

ABSTRACT
Sirtuin 6 (SIRT6) has the function of regulating autophagy. The aim of this study was to investigate the mechanism through which SIRT6 relieved acute kidney injury (AKI) caused by sepsis. The AKI model was established with lipopolysaccharides (LPS) using mice. Hematoxylin-eosin (HE) staining and streptavidin-perosidase (SP) staining was used to observe kidney tissue and test SIRT6 and LC3B proteins in kidney. Enzyme-linked immunosorbent assay (ELISA) was performed to detected the tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) concentrations. Cell counting kit-8 (CCK-8) assay and flow cytometry were carried out to test the cell viability and apoptosis rate respectively. Protein and mRNA were determined by Western blot and quantitative real-time polymerase chain reaction (qRT-PCR). AKI induced by LPS had self-repairing ability. At 12 h after modeling, the expression levels of TNF-α, IL-6, SIRT6 and LC3B-II/LC3B-I were first significantly increased and were then significantly decreased at 48 h after modeling. LPS inhibited the growth of HK-2 cells and promoted the expressions of TNF-α, IL-6, SIRT6 and LC3B. Overexpression of SIRT6 down-regulated the secretion of TNF-α and IL-6 induced by LPS. SIRT6 overexpression inhibited apoptosis induced by LPS and promoted autophagy in HK-2 cells. Silencing of the SIRT6 gene not only promoted the secretion of TNF-α and IL-6 by HK-2 cells, but also promoted apoptosis and reduced autophagy. LPS up-regulated the expression of SIRT6 gene in HK-2 cells. Overexpression of the SIRT6 gene could inhibit apoptosis and induce autophagy, which might be involved in repairing kidney damage caused by LPS.
KEYWORDS: Sirtuin 6, acute kidney injury, apoptosis, autophagy
Introduction
Sepsis refers to the systemic inflammatory response syndrome (SIRS) caused by infection, and it is a common complication of burns, trauma and infectious diseases [1]. Although new antibiotics have been successfully marketed in recent years, early fluid resuscitation, multi-organ function support and other technologies have been discovered, the incidence and mortality of sepsis have not been accordingly reduced [2]. Severe sepsis and septic shock have become one of the leading causes to death in ICU patients [3].
Further development of sepsis can cause multiple organ dysfunction syndromes, among which acute kidney injury (AKI) is one of the most common complications of sepsis [4]. Endothelial dysfunction and microcirculation disturbance caused by endothelial cell apoptosis is a key link in the pathogenesis of sepsis, and cell apoptosis also plays a key role in AKI [5,6]. Previous studies have found that autophagy was activated in multiple organ damages caused by sepsis [7–9]. However, the role of autophagy on target cells and target organs is still controversial.
Autophagy is an intracellular bulk degradation system, it has the function of regulating and maintaining the homeostasis of cells and tissues. The formation of autophagosome is the main process of autophagy, and the fusion of autophagosomes and lysosomes can degrade the contents in the autophagosome [10,11]. LC3B, which belongs to microtubule-associated protein 1 light chain 3 (LC3) family, is a homolog of atg8 gene in mammalian cells and is currently recognized as an autophagy-related protein. LC3B localizes on the surface of autophagic vesicle membrane and participates in autophagosome formation, and the expression intensity of LC3B is closely related to autophagy activity [12–14].
Sirtuins are a family of NAD+-dependent histone deacetylases, which are involved in inflammation, energy metabolism, stress resistance and cancer [15]. Sirtuin 6 (SIRT6) is closely related to the autophagy of cells [16], and study has shown that the expression level of Sirt6 is reduced in diabetic nephropathy mice, while only few researches were conducted on SIRT6 and autophagy in AKI [17]. Recent study has shown that SIRT6 overexpression could attenuate cisplatin-induced AKI [18]. We therefore explored the role of SIRT6 in sepsis-induced acute kidney injury.
This research mainly studies the expression characteristics of SIRT6 in AKI caused by sepsis, and explores the effect of SIRT6 overexpression or low expression on AKI and its mechanism of action. The study provides a new approach to a better clinical treatment of AKI.
Materials and methods
Animals and modeling methods
C57BL/6 mice (22 g–26 g, SFP) were purchased from Laboratory Animal Secter (China). Modeling and follow-up experimental programs had been approved by the Institutional Animal Care and Use Committee and China Council on Animal Care.
60 mice were randomly divided into 2 groups: control group (n = 12) and LPS group (n = 48). The mice in LPS group were injected with LPS (20mg/kg) intraperitoneally (i.p.), while the mice in control group were injected with phosphate buffer saline (PBS) (i.p.).
Sample collection
Twelve mice were taken at 1h (LPS-1 h group), 12h (LPS-12 h group), 24h (LPS-24 h group) and 48h (LPS-48 h group) after LPS injection, and then anesthetized with 1% sodium pentobarbital (50 mg/kg). The thoracic cavity was cut, and blood samples of the 12 mice in each group were taken by heart puncture and maintained in anticoagulation tubese. After being solidified, the serum was collected by centrifugation (3500 rps, 10 min). The renal arteries and veins were ligated using vascular clamps, and the two kidneys in the 5 groups were collected with excess fat being removed. One kidney was fixed in 10% neutral formaldehyde for hematoxylin-eosin (HE) staining and Streptavidin-perosidase (SP) staining, while the other was stored under −80°C for Western blot and quantitative real-time polymerase chain reaction (qRT-PCR) detection.
