

ABSTRACT
Parkinson’s disease (PD) is characterized by the degeneration of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies (LB) in neurons. α-Synuclein (αSyn) is a major component of LB and promote the PD pathogenesis via its accumulation by the impaired proteasomal or autophagic clearance. Numerous studies have revealed that the reduction of proteasome activity and autophagy is accelerated by cellular senescence. Leucine-rich repeat kinase 2 (LRRK2) contributes to PD progression and its most prevalent mutation, G2019S LRRK2, increases its activity. Our previous report has shown that the G2019S LRRK2 mutant promoted p53-induced p21 expression and neuronal cytotoxicity. The p53-p21 pathway plays a role in cellular senescence. We hypothesized that the loss of dopaminergic neurons by the stimulated p53-p21 pathway via the G2019S LRRK2 mutation might be associated with cellular senescence, thereby promoting the accumulation of αSyn. We confirmed that the ectopic expression of the phosphomimetic p53 mutant, p21, or G2019 in differentiated SH-SY5Y cells increased the following: 1) the expression of β-galactosidase, a marker of cellular senescence, and the activity of senescence-associated β-galactosidase, 2) endogenous αSyn protein level, but not its mRNA level, and 3) αSyn fibril accumulation in dSH-SY5Y via low proteasome and cathepsin D activities. Elevated oligomeric αSyn and the increase in β-galactosidase with induced p21 were observed in brain lysates of G2019S transgenic mice. Our results suggest that cellular senescence is promoted via the p53-p21 pathway due to the G2019S LRRK2 mutation. Eventually, decreased protein degradation by G2019S-mediated senescence could accelerate αSyn aggregate formation.
KEYWORDS: Parkinson’s disease, LRRK2, cellular senescence, α-synuclein
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease. The primary symptoms of PD include rigidity, resting tremors, and bradykinesia, which occur due to a progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). The formation of massive protein aggregates, including Lewy bodies (LB) and Lewy neurites (LN), has been identified as a pathogenic marker of PD in post-mortem studies. α-Synuclein (αSyn) is a major component of LB and LN and the accumulation of αSyn by impaired protein quality control systems (QCSs), such as the ubiquitin-proteasome or autophagy-lysosome pathway, is a well-defined mechanism of PD pathology [1]. The progression of PD is related to aging, and deteriorated protein QCSs are one consequence of aging [2]. The phenomenon of aging at the cellular level is called cellular senescence, which refers to irreversible cell cycle arrest. Cellular senescence results in reduced proteasome activity, abnormal autophagy, and decreased lysosomal cathepsin activity in various cell lines [3–6]. These symptoms of senescence are similar to the causes of death of dopaminergic neurons in PD. In addition, restored lysosomes by anti-aging treatments have resulted in decelerated propagation of synucleinopathy in Caenorhabditis elegans [7].
The leucine-rich repeat kinase 2 (LRRK2) gene is a PD-causative gene and its familial mutant G2019S, which harbors increased kinase activity, manifests in late-onset progression of PD. The penetration of G2019S LRRK2 in sporadic PD is estimated to increase incrementally from 28% at 59 y of age to 74% at 79 y of age [8]. This evidence suggests that hyperactive LRRK2 may be associated with aging progression. Our previous report revealed that LRRK2 phosphorylated the T304/377 site of p53, a well-known tumor suppressor, thereby increasing p21Waf/Cip1 (p21) expression and cytotoxicity in differentiated SH-SY5Y (dSH-SY5Y) cells, a dopaminergic neuron-like cell line, and rat primary cortical neurons [9]. This p53-p21 induction plays a critical role in cellular senescence [10], while the role of cellular senescence in the loss of dopaminergic neurons and the accumulation of αSyn remains elusive. We proposed that aging-related PD-pathogenic G2019S could promote cellular senescence and aggravate protein QCSs in neurons, because LB-positive cases in G2019S LRRK2 PD patients were reported in previous studies [11]. Specifically, we hypothesized that G2019S-mediated phosphorylated p53 could provoke p21-derived cellular senescence, resulting in the accumulation of αSyn.
