

Abstract
Background
Epithelial cell transformation sequence 2 (ECT2) is upregulated in glioma and promotes glioma cell proliferation. A preliminary experiment showed a positive correlation between ECT2 and pituitary tumor-transforming gene 1 (PTTG1). The aim of this study was to explore how ECT2 affects PTTG1 to influence the proliferation of glioma cells.
Methods
The expression of ECT2 in glioma was detected by western blot and reverse transcription PCR. The effect of ECT2 on glioma proliferation was examined using cell proliferation–related assays and in vivo experiments. The effect of ECT2 on the stability of E2F transcription factor 1 (E2F1) and the expression of PTTG1 were examined by western blot, co-immunoprecipitation, and in vivo ubiquitination assays.
Results
ECT2 was upregulated in gliomas and was negatively correlated with prognosis; its downregulation inhibited glioma cell proliferation. Furthermore, ECT2 regulated PTTG1 expression by affecting the stability of E2F1, thereby affecting the glioma cell proliferation. In addition, the deubiquitinating enzyme proteasome 26S subunit, non-ATPase 14 (PSMD14) affected the degradation of E2F1 and regulated the stability of E2F1. Interestingly, ECT2 regulated the expression of PSMD14.
Conclusion
In this study, we clarify a new mechanism by which ECT2 regulates the expression of PTTG1 and thus affects the proliferation of glioma cells: ECT2 influences the stability of E2F1 by regulating the expression of the deubiquitinating enzyme PSMD14, thereby affecting the expression of PTTG1. Intensive and extensive understanding of the mechanism of ECT2 in glioma proliferation may provide an opportunity for the development of new molecular therapeutic targets for glioma treatment.
Keywords: ECT2, PSMD14, PTTG1, ubiquitination, proliferation
Key Points.
We clarified the existence of the ECT2/PSMD14/E2F1/PTTG1 pathway, a new mechanism that affects the proliferation of glioma.
This may provide an opportunity to develop new molecular therapeutic targets for glioma.
Importance of the Study.
ECT2 is highly expressed in glioma, but its mechanism in the development of glioma remains largely unknown. In this study, we clarified the existence of the ECT2/PSMD14/E2F1/PTTG1 pathway, a new mechanism that affects the proliferation of glioma. Extensive and intensive understanding of the role of ECT2 in the development of gliomas may provide an opportunity to develop new molecular therapeutic targets for glioma.
Epithelial cell transformation sequence 2 (ECT2) is a guanine nucleotide exchange factor encoded by the human ECT2 gene.1 The protein is expressed during the interval from cell division to mitosis, co-localizes with the central spindle, and activates Ras homolog family member A (RhoA),2–4 thereby promoting the formation of the mitotic sulcus and cleavage. ECT2 is upregulated in many human cancers and functions as an oncogene.5,6
Ubiquitination is a reversible posttranslational modification that is the covalent binding of one or more ubiquitin molecules to a substrate protein under the action of a series of enzymes.7–10 Ubiquitination plays key roles in various signal transduction cascades and in determining protein stability.11–13 Deubiquitination is a reversal of the ubiquitination process mediated by deubiquitinating enzymes.13 A number of deubiquitinating enzymes have been identified in eukaryotic cells which cleave thioester bonds between ubiquitin and substrate proteins, thereby releasing the substrate from the ubiquitin complex and avoiding degradation via proteases.14–17 The maintenance of the ubiquitin system’s homeostasis relies on the mutual coordination of ubiquitination and deubiquitinating enzyme components and functions. Breaking this balance for any reason can lead to the occurrence of different diseases, including cancer.18,19
Our group has verified that pituitary tumor-transforming gene 1 (PTTG1) is highly expressed in glioma and can promote glioma cell proliferation.20 In 2017, Wondergem et al reported that PTTG1 can affect the activation of small G protein RhoA by affecting the expression of ECT2 in renal cancer cells, thereby affecting cell aggression.21 We confirmed through experiments that this pathway does not exist in glioma; however, our analyses of databases and clinical tissue samples suggest a clear positive correlation between ECT2 and PTTG1, and subsequent cell experiments confirmed that ECT2 can affect the expression of PTTG1, which further influences glioma cell proliferation. Afterward, we confirmed that ECT2 could influence the stability of the transcription factor E2F1 and further influenced the expression of PTTG1 by affecting the ubiquitination and degradation of E2F1. Additionally, we discussed the mechanism by which ECT2 affects E2F1 degradation. We confirmed that ECT2 influences the degradation of E2F1 by regulating the expression of the deubiquitinating enzyme proteasome 26S subunit, non-ATPase 14 (PSMD14). In summary, we demonstrated the presence of ECT2/PSMD14/E2F1/PTTG1, a novel pathway that promotes glioma cell proliferation.
Materials and Methods
Details of the materials and methods are provided in the Supplementary Material.
Ethics Statement
This study was conducted in accordance with institutional ethical standards, the Declaration of Helsinki, and national and international guidelines. Informed consent was obtained from all patients involved in this study, and the study protocol was approved by the Clinical Research Ethics Committee of Nanjing Medical University. Animal experiments and experimental procedures were in accordance with the Animal Management Rule of the Chinese Ministry of Health (documentation 55, 2001) and were approved by the Nanjing Medical University Animal Experimental Ethics Committee.
Public Datasets
See the Supplementary Material for details.
Human Tissue Samples
Human glioma samples and non-tumorous brain tissues (obtained from the cortex of decompressive surgery patients with brain trauma or hypertensive intracerebral hemorrhage) were consecutively recruited between December 2014 and December 2016 from the Department of Neurosurgery of the First Affiliated Hospital of Nanjing Medical University. All patients with glioma underwent surgery for the first time and had not previously received radiotherapy or chemotherapy. Pathological diagnoses were based on the 2007 World Health Organization classification of tumors of the central nervous system. The pathological grades of 19 glioma specimens were: 8 cases of grade I, 3 of grade II, 3 of grade III, and 5 of grade IV.
Cell Culture
The human glioblastoma (GBM) cell lines A172, U251, U87, U118, H4, and LN229 were identified by American Type Culture Collection using the short tandem repeat genotyping method and cultured according to the manufacturer’s recommendations. The human primary GBM cell line N3 was donated by the Beijing Institute of Neurosurgery, Capital Medical University. Cells were maintained in Dulbecco’s modified Eagle’s medium (HyClone) supplemented with 10% fetal bovine serum. Normal human astrocyte (NHA) cells were purchased from Lonza and cultured in astrocyte growth medium supplemented with recombinant human epidermal growth factor (rhEGF), insulin, ascorbic acid, GA-1000, L-glutamine, and 5% fetal bovine serum. All cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
Western Blot Analysis
All western blot analyses were performed according to our previous study.22 See the Supplementary Material for details.
Small Interfering RNAs, Plasmid and Lentivirus Construction, and Transfection
See the Supplementary Material for details on small interfering (si)RNAs and lentivirus construction, transfection, and stable cell establishment.
RNA Isolation and Semi-Quantitative Reverse Transcription PCR
Total RNA from cells was extracted using TRIzol Reagent (Life Technologies) following the manufacturer’s protocol. Semi-quantitative reverse transcription (RT) PCR was performed according to our previous study.23 See Supplementary Material for details.