HE staining
The fixed kidney tissue was embedded in paraffin. The sample was cut into 4μm thick uniform flakes and placed on APES-coated glass slides. The samples were deparaffinized using xylene and hydrated and stained using HE reagent (Sigma, USA). Hematoxylin was added and the sample was incubated at room temperature for 5 min. After being washed, eosin was added and the sample was incubated at room temperature for about 2 min. The alveolar morphology was observed under a microscope.
Immunohistochemistry (IHC) technique
SP staining was performed to detect the LC3B and SIRT6 proteins in kidney. Specimens were cut in 4-μm-thick section and deparaffinezed in xylene. 0.01 mol/L citrate buffer solution was used for antigen retrieval and 50 μL of perosidase blocking solution was added to block endogenous perosidase activity. The primary antibody (anti-LC3B, ab48394, Abcam anti-SIRT6, ab62739, Abcam) was added according to the instructions and incubated at 4°C for 12 h. The secondary antibody (goat Anti-rabbit HRP, ab205718, Abcam) was added and incubated at room temperature for 10 min. 100 μL of DAB was added and counterstained for 5 min, and the staining was observed under a microscope. The expression level of each group relative to the control group was expressed using a semi-quantitative method.
Cells culture and transfection
The human tubular epithelial cell line HK-2 was purchased from ATCC (USA). The cells were cultured in RPMI 1640 medium containing 10% FBS at 37°C in an incubator with 5% CO2. Culture-related reagents were purchased from GIBCO invitrogen (USA). The cells were divided into 4 groups and respectively cultured with 0, 5, 10 and 50 μg/mL LPS for 48 h.
The SIRT6 coding sequences were subcloned into pcDNA3.1 (Sangon Biotech, China) to construct pcDNA expression vectors. SIRT6 transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the instruction. siSIRT6 was purchased from GenePharma (China) and the siRNA transfections were performed by Lipofectamine TM 2000. The empty plasmid was used as control.
Cell counting kit-8 (CCK-8) assay
The CCK-8 assay was carried out to test the cell viability and the kit was purchased from Tongren (Japan). The cells were pre-incubated at 37°C in 5% CO2 atmosphere, and CCK-8 reagent were then added and cultured at 37°C in 5% CO2 for 4 h. The optical density (OD) of each well at 450 nm was measured using a microplate reader (ELX 800, Bio-Teck, USA). All the reactions were repeated for 3 times.
Enzyme-linked immunosorbent assay (ELISA) and chemical colorimetry
The concentrations of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) in serums and in cell culture medium were detected using ELISA. The kit was purchased from Cusabio Biotech Co., Ltd. (Wuhan, China) and the process was conducted following the manufacturer’s instructions. All the reactions were repeated for 3 times.
Biochemical indicators
The concentrations of creatinine (Cr), serum urea nitrogen (BUN) and cystatin C (Cys C) in serum and in cell culture medium were measured by using biochemical kits purchased from the Institute of Bioengineering (Nanjing, China). All the reactions were repeated for 3 times
QRT-PCR
QRT-PCR was carried out to detect the LC3B and SIRT6 mRNA expression levels. The cells were triturated and lysed, and then the RNA was extracted by using RNA extraction kit (Promega, Beijing, China). Reverse transcription kit (TaKaRa, Japan) was used to synthesize cDNA. Reverse transcription reactionr reaction conditions was set at 37°C for 15 min and reverse transcriptase inactivation condition was set at 85°C for 15 s. QRT-PCR was performed with the QRT-PCR kit (TaKaRa, Japan). PCR was performed by activating the DNA polymerase at 95°C for 5 min, followed by 40 cycles of two-step PCR (at 95°C for 10 s and t 60°C for 30 s) and a final extension at 75°C for 10 min and held at 4°C. RNase-free water was used as the templates of negative control. All primers were obtained from Genewiz (Suzhou, Jiangsu China) and listed in Table 1. The formula 2−ΔΔCT was applied to analyze the mRNA expression levels. All the reactions were repeated for 3 times.
Table 1.
The sequences of primers.
| Primer name | Sequence (5ʹ-3ʹ) | Product size (bp) |
|---|---|---|
| SIRT6-Forward | CCCGGATCAACGGCTCTATC | |
| SIRT6-Reverse | GCCTTCACCCTTTTGGGGG | 214 |
| LC3B-Forward | GGCCTTCTTCCTGTTGGTCA | |
| LC3B-Reverse | TCTCCTGGGAGGCATAGACC | 122 |
| GAPDH-Forward | ATGGGGAAGGTGAAGGTCG | |
| GAPDH-Reverse | GGGGTCATTGATGGCAACAATA | 106 |
Western blot
LC3B-I, LC3B-II and SIRT6 proteins were tested by Western blot. The cells was added to protein lysis buffer (RIPA; Cell Signaling Technology, Inc., Danvers, MA, USA), centrifuged at 12,000 x g for 30 minutes at 4°C, then supernatant was collected. The protein concentration was tested by the BCA protein kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). SDS-PAGE gel was prepared and applied to electrophoresis. The PVDF membrane (Bio-Rad, USA) was transferred by a Trans-Blot Transfer Slot (Bio-Rad, USA) and blocked with 5% fat-free milk for 2 h at room temperature. The primary antibody (anti-LC3B, Abcam, ab51520, dilution: 1:800; anti-SIRT6, Abcam, ab62739, dilution: 1:800) was added according to the kit instruction, and the samples was shaken at room temperature for 2 h and incubated at 4°C for 12 h. The secondary antibody (mouse anti-human IgG, Abcam, ab1927, dilution: 1:10,000; rabbit anti-human IgG, Abcam, ab6759, dilution: 1:8000; rabbit anti-goat IgG, Abcam, ab6741, dilution: 1:10,000) was added and incubated at room temperature for 1.5 h. Chemiluminescence detection was carried out use ECL reagent (Huiying, Shanghai, China). All the reactions were repeated for 3 times.