Materials and methods
Plasmids, antibodies, and reagents
Construction of Myc-G2019S, Flag-p21, HA-p53 wild-type (WT), and the indicated mutant plasmids were described previously [9]. The following antibodies were used in this study: monoclonal β-galactosidase (Santa Cruz, sc-377257), monoclonal hemagglutinin (HA)-tag [C29F4] (CST, #3724), monoclonal αSyn (BD Biosciences, 610786), monoclonal α-tubulin [DM1A] (Sigma, T9026), monoclonal β-actin (Santa Cruz, sc-47778), monoclonal myc [9E10] (Santa Cruz, sc-40), polyclonal p21 [C-19] (Santa Cruz, sc-397) antibody, goat anti-rabbit or anti-mouse IgG conjugated with horseradish peroxidase (Jackson ImmunoResearch, 111-035-003 or 115-035-003), Goat anti-mouse Alexa Fluor 488 antibody (A-11001), and anti-rabbit Texas Red antibody (T-2767). All-trans retinoic acid (R2625), poly-L-lysine (P8920), and Hoechst 33342 (14533) were purchased from Sigma-Aldrich.
Western blot analysis
SH-SY5Y cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in an incubator with 5% CO2. SH-SY5Y cells (1 × 105) were differentiated for four days by the addition of 10 μM retinoic acid. On day 5, the dSH-SY5Y cells were transfected with 1 μg of each DNA using Lipofectamine LTX (ThermoFisher, 15338100) for the indicated times. Cells were harvested with 1× sample buffer and the lysates were applied to nitrocellulose membranes following sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The rest of the process was performed as described previously [9].
Immunofluorescence analysis and senescence-associated GlycoGreen assay
dSH-SY5Y cells were transfected with the indicated plasmid DNA for 48 h. The protocol for immunofluorescence was described in our previous study [9]. For the staining of senescence-associated β-galactosidase activity, 1 μM senescence-associated GlycoGreen (Goryo Chemical, GC611) in in growth media was incubated for 4 h. Images were acquired using FLoid (Invitrogen) and analyzed by Zen (Zeiss). The present images of GlycoGreen was captured with LSM700 confocal microscope (Zeiss) to show in the high resolution.
Fibrillization and treatment of αSyn
Recombinant human His6-αSyn protein (Boston Biochem, Cat# SP-480, Lot# DBQA0116051) was purchased. αSyn monomers (250 μM) were incubated in hydroxyethyl piperazineethanesulfonic acid (HEPES, 50 mM, pH 7.5) with 100 mM NaCl for one wk in an incubator at 37°C, sonicated for 30 s at 20% amplification using Vibra-Cell Ultrasonic Liquid Processors (Sonics & Materials Inc., VCX 130), and further incubated for 1.5 wk at 37°C. To obtain αSyn fibrils, the sample was ultracentrifuged at 100,000 × g for 1 h in a Beckman Optima TLX tabletop ultracentrifuge (Beckman Coulter). The pellet was reconstituted in the same buffer as that used for the fibrils. αSyn fibrils (5 μM) were placed in 50 mM HEPES (pH 7.5) and 100 mM NaCl and added to the dSH-SY5Y cells for 24 h. To remove membrane-bound αSyn fibrils, the cells were washed with 2 N HCl and subsequently with cold Dulbecco’s phosphate-buffered saline. Then, the cells were lysed in 1× sample buffer (50 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, 1% β-mercaptoethanol, and 0.02% bromophenol blue) and analyzed by western blot.
Circular dichroism
To validate the αSyn fibrils, circular dichroism of diluted (35 μM) αSyn monomers or fibrils was performed from 190 to 250 nm at a bandwidth of 0.2 nm steps using a J-810 Spectropolarimeter (Jasco).
Proteasome activity assay
Each transfected set of 2 × 106 dSH-SY5Y cells was collected and 0.5% NP-40 in phosphate-buffered saline was added to prepare the lysates used in the Proteasome Activity Fluorometric Assay Kit (Biovision Inc., #K245-100). The substrate reaction steps were followed according to the manufacturer’s instructions. The plate was read at Ex/Em = 328/460 nm in a fluorescence spectrometer (LS55, Perkin Elmer).
Cathepsin D activity assay
dSH-SY5Y cells (5 × 106) transfected with the indicated plasmids were lysed with the lysis buffer provided in the Cathepsin D Activity Fluorometric Assay Kit (Biovision Inc., #K143-100). After performing the reactions according to the manufacturer’s instructions, the amount of fluorometric byproduct from the substrate resulting from cathepsin D was measured at Ex/Em = 328/460 nm using a fluorescence spectrometer.
