

ABSTRACT
Resistance to cell death and evasion of immunosurveillance are major causes of cancer persistence and progression. Tumor cell-intrinsic activation of the RNA receptor retinoic acid-inducible gene-I (RIG-I) can trigger an immunogenic form of programmed tumor cell death, but its impact on antitumor responses remains largely unexplored. We show that activation of intrinsic RIG-I signaling induces melanoma cell death that enforces cross-presentation of tumor-associated antigens by bystander dendritic cells. This results in systemic expansion and activation of tumor-antigen specific T cells in vivo with subsequent regression of pre-established melanoma. These processes were dependent on the signaling hub MAVS and type I interferon (IFN-I) signaling in the host cell. Using melanoma cells deficient for the transcription factors IRF3 and IRF7, we demonstrate that RIG-I-activated tumor cells used as a vaccine are a relevant source of IFN-I during T cell cross-priming in vivo. Thus, our findings may facilitate translational development of personalized anticancer vaccines.
KEYWORDS: Immunogenic cell death, pattern recognition receptors, nucleic acid receptors, RIG-I, anticancer vaccine, dendritic cells, tumor immunotherapy, personalized medicine
Introduction
Next-generation sequencing and novel bioinformatic algorithms have led to the identification of somatic tumor mutations giving rise to tumor-specific neoantigens that can drive antitumor immunity. However, the development of spontaneous immune responses is often compromised by the immunosuppressive tumor milieu or insufficient levels of tumor antigen to reach the threshold for T cell recognition.1 A prerequisite for tumor-specific adaptive immune responses is that specialized antigen presenting cells (APCs) – particularly dendritic cells (DCs) – take up, process and present tumor-associated antigens to cytotoxic T cells. Such cross-priming of tumor-specific T cells has been shown to be dependent on DC maturation mediated by type I IFN (IFN-I, that is IFN-α and IFN-β) signaling in DCs.2,3 In contrast to microbial infections, the tumor microenvironment often lacks proinflammatory signals resulting in suboptimal DC activation.
Under certain circumstances, tumor cells can undergo a special form of programmed tumor cell death that favors recognition and elimination by the immune system. Such immunogenic cell death (ICD) has been shown in response to treatment with certain chemotherapeutic agents (oxaliplatin, doxorubicin) or radiation.4 One of the characteristics of ICD seems to be the release of proinflammatory factors called danger-associated molecular patterns (DAMPs) that can lead to DC maturation via stimulation of innate pattern recognition receptors. In the context of immunogenic chemotherapy, the secretion of ATP and HMGB1 as well as exposure of calreticulin on the outer membrane leaflet have been suggested to contribute to the immunogenicity of ICD amongst others.4 However, at this stage, it is unclear whether these mechanisms play a role in the treatment efficacy of chemotherapy used in human patients.
Increasing numbers of studies now harness different pathways to therapeutically induce ICD to improve the immunogenicity of cancer cells in order to use them as antigen source in the treatment of malignant disease.5 The RNA receptor family of RIG-I-like helicases has recently been associated with ICD in pancreatic carcinoma.6 Its eponymous member RIG-I is frequently expressed in the cytosol of most nucleated cells including malignant tumor cells.7 Tumor-intrinsic RIG-I activation by the specific ligand 5ʹ-triphosphorylated RNA (3pRNA) and subsequent signaling via the mitochondria-located adapter molecule MAVS has been found to trigger strong, cell autonomous apoptosis induction.8,9 Hereby, RIG-I activation induces the pro-apoptotic BH3-only proteins Puma and Noxa, which results in programmed cell death by the executioner caspase-3. This pathway was found to be particularly active in malignant cells and to result in an immunogenic form of tumor cell death, associated with potent cross-priming of tumor-specific cytotoxic T cells in vitro.6 Furthermore, RIG-I activation and downstream MAVS signaling in immune – and to a lesser extent in tumor cells – can induce NFκB-mediated pro-inflammatory cytokine production, ASC-containing inflammasome formation10–12 and IFN-I release triggered by the transcription factors IRF3 and IRF7.13–14 However, the in vivo relevance of RIG-I-mediated tumor cell death and the factors that mediate its immunogenicity remain to be determined.
We here demonstrate that targeting RIG-I within melanoma cells in vitro results in immunogenic cell death, turning melanoma cells into a cellular antitumor vaccine that activates host MAVS and IFN-I signaling in recipient animals.