Statistical analysis
All the experimental data were shown as mean ± standard deviation (SD). Statistical analysis used SPSS 20 (SPSS, Inc., Chicago, IL, USA). The one-way analysis of variance (ANOVA) following Tukey’s multiple comparison n was carried out to analyze differences between the experimental groups. The statistical significant was expressed as P < 0.05.
Result
Effects of LPS on mouse kidney
The shape and arrangement of the renal tissue cells in the control group were normal. After 1 hour of modeling with LPS, mild tubular epithelial cell shedding occurred and mild vacuolar degeneration occurred. After 12 hours of modeling, a significant vacuolar degeneration of focal tubular epithelial cells was observed, and the edges of the brush margins fell off. After 24 hours of modeling, the renal tissues of the mice showed significant pathological changes, and severe vacuolar degeneration of renal tubular epithelial cells, detachment of the tubular epithelial cells and inflammatory cell infiltration were identified. After being modeled for 48 h, the histopathological features of the kidney were significantly improved, the renal tubular epithelial cells showed a repair state and only a few brush-like edges fall off. Renal tubular epithelial cells were regenerated in many places, however, inflammatory cell infiltration was still visible in renal interstitial (Figure 1(a)). This suggested that LPS could cause acute kidney injury in mice and this injury could be relieved after 48 h.
Figure 1.
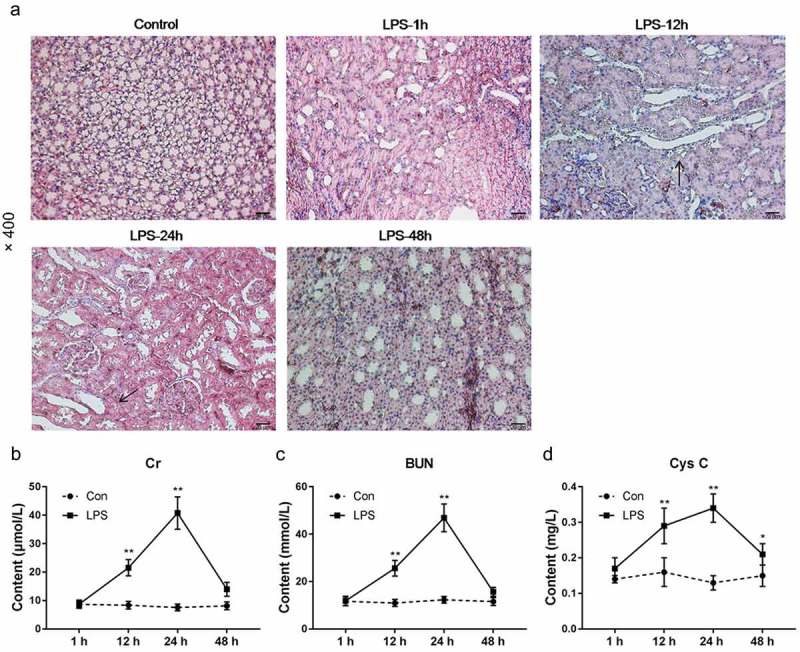
Effects of LPS on mouse kidney. (a) HE staining was used to detect kidney tissue morphology. (b–d) Cr, BUN and Cys C in the serum of mice were measured. *P< 0.05,**P< 0.01, versus 1 h. (biological replicate = 12, technical replicate = 3).
By measuring the serum biochemical parameters of mice, it was found that Cr, BUN and Cys C in the serum of mice increased significantly within 24 h, and that the indexes decreased significantly at 48 h (Figure 1(b–d)). This suggested that in mice, LPS could cause renal function damage, which was significantly restored at 48 h. This indicated that the mouse kidney had the ability to self-repair kidney damage.
Effects of LPS on inflammatory factors, SIRT6 and autophagy-related proteins
To further demonstrate that the AKI model was successfully established, the levels of TNF-α and IL-6 in the serum of mice were tested. It was found that TNF-α was significantly up-regulated at 1 h and then decreased within 48 h. IL-6 increased significantly at 12 h and then decreased (Figure 2(a–b)). This suggested that the AKI model was successfully established.
Figure 2.

Effects of LPS on inflammatory factors, SIRT6 and autophagy-related proteins. (a–b) ELISA was used to test the concentration of TNF-α and IL-6 in serum. (c–g) QRT-PCR and Western blot were applied to tested mRNAs and proteins of SIRT6 and LC3B. *P< 0.05,**P< 0.01, versus control group. (biological replicate = 12, technical replicate = 3).