EdU Incorporation Assay
Transfected cells were seeded into 24-well plates at 5 × 104 cells per well. After 24 h, the cells were exposed to 10 μM EdU (5-ethynyl-2'-deoxyuridine; Life Technologies) for 2 h, fixed, permeabilized, and stained with both the Alexa Fluor reaction cocktail for EdU and Hoechst for cell nuclei, according to the manufacturer’s protocol. Finally, cells were visualized under a fluorescent microscope.
Cell Viability Assay
Cell viability was measured in duplicate samples with a Cell Counting Kit 8 (CCK-8) in 3 independent experiments. Stably transfected glioma cells were seeded in 96-well plates at a density of 1 × 104 cells per well; cultured for 24, 48, 72, or 96 h before adding 10 μL of CCK-8 reagent in each well; and incubated for another 2 h. The absorbance at a wavelength of 450 nm (optical density 450) was measured using a microplate reader (ThermoFisher Scientific).
Chromatin Immunoprecipitation and Dual-Luciferase Reporter Assays
See the Supplementary Material for details.
Nude Mouse Model of Intracranial Glioma
An intracranial model of glioma in nude mice was used as described previously.22 See the Supplementary Material for details.
Immunohistochemistry
All immunohistochemistry experiments were performed according to methods described in our previous study.22 See the Supplementary Material for details.
Statistical Analysis
The presented results are representative of experiments repeated at least 3 times. Data were analyzed with GraphPad Prism 7. Student’s t-tests and ANOVA were used to detect the differences in the results between groups. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology analysis were performed via DAVID (Database for Annotation, Visualization and Integrated Discovery) (http://david.abcc.ncifcrf.gov/). Correlation between the expression profiles of 2 genes was analyzed using Spearman’s rank test. Overall survival curves were calculated using the Kaplan–Meier method and compared using the log-rank test (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
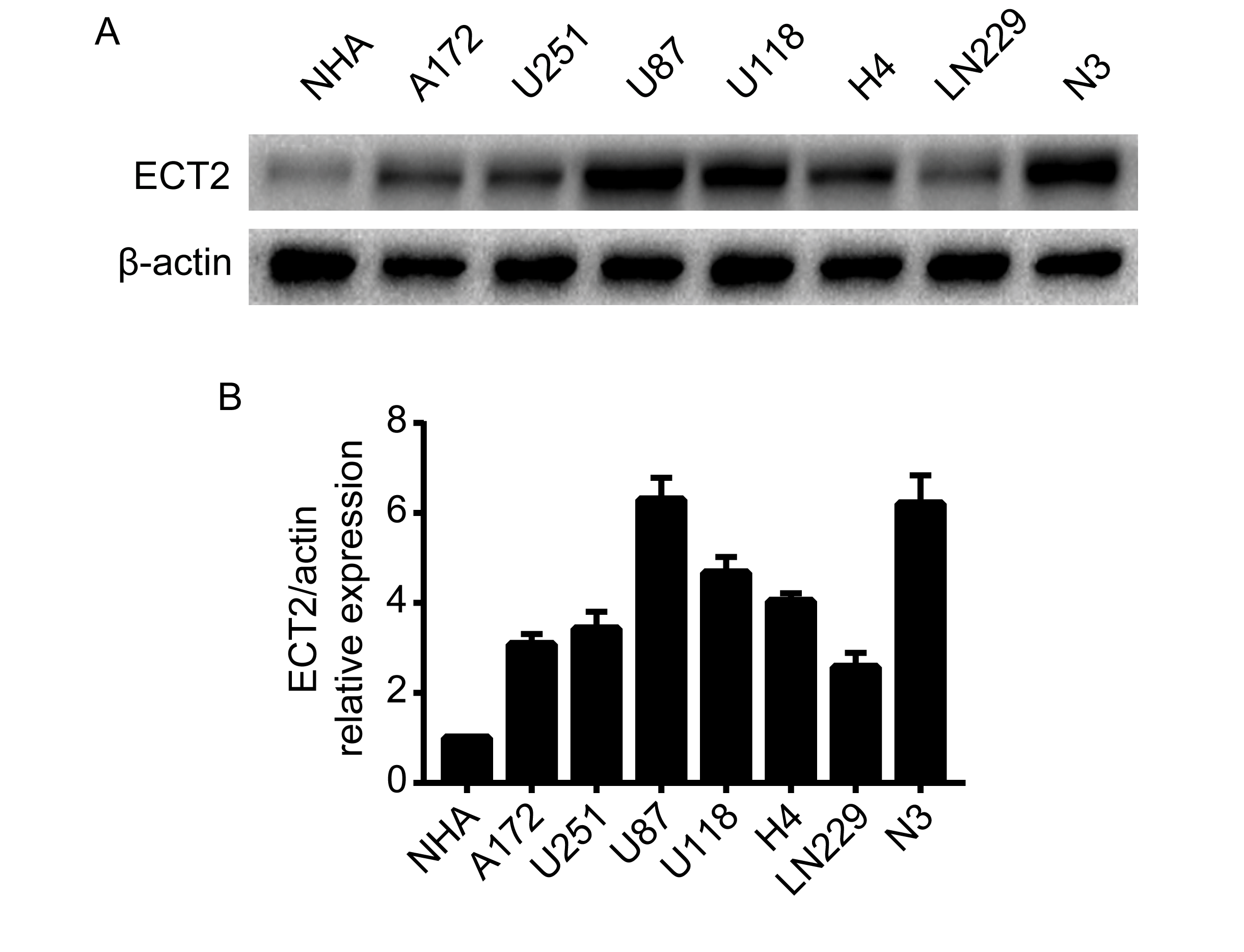
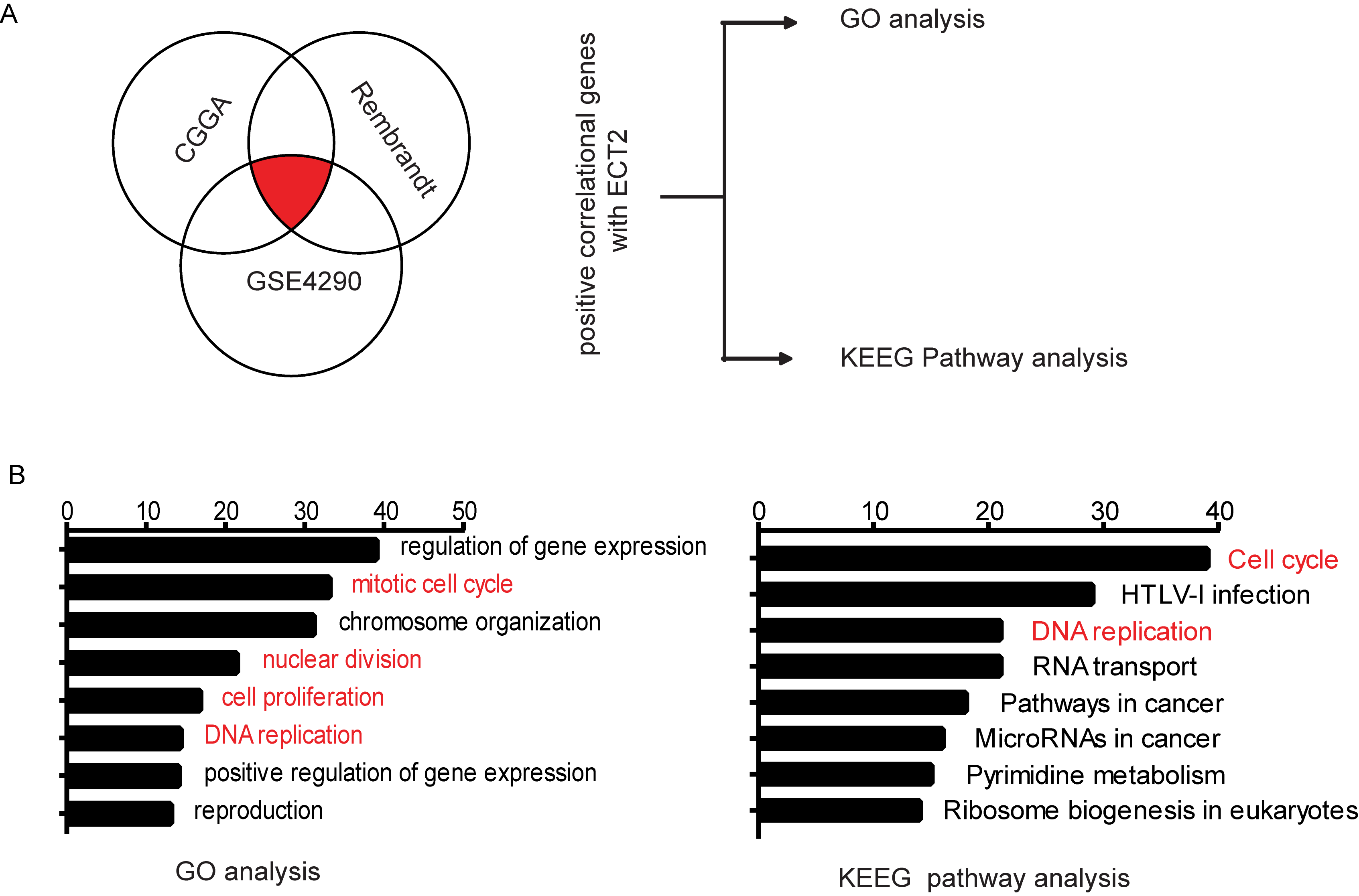
ECT2 is highly expressed in glioma and promotes the proliferation of glioma cells. To elucidate the role and clinical significance of ECT2 in glioma, we first analyzed ECT2 expression and the associated clinical prognosis in glioma using 4 databases: the Chinese Glioma Genome Atlas (CGGA), The Cancer Genome Atlas (TCGA), GSE16011, and the Repository of Molecular Brain Neoplasia Data (REMBRANDT). The results showed that the expression of ECT2 in high-grade glioma or GBM was significantly higher than that in low-grade glioma (Fig. 1A), and the increased ECT2 expression was associated with a shorter survival period except in the database of TCGA (Supplementary Figure 1). We then performed a western blot analysis on protein extracted from the clinical samples, and we found that the expression pattern of ECT2 was consistent