Results and discussion
RIG-I signaling in melanoma cells triggers immunogenic cell death with potent CD8+ T cell activation and subsequent antitumor immunity
To address the immunogenicity of tumor-intrinsic RIG-I signaling in melanoma, we used the B16 cell line expressing the model antigen ovalbumin (B16.OVA). Targeting the RIG-I pathway in melanoma cells by transfection of a specific ligand (in vitro transcribed and purified 5ʹ-triphosphorylated-RNA, 3pRNA) induced rapid induction of apoptosis with surface expression of annexin-V on the plasma membrane and subsequent tumor cell death in vitro (Figure 1a). Cell death induction by 3pRNA but not the chemotherapeutic agent oxaliplatin was abolished in melanoma cells that are genetically deficient for Ddx58 encoding RIG-I (RIG-I-/-) (Figure 1b). Furthermore, RIG-I-mediated cell death but not oxaliplatin treatment induced potent cross-presentation of tumor-associated antigens by co-cultured bone marrow-derived dendritic cells in vitro (Figure 1c). Immunization of mice with B16.OVA cells undergoing RIG-I-mediated cell death following in vitro transfection with 3pRNA (termed “3p-B16”) resulted in systemic expansion and activation of tumor-antigen specific cytotoxic T cells in vivo (Figure 1(d–e)).
Figure 1.
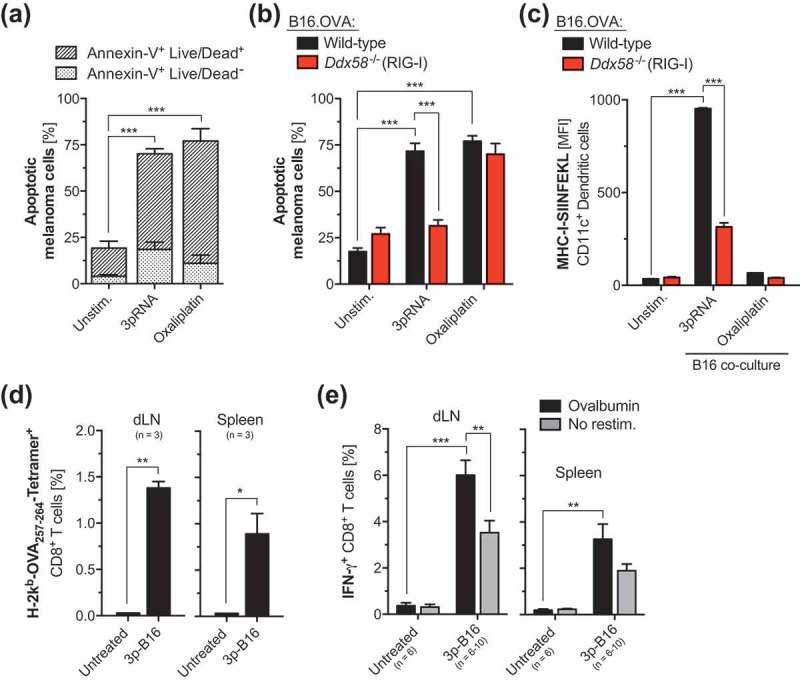
RIG-I signaling in melanoma cell death triggers immunogenic cell death with potent CD8+ T cell cross-priming in vivo. (a) Wild-type B16.OVA melanoma cells were transfected with a specific RIG-I ligand (5ʹ-triphosphorylated-RNA, 3pRNA) or were treated with the chemotherapeutic agent oxaliplatin for 48 h. Induction of melanoma cell death was assessed by Annexin-V and a Life/Dead marker (7-Aminoactinomycin D, 7-AAD) staining. Bars are divided in Annexin-V+ Life/Dead− (early apoptotic) and Annexin-V+ Life/Dead+ (late apoptotic, secondary necrotic) cell fractions. (b) WT and RIG-I-deficient (Ddx58-/-) B16.OVA cells were treated as described above. The frequency of apoptotic cells was determined as described above. (c) WT and RIG-I-deficient (Ddx58-/-) B16.OVA cells were treated as described, were extensively washed and subsequently co-cultured with bone marrow-derived dendritic cells (BM-DCs). After 24 h exposure to tumor cells, cross-presentation of the processed OVA peptide-epitope SIINFEKL in the context of MHC-I by CD11c+ conventional DCs was analyzed by flow cytometry. (d-e) WT B16.OVA cells were transfected with 3pRNA in vitro as described above. After 48 h, non-adherent cells (3p-B16) were harvested, washed and were repeatedly injected s.c. in WT recipient mice. 7 days after the second immunization, (d) the frequency of H-2Kb-SIINFEKL Tetramer+ CD8+ T cells in draining lymph nodes (dLNs) and spleen was determined. (e) Complete lymph node cells or splenocytes that were harvested from mice treated as described above were ex vivo restimulated with ovalbumin and IFN-γ release by CD8+ T cells was analyzed by flow cytometry. Data give mean ± S.E.M. frequency of IFN-γ+ cytotoxic T cells of n = 6–10 individual mice per group. All in vitro data show mean ± S.E.M. of at least triplicate samples. All data are pooled from or are representative of at least two independent experiments. MFI, mean fluorescence intensity. Unstim, Unstimulated. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To test whether tumor cell-intrinsic RIG-I signaling induces bona fide ICD, we injected such immunized mice with living B16.OVA melanoma cells. Indeed, immunization of mice with B16.OVA cells undergoing RIG-I-mediated cell death largely protected recipient animals from subsequent melanoma challenge (Figure 2a) with 7 out of 8 mice being tumor-free at data census. Consistent with this, tumor antigen-specific immunity induced by a RIG-I-activated 3p-B16 cellular vaccine also translated into strong regression of pre-established melanoma (Figure 2b). Depletion experiments showed that 3p-B16-induced antitumor immunity was mediated by both CD8+ cytotoxic T cells and NK1.1+ NK cells. The latter is in line with previous work demonstrating that therapeutic targeting of RIG-I can result in NK cell-mediated melanoma cell killing.9 Importantly, we found that the immunogenicity of RIG-I-induced tumor cell death was not dependent on the presence of the model antigen OVA. Immunization of mice with poorly immunogenic B16-F10 melanoma cells undergoing RIG-I-induced cell death partially protected recipients from subsequent B16-F10 melanoma challenge, associated with strongly reduced tumor growth in this aggressive model and 33% of mice being tumor-free at data census (Figure 2c). Taken together, these data show that RIG-I signaling in melanoma cells induces ICD with potent cross-priming of tumor antigen-specific CD8+ T cells and subsequent anti-tumor immunity.