To further investigate whether autophagy was involved in the repair of kidney injury, SIRT6 and LC3B mRNA and protein were detected by performing qRT-PCR and Western blot, and SP-staining was used to detect the expression levels of SIRT6 and LC3B proteins in kidney tissue. The results showed that at 12 h after modeling, the expression levels of SIRT6 and LC3B-II/LC3B-I were significantly increased, followed by a slight down-regulation (Figures 2(c–g), 3(a–b)). This indicated that SIRT6 and LC3B-II/LC3B-I were highly expressed after receiving LPS and kidney injury, suggesting that autophagy might be involved in the repair of kidney injury.
Figure 3.

Effects of LPS on autophagy-related proteins. (a–b) SP staining was performed to detect the LC3B and SIRT6 proteins in kidney. *P< 0.05,**P< 0.01, versus control group. (biological replicate = 12).
Effects of LPS on HK-2 cells
To further explore the role of autophagy in renal injury, we first investigated the effect of LPS on HK-2 cells. The tubular epithelial cell line was a main cell affected by LPS-induced kidney injury, thus, HK-2 was selected for study. CCK-8 assay showed that LPS inhibited the growth of HK-2 cells (Figure 4(a)). ELISA results showed that TNF-α and IL-6 in culture fluid also increased as LPS increased (Figure 4(b–c)). LPS upregulated the expression levels of SIRT6 and LC3B-II/LC3B-I mRNA and protein in a dose-dependent manner (Figure 4(d–h)). This suggested that LPS could also inhibit the growth of HK-2 at the cellular level and affect the autophagy of cells.
Figure 4.

Effects of LPS on HK-2 cells. (a) The CCK-8 assay was applied to detect cell viability. (b–c) ELISA was used to test the concentration of TNF-α and IL-6 in culture. (d–h) SIRT6 and LC3B mRNAs and proteins were detected by QRT-PCR and Western blot. *P< 0.05,**P< 0.01, versus 0 h. (biological replicate = 12, technical replicate = 3).
Effects of SIRT6 overexpression on HK-2 cells
To further investigate the effects of SIRT6 on apoptosis and autophagy in renal injury, SIRT6 overexpressing HK-2 cells were established. The CCK-8 assay and ELISA results showed that overexpression of SIRT6 had no significant effect on HK-2 cell viability or TNF-α or IL-6 secretion, whereas overexpression of SIRT6 could significantly relieved cell proliferation inhibition and secretion of TNF-α and IL-6 induced by LPS (Figure 5(a–c)).
Figure 5.
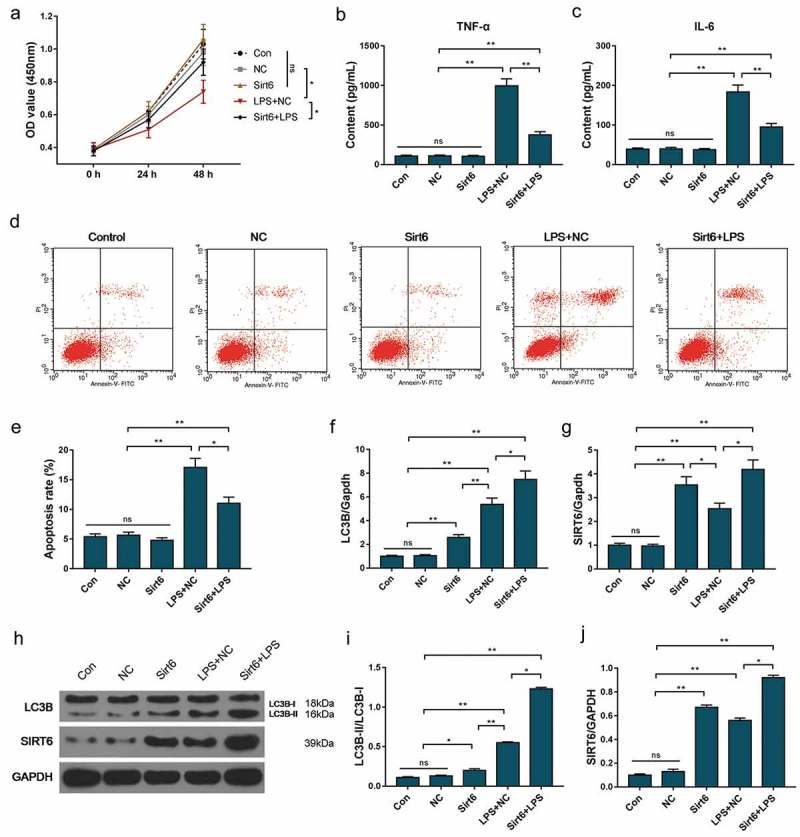
Effects of SIRT6 overexpression on HK-2 cells. (a) The CCK-8 assay was applied to detect cell viability. (b–c) ELISA was used to test the concentration of TNF-α and IL-6 in culture. (d–e) Apoptosis rate was detected using flow cytometry. (f–j) SIRT6 and LC3B mRNAs and proteins were detected by QRT-PCR and Western blot. *P< 0.05,**P< 0.01. (biological replicate = 12, technical replicate = 3).
Flow cytometry results showed that LPS caused an increase in apoptosis rate. Overexpression of SIRT6 had no significant effect on apoptosis of HK-2 cells, whereas SIRT6 overexpressing could inhibit apoptosis induced by LPS (Figure 5(d–e)). Further studies had also shown that up-regulation of SIRT6 gene expression induced LC3B-II/LC3B-I level (Figure 5(f–j)). This suggested that overexpression of SIRT6 could inhibit apoptosis of HK-2 cells and further induce autophagy. This also demonstrated that overexpression of SIRT6 was involved in repairing kidney damage caused by LPS.