with the results from our database analyses (Fig. 1B, C). We further examined the expression of ECT2 in glioma cell lines and NHAs by western blot. The results showed that the expression of ECT2 was higher in glioma cells than in NHAs and was most pronounced in U87 and N3 cells (Supplementary Figure 2A, B). In summary, ECT2 is highly expressed in glioma and is positively correlated with pathological grades. To explore the effect ECT2 might have on glioma properties, a Pearson correlation analysis was implemented using MEV (MultiExperiment Viewer) software to identify target genes that were positively associated with ECT2 (r > 0.4). In total, 551 upregulated genes were identified (Supplementary Figure 3A). To clarify the associations between these genes, the DAVID web tool was used for gene ontology enrichment analysis and KEGG pathway analysis. The upregulated genes were mainly enriched in the terms, positive regulation of cell cycle, and cell proliferation (Supplementary Figure 3B).
Fig. 1.

ECT2 is highly expressed in glioma and promotes the proliferation of glioma cells. (A) The transcriptional levels of ECT2 were analyzed in the CGGA, TCGA, GSE16011, and REMBRANDT glioma databases. (B, C) The expression levels of ECT2 in 7 nontumor brain specimens, 11 low-grade glioma tissues, and 8 high-grade glioma tissues were measured using western blot; β-actin served as a loading control. The CCK-8 assay was used to detect the changes in cell proliferation at 24, 48, 72, and 96 h after downregulation of ECT2 in U87 and N3 cells (D) or overexpression of ECT2 in LN229 and A172 cells (E) (by ANOVA analysis, n = 3). An EdU assay was used to evaluate the proliferation of glioma cells after the downregulation of ECT2 in U87 cells (F) or overexpression of ECT2 in LN229 cells (G). Representative images are shown (original magnification, 200×). Scale bar: 100 μm.
To confirm that ECT2 affects cell proliferation in glioma, we performed in vitro proliferation-related experiments. We seeded U87 or N3 glioma cells into 96-well plates at a density of 1000 cells per well. The following day we transfected the cells with ECT2 siRNA; the downregulatory effect on ECT2 is shown in Supplementary Figure 4A, B, and the interference effect of siECT2-2 is the most obvious. CCK-8 was then used to continuously monitor changes in cell proliferation at 0, 24, 48, 72, and 96 h after transfection. We found that proliferation was significantly reduced after ECT2 downregulation (Fig. 1D). EdU experiments further confirmed these results (Fig. 1F, Supplementary Figure 4E, G). At the same time, we employed the same experiments to detect variation in cell proliferation after overexpression of ECT2 (Supplementary Figure 4C, D), and the results indicated that cell proliferation was significantly increased after ECT2 upregulation (Fig. 1E, G, Supplementary Figure 4F, H). In summary, we demonstrate that ECT2 can promote the proliferation of glioma cells.