Figure 2.
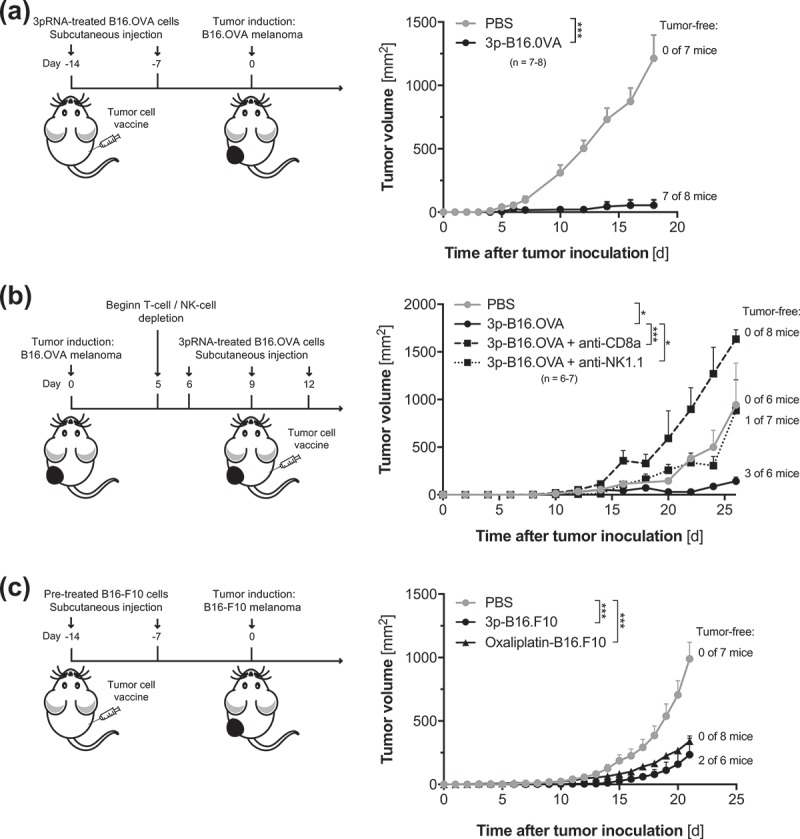
RIG-I-mediated ICD induces strong antitumor immunity. (a) B16.OVA cells were transfected with 3pRNA in vitro. After 48 h, non-adherent cells (3p-B16.OVA) were harvested washed and were repeatedly injected s.c. into recipient mice as described for Figure 1d. Seven days after the second immunization, mice were injected with 105 viable, untreated B16.OVA melanoma cells in the right flank. Data give mean tumor growth ± S.E.M. of n = 7–8 individual mice per group. (b) Mice were implanted with 1 × 105 viable, untreated B16.OVA cells in the right flank. When tumors were readily visible, recipient animals were s.c. injected on day 6, 9 and 12 with 3pRNA-pretreated B16 OVA cells (3p-B16.OVA, as described above). Some mice were additionally treated with T-cell (anti-CD8a) or NK-cell (anti-NK1.1) depleting antibodies. Data give mean tumor growth ± S.E.M. of n = 6–7 individual mice per group. (c) Poorly immunogenic B16-F10 melanoma cells were either transfected with 3pRNA or were treated with oxaliplatin in vitro. After 48 h, non-adherent cells (3p-B16.F10) were harvested washed and were repeatedly injected s.c. into the left flank of recipient mice as described for Figure 3a. Seven days after the second immunization, mice were injected with 105 viable untreated B16-F10 melanoma cells in the right flank. Data give mean tumor growth ± S.E.M. of n = 6–8 individual mice per group. All data are pooled from at least two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Additionally, immunization with oxaliplatin-treated melanoma cells resulted in systemic anti-tumor immunity associated with delayed tumor progression (Figure 2c). However, this is not reflected by our in vitro data in which oxaliplatin-induced melanoma cell death failed to enhance tumor antigen cross-presentation by dendritic cells (Figure 1c). The latter findings are in line with previous studies in pancreatic carcinoma, in which the authors were able to detect enhanced maturation of DCs (as determined by upregulation of the co-stimulatory molecule CD86) and enhanced endosomal uptake of fluorescently labeled tumor antigen in vitro only in response to RIG-I-treated but not oxaliplatin-treated tumor cells.6 Chemotherapy-induced ICD was initially described in the CT26 colon carcinoma and MCA205 fibrosarcoma cell lines. Whether the apparent lack of oxaliplatin-induced ICD in some of our assays is related to our experimental system or whether there are indeed differences in the susceptibility to oxaliplatin-induced ICD among different cancer cell lines and thus cancer entities, remains to be investigated.