The measurement of mRNA level αSyn
RNeasy Plus Mini Kit (Qiagen, Cat# 74134) was used for the isolation of mRNA. Synthesis of cDNAs from 300 ng of mRNA was performed using SuperScript II Reverse Transcriptase (Invitrogen, 18064014). The PCR products were amplified using the following primers; αSyn (F: TGTAGGCTCAAAACCAAG, R: GCTGGGTAGAGAATGGATGAACA), β-actin (F: CGCCGCCAGCTCACCATG, R: CACGAT GGAGGGGAAGACGG), HA-tagged p53 (F: GGCTACCCATACGATGTTCCAG, R: TCTGGCATTCTGGGAGCTTCATC), Flag-p21 (F: GACTACAAAGACGATGACGATAAAGC, R: GGTACAAGACAGTGACAGGTC), myc-G2019S (F: GTCATGCTGGTATTGGGC, R: CTTCTGAGATGAGTTTTTGTTCCTCGAC). Final products were subjected to 1.5% agarose gels electrophoresis, and bands on the gel were captured with G:Box (Syngene).
Animal care and western blot and native gel analysis of brain lysate
Animal care and brain sample preparation procedures were described in our previous study [12]. Brain lysates mixed in Laemmli sample buffer were subjected to western blot. For native gel analysis, brain lysates mixed with a non-denaturation conditioned sample buffer (321.5 mM Tris-HCl at pH 6.8, 50% glycerol, 0.5% bromophenol blue) were subjected to electrophoresis on a native gel (Invitrogen, BN1004BOX). The proteins were then transferred onto polyvinylidene fluoride membranes (Amersham, 45-004-110) and subjected to western blotting with specific antibodies.
Statistics analysis
The digitized bands of the western blots and mRNA analysis gel were quantified using Multi Gauge V3.0 (Fujifilm). Statistical analysis was performed and all graphs (mean with standard error of the mean) were prepared using Prism 6.0 (GraphPad Software Inc.). Statistical significance is indicated in the graphs as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Results
Up-regulated β-galactosidase protein levels in G2019S-mediated phosphomimetic p53 and overexpression of p21 or G2019S LRRK2
In our previous study, we observed the phosphorylation of the T304/377 site of p53 by LRRK2 and the induction of p21 expression in dSH-SY5Y cells and primary cortical neurons [9]. The up-regulation of β-galactosidase (β-gal) protein levels and the activation of senescence-associated β-gal via the elevation of p21 expression in fibroblasts, breast cancer cell lines, and NIH3T3 cells have also been reported [13,14]. The most common marker of cellular senescence is the increase in β-gal protein level [15]. We observed a significant increase in β-gal protein level with the expression of T304/377D p53 (TD-p53) compared to that in the vector and WT-p53 in dSH-SY5Y cells (Figure 1(a)). In addition, the overexpression of p21 and G2019S LRRK2 in dSH-SY5Y cells resulted in significant increases in β-gal protein expression (Figure 1(b,c)). The increase in β-gal fluorescence by the transient expression of TD-p53, but not phospho-dead mutant T304/377A p53(TA-p53), was significant compared to that of WT-p53 (Figure 2(a)). Moreover, TD-p53-expressing dSH-SY5Y cells exhibited increases in β-gal puncta, which seemed to be located in the lysosome because senescence-associated β-gal is regarded as lysosomal [16]. We evaluated the activity of senescence-associated β-gal using GlycoGreen dye, a synthesized β-gal substrate, and found that ectopic expressions of either red fluorescence protein (RFP)-TD-p53 or RFP-p21 elevated GlycoGreen fluorescence by 2–3 folds (Figure 2(b)). These data suggest that G2019S LRRK2 promoted cellular senescence via phosphorylated p53-mediated p21 induction.
Figure 1.
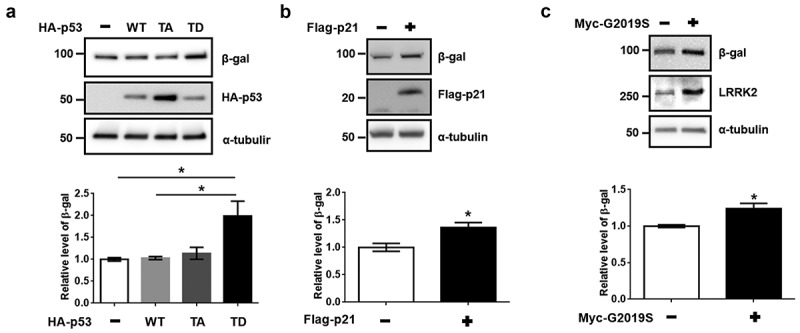
Induction of β-galactosidase by transfection of phosphomimetic p53, p21, and G2019S LRRK2. dSH-SY5Y cells on 12-well plate were transfected with 1 μg of indicated DNA for (a) 48 h, (b) 30 h, or (c) 72 h (N = 3). Cell lysates were evaluated by western blot under identical conditions, using antibodies against HA and β-gal. The expression of β-gal was normalized to that of α-tubulin. Statistical significances were analyzed by one-way analysis of variance (ANOVA), Bonferroni’s (a), or Student’s t-test (b, c).
