bagasra O, Kajdacsy-Balla A, Nonn L. miR-183-96-182 cluster is overexpressed in prostate tissue and regulates zinc homeostasis in prostate cells. J Biol Chem. 2011;286:44503–44511. doi: 10.1074/jbc.M111.262915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu J, Li A, Hong SM, Hruban RH, Goggins M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin Cancer Res. 2012;18:981–992. doi: 10.1158/1078-0432.CCR-11-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang X, Zheng D, Hu P, Zeng Z, Li M, Tucker L, Monahan R, Resnick MB, Liu M, Ramratnam B. Glycogen synthase kinase 3 beta inhibits microRNA-183-96-182 cluster via the beta-Catenin/TCF/LEF-1 pathway in gastric cancer cells. Nucleic Acids Res. 2014;42:2988–2998. doi: 10.1093/nar/gkt1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, Haura EB, Schabath MB, Coppola D, Wei S, et al. TGF-beta-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci USA. 2014;111:4203–4208. doi: 10.1073/pnas.1319269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu WH, Chang LS. Suppression of Akt/Foxp3-mediated miR-183 expression blocks Sp1-mediated ADAM17 expression and TNFalpha-mediated NFkappaB activation in piceatannol-treated human leukemia U937 cells. Biochem Pharmacol. 2012;84:670–680. doi: 10.1016/j.bcp.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka H, Sasayama T, Tanaka K, Nakamizo S, Nishihara M, Mizukawa K, Kohta M, Koyama J, Miyake S, Taniguchi M, et al. MicroRNA-183 upregulates HIF-1alpha by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J Neurooncol. 2013;111:273–283. doi: 10.1007/s11060-012-1027-9. [DOI] [PubMed] [Google Scholar]
- 20.Qiu M, Liu L, Chen L, Tan G, Liang Z, Wang K, Liu J, Chen H. microRNA-183 plays as oncogenes by increasing cell proliferation, migration and invasion via targeting protein phosphatase 2A in renal cancer cells. Biochem Biophys Res Commun. 2014;452:163–169. doi: 10.1016/j.bbrc.2014.08.067. [DOI] [PubMed] [Google Scholar]
- 21.Sarver AL, Li L, Subramanian S. MicroRNA miR-183 functions as an oncogene by targeting the transcription factor EGR1 and promoting tumor cell migration. Cancer Res. 2010;70:9570–9580. doi: 10.1158/0008-5472.CAN-10-2074. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Quan H, Wang S, Li X, Che X. MiR-183 promotes growth of non-small cell lung cancer cells through FoxO1 inhibition. Tumour Biol. 2015;36:8121–8126. doi: 10.1007/s13277-015-3550-8. [DOI] [PubMed] [Google Scholar]
- 23.Djeu JY, Jiang K, Wei S. A view to a kill: signals triggering cytotoxicity. Clin Cancer Res. 2002;8:636–640. [PubMed] [Google Scholar]
- 24.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. doi: 10.1038/nrc.2015.5. [DOI] [PubMed] [Google Scholar]
- 25.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. [DOI] [PubMed] [Google Scholar]
- 26.Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, Phillips JH. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 1999;285:730–732. [DOI] [PubMed] [Google Scholar]
- 27.Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci USA. 1999;96:6879–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutherland CL, Rabinovich B, Chalupny NJ, Brawand P, Miller R, Cosman D. ULBPs, human ligands of the NKG2D receptor, stimulate tumor immunity with enhancement by IL-15. Blood. 2006;108:1313–1319. doi: 10.1182/blood-2005-11-011320. [DOI] [PubMed] [Google Scholar]
- 29.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–441. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carayannopoulos LN, Naidenko OV, Kinder J, Ho EL, Fremont DH, Yokoyama W. Ligands for murine NKG2D display heterogeneous binding behavior. Eur J Immunol. 2002;32:597–605. doi: 10.1002/1521-4141(200203)32:3<597::AID-IMMU597>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 31.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1:119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 32.Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. 2015;3:575–582. doi: 10.1158/2326-6066.CIR-15-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujita H, Hatanaka Y, Sutoh Y, Suzuki Y, Oba K, Hatanaka KC, Mitsuhashi T, Otsuka N, Fugo K, Kasahara M, et al. Immunohistochemical validation and expression profiling of NKG2D ligands in a wide spectrum of human epithelial neoplasms. J Histochem Cytochem. 2015;63:217–227. doi: 10.1369/0022155414563800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okita R, Yukawa T, Nojima Y, Maeda A, Saisho S, Shimizu K, Nakata M. MHC class I chain-related molecule A and B expression is upregulated by cisplatin and associated with good prognosis in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2016;65:499–509. doi: 10.1007/s00262-016-1814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, Raulet DH. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015;348:136–139. doi: 10.1126/science.1258867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 37.Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol. 2002;169:4098–4102. doi: 10.4049/jimmunol.169.8.4098. [DOI] [PubMed] [Google Scholar]
- 38.Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, Weller M, Friese MA. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain. 2006;129:2416–2425. doi: 10.1093/brain/awl205. [DOI] [PubMed] [Google Scholar]