Tumor-derived IFN-I contributes to the antitumor responses induced by a RIG-I-activated, cellular antitumor vaccine
We next addressed the molecular pathways that are involved in 3p-B16 cellular vaccine-induced immune responses. Generally, the importance of host IFN-I signaling – particularly within DCs – for the induction of T cell-based antitumor immunity has been widely acknowledged.2,3 A contribution of tumor IFN-I has been suggested by an in vitro study showing that tumor cell-derived IFN-I can potentially mediate DC maturation following interaction with RIG-I-stimulated pancreatic tumor cells.6 However, ultimate in vivo experimental evidence is lacking. To address this, we generated melanoma cell lines genetically deficient for the RIG-I downstream transcription factors IRF3 and IRF7 (IRF3/7-/- B16.OVA cells). We found that activation of the RIG-I pathway within melanoma cells (which eventually results in programmed cell death) triggers rapid production of tumor IFN-I via activation of IRF3 and IRF7 (Figure 3a). Following co-culture with B16.OVA melanoma cells undergoing RIG-I-induced programmed tumor cell death, maturation as well as cross-presentation of tumor-associated antigen was largely abolished in DCs deficient for IFN-I receptor signaling (Figure 3(b–c)). Furthermore, tumor antigen-specific T-cell responses induced by a RIG-I-activated 3p-B16 cellular vaccine in vivo were strongly diminished in recipient mice with blocked IFN-I receptor signaling (Figure 3d).
Figure 3.
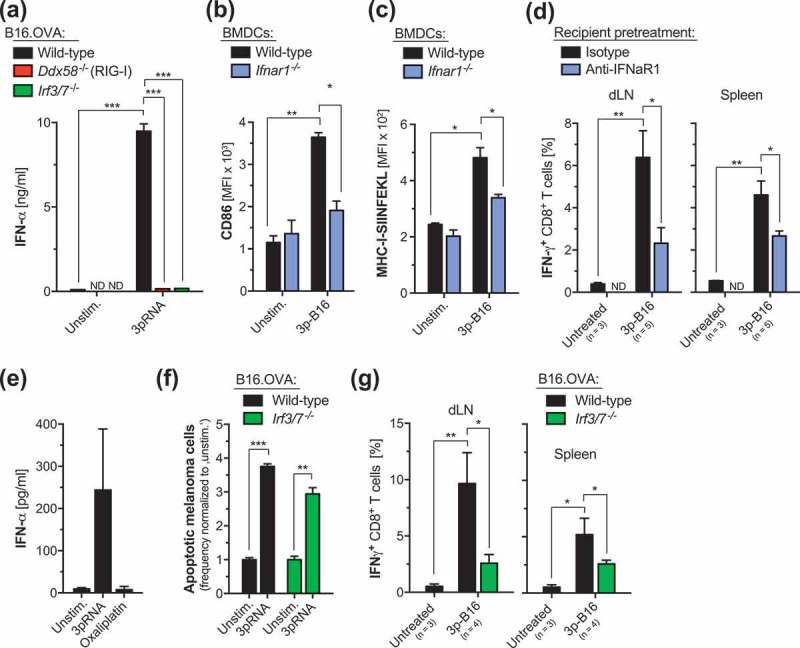
Tumor-derived IFN-I contributes to the antitumor responses induced by a RIG-I-activated, cellular antitumor vaccine. (a) Wild-type, RIG-I- (Ddx58-/-) and IRF3/7-deficient (Irf3/7-/-) B16.OVA cells were transfected with 3pRNA in vitro as described for Figure 1a. After 48 h, cumulated tumor cell-derived IFN-α was determined by ELISA. (b-c) 3pRNA-treated B16.OVA cells were extensively washed and were subsequently co-cultured with BM-DCs harvested from wild-type or IFNaR1-deficient (Ifnar1-/-) donor mice. After 24 h exposure to tumor cells, (b) expression of the costimulatory molecule CD86 and (c) cross-presentation of the processed OVA peptide-epitope SIINFEKL in the context of MHC-I by CD11c+ conventional DCs was analyzed by flow cytometry. (d) WT B16.OVA cells were transfected with 3pRNA in vitro and were repeatedly injected s.c. in WT recipient mice as described for Figure 1d. Some mice were additionally treated with anti-IFNaR1 antibodies, beginning two days prior to the immunization. Complete draining lymph node (dLN) cells and splenocytes were harvested and the frequency of IFN-γ+ CD8+ T cells was analyzed by flow cytometry after ex vivo restimulation with ovalbumin. Data give the mean value ± S.E.M. of individual mice. (e) B16.OVA cells were transfected with 3pRNA in vitro as described. After 48 h, culture supernatant was discharged and cells were reseeded in fresh medium. Additional 24 h later, IFN-α levels were determined by ELISA. (f) Wild-type and IRF3/7-deficient (Irf3/7-/-) B16.OVA cells were transfected with 3pRNA and induction of cell death was assessed by