ECT2 Promotes the Expression of PTTG1
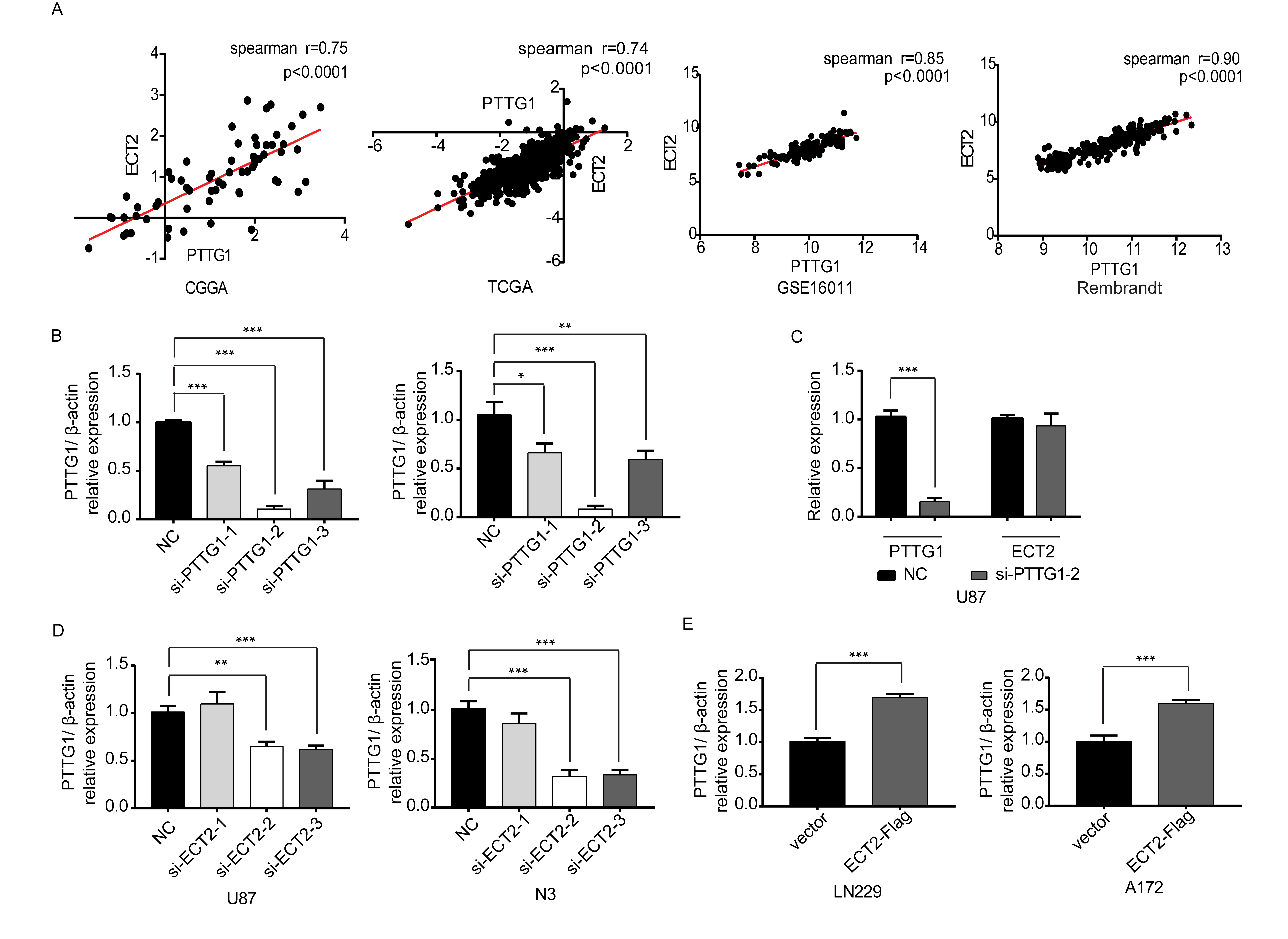
We have reported that PTTG1 can promote the proliferation of glioma cells; in addition, PTTG1 alters the proliferation of renal cancer cells by regulating ECT2 expression. To determine whether PTTG1 affects ECT2 expression in glioma, U87 and N3 cells were transfected with siPTTG1. We extracted the protein from these cells 48 h after transfection and used western blot to detect the levels of PTTG1 and ECT2. We found that the second PTTG1 siRNA produced the greatest interference in U87 and N3 glioma cells (Fig. 2A, Supplementary Figure 5B). We did not observe a significant decrease in ECT2 after PTTG1 downregulation (Fig. 2B, Supplementary Figure 5C); however, we found a significant positive correlation between ECT2 and PTTG1 after analyzing the glioma databases (Supplementary Figure 5A) and clinical tissues by western blot (Spearman’s correlation r = 0.55) (Fig. 2C–E). Therefore, we proposed a hypothesis that ECT2 may be located upstream of PTTG1 and regulates the expression of PTTG1. To prove this hypothesis, U87 and N3 glioma cells were transfected with siECT2. After interference was verified (Fig. 2F), western blot and RT-PCR were employed to detect changes of protein and mRNA levels, and our results showed that the protein and mRNA levels of PTTG1 were significantly reduced after ECT2 interference (Fig. 2F, H, Supplementary Figure 5D). Similarly, ECT2 was upregulated in the LN229 and A172 cell lines; western blot and RT-PCR results showed that PTTG1 protein and mRNA levels were significantly increased after the overexpression of ECT2 (Fig. 2G, I, Supplementary Figure 5E). In summary, we conclude that ECT2 can indeed regulate the expression of PTTG1.
Fig. 2.

ECT2 promotes the expression of PTTG1. (A) Downregulation of PTTG1 in U87 and N3 cells was confirmed using western blot. (B) The protein level of ECT2 was unchanged after the downregulation of PTTG1 using western blot. (C, D) The expression of PTTG1 and ECT2 in clinical tissues was measured using western blot; β-actin served as a loading control. (E) Spearman correlation analysis of the relationship between ECT2 and PTTG1 in identical clinical tissues (r = 0.55, P = 0.0033). (F, G) Western blot was used to detect the changes in PTTG1 protein levels after interference or overexpression of ECT2 in glioma cells. (H, I) RT-PCR was used to detect the mRNA levels of PTTG1 and E2F1 after downregulation or upregulation of ECT2 in glioma cells. Data represent the mean ± SD (*P < 0.05, **P < 0.01).
ECT2 Regulates the Expression of PTTG1 by Affecting the Ubiquitination and Degradation of E2F1
We have shown that ECT2 can regulate the expression of PTTG1. Transcription factors are involved in the formation of transcription initiation complexes and therefore play a crucial role in the regulation of gene expression. In 2009, Zhou et al reported that the transcription factor E2F1 could promote the expression of PTTG1 in pituitary tumors.24 We then analyzed the correlation between ECT2 and PTTG1 genes and E2F1 in the databases and found that E2F1 is positively correlated with both genes (Supplementary Figure 6A, B). This allows E2F1 to act as a bridge between ECT2 and PTTG1. Furthermore, we also identified possible binding sites for E2F1 in the promoter region of PTTG1 through the JASPAR database (Supplementary Figure 7A). To verify whether E2F1 can regulate the expression of PTTG1 in glioma, luciferase reporter assays and chromosomal co-immunoprecipitation experiments were performed (Supplementary Figures 6D, 7B). In addition, using western blot and RT-PCR we found that the protein and mRNA levels of PTTG1 were decreased after downregulation of E2F1 (Fig. 3A, C, Supplementary Figure 7E). Similarly, we overexpressed E2F1 in A172 and LN229 cells. Western blot results showed that PTTG1 protein levels were significantly increased after E2F1 overexpression (Fig. 3B, Supplementary Figure 7F). In summary, we demonstrated that E2F1 altered PTTG1 expression in glioma.
Fig. 3.