Effect of SIRT6 silencing on HK-2 cells
Plasmid transfection was used to reduce the expression of SIRT6 in HK-2 cells. The CCK-8 assay and ELISA results showed that SIRT6 gene silencing had no significant effect on HK-2 cell viability and TNF-α and IL-6 secretion, whereas SIRT6 gene silencing inhibited cell proliferation and promoted TNF-α and IL-6 secretion (Figure 6(a–c)).
Figure 6.
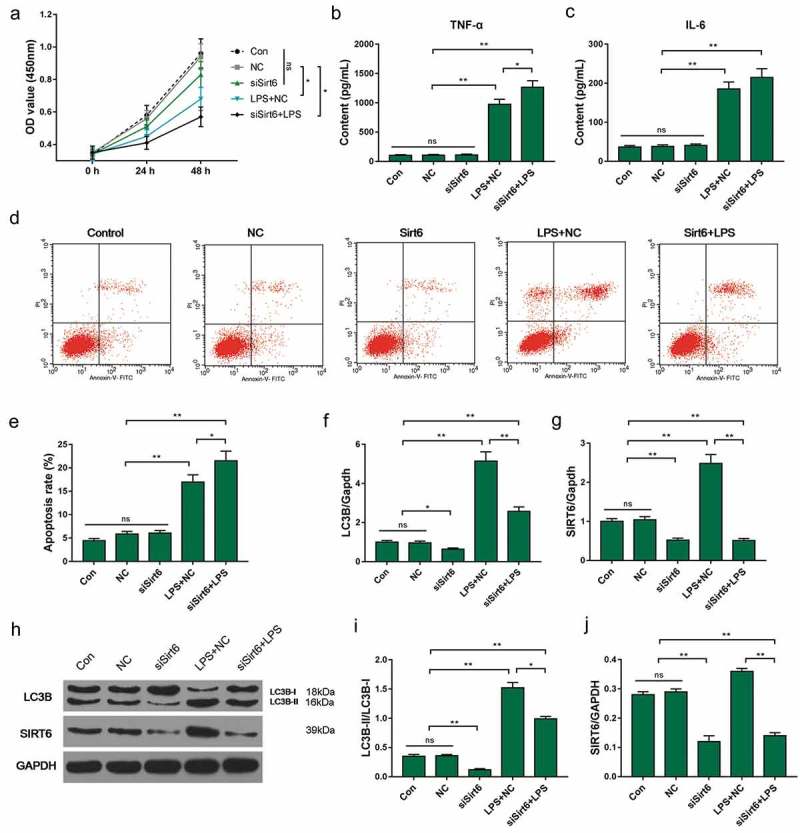
Effect of SIRT6 silencing on HK-2 cells. (a) The CCK-8 assay was applied to detect cell viability. (b–c) ELISA was used to test the concentration of TNF-α and IL-6 in culture. (d–e) Apoptosis rate was detected using flow cytometry. (f–j) SIRT6 and LC3B mRNAs and proteins were detected by QRT-PCR and Western blot. *P< 0.05,**P< 0.01. (biological replicate = 12, technical replicate = 3).
Flow cytometry results showed that LPS caused an increase in apoptosis rate. Low expression of SIRT6 gene had no significant effect on apoptosis and LC3B-II/LC3B-I of HK-2 cells. Down-regulation of SIRT6 gene expression levels could induce apoptosis induced by LPS (Figure 6(d–e)). Further studies had also shown that SIRT6 gene silencing down-regulated LC3B-II/LC3B-I levels (Figure 6(f–j)). This also suggested that SIRT6 participated in the process of relieving kidney damage.
Discussion
Sepsis is a systemic inflammatory response caused by various infections, to which pneumonia is a leading cause of sepsis [19,20]. Sepsis is further developed to cause damage to multiple organs, which mainly affect the kidney and target endothelial cells a [21]. Epithelial cells also participate in the inflammatory response [22]. Epithelial cells can release inflammatory factors such as TNF-α and IL-6, and recruit a large number of neutrophils to cause inflammatory damage. TNF-α and IL-6 are considered as the determining factors of the poison [23,24]. Therefore, TNF-α and IL-6 were used to assess kidney damage in mice with sepsis.
Gram-negative (G−) bacterial infection is one of the important causes to sepsis, and LPS acts as an outer layer of G-cell wall that can cause acute inflammation [25]. The results of this study showed that the concentration of TNF-α and IL-6 in serum of the mice was significantly increased when intraperitoneal injection of LPS for 12 h was implemented, suggesting that LPS could cause severe inflammation. At 24 h after modeling, vacuolar degeneration and detachment occurred in renal tubular epithelial cells, and Cr, BUN and Cys C levels in the serum of mice also increased. This demonstrated that kidney function damage occurred during modeling for 24 h, suggesting that the AKI model was successfully modeled. Further research showed that the inflammatory infiltration of kidney tissue was significantly reduced at 48 h after modeling, and kidney function indicators returned to normal levels. This suggests that within 48 h of modeling, mice could resist kidney injury induced by LPS through certain mechanisms.