Annexin-V and Life/Dead marker staining. (g) WT and Irf3/7-/- B16.OVA cells were transfected with 3pRNA in vitro and were repeatedly injected s.c. in WT recipient mice as described for Figure 1d. Complete draining lymph node cells and splenocytes were harvested, and IFN-γ release by CD8+ T cells was analyzed by flow cytometry after ex vivo restimulation with ovalbumin. All results are representative of at least two independent experiments. Unstim, Unstimulated. ND, not determined. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
From these data, we hypothesized that IRF3/7-mediated IFN-I release by melanoma cells during the initiation of RIG-I-induced programmed cell death contributes to its immunogenicity. In this, a 3p-B16 cellular vaccine of melanoma cells undergoing immunogenic cell death may be a relevant source of IFN-I for subsequent cross-priming of CD8+ T cells, once transferred into tumor-bearing animals. Indeed, we found that B16.OVA cells undergoing RIG-I-mediated programmed cell death 48 h after in vitro transfection with 3pRNA, still produced significant levels of IFN-I when they were used as a cellular vaccine in recipient mice (Figure 3e). Importantly, apart from defective IFN-I production, IRF3/7-/- B16.OVA cells were still susceptible to RIG-I-mediated cell death (Figure 3f). Nevertheless, in vivo immunization with 3pRNA-pretreated IRF3/7-/- melanoma cells undergoing programmed cell death failed to induce CD8+ T-cell activation in recipient mice (Figure 3g). In sum, these data show that IRF3/7-mediated IFN-I production within melanoma cells does not contribute to the induction of RIG-I-induced apoptosis. However, IFN-I release from tumor cells undergoing such in vitro induced programmed tumor cell death is critical for the immunogenicity of this process.
3p-cancer vaccines target host MAVS but not STING or NLRP3 inflammasome signaling
Different DAMPs can be involved in immunogenic cell death induced by chemotherapeutic agents. While IFN-I production by tumor cells has been associated with the efficacy of the immunogenic anthracycline doxorubicin,15 ATP release from dying tumors cells and subsequent activation of the NLRP3 inflammasome seems to be a critical factor for the immune system to perceive cell death as immunogenic in the context of oxaliplatin treatment.16 We next addressed the latter pathways’ role in RIG-I-induced immunogenic cell death. We hereby found, that – similar to oxaliplatin treatment – 3pRNA-mediated RIG-I activation of B16 melanoma cells resulted in i) the secretion of the DAMPs ATP and HMGB1 as well as ii) exposure of calreticulin on the outer membrane leaflet (Figure 4a). However, CD8+ T-cell activation following administration of a RIG-I-activated 3p-B16 cellular vaccine in vivo was independent of NLRP3 or its adapter molecule ASC in host cells (Figures 4(b–c)). This is in line with findings from a previous in vitro study in pancreatic carcinoma cells lines showing that RIG-I-induced ICD occurs independent of the interaction of ATP with the purinergic receptor P2X7 or NLRP3 as well as HMGB1 detection by TLR4 and RAGE, respectively.6
Figure 4.