Figure 2.
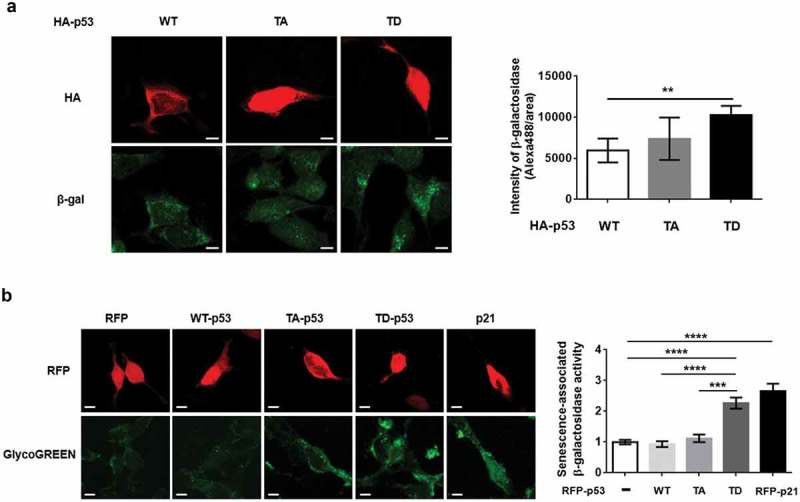
The activation of senescence-associated β-galactosidase by phosphomimetic p53 and p21. (a) dSH-SY5Y cells transfected with indicated DNA were analyzed by levels of β-gal immunostaining in HA-positive cells (N = 4; cell numbers of WT-p53 = 8, TA-p53 = 12, and TD-p53 = 11). (b) The intensity of GlycoGreen levels RFP-expressed dSH-SY5Y cells was analyzed to measure senescence-associated β-galactosidase activities (N = 4; cell numbers of RFP = 51, RFP WT-p53 = 25, RFP TA-p53 = 32, RFP TD-p53 = 32, and RFP p21 = 50). One-way ANOVA with Tukey’s post-hoc analysis was performed for statistical significance.
Decreased proteasome and cathepsin D activity in senescent cell conditions
Chondrogianni et al. reported that senescence associated events decreased proteasome activity in human fibroblasts [3]. Lysosomal cathepsins, whose activities are related to aging and neurodegeneration, have been associated with senescence [17,18]. To determine the senescent effects on protein quality control in post-mitotic dSH-SY5Y cells, we investigated proteasome and cathepsin D activities. Any overexpression of TD-p53, p21, or G2019S resulted in significant reduction of proteasome activity compared to that of the vector, WT-p53, or TA-p53 (Figure 3(a)). In the cathepsin D activity assay, expression of p21 and G2019S showed significantly decreased activity compared to that in the vector, WT-p53, and TA-p53. TD-p53 showed a slight decrease, although the difference was not significant (Figure 3(b)). Taken together, these results indicate that the reduced QCS in aged dopaminergic neurons might be due to the reduction in both proteasome and lysosomal cathepsin D activity in cellular senescence.
Figure 3.
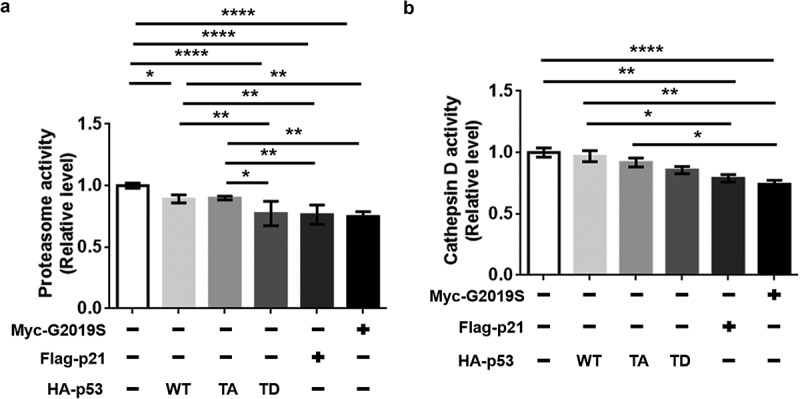
Reduced proteasome and cathepsin D activity by ectopic expression of TD-p53, p21, or G2019S. dSH-SY5Y cells on (a) 100-mm and (b) 60-mm dishes were transfected with the indicated DNAs (1 μg) for 48 h (vector, WT-p53, TA-p53, and TD-p53), 30 h (Flag-p21), or 72 h (myc-G2019S). Assays were performed with duplicates of each sample (N = 3). One-way ANOVA and Tukey’s multiple comparison tests were performed for statistical analysis.