The pathogenesis of AKI caused by sepsis still remains unclear, though the theory of apoptosis has been widely accepted [26]. Autophagy exists in normal physiological and pathological conditions and plays an important role in the process of metabolizing waste and removing pathogenic bacteria [27]. Autophagy is one of the main ways to cell death and the main mechanism for maintaining and promoting cell survival and metabolism [28,29]. Previous study suggested that autophagy may help maintain the stability of the intracellular environment and remove harmful substances. As our understanding deepened on autophagy mechanism in recent years, it has been found that the role of autophagy varied in terms of specific disease models, cells and even development stages of the same disease. Carchman et al. [30] found that autophagy of mitochondria was involved in repair of sepsis hepatocyte damage through TLR-4 and TLR-9. Unuma et al. [31] found that sepsis-induced myocardial damage could increase autophagy, which could help to inhibit cell damage caused by oxidative stress and promote cell function recovery. However, Watanabe’ study [32] showed that in the hepatocyte damage caused by sepsis, the increase of autophagy was not determined to be an adaptive protective response of hepatocytes. However, to the best of our knowledge, only few researches were conducted on autophagy in AKI.
The results of this study showed that the expression levels of SIRT6 and LC3B-II/LC3B-I mRNA and protein in the kidney tissue of the mice increased significantly at 12 h after modeling. At 24 h and 48 h after modeling, a decrease at the level was observed. The results of SP assay also obtained similar results. Recent study has shown that SIRT6 was closely related to the normal physiological function of the kidney [33]. Deletion of SIRT6 expression in mouse kidney could lead to progressive nephritis, glomerular hypertrophy, loss of podocyte structure and fibrosis. This is for the first time that researchers demonstrated that SIRT6 plays a crucial role in maintaining glomerular function [17]. SIRT6 is also involved in autophagy of cells. It has been found that up-regulation of SIRT6 expression down-regulated the level of autophagy by inhibiting the signaling of IGF-Akt-mTOR [34]. There are two methods for detecting autophagy in cells, one is to detect the number of autophagosomes, and the number of autophagosomes can directly reflect the intensity of autophagy formation [35], the other is to detect autophagy-related proteins. LC3B is the most recognized autophagy-related protein in mammalian cells. LC3B localizes to the surface of the autophagic vesicle membrane and directly participates in the formation of autophagosomes. LC3B is classified into type I and type II. LC3B appears to be soluble LC3B-I before autophagy. When autophagy is initiated, LC3B-I will combine with phosphatidyl alcohol on autophagic membrane by ubiquitination and evolve into LC3B-II [36–38]. Therefore, the level of LC3B-II/LC3B-I is proportional to the number of autophagic vacuoles. To further investigate the mechanism by which mice resist LPS-induced kidney injury, human tubular epithelial cell line HK-2 cells were used as subject to investigate the effects of SIRT6 on autophagy and AKI.
The results showed that LPS inhibited the growth of HK-2 cells and promoted the secretion of TNF-α and IL-6. LPS also up-regulated the expression levels of SIRT6 and LC3B-II/LC3B-I mRNA and protein in HK-2 cells. The effects of SIRT6 gene overexpression and low-expression on HK-2 cell apoptosis and autophagy were studied by plasmid transfection. The results showed that overexpression of SIRT6 attenuated the inhibitory effect of LPS on the growth of HK-2 cells and decreased the expression of inflammatory factors. Overexpression of SIRT6 relieved LPS-induced apoptosis and significantly up-regulated the levels of SIRT6 and LC3B-II/LC3B-I mRNA and protein in HK-2 cells to promote autophagy. The effects of SIRT6 low-expression on HK-2 cell apoptosis and autophagy was opposite to those shown by SIRT6 overexpression. Yanai’s study [39] found that autophagy in septic macrophage knockout HMGB1 gene was significantly inhibited, and that mice, which were knocked out of HMGB1, were prone to develop sepsis, suggesting that HMGB1-mediated autophagy played a cytoprotective role in sepsis. This experiment indicated that SIRT6 was overexpressed in the kidney tissue of AKI induced by LPS. Overexpression of SIRT6 promoted autophagy and reduced apoptosis in HK-2 cells.It was found that DNA-PKcs and CtIP deacetylation may be used as crucial for SIRT6-mediated DNA repair, which provides the rationale for the clinical evaluation of SIRT6 modulators in the treatment of leukemia [40]. There are also studies suggesting that the modulation of SIRT6 activity may be a potential new therapeutic method for diabetic atherosclerotic and pancreatic cancer [41,42].
In summary, overexpression of SIRT6 can protect kidney epithelial cell damage of AKI by inhibiting apoptosis and inducing autophagy, which might become a new approach to the treatment of AKI.