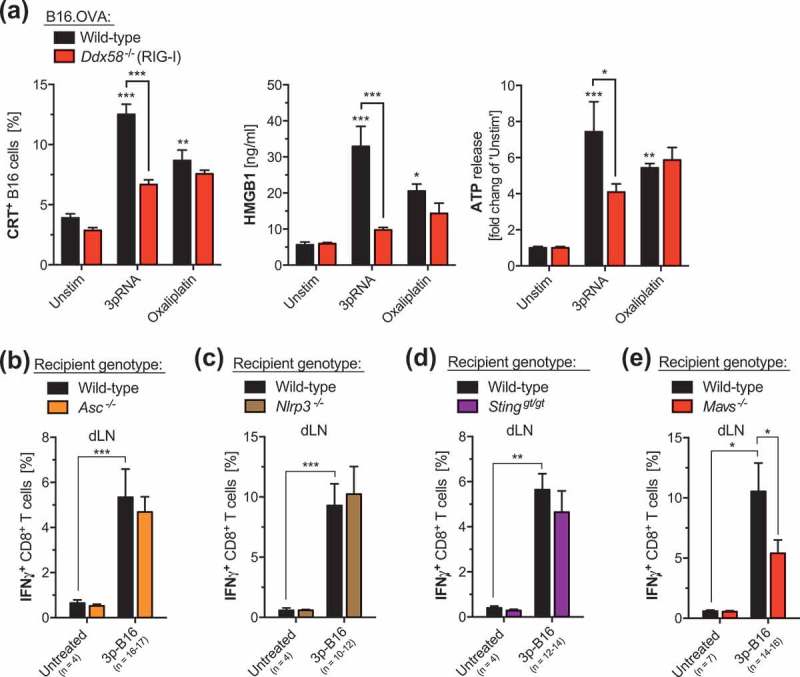
3p-cancer vaccines target host MAVS but not STING or NLRP3 inflammasome signaling. (a) Wild-type (WT) and RIG-I-deficient (Ddx58-/-) B16.OVA melanoma cells were transfected in vitro with 3pRNA or were treated with oxaliplatin as described for Figure 1. Cell culture supernatants were analyzed for the concentration of ATP and HMGB1 by ELISA. The frequency of B16 melanoma cells with calreticulin (CRT) exposed on the outer plasma membrane leaflet was determined by flow cytometry. Data show mean ± S.E.M. of at least n = 7 biological replicates pooled from two independent experiments. (b-e) Mice were immunized with 3pRNA-stimulated B16.OVA melanoma cells (3p-B16) as described for Figure 1d. Complete draining lymph node cells were harvested from immunized wild-type, (b) ASC-deficient (Asc-/-), (c) NLRP3-deficient (Nlrp3-/-), (d) STING-deficient (Stinggt/gt) and (e) MAVS-deficient (Mavs-/-) mice, and were ex vivo restimulated with ovalbumin. IFN-γ release by CD8+ T cells was analyzed by flow cytometry. Data give mean ± S.E.M. of the indicated number of individual mice per group that were pooled from at least three independent experiments. Unstim, Unstimulated. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We also found that the induction of these ICD ‘hallmark’ DAMPs following oxaliplatin treatment was independent from tumor cell-intrinsic RIG-I signaling (Figure 4a). In this context, a recent study suggested that the release of immunostimulatory ATP and HMGB1 upon oxaliplatin treatment can be facilitated by RIPK3- and MLKL-mediated necroptotic signaling.17 Hereby, it is important to mention that the immunogenicity of cell death is not only dictated by the form of cell death (apoptosis, necroptosis, pyroptosis, necrosis, etc.), but rather has been suggested to be dependent on specific (pro-inflammatory) signaling events triggered in dying cells.18 Therefore, our data show that RIG-I-induced and chemotherapy-induced ICD rely on different molecular mechanisms.
Host nucleic acid receptor systems have been associated with pro-inflammatory signaling and the development of antitumor T cell immunity. Detection of tumor-derived DNA via the cGAS/STING pathway within host DCs has been suggested to mediate spontaneous antitumor immunity in an immunogenic model of malignant melanoma.19 We found that 3p-B16 cellular vaccine-mediated activation of tumor antigen-specific CD8+ T cells was not compromised in STING-deficient animals (Figure 4d). In contrast, we found that 3p-B16 immunization efficacy was reduced in recipient animals deficient for the adaptor molecule MAVS and thus RIG-I downstream signaling in non-malignant host cells (Figure 4e). The latter is consistent with previous findings that RIG-I receptor activation has the potential to facilitate antigen-specific T-cell cross-priming in the context of antiviral immunity.20 Yet, the identity of possible RIG-I ligands released in vivo by melanoma cells undergoing 3pRNA-induced cell death remain to be determined.
One limitation of our study is that the cellular vaccine may leak the in vitro transfected 3pRNA to – at least partly – facilitate host RIG-I/MAVS signaling in vivo. In addition, the finding that 3p-B16-mediated induction of antitumor immunity was independent of STING signaling in host cells is unexpected. Perhaps this could be explained by redundant activation of the RIG-I/MAVS and cGAS/STING pathways in host DCs in this context. Likewise, in vivo application of purified 3pRNA – as performed in a phase I/II clinical trial with intralesional 3pRNA administration in solid tumors and lymphomas (RGT100–001; ClinicalTrials.gov Identifier: NCT03065023) – may address different host pathways as compared to the in vitro generated “cellular 3p-B16 vaccine” described in our study.
Together, we here demonstrate that targeting RIG-I within melanoma cells in vitro results in immunogenic cell death, turning melanoma cells into an IFN-I releasing cellular vaccine. This approach may serve as the basis for the translational development of personalized anticancer vaccines from autologous tumor cells. However, new anticancer vaccines, when progressing from the preclinical phase to clinical testing, often fail to maintain immunogenicity when being produced in accordance with good manufacturing procedure (GMP) regulatory criteria. Thus, further studies will have to clarify whether RIG-I-induced cellular vaccines are still potent once they had been avitalized by physical cell death modalities to ensure complete cancer cell death prior to patient re-transplantation.