ECT2 regulates the expression of PTTG1 by affecting the ubiquitination and degradation of E2F1. (A, B) Western blot was used to detect the protein level of E2F1 and PTTG1 after downregulation or overexpression of E2F1 in glioma cells. (C) RT-PCR was performed to detect the mRNA level of E2F1, PTTG1, and ECT2 after downregulation of E2F1 in U87 and N3 glioma cells. (D) Cells overexpressing E2F1-Flag or control cells were transfected with NC or siECT2-2 for 48 h. Cell lysates were analyzed by western blot using the indicated antibodies. (E, F) U87 cells were pretreated with or without MG132 or chloroquine and then cells were treated with cycloheximide (100 μg/mL) for an additional 0.5, 1, 2, 4, and 8 h. Cell lysates were analyzed by western blot with antibodies against E2F1 and β-actin. (G) U87 cells were transfected with either NC or siECT2-2 for 48 h, followed by cycloheximide (100 μg/mL) treatment for the indicated times. The cell extracts were analyzed by western blot. (H) LN229 cells with or without stable expression of ECT2-Flag were treated with cycloheximide (100 μg/mL) for the indicated timepoints. The cell lysates were examined by western blot. (I, J) Forty-eight hours after downregulation of ECT2 in U87 cells or upregulation of ECT2 in LN229 cells, an in vivo ubiquitination assay was performed with modified immunoprecipitation. Data represent the mean ± SD (**P < 0.01).
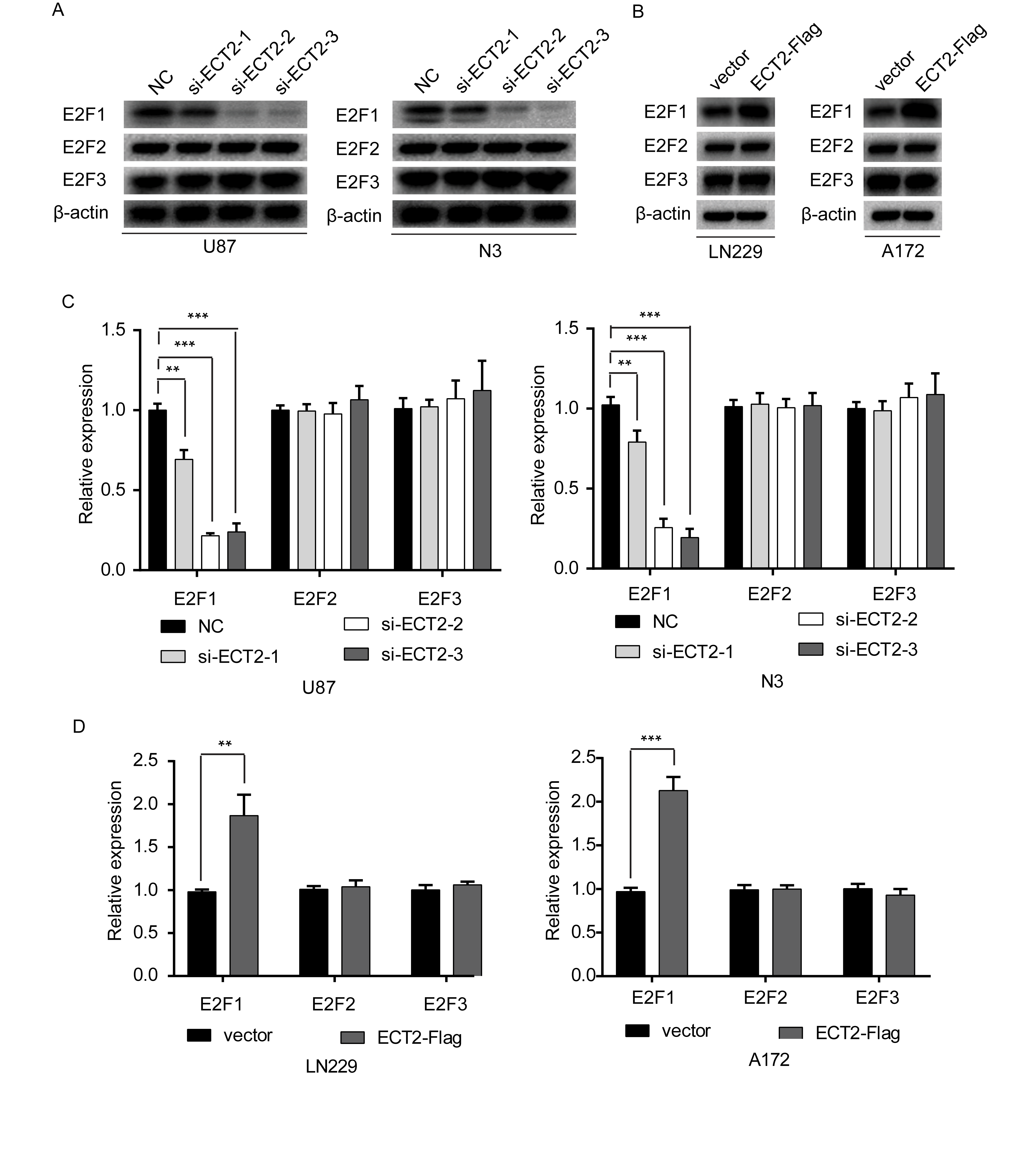
The previous results showed that ECT2 and E2F1 were positively correlated, and both ECT2 and E2F1 alter PTTG1 expression. Thus, we investigated whether ECT2 affects the expression of PTTG1 through the transcription factor E2F1. Using western blot and RT-PCR we measured the E2F1 levels after downregulation or overexpression of ECT2, and found that the E2F1 protein level was decreased after interference of ECT2 (Supplementary Figure 8A, B) or increased after overexpression of ECT2 in glioma cells (Supplementary Figure 8C, D). However, we did not observe any change in E2F1 mRNA levels after ECT2 overexpression or interference in glioma cells (Fig. 2H, I). To determine whether ECT2 specifically regulated E2F1 levels, we examined levels of the other two E2F family members: E2F2 and E2F3. ECT2 regulated E2F1 protein levels without altering E2F2 and E2F3 abundance (Supplementary Figure 8A–D). To further confirm that ECT2 indeed regulates the expression of PTTG1 through E2F1, we transfected siECT2 together with E2F1-Flag plasmids in U87 and N3 glioma cells. Western blot was employed to detect the protein level of ECT2, E2F1, and PTTG1 using corresponding antibodies. Overexpression of E2F1 reversed the inhibition of PTTG1 levels caused by the downregulation of ECT2 (Fig. 3D). In short, we demonstrated that ECT2 regulated the expression of PTTG1 through E2F1.