Funding Statement
This work was supported by the Xuzhou Science and Technology Project(study on the mechanism of action of autophagy in the diagnosis and treatment of acute kidney injury) under Grant [KC16SH020]; the General Program of National Natural Science Foundation in China, “the regulation of ERK potassium channel on the secretion of H+ in distal nephron sputum cells” under Grant [31571187]; the Jiangsu University Medical Clinical Science and Technology Development Fund Project (The mechanism of action and clinical diagnosis and treatment of autophagy in acute kidney injury caused by sepsis) under Grant [JLY20160127]; the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions under Grant [PPZY2015B161]; the IN 2018 the Fifth Phase of “333 Project” Scientific Research Project in Jiangsu Province under Grant [BRA2018274].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Dellinger RP, Levy MM, Rhodes A, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013. February;41(2):580–637. PubMed PMID: 23353941; eng. [DOI] [PubMed] [Google Scholar]
- [2].Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med. 2017. March;45(3):486–552. PubMed PMID: 28098591; eng. [DOI] [PubMed] [Google Scholar]
- [3].Sossdorf M, Otto GP, Menge K, et al. Potential effect of physiotherapeutic treatment on mortality rate in patients with severe sepsis and septic shock: a retrospective cohort analysis. J Crit Care. 2013. December;28(6):954–958. PubMed PMID: 23958242; eng. [DOI] [PubMed] [Google Scholar]
- [4].Poukkanen M, Wilkman E, Vaara ST, et al. Hemodynamic variables and progression of acute kidney injury in critically ill patients with severe sepsis: data from the prospective observational FINNAKI study. Crit Care. 2013. December 13;17(6):R295 PubMed PMID: 24330815; PubMed Central PMCID: PMCPMC4056430. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Basile DP, Yoder MC.. Renal endothelial dysfunction in acute kidney ischemia reperfusion injury. Cardiovasc Hematol Disord Drug Targets. 2014;14(1):3–14. PubMed PMID: 25088124; PubMed Central PMCID: PMCPMC4215733. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gui D, Huang J, Liu W, et al. Astragaloside IV prevents acute kidney injury in two rodent models by inhibiting oxidative stress and apoptosis pathways. Apoptosis. 2013. April;18(4):409–422. PubMed PMID: 23325448; eng. [DOI] [PubMed] [Google Scholar]
- [7].Hsieh CH, Pai PY, Hsueh HW, et al. Complete induction of autophagy is essential for cardioprotection in sepsis. Ann Surg. 2011. June;253(6):1190–1200. PubMed PMID: 21412148; eng. [DOI] [PubMed] [Google Scholar]
- [8].Yen YT, Yang HR, Lo HC, et al. Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery. 2013. May;153(5):689–698. PubMed PMID: 23434181; eng. [DOI] [PubMed] [Google Scholar]
- [9].Lee S, Lee SJ, Coronata AA, et al. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal. 2014. January 20;20(3):432–442. PubMed PMID: 23971531; PubMed Central PMCID: PMCPMC3894711. eng DOI: 10.1089/ars.2013.5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mizushima N, Komatsu M.. Autophagy: renovation of cells and tissues. Cell. 2011. November 11;147(4):728–741. PubMed PMID: 22078875; eng. [DOI] [PubMed] [Google Scholar]
- [11].Liu S, Lu B. Reduction of protein translation and activation of autophagy protect against PINK1 pathogenesis in drosophila melanogaster. PLoS Genet. 2010. December 9;6(12):e1001237 PubMed PMID: 21151574; PubMed Central PMCID: PMCPMC3000346. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010. November 2;107(44):18880–18885. PubMed PMID: 20956295; PubMed Central PMCID: PMCPMC2973911. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang J, Kang R, Huang H, et al. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy. 2014. May;10(5):766–784. PubMed PMID: 24589849; PubMed Central PMCID: PMCPMC5119055. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen S, Jiang YZ, Huang L, et al. The residual tumor autophagy marker LC3B serves as a prognostic marker in local advanced breast cancer after neoadjuvant chemotherapy. Clin Cancer Res. 2013. December 15;19(24):6853–6862. PubMed PMID: 24141623; eng. [DOI] [PubMed] [Google Scholar]
- [15].Ng F, Tang BL. Sirtuins’ modulation of autophagy. J Cell Physiol. 2013. December;228(12):2262–2270. PubMed PMID: 23696314; eng. [DOI] [PubMed] [Google Scholar]
- [16].Lee IH, Yun J, Finkel T. The emerging links between sirtuins and autophagy. Methods Mol Biol. 2013;1077:259–271. PubMed PMID: 24014412; PubMed Central PMCID: PMCPMC4167386. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu M, Liang K, Zhen J, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting notch signaling. Nat Commun. 2017. September 4;8(1):413 PubMed PMID: 28871079; PubMed Central PMCID: PMCPMC5583183. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li Z, Xu K, Zhang N, et al. Overexpressed SIRT6 attenuates cisplatin-induced acute kidney injury by inhibiting ERK1/2 signaling. Kidney Int. 2018. April;93(4):881–892. PubMed PMID: 29373150; eng. [DOI] [PubMed] [Google Scholar]