Accumulated endogenous and transmitted αSyn by expression of senescent genetic factors
Several reports have suggested that aging-mediated impairment of protein homeostasis could enhance the aggregation of αSyn in dopaminergic neurons [2,19]. To verify the effect of senescence on the accumulation of αSyn in a dopaminergic neuron cell line, we measured the protein levels of endogenous αSyn (endo-αSyn) in dSH-SY5Y cells. A significant increase in endo-αSyn level was observed following the transfection of TD-p53 compared to that of the vector and WT-p53 (Figure 4(a)). Significantly increased endo-αSyn levels due to p21 and G2019S expression were also observed (Figure 4(b,c)). However, the levels of αSyn mRNA showed slight, non-significant increases following transfection with TD-p53, p21, and G2019S compared to those of the vector or WT-p53 (Figure 4(d)). To elucidate the clearance of transmitted αSyn by the expression of ectopic TD-p53, p21, and G2019S, we treated dSH-SY5Y cells with αSyn fibrils for 24 h. In the western blot analysis, the generated αSyn fibrils showed a high-molecular-weight band that was not observed in the monomers (Figure 5(a)). The secondary structure of the αSyn fibrils shifted to β-sheets from the random-coil structure of their monomeric component in the circular dichroism analysis (Figure 5(b)), thus confirming the fibrillization of αSyn. Interestingly, we observed a significant increase in the levels of endo-αSyn and αSyn fibrils with transient expression of TD-p53, p21, and G2019S (Figure 5(c)). Conclusively, G2019S-mediated senescence via induced p21 expression, as a downstream target of TD-p53, contributed to the accumulation of αSyn in dopaminergic neurons.
Figure 4.
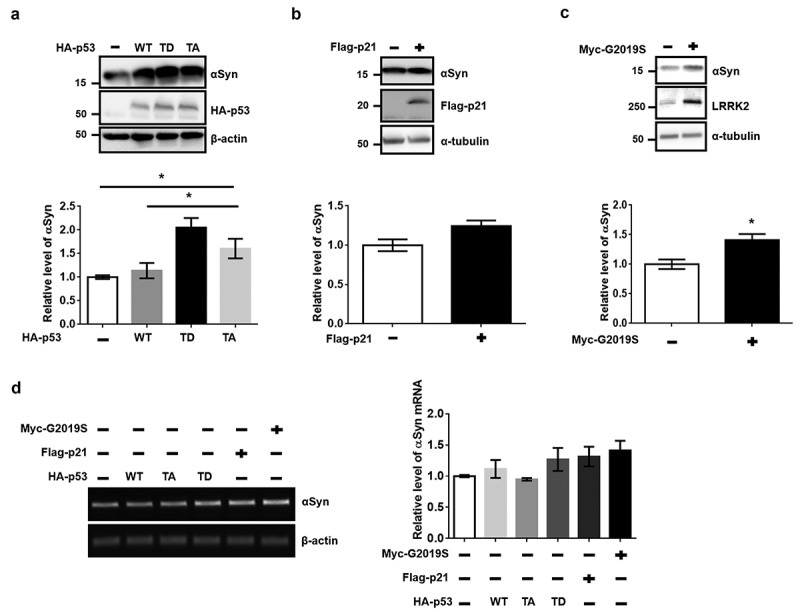
Increases in endogenous αSyn levels by overexpression of TD-p53, p21, or G2019S. dSH-SY5Y cells on 12-well plates were transfected with 1 μg of the indicated DNAs for (a) 48 h, (b) 30 h, or (c) 72 h. The level of endo-αSyn was normalized to that of β-actin or α-tubulin. Statistical significance was assessed by one-way ANOVA and Bonferroni’s multiple comparison tests, N = 4. (d) The mRNA level of endogenous αSyn was analyzed by reverse-transcript PCR. The relative levels of αSyn mRNA were normalized according to β-actin mRNA. The expression of tagged proteins was confirmed by PCR product using each pair of specific priemrs. One-way ANOVA, Bonferroni’s multiple comparison test (N = 3) was used for the statistical analysis.