Previously, we confirmed that ECT2 affected the protein level of E2F1 but not its mRNA level. Therefore, we posited that ECT2 affected mainly the stability of the E2F1 protein in glioma. It is well known that the degradation of intracellular proteins mainly depends on one of 2 pathways: the lysosomal or the ubiquitin-proteasome pathway. About 80% of proteins are degraded by the latter. Thus, we investigated which pathway is the main mechanism for degradation of E2F1 in glioma. We treated U87 cells with MG132 (20 μΜ), a protease inhibitor, or chloroquine (20 μΜ), a lysine autophagy inhibitor. After adding cycloheximide (CHX; 100 μg/mL) for 0.5, 1, 2, 4, and 8 h, western blot was performed to analyze the cellular extracts. The results showed that E2F1 degradation was inhibited by a proteasome inhibitor (MG132), but not by a lysosome/autophagy inhibitor (chloroquine) (Fig. 3E, F, Supplementary Figure 9A, B), suggesting that E2F1 degradation occurs mainly in glioma cell proteasomes. To verify whether ECT2 can affect the degradation of E2F1, U87 and N3 cells transfected with siECT2 were treated with CHX to observe the degradation rate of E2F1. We collected proteins after treatment with CHX at specific timepoints (0, 0.5, 1, 2, 4, and 8 h); western blot was then applied to detect changes in E2F1 protein levels. The results showed that the degradation of E2F1 accelerated after downregulation of ECT2 (Fig. 3G, Supplementary Figure 9C, E, F). Meanwhile, E2F1 protein degrades more slowly after ECT2 overexpression (Fig. 3H, Supplementary Figure 9D, G, H). Together, these data indicate that ECT2 can influence the degradation of E2F1 through the proteasome pathway. To further verify this conclusion, in vivo ubiquitination experiments were used to examine the ubiquitination level of E2F1 after downregulation or overexpression of ECT2. Our results showed that the ubiquitination level of E2F1 increased after ECT2 interference (Fig. 3I), while the level decreased after ECT2 overexpression (Fig. 3J). In summary, we conclude that ECT2 inhibits the degradation of E2F1 through the ubiquitin-proteasome pathway.
PSMD14 Deubiquitinates and Stabilizes E2F1 to Regulate PTTG1 Expression
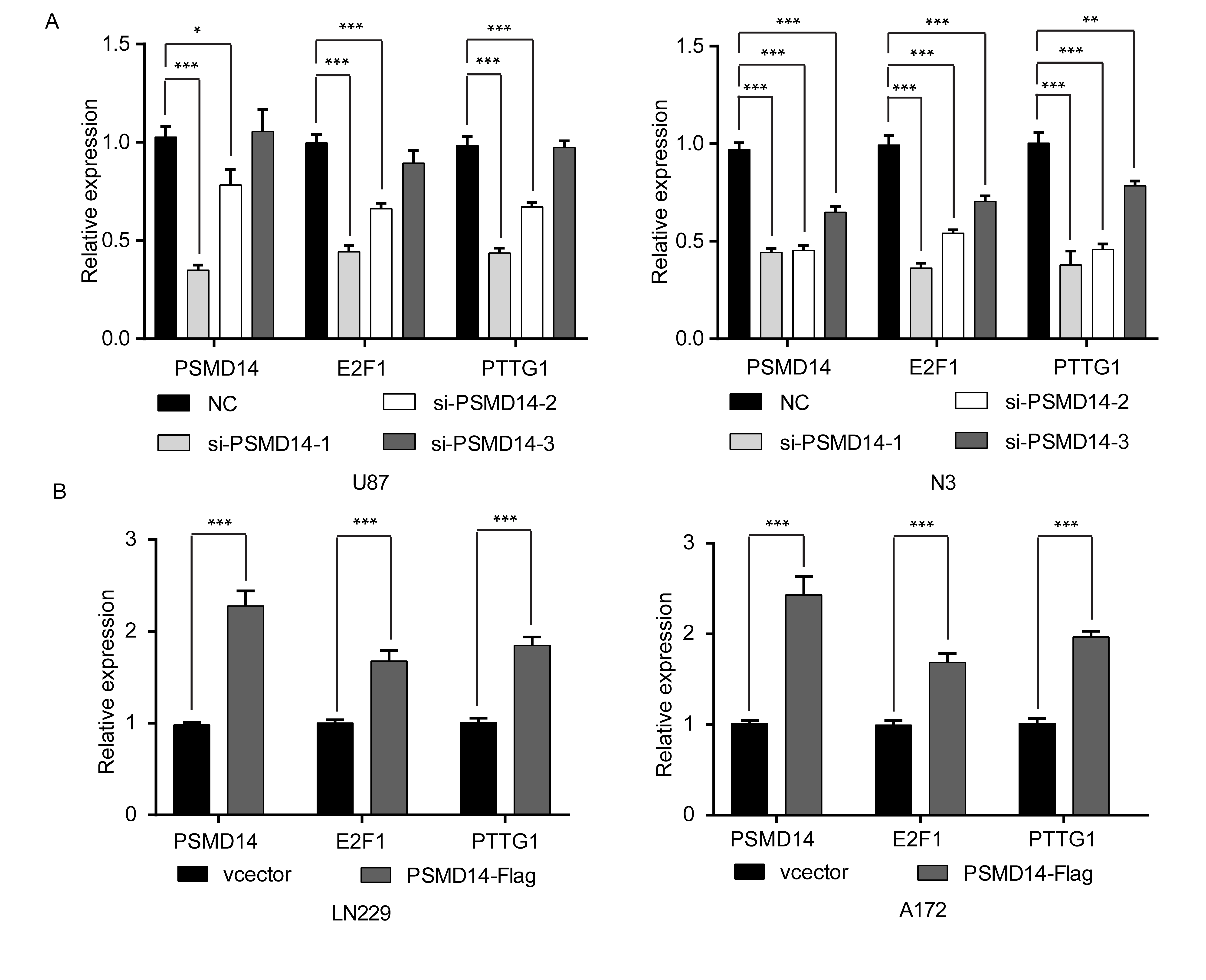
We have shown that ECT2 influences the expression of PTTG1 by affecting the ubiquitin degradation of E2F1. The ubiquitination state of proteins is regulated by both the ubiquitination and the deubiquitination system. Recently, it has been reported that the deubiquitination enzyme PSMD14 can affect the ubiquitylation state of E2F1 in hepatoma cells.25 To explore whether PSMD14 could also affect the ubiquitylation state of E2F1 in glioma, co-immunoprecipitation experiments were performed to detect the relationship between PSMD14 and E2F1, and the results showed that PSMD14 could indeed co-precipitate with E2F1 in U87 and N3 cells (Fig. 4A). Furthermore, in vivo ubiquitination assays were used to examine the ubiquitination level of E2F1 after downregulation or overexpression of PSMD14, and our results showed that the ubiquitination level of E2F1 increased after PSMD14 interference (Fig. 4B), while these decreased after PSMD14 overexpression (Fig. 4C). These results indicate that PSMD14 may alter the ubiquitination of E2F1. Next, we investigated what impact PSMD14 has on the protein levels of E2F1 and PTTG1. Protein was extracted 48 h after interference or overexpression of PSMD14 and subjected to western blot assays to detect changes in E2F1 and PTTG1 expression levels using corresponding antibodies. Our results showed that the level of E2F1 and PTTG1 decreased after PSMD14 interference (Fig. 4D, Supplementary Figure 10A), while these levels increased after overexpression of PSMD14 (Fig. 4E, Supplementary Figure 10B). In conclusion, PSMD14 can indeed affect PTTG1 expression in glioma by affecting the ubiquitination-mediated degradation of E2F1.
Fig. 4.

The deubiquitinase PSMD14 stabilizes E2F1 and further promotes the expression of PTTG1. (A) Endogenous E2F1 was found to bind PSMD14 in U87 and N3 cells using co-immunoprecipitation assays. (B, C) Forty-eight hours after downregulation of PSMD14 in U87 and N3 cells or upregulation of PSMD14 in LN229 and A172 cells, an in vivo ubiquitination assay was performed with modified immunoprecipitation. (D) The protein levels of PSMD14, E2F1, and PTTG1 were detected by western blot after downregulation of PSMD14 in U87 and N3 cells. (E) Western blot was employed to detect the protein levels of PSMD14, E2F1, and PTTG1 after overexpression of PSMD14 in LN229 and A172 cells. β-actin served as the loading control.