- 39.Castro D, Moreira M, Gouveia AM, Pozza DH, De Mello RA. MicroRNAs in lung cancer. Oncotarget. 2017;8:81679–81685. doi: 10.18632/oncotarget.20955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 41.Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155–168. [PubMed] [Google Scholar]
- 42.Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown BD, Sitia G, Annoni A, Hauben E, Sergi LS, Zingale A, Roncarolo MG, Guidotti LG, Naldini L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood. 2007;109:2797–2805. doi: 10.1182/blood-2006-10-049312. [DOI] [PubMed] [Google Scholar]
- 44.Korkolopoulou P, Kaklamanis L, Pezzella F, Harris AL, Gatter KC. Loss of antigen-presenting molecules (MHC class I and TAP-1) in lung cancer. Br J Cancer. 1996;73:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–244. [DOI] [PubMed] [Google Scholar]
- 46.Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH, Mihelich BL, Khramtsova EA, Arva N, Vaishnav A, Johnson DN, et al. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391:703–707. doi: 10.1038/35642. [DOI] [PubMed] [Google Scholar]
- 47.Cantoni C, Bottino C, Vitale M, Pessino A, Augugliaro R, Malaspina A, Parolini S, Moretta L, Moretta A, Biassoni R. NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J Exp Med. 1999;189:787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lanier LL, Corliss B, Wu J, Phillips JH. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity. 1998;8:693–701. [DOI] [PubMed] [Google Scholar]
- 49.Orbelyan GA, Tang F, Sally B, Solus J, Meresse B, Ciszewski C, Grenier J-C, Barreiro LB, Lanier LL, Jabri B. Human NKG2E is expressed and forms an intracytoplasmic complex with CD94 and DAP12. J Immunol. 2014;193:610–616. doi: 10.4049/jimmunol.1400556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marrack P, Rubtsova K, Scott-Browne J, Kappler JW. T cell receptor specificity for major histocompatibility complex proteins. Curr Opin Immunol. 2008;20:203–207. doi: 10.1016/j.coi.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, Mandelboim M, Mandelboim O. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol. 2008;9:1065–1073. doi: 10.1038/ni.1642. [DOI] [PubMed] [Google Scholar]
- 52.Kishikawa T, Otsuka M, Yoshikawa T, Ohno M, Takata A, Shibata C, Kondo Y, Akanuma M, Yoshida H, Koike K. Regulation of the expression of the liver cancer susceptibility gene MICA by microRNAs. Sci Rep. 2013;3:2739. doi: 10.1038/srep02739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siu MK, Tsai YC, Chang YS, Yin JJ, Suau F, Chen WY, Liu Y-N. Transforming growth factor-beta promotes prostate bone metastasis through induction of microRNA-96 and activation of the mTOR pathway. Oncogene. 2015;34:4767–4776. doi: 10.1038/onc.2014.414. [DOI] [PubMed] [Google Scholar]
- 54.Viel S, Besson L, Marotel M, Walzer T, Marcais A. Regulation of mTOR, metabolic fitness, and effector functions by cytokines in natural killer cells. Cancers (Basel). 2017;9. doi: 10.3390/cancers9100132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, Degouve S, Djebali S, Sanlaville A, Charrier E, et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. 2016;9:ra19. doi: 10.1126/scisignal.aad1884. [DOI] [PubMed] [Google Scholar]
- 56.Kye MJ, Niederst ED, Wertz MH, Goncalves Ido C, Akten B, Dover KZ, Peters M, Riessland M, Neveu P, Wirth B, et al. SMN regulates axonal local translation via miR-183/mTOR pathway. Hum Mol Genet. 2014;23:6318–6331. doi: 10.1093/hmg/ddu350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yadav D, Ngolab J, Lim RS, Krishnamurthy S, Bui JD. Cutting edge: down-regulation of MHC class I-related chain A on tumor cells by IFN-gamma-induced microRNA. J Immunol. 2009;182:39–43. doi: 10.4049/jimmunol.182.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moretta L, Pietra G, Vacca P, Pende D, Moretta F, Bertaina A, Mingari MC, Locatelli F, Moretta A. Human NK cells: from surface receptors to clinical applications. Immunol Lett. 2016;178:15–19. doi: 10.1016/j.imlet.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 59.Llano M, Lee N, Navarro F, Garcia P, Albar JP, Geraghty DE, López-Botet M. HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: preferential response to an HLA-G-derived nonamer. Eur J Immunol. 1998;28:2854–2863. doi: 10.1002/(SICI)1521-4141(199809)28:09<2854::AID-IMMU2854>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 60.Wieten L, Mahaweni NM, Voorter CE, Bos GM, Tilanus MG. Clinical and immunological significance of HLA-E in stem cell transplantation and cancer. Tissue Antigens. 2014;84:523–535. doi: 10.1111/tan.12478. [DOI] [PubMed] [Google Scholar]
- 61.Ho JW, Hershkovitz O, Peiris M, Zilka A, Bar-Ilan A, Nal B, Chu K, Kudelko M, Kam YW, Achdout H, et al. H5-type influenza virus hemagglutinin is functionally recognized by the natural killer-activating receptor NKp44. J Virol. 2008;82:2028–2032. doi: 10.1128/JVI.02065-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brusilovsky M, Radinsky O, Cohen L, Yossef R, Shemesh A, Braiman A, Mandelboim O, Campbell KS, Porgador A. Regulation of natural cytotoxicity receptors by heparan sulfate proteoglycans in -cis: A lesson from NKp44. Eur J Immunol. 2015;45:1180–1191. doi: 10.1002/eji.201445177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silva-Santos B, Serre K, Norell H. gammadelta T cells in cancer. Nat Rev Immunol. 2015;15:683–691. doi: 10.1038/nri3904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.