Figure 5.
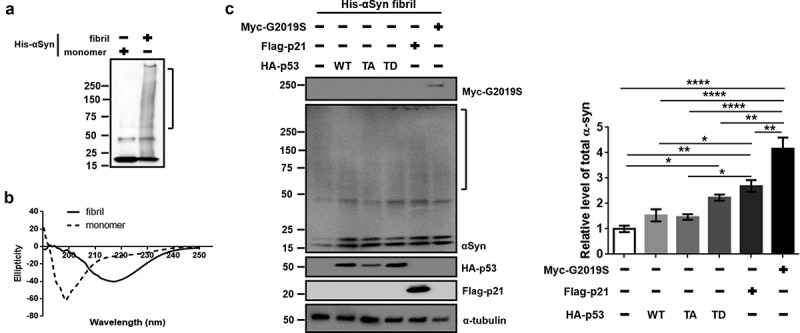
Accumulation of αSyn fibrils in dSH-SY5Y by TD-p53, p21, or G2019S expression. (a) Western blot of His-tagged recombinant αSyn monomers and fibrils. (b) Secondary conformations of His-tagged recombinant αSyn monomers and fibrils were analyzed by circular dichroism. (c) After transfection of dSH-SY5Y cells with the indicated DNAs (0.5 μg) for 24 h (vector, WT-p53, TA-p53, and TD-p53), 6 h (Flag-p21), or 48 h (myc-G2019S) in a 24-well plate, 5 μM αSyn fibrils were added and incubated for 24 h. The membrane-bound αSyn fibrils were washed with 2 N HCl. Total αSyn of the cell lysates was quantified with a column box, including the high-molecular-weight band (square bracket), to the 20-kDa His αSyn monomer and endo-αSyn monomer. The level of total αSyn was normalized to that of α-tubulin. Statistical significance was assessed by one-way ANOVA and Bonferroni’s multiple comparison tests, N = 3.
To elucidate the enhancement of senescence by G2019S LRRK2 in vivo, we analyzed whole-brain lysates of G2019S transgenic (G2019S TG) mice. G2019S TG mice showed a significant increase in β-gal levels along with a significant induction of p21 (Figure 6(a)). To evaluate the accumulation of oligomeric αSyn in the brain of G2019 TG mice, we carried out a native gel analysis with the mouse brain lysates. The amount of αSyn aggregates was elevated in the brain of G2019S TG mice (Figure 6(b)). Accumulated αSyn in G2019S TG mice was also observed by western blot and dot blot assay using αSyn conformation-specific antibodies [20].
Figure 6.
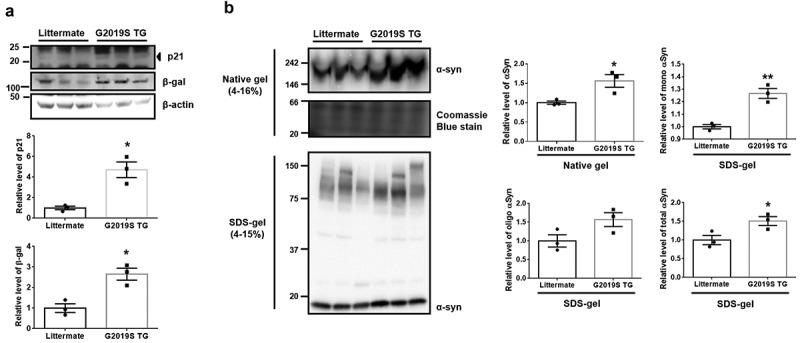
Elevation of αSyn aggregates via the induction of β-gal and p21 levels in the brain of G2019S transgenic mice. (a) β-gal and p21 levels in littermate and G2019S TG brain lysates were detected by western blot. (b) To identify the presence of or increase in oligomeric αSyn of littermate or transgenic mouse brains, non-denaturated brain lysates were analyzed using a native gel. Statistical significance was assessed by one-way analysis of variance and Bonferroni’s multiple comparison tests, N = 3.