- [19].Kellum JA, Kong L, Fink MP, et al. Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) study. Arch Intern Med. 2007. August 13–27;167(15):1655–1663. PubMed PMID: 17698689; PubMed Central PMCID: PMCPMC4495652. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rautanen A, Mills TC, Gordon AC, et al. Genome-wide association study of survival from sepsis due to pneumonia: an observational cohort study. Lancet Respir Med. 2015. January;3(1):53–60. PubMed PMID: 25533491; PubMed Central PMCID: PMCPMC4314768. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Alobaidi R, Basu RK, Goldstein SL, et al. Sepsis-associated acute kidney injury. Semin Nephrol. 2015. January;35(1):2–11. PubMed PMID: 25795495; PubMed Central PMCID: PMCPMC4507081. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zheng S, Pan Y, Wang C, et al. HMGB1 turns renal tubular epithelial cells into inflammatory promoters by interacting with TLR4 during sepsis. J Interferon Cytokine Res. 2016. January;36(1):9–19. PubMed PMID: 26312770; eng. [DOI] [PubMed] [Google Scholar]
- [23].Jekarl DW, Lee SY, Lee J, et al. Procalcitonin as a diagnostic marker and IL-6 as a prognostic marker for sepsis. Diagn Microbiol Infect Dis. 2013. April;75(4):342–347. PubMed PMID: 23391607; eng. [DOI] [PubMed] [Google Scholar]
- [24].Baghel K, Srivastava RN, Chandra A, et al. TNF-alpha, IL-6, and IL-8 cytokines and their association with TNF-alpha-308 G/A polymorphism and postoperative sepsis. J Gastrointest Surg. 2014. August;18(8):1486–1494. PubMed PMID: 24944154; eng. [DOI] [PubMed] [Google Scholar]
- [25].Roger T, Froidevaux C, Le Roy D, et al. Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci U S A. 2009. February 17;106(7):2348–2352. PubMed PMID: 19181857; PubMed Central PMCID: PMCPMC2650125. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kockara A, Kayatas M. Renal cell apoptosis and new treatment options in sepsis-induced acute kidney injury. Ren Fail. 2013;35(2):291–294. PubMed PMID: 23181751; eng. [DOI] [PubMed] [Google Scholar]
- [27].Tan YQ, Zhang J, Zhou G. Autophagy and its implication in human oral diseases. Autophagy. 2017. February;13(2):225–236. PubMed PMID: 27764582; PubMed Central PMCID: PMCPMC5324841. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015. March;22(3):367–376. PubMed PMID: 25257169; PubMed Central PMCID: PMCPMC4326571. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Oehme I, Linke JP, Bock BC, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A. 2013. July 9;110(28):E2592–E601. PubMed PMID: 23801752; PubMed Central PMCID: PMCPMC3710791. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Carchman EH, Whelan S, Loughran P, et al. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. Faseb j. 2013. December;27(12):4703–4711. PubMed PMID: 23982147; PubMed Central PMCID: PMCPMC3834775. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Unuma K, Aki T, Funakoshi T, et al. Cobalt protoporphyrin accelerates TFEB activation and lysosome reformation during LPS-induced septic insults in the rat heart. PLoS One. 2013;8(2):e56526 PubMed PMID: 23457579; PubMed Central PMCID: PMCPMC3574118. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Watanabe E, Muenzer JT, Hawkins WG, et al. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab Invest. 2009. May;89(5):549–561. PubMed PMID: 19188912; PubMed Central PMCID: PMCPMC3822608. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang W, Liu H, Zhu S, et al. Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging (Albany NY). 2017. March 28;9(3):1069–1083. PubMed PMID: 28351995; PubMed Central PMCID: PMCPMC5391219. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Takasaka N, Araya J, Hara H, et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol. 2014. February 1;192(3):958–968. PubMed PMID: 24367027; eng. [DOI] [PubMed] [Google Scholar]
- [35].Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. PubMed PMID: 21548784; eng. [DOI] [PubMed] [Google Scholar]
- [36].Wu J, Dang Y, Su W, et al. Molecular cloning and characterization of rat LC3A and LC3B–two novel markers of autophagosome. Biochem Biophys Res Commun. 2006. January 6;339(1):437–442. PubMed PMID: 16300744; eng. [DOI] [PubMed] [Google Scholar]
- [37].Shvets E, Abada A, Weidberg H, et al. Dissecting the involvement of LC3B and GATE-16 in p62 recruitment into autophagosomes. Autophagy. 2011. July;7(7):683–688. PubMed PMID: 21460636; eng. [DOI] [PubMed] [Google Scholar]
- [38].Koukourakis MI, Kalamida D, Giatromanolaki A, et al. Autophagosome proteins LC3A, LC3B and LC3C have distinct subcellular distribution kinetics and expression in cancer cell lines. PLoS One. 2015;10(9):e0137675 PubMed PMID: 26378792; PubMed Central PMCID: PMCPMC4574774. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yanai H, Matsuda A, An J, et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci U S A. 2013. December 17;110(51):20699–20704. PubMed PMID: 24302768; PubMed Central PMCID: PMCPMC3870753. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cagnetta A, Soncini D, Orecchioni S, et al. Depletion of SIRT6 enzymatic activity increases acute myeloid leukemia cells’ vulnerability to DNA-damaging agents. Haematologica. 2018. January;103(1):80–90. PubMed PMID: 29025907; PubMed Central PMCID: PMCPMC5777193. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Balestrieri ML, Rizzo MR, Barbieri M, et al. Response to comment on Balestrieri et al. Sirtuin 6 expression and inflammatory activity in diabetic atherosclerotic plaques: effects of incretin treatment. Diabetes. 2015;64:1395–1406. Diabetes. 2015 May;64(5):e6 PubMed PMID: 25908881; eng. [DOI] [PubMed] [Google Scholar]
- [42].Demir IE, Ceyhan GO, Friess H. Epigenomic therapies: the potential of targeting SIRT6 for the treatment of pancreatic cancer. Expert Opin Ther Targets. 2017. January;21(1):1–3. PubMed PMID: 27885875; eng. [DOI] [PubMed] [Google Scholar]