Material and methods
Mice
Female C57BL/6J mice were purchased from Janvier. Nlrp3-/- and Asc-/- mice were a gift from J. Tschopp. Ifnar1-/- and Mavs-/- mice were provided by U. Kalinke (Hannover, Germany). Sting-deficient mice (Tmem173gt/gt) were from the Jackson Laboratory. All mouse strains have been described previously.21–24 Mice were at least six weeks of age at the onset of experiments and were maintained in specific pathogen-free conditions. Animal studies were approved by the local regulatory agency (Regierung von Oberbayern, Munich, Germany).
Media and reagents
RPMI-1640 medium (Invitrogen) and DMEM (Invitrogen) were supplemented with 10% (v/v) FCS (Hyclone), 3 mM L-glutamine, 100 U/ml of penicillin and 100 μg/ml of streptomycin (all from Sigma-Aldrich). OptiMEM reduced serum medium was from Invitrogen. Double-stranded in vitro-transcribed 3pRNA (sense, 5′- UCA AAC AGU CCU CGC AUG CCU AUA GUG AGU CG −3′) was generated as described.9
Cell lines and CRISPR-cas9-mediated genome editing
The B16-F10 murine melanoma cell line expressing full-length chicken ovalbumin (here referred to as B16.OVA) was cultured in complete DMEM medium supplemented with 400 μg/ml G418 (from Sigma-Aldrich). Gene deficient B16.OVA cells were engineered using the CRISPR-Cas9 system as previously described.25 In brief, B16.OVA cells were genetically edited using the Streptococcus pyogenes nuclease Cas9, together with different guide RNAs (Target sequences: Ddx58 5′-GGCTGATGAGGATGATGGAGCGG-3′, Irf3 5′-GCATGGAAACCCCGAAACCG-3′, Irf7 5′-CTACGACCGAAATGCTTCCA-3′). The guide RNAs were cloned into pSpCas9(BB)-2A-GFP (pX458, a gift from Feng Zhang; Addgene plasmid #48138), a bicistronic expression vector expressing Cas9 and a sgRNA. B16.OVA were transfected with Lipofectamin 2000, and GFP-expressing single cell clones were isolated by FACS 24 h after transfection. Gene deficient clones were identified by immunoblotting.
Cell culture experiments
In vitro, B16.OVA cells were transfected with 3pRNA (3 µg/ml) complexed in Lipofectamin 2000 (Life Technologies, Darmstadt, Germany) according to the manufacturer’s protocol using Gibco® Opti-MEM. Alternatively, B16 cells were treated with 30 μg/ml oxaliplatin. 48 hours later, induction of cell death was assessed by staining with Annexin-V (BD Bioscience) and 7-AAD (BioLegend). ATP or HMBG1 release was determined using the ATP Assay Kit (Abcam) or a commercial ELISA (IBL) following the manufacturer’s protocol, respectively. Levels of IFN-I were determined by self-established ELISAs as described earlier.26 Bone marrow-derived dendritic cells (BMDCs) were generated by culturing bone marrow cells in complete RPMI medium supplemented with 20 ng/ml GM-CSF (from Immunotools, Friesoythe, Germany). For co-culture experiments, B16.OVA were treated as described above. After 48 h, B16.OVA cells were washed twice in PBS, and were then co-cultured with BM-DCs for 24 h.
Flow cytometry
Cell suspensions were stained in PBS with 3% FCS. Fluorochrome-coupled antibodies were purchased from eBioscience or BioLegend. Anti-calreticulin (ab2907) was purchased from Abcam. The anti-mouse OVA257–264 (SIINFEKL) peptide bound to H-2Kb-antibody (clone 25-D1.16) was purchased from eBioscience. The iTAg MHC-I murine tetramers detecting SIINFEKL-specific CD8+ T cells were from MBL (Woburn, MA). For intracellular cytokine staining the Foxp3 Transcription Factor Fixation/Permeabilization Kit (eBioscience) was used. Data were acquired on a FACSCanto II (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Immunization with 3pRNA-treated B16.OVA cells
For each mouse, 106 B16.OVA cells were transfected in vitro with 3pRNA. After 48 h, all cells which were easily detached by rinsing the culture flask were extensively washed and were injected subcutaneously (s.c.) in the right hock. The therapy was repeated at day 7. At day 14 mice were sacrificed and draining lymph nodes and the spleen were removed. Single cell suspensions of these organs were cultured in the presence of 1 µg/ml OVA protein in complete RPMI medium for 72 h and IFN-γ levels of CD8+ T cells were analyzed by flow cytometry. In some experiments, mice were pre-treated intraperitoneally (i.p.) with 400 µg anti-murine IFNaR1 antibody (clone MAR1–5A3, BioXCell, West Lebanon, NH) one day prior to the above immunization.