Discussion
Senescent cell cycle arrest events present multiple cellular features, including altered gene expression, dysregulated autophagy, abnormal metabolism, and cell death, in numerous cell lines, including cancer cells, fibroblasts, and other proliferating-competent cells. Developed cellular senescence in endothelial cells acquires induction of senescence-associated secretory phenotype (SASP) factors such as pro-inflammatory cytokines, growth factors, proteases, soluble receptors, and reactive oxygen species [21]. The SASP factors, matrix metalloproteinase 3 and p16INK4, are induced in the astrocytes of aged human brain tissue in autopsied and cortical brain tissue of patients with Alzheimer’s disease (AD) compared to age-matched controls [22]. Moreover, increased numbers of senescent astrocytes were observed in autopsied SNpc of PD patients relative to age-matched controls, and elevated levels of p16INK4 protein and γH2AX foci, a marker of DNA damage, were also detected in the SNpc of PD patients [23,24]. Our previous study showed that the expression of TD-p53, a phosphomimetic form of LRRK2-mediated p53, increased the expression and release of the pro-inflammatory cytokine tumor necrosis factor α (TNFα), which is one of the SASP factors, in a BV2 murine microglial cell line [25]. However, we did not test p21, p16INK4, and β-gal induction at that time. Surprisingly, anti-TNFα treatment attenuated the induction of SASP in human umbilical vein endothelial cells and MCF-7 cells [26]. The results of a recent study revealed that post-mitotic neurons underwent p21-dependent senescence, which was derived by DNA damage response [27]. These reports support that senescent astroglia and neurons may be involved in neurodegenerative diseases, including AD, PD, Huntington’s disease, and amyotrophic lateral sclerosis.
LRRK2 is a critical gene for the pathogenesis of PD. Although the cellular function of LRRK2 is not fully understood, the G2019S pathogenic mutant, which harbors upregulated kinase activity, manifests the degeneration of neurite, mitochondrial dysfunction, apoptotic death, abnormal gene transcription, defective protein quality control, and acceleration of protein aggregates in dopaminergic neurons [11,20,28–30]. These features are related to the cellular senescence, and moreover, our findings, that are extended from our previous report [9], support the senescent state initiated by LRRK2. Especially, G2019S mutant was majorly detected in late-onset sporadic PD patients [31], and senescence has been regarded as a major symptom of ageing [10]. These similarities between LRRK2-mediated neuronal damage and cellular senescence could be critical for the pathology of PD. In addition, a recent study has revealed that the cellular senescence in parkinsonian SNpc is induced by paraquat [32], which is known as a PD-toxin acting via the cellular function of LRRK2 [33].
Previous studies have reported the association between decreased proteasome or cathepsin D activity and cellular senescence [3,34]. The degradation of αSyn is accomplished by both the ubiquitin-proteasome and autophagy-lysosome pathways [35]. Cathepsin D is a critical lysosomal enzyme involved in the clearance of αSyn [36]. The introduction of anti-aging genetic factors or pharmacological treatment in C. elegans decelerated the propagation of αSyn aggregates and rescued the life span of damaged worms by αSyn, via the reactivated lysosome biogenesis [7]. We observed reduced proteasome and cathepsin D activity in dSH-SY5Y cells with elevated β-gal levels (Figures 1, 2, and 3), and these cells also exhibited accumulated αSyn monomers and fibrils (Figures 4 and 5). In addition, G2019S TG mice showed increases in αSyn aggregates with the induction of β-gal and p21 levels (Figure 6). Our results suggest that cellular senescence mediated by G2019S LRRK2 in post-mitotic dopaminergic neurons might enhance the accumulation of αSyn by reducing proteasome and cathepsin D activity.
Conclusion
In our previous study, LRRK2 phosphorylated p53 at T304/377 and promoted p21 expression so that neuronal cell death was provoked. In this study, we observed that phosphorylation of T304/377 in the p53-p21 induction pathway was involved in cellular senescence. The reduction of proteasome and cathepsin D activities could aggravate the accumulation of misfolded αSyn, thereby accelerating the aggregation of αSyn in neuron and the propagation of synucleinopathy. These novel findings suggest that the relationship between cellular senescence and LRRK2 activity is responsible for the age-related propagation of αSyn aggregates in PD pathogenesis.