ECT2 Promotes the Ubiquitination and Degradation of E2F1 by Regulating the Expression of PSMD14
ECT2 affects the ubiquitination of E2F1, and the deubiquitinating enzyme PSMD14 can also affect the ubiquitination of E2F1. We propose the following hypothesis: ECT2 affects the ubiquitination of E2F1 by regulating PSMD14. Western blot was employed to verify the above hypothesis. As a result, we found that the protein level of PSMD14 was decreased after downregulation of ECT2 in U87 and N3 glioma cells (Fig. 5A, Supplementary Figure 11A), while its level increased after overexpression of ECT2 in A172 and LN229 glioma cells (Fig. 5B, Supplementary Figure 11B). In 2017, Wang et al reported that the deubiquitinating enzyme USP11 can affect the repair of lung epithelial cell damage by altering the expression of the Peg10 gene through the ubiquitination-mediated degradation of E2F1.26 However, we found no significant changes in the protein levels of USP11 after ECT2 interference or overexpression (Fig. 5A, B, Supplementary Figure 11A, B). Therefore, we posit that ECT2 specifically regulates PSMD14, which further affects the ubiquitination and degradation of E2F1 in glioma. To further verify this hypothesis, co-immunoprecipitation was used to detect the binding of PSMD14 and E2F1 after downregulation or upregulation of ECT2 in glioma cells. Our results showed that the binding of PSMD14 with E2F1 decreased after the interference of ECT2 in U87 and N3 cells (Fig. 5C), while binding increased after overexpression of ECT2 in A172 and LN229 cells (Fig. 5D). To confirm that ECT2 regulates E2F1 and PTTG1 through PSMD14, we transfected siECT2 together with PSMD14-Flag plasmids in U87 and N3 glioma cells. Western blot using corresponding antibodies was employed to detect the protein levels of ECT2, PSMD14, E2F1, and PTTG1. Overexpression of PSMD14 reversed the inhibition of E2F1 and PTTG1 protein levels caused by the downregulation of ECT2 (Fig. 5E). Since ECT2 affects the protein level of PSMD14, we investigated the effect of ECT2 on PSMD14 mRNA levels. Using RT-PCR we found that the mRNA level of PSMD14 decreased after the interference of ECT2 in U87 and N3 cells (Fig. 5F), while PSMD14 levels increased after overexpression of ECT2 in A172 and LN229 cells (Fig. 5G). In summary, we posit that ECT2 alters the ubiquitination of E2F1 by affecting the expression of PSMD14 in glioma, thereby affecting the expression of PTTG1 and further altering glioma cell proliferation.
Fig. 5.

ECT2 promotes the ubiquitination and degradation of E2F1 by regulating the expression of PSMD14. (A, B) The protein levels of PSMD14 and USP11 were detected using western blot after downregulation or overexpression of ECT2 in glioma cells. β-actin served as the loading control. (C, D) After interference or overexpression of ECT2, cell lysates were extracted and analyzed by immunoprecipitation with corresponding antibodies. (E) Cells overexpressing PSMD1 4-Flag or control were transfected with NC or siECT2-2 for 48 h. Cell lysates were analyzed by western blot. (F, G) RT-PCR was used to detect the mRNA level of PSMD14 after downregulation or overexpression of ECT2 in glioma cells.
ECT2 Promotes Tumor Growth In Vivo
In vitro experiments show that ECT2 can promote glioma cell proliferation, so we designed in vivo experiments to further verify the effect of ECT2 on the proliferation of glioma cells. We transfected lentivirus containing short hairpin (sh) nucleocapsid (NC) or shECT2 in U87 cells stably expressing luciferase, and the knockdown effect was verified using western blot (Supplementary Figure 12A). These U87 cells were then implanted into the left caudate putamen in nude mice to create intracranial orthotopic tumor models. The size of the intracranial tumors in nude mice was dynamically observed and recorded using an In Vivo Imaging System. Statistical analysis revealed that the growth of intracranial tumors was significantly inhibited in the shECT2 group (Fig. 6A, B). Kaplan–Meier survival analysis revealed that the survival time of nude mice was significantly prolonged after downregulation of ECT2 (Fig. 6C). Moreover, hematoxylin and eosin staining showed that the tumor volume of the shECT2 group was significantly smaller than that of the control group (Fig. 6D, E). In addition, the results of immunohistochemistry showed that the expression of PSMD14, E2F1, PTTG1, and Ki-67 in the in situ tumors was significantly reduced after interference with ECT2 expression (Supplementary Figure 12B, C). The above experiments further confirmed that ECT2 could promote the proliferation of glioma cells in vivo and proved that ECT2 altered the protein expression levels of PSMD14, E2F1, and PTTG1. These results were consistent with our in vitro experimental results.
Fig. 6.

ECT2 promotes tumor growth in vivo. (A, B) U87 cells pretreated with shNC or shECT2 lentivirus and a lentivirus containing luciferase were implanted into the brains of nude mice, and tumor formation was assessed using bioluminescence imaging at days 0, 6, 12, 18, 24, and 30 after implantation. (C) Overall survival was determined using Kaplan–Meier survival curves, and a log-rank test was used to assess the statistical significance of the differences. (D, E) Tumor volume in the shECT2 group was significantly diminished based upon hematoxylin and eosin histology.
Discussion
The human ECT2 gene was originally identified as an oncogene capable of transforming NIH/3T3 fibroblasts.27 Many reports have shown that ECT2 promotes malignant transformation in many cancers.28,29 ECT2-deficient glioma cells appear to be multinucleated, which is a hallmark feature of cytokinesis,1 indicating that the decrease in proliferation observed in ECT2 knockout glioma cells is due to the inability of ECT2-deficient cells to undergo effective cell division. However, there are few reports on the specific molecular mechanisms of the effect of ECT2 on the proliferation of glioma cells.
Our group has reported that PTTG1 is highly expressed in glioma and has a positive correlation with pathological grades.20 Therefore, we sought to explore the specific mechanisms of PTTG1 effects upon the proliferation of glioma cells. Wondergem et al reported that PTTG1 can affect the activation of RhoA by regulating the expression of ECT2 in renal cancer cells,21 further affecting cell aggression. We have confirmed through experiments that this pathway does not exist in glioma. However, analysis of the commonly used glioma databases showed that the expression profiles of ECT2 and PTTG1 showed a significant positive correlation. Therefore, we propose the assumption that ECT2 is located upstream of PTTG1. Later experiments confirmed our hypothesis. This study focused mainly on how ECT2 affected the expression of PTTG1 and thus influenced the proliferation of glioma cells.