Tumor challenge and treatment
For tumor challenge, mice were injected s.c. in the right flank with 105 untreated B16.OVA cells on day 0. When tumors were readily visible, 106 3pRNA-treated B16.OVA cells were injected s.c. in the right hock as described above. Treatment was repeated on day 3 and 6. Optional treatment with anti-CD8a (clone 2.43, BioXCell) or anti-NK1.1 (clone PK136) depleting antibodies was initiated one day prior to vaccination (100 µg i.p.) and was repeated twice weekly (50 µg i.p.). For the prophylactic models, mice were vaccinated twice with in vitro pre-treated B16 melanoma cells on days −14 and −7 as described above, before s.c. tumor induction with 105 B16.OVA or B16-F10 cells on day 0. Mice were euthanized when the maximum tumor diameter exceeded 15 mm according to standard legal procedure (responsible state office Regierung von Oberbayern).
Statistics
All data are presented as mean ± S.E.M. Statistical significance of single experimental findings was assessed with the independent two-tailed Student’s t-test. For multiple statistical comparison of a data set the one-way ANOVA test with Bonferroni post-test was used. Significance was set at P values < 0.05, p < 0.01, and p < 0.001 and was then indicated with an asterisk (*, ** and ***). All statistical calculations were performed using Prism (GraphPad Software).
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [Project 360372040 - SFB 1335]; Deutsche Forschungsgemeinschaft [PO 1575/3-1]; Deutsche Krebshilfe [111620]; Else Kröner-Fresenius-Stiftung [2015_A06]; Else Kröner-Fresenius-Stiftung [2012_A61]; Else Kröner-Fresenius-Stiftung [Forschungskolleg TUM]; European Hematology Association; DKMS [Mechtild Harf Research Grant]; Carl Maximilian and Carl Manfred Bayer-Foundation.
Acknowledgments
S.B., S.H., T.H., and H.P. designed the research, analyzed and interpreted the results. S.B., F.S., A.W., T.N., and D.F.R.B. did the experiments. J.C.F. provided substantial intellectual input. S.H. and H.P. prepared the manuscript and guided the study. H. P. is supported by the EMBO Young Investigator Program. This work is part of the doctoral thesis of S.B. and F.S. at the Technical University of Munich.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hu Z, Ott PA, Wu CJ.. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18:168–182. doi: 10.1038/nri.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF.. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 5.Montico B, Nigro A, Casolaro V, Dal Col J. Immunogenic apoptosis as a novel tool for anticancer vaccine development. Int J Mol Sci. 2018;19:594. doi: 10.3390/ijms19020594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duewell P, Steger A, Lohr H, Bourhis H, Hoelz H, Kirchleitner SV, Stieg MR, Grassmann S, Kobold S, Siveke JT, et al. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8(+) T cells. Cell Death Differ. 2014;21:1825–1837. doi: 10.1038/cdd.2014.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barchet W, Wimmenauer V, Schlee M, Hartmann G. Accessing the therapeutic potential of immunostimulatory nucleic acids. Curr Opin Immunol. 2008;20:389–395. doi: 10.1016/j.coi.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest. 2009;119:2399–2411. doi: 10.1172/JCI37155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, et al. 5ʹ-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14:1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 10.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschläger N, Schlee M, Rothenfusser S, Barchet W, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 11.Franchi L, Eigenbrod T, Munoz-Planillo R, Ozkurede U, Kim YG, Chakrabarti A, Gale M, Silverman RH, Colonna M, Akira S, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol. 2014;193:4214–4222. doi: 10.4049/jimmunol.1400582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, Vidal SM, Pekosz A. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013;9:e1003256. doi: 10.1371/journal.ppat.1003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5ʹ-diphosphates. Nature. 2014;514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann -K-K, Schlee M, et al. 5ʹ-Triphosphate RNA is the ligand for RIG-I. Sci. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 15.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–1309. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 16.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 17.Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, Pietrocola F, Vacchelli E, Souquere S, Sauvat A, et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunol. 2016;5:e1149673. doi: 10.1080/2162402X.2016.1149673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis E Sousa C, Green DR, Oberst A, Albert ML. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8⁺ T cells. Sci. 2015;350:328–334. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hochheiser K, Klein M, Gottschalk C, Hoss F, Scheu S, Coch C, Hartmann G, Kurts C. Cutting edge: the RIG-I ligand 3pRNA potently improves CTL cross-priming and facilitates antiviral vaccination. J Immunol. 2016;196:2439–2443. doi: 10.4049/jimmunol.1501958. [DOI] [PubMed] [Google Scholar]
- 21.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 22.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Sci. 1994;264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 23.Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 25.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourquin C, Hotz C, Noerenberg D, Voelkl A, Heidegger S, Roetzer LC, Storch B, Sandholzer N, Wurzenberger C, Anz D, et al. Systemic cancer therapy with a small molecule agonist of toll-like receptor 7 can be improved by circumventing TLR tolerance. Cancer Res. 2011;71:5123–5133. doi: 10.1158/0008-5472.CAN-10-3903. [DOI] [PubMed] [Google Scholar]