Acknowledgments
This paper was supported by Wonkwang University in 2018.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Pan T, Kondo S, Le W, et al. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–1978. [DOI] [PubMed] [Google Scholar]
- [2].Douglas PM, Dillin A.. Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010;190:719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chondrogianni N, Stratford FL, Trougakos IP, et al. Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J Biol Chem. 2003;278:28026–28037. [DOI] [PubMed] [Google Scholar]
- [4].García-Prat L, Martínez-Vicente M, Perdiguero E, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37–42. [DOI] [PubMed] [Google Scholar]
- [5].Sitte N, Merker K, Von Zglinicki T, et al. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part II–aging of nondividing cells. FASEB J. 2000;14:2503–2510. [DOI] [PubMed] [Google Scholar]
- [6].Sitte N, Merker K, Von Zglinicki T, et al. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part I–effects of proliferative senescence. FASEB J. 2000;14:2495–2502. [DOI] [PubMed] [Google Scholar]
- [7].Kim DK, Lim HS, Kawasaki I, et al. Anti-aging treatments slow propagation of synucleinopathy by restoring lysosomal function. Autophagy. 2016;12:1849–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ho DH, Kim H, Kim J, et al. Leucine-rich repeat kinase 2 (LRRK2) phosphorylates p53 and induces p21(WAF1/CIP1) expression. Mol Brain. 2015;8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Munoz-Espin D, Serrano M.. Cellular senescence: from physiology to pathology [review]. Nat Rev Mol Cell Biol. 2014;15:482–496. [DOI] [PubMed] [Google Scholar]
- [11].Ross OA, Toft M, Whittle AJ, et al. Lrrk2 and lewy body disease. Ann Neurol. 2006;59:388–393. [DOI] [PubMed] [Google Scholar]
- [12].Ho DH, Je AR, Lee H, et al. LRRK2 kinase activity induces mitochondrial fission in microglia via Drp1 and modulates neuroinflammation. Exp Neurobiol. 2018;27:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Capparelli C, Chiavarina B, Whitaker-Menezes D, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen X, Zhang W, Gao YF, et al. Senescence-like changes induced by expression of p21Waf1/Cip1 in NIH3T3 cell line. Cell Res. 2002;12:229–233. [DOI] [PubMed] [Google Scholar]
- [15].Kurz DJ, Decary S, Hong Y, et al. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113:3613–3622. [DOI] [PubMed] [Google Scholar]
- [16].Lee BY, Han JA, Im JS, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging cell. 2006;5:187–195. [DOI] [PubMed] [Google Scholar]
- [17].Kraus S, Bunsen T, Schuster S, et al. Cellular senescence induced by cathepsin X downregulation. Eur J Cell Biol. 2011;90:678–686. [DOI] [PubMed] [Google Scholar]
- [18].Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev. 2016;32:22–37. [DOI] [PubMed] [Google Scholar]
- [19].Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev. 2011;10:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ho DH, Kim H, Nam D, et al. LRRK2 impairs autophagy by mediating phosphorylation of leucyl-tRNA synthetase. Cell Biochem Funct. 2018;36:431–442. [DOI] [PubMed] [Google Scholar]
- [21].Coppé J-P, Desprez P-Y, Krtolica A, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Ann Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bhat R, Crowe EP, Bitto A, et al. Astrocyte senescence as a component of Alzheimer’s disease. PLOS ONE. 2012;7:e45069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chinta SJ, Woods G, Rane A, et al. Cellular senescence and the aging brain. Exp Gerontol. 2015;68:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rodier F, Muñoz DP, Teachenor R, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2010;124:68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ho DH, Seol W, Eun JH, et al. Phosphorylation of p53 by LRRK2 induces microglial tumor necrosis factor α-mediated neurotoxicity. Biochem Biophys Res Commun. 2017;482:1088–1094. [DOI] [PubMed] [Google Scholar]
- [26].Prattichizzo F, Giuliani A, Recchioni R, et al. Anti-TNF-α treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget. 2016;7:11945–11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging cell. 2012;11:996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang X, Yan MH, Fujioka H, et al. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012;21:1931–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Winner B, Melrose HL, Zhao C, et al. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis. 2011;41:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hébert S, Dorval V. LRRK2 in transcription and translation regulation: relevance for Parkinson’s disease. Front Neurol. 2012;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Infante J, Rodríguez E, Combarros O, et al. LRRK2 G2019S is a common mutation in Spanish patients with late-onset Parkinson’s disease. Neurosci Lett. 2006;395:224–226. [DOI] [PubMed] [Google Scholar]
- [32].Chinta SJ, Woods G, Demaria M, et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 2018;22:930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Imai Y, Gehrke S, Wang HQ, et al. Phosphorylation of 4E‐BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. Embo J. 2008;27:2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Su S, Zhu X, Lin L, et al. Lowering endogenous cathepsin D abundance results in reactive oxygen species accumulation and cell senescence. Mol Cell Proteomics. 2017;16:1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Webb JL, Ravikumar B, Atkins J, et al. Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. [DOI] [PubMed] [Google Scholar]
- [36].Sevlever D, Jiang P, Yen SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry. 2008;47:9678–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]