Transcription factors play a crucial role in the regulation of gene expression. E2F1 reportedly affects PTTG1 expression in pituitary tumors.24 We found possible binding sites for E2F1 in the promoter region of the PTTG1 gene using JASPAR software. Using western blot, RT-PCR, and co-immunoprecipitation we verified that ECT2 can indeed regulate the expression of PTTG1 by affecting transcription factor E2F1 in glioma. In addition, we found that ECT2 could affect only the protein level of E2F1 without affecting its mRNA level. Therefore, we posited that ECT2 affected mainly the stability of E2F1. Protein stability is related to the degradation of proteins, and there are 2 protein degradation pathways in human cells: the lysosomal/autophagic pathway and the ubiquitin-proteasome pathway.30 To verify the main pathway through which E2F1 is degraded, glioma cells were treated with the lysosomal inhibitor chloroquine and the proteasome inhibitor MG132. Finally, we confirmed that the degradation of E2F1 in glioma was mainly through the ubiquitin-proteasome pathway. Using western blot and in vivo ubiquitination assays, we further confirmed that ECT2 can indeed affect the expression of PTTG1 by affecting the ubiquitination of E2F1.
It has been reported that the deubiquitinating enzymes PSMD14 and USP11 are closely related to the ubiquitination and degradation of E2F1.25,26 Using western blot, co-immunoprecipitation, and in vivo ubiquitination assays, we confirmed that ECT2 affects the stability of E2F1 by PSMD14 instead of USP11, further altering the expression of PTTG1. As for ECT2 affecting PSMD14, RT-PCR experiments indicated that ECT2 could regulate the expression of PSMD14. In summary, we posit that ECT2 in glioma affects the ubiquitination degradation of E2F1 by regulating the expression of PSMD14, further altering the expression of PTTG1 and thus cell proliferation. A previous study reported that PTTG1 affected the expression of ECT2 in renal cancer,21 but our study confirmed that ECT2 altered the expression of PTTG1 in glioma. We posit that this contradiction is due to the different backgrounds of these studies. Our study clarified the expression of ECT2 in glioma, its influence on the prognosis of patients, and its effect on the proliferation of glioma cells and the related mechanisms. Our results provide a new perspective for the study of the development of glioma and provide a theoretical basis for the development of novel anti-proliferative cancer therapeutic drugs by targeting ECT2. However, much research remains to be continued, such as exploring the specific mechanism by which ECT2 affects the expression of PSMD14, and how PTTG1 affects the proliferation of glioma cells.
Funding
This work was supported by grants from the National Key Research and Development Plan (2016YFC0902500), National Natural Science Foundation of China (81772682, 81772679, 81672501), Jiangsu Province’s Natural Science Foundation (20170108, 20151585), the Program for Advanced Talents within Six Industries of Jiangsu Province (2015-WSN-036,2016-WSW-013), Jiangsu Province’s Key Discipline of Medicine (ZDXKA2016001), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Trent Rogers, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Conflict of interest statement.
No conflicts declared.
References
- 1. Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol. 1999;147(5):921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen J, Xia H, Zhang X, et al. ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. J Hepatol. 2015;62(6):1287–1295. [DOI] [PubMed] [Google Scholar]
- 3. Morin P, Flors C, Olson MF. Constitutively active RhoA inhibits proliferation by retarding G(1) to S phase cell cycle progression and impairing cytokinesis. Eur J Cell Biol. 2009;88(9):495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eguchi T, Takaki T, Itadani H, Kotani H. RB silencing compromises the DNA damage-induced G2/M checkpoint and causes deregulated expression of the ECT2 oncogene. Oncogene. 2007;26(4):509–520. [DOI] [PubMed] [Google Scholar]
- 5. Fields AP, Justilien V. The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv Enzyme Regul. 2010;50(1):190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene. 2009;28(41):3597–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. [DOI] [PubMed] [Google Scholar]
- 8. Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. [DOI] [PubMed] [Google Scholar]
- 9. Peng J, Schwartz D, Elias JE, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21(8):921–926. [DOI] [PubMed] [Google Scholar]
- 10. Meierhofer D, Wang X, Huang L, Kaiser P. Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J Proteome Res. 2008;7(10):4566–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137(1):133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24(19):3353–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: beyond the Usual Suspects’ review series. EMBO Rep. 2008;9(6):536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Graner E, Tang D, Rossi S, et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell. 2004;5(3):253–261. [DOI] [PubMed] [Google Scholar]
- 15. Popov N, Wanzel M, Madiredjo M, et al. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol. 2007;9(7):765–774. [DOI] [PubMed] [Google Scholar]
- 16. Stegmeier F, Rape M, Draviam VM, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446(7138):876–881. [DOI] [PubMed] [Google Scholar]
- 17. Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424(6950):793–796. [DOI] [PubMed] [Google Scholar]
- 18. Murali R, Wiesner T, Scolyer RA. Tumours associated with BAP1 mutations. Pathology. 2013;45(2):116–126. [DOI] [PubMed] [Google Scholar]
- 19. Oliveira AM, Chou MM. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol. 2014;45(1):1–11. [DOI] [PubMed] [Google Scholar]
- 20. Zhi T, Jiang K, Xu X, et al. MicroRNA-520d-5p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting PTTG1. Am J Transl Res. 2017;9(11):4872–4887. [PMC free article] [PubMed] [Google Scholar]
- 21. Wondergem B, Zhang Z, Huang D, et al. Expression of the PTTG1 oncogene is associated with aggressive clear cell renal cell carcinoma. Cancer Res. 2012;72(17):4361–4371. [DOI] [PubMed] [Google Scholar]
- 22. Zhi T, Jiang K, Zhang C, et al. MicroRNA-1301 inhibits proliferation of human glioma cells by directly targeting N-Ras. Am J Cancer Res. 2017;7(4):982–998. [PMC free article] [PubMed] [Google Scholar]
- 23. Meng Q, Zhi T, Chao Y, et al. Bex2 controls proliferation of human glioblastoma cells through NF-κB signaling pathway. J Mol Neurosci. 2014;53(2):262–270. [DOI] [PubMed] [Google Scholar]
- 24. Zhou C, Wawrowsky K, Bannykh S, Gutman S, Melmed S. E2F1 induces pituitary tumor transforming gene (PTTG1) expression in human pituitary tumors. Mol Endocrinol. 2009;23(12):2000–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang B, Ma A, Zhang L, et al. POH1 deubiquitylates and stabilizes E2F1 to promote tumour formation. Nat Commun. 2015;6:8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang D, Zhao J, Li S, et al. Phosphorylated E2F1 is stabilized by nuclear USP11 to drive Peg10 gene expression and activate lung epithelial cells. J Mol Cell Bio. 2018;10(1):60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miki T, Smith CL, Long JE, Eva A, Fleming TP. Oncogene ect2 is related to regulators of small GTP-binding proteins. Nature. 1993;362(6419):462–465. [DOI] [PubMed] [Google Scholar]
- 28. Saito S, Liu XF, Kamijo K, et al. Deregulation and mislocalization of the cytokinesis regulator ECT2 activate the Rho signaling pathways leading to malignant transformation. J Biol Chem. 2004;279(8):7169–7179. [DOI] [PubMed] [Google Scholar]
- 29. Frederick LA, Matthews JA, Jamieson L, et al. Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene. 2008;27(35):4841–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kirkin V, Dikic I. Ubiquitin networks in cancer. Curr Opin Genet Dev. 2011;21(1):21–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.