

ABSTRACT
Esophageal squamous cell carcinoma (ESCC) is a serious malignancy with limited options for targeted therapy. The exploration of novel targeted therapies for combating ESCC is urgently needed. Cyclin-dependent kinases (CDKs) play important roles in the progression of cancers; however, the function of CDK11p110 (cyclin-dependent kinase 11p110) in ESCC is still unknown. Here, we investigated the effects and molecular mechanisms of CDK11p110 in the proliferation and growth of ESCC by examining the expression of CDK11p110 in ESCC tissues and by detecting phenotypic changes in ESCC cells after CDK11p110 knockdown or overexpression in vitro and in vivo. According to the tissue microarray analysis, compared with its expression level in normal tissues, the expression level of CDK11p110 was significantly elevated in ESCC tissues; this result was in concordance with the data in TCGA (The Cancer Genome Atlas) datasets. In addition, RNAi-mediated CDK11p110 silencing exerted a substantial inhibitory effect on the proliferation, clonogenicity and migration ability of ESCC cells. Further study indicated that CDK11p110 knockdown arrested ESCC cells in the G2/M phase of the cell cycle and induced cell apoptosis. Moreover, stable shRNA-mediated CDK11p110 knockdown inhibited tumor growth in an ESCC xenograft model, and overexpression of CDK11p110 enhanced tumor growth. In addition, the Ki67 proliferation index was closely associated with the elevation or depletion of CDK11p110 in vivo. In summary, this study provides evidence that CDK11p110 play a critical role in the tumorigenicity of ESCC cells, which suggests that CDK11p110 may be a promising therapeutic target in ESCC.
Abbreviations: CDKs: cyclin-dependent kinases; CDK11: Cyclin-dependent kinase 11; CDK11p110: Cyclin-dependent kinase 11p110, the larger isomer of cyclin-dependent kinase 11; ESCC: esophageal squamous cell carcinoma; FACS: fluorescence-activated cell sorting; FDA: the Food and Drug Administration; TCGA: The Cancer Genome Atlas; TMA: tissue microarray.
KEYWORDS: CDK11p110, kinase, target, ESCC, tumorigenicity
Introduction
Esophageal cancer is a serious malignancy with high mortality and poor prognosis [1,2], it ranks as the eighth most prevalent diagnosed cancer and as the sixth leading cause of cancer death worldwide. Esophageal squamous cell carcinoma (ESCC) accounts for approximately 90% of the 456,000 incident esophageal cancers each year and has a high incidence in areas such as Eastern to Central Asia, along the Rift Valley in East Africa, and into South Africa [3,4]. Despite the gold-standard trimodality treatment, which includes neoadjuvant chemoradiation followed by surgery, and the many advances in diagnosis and treatment during the past decades [5], the overall 5-year survival rate of patients with ESCC is only approximately 20%, partly due to the lack of effective treatment strategies and corresponding drug targets [4].
Cyclin-dependent kinases (CDKs), which are a family of serine/threonine kinases that are involved in the regulation of the cell cycle, transcription, and RNA splicing, are frequently overexpressed and/or constitutively activated in cancer [6,7]. Emerging studies have indicated that the dysregulation of CDKs plays crucial roles in tumor formation and development, as well as in therapy resistance. Hence, targeting CDKs has become a promising therapeutic strategy for cancer [8–10]. Recently, three CDK4/6 inhibitors, namely, palbociclib, ribociclib and abemaciclib have been approved by the Food and Drug Administration (FDA) for the treatment of advanced HR-positive and HER2-negative breast cancer [11–13]. Additionally, a number of inhibitors targeting CDKs, including CDK1, CDK2, CDK4, CDK5, CDK6, CDK7, CDK8 and CDK9, are in clinical trials against malignancies such as breast cancer, leukemia, myeloma, lymphoma, sarcoma, and non-small cell lung cancer [14,15]. These promising clinical trials will facilitate the development of more potent CDK-based therapeutic strategies.
CDK11 (formerly named PITSLRE) is a serine/threonine protein kinase in the CDK family, which plays diverse roles in controlling RNA splicing, apoptosis, and cytokinesis [16,17]. CDK11 is encoded by two highly homologous genes, namely, CDK11A and CDK11B, and includes three major isoforms, namely, CDK11p110, CDK11p58 and CDK11p46. The larger CDK11p110 protein is encoded by the full-length CDK11 mRNA and is highly expressed in cancer cells throughout the cell cycle; CDK11p110 leads to the generation of the CDK11p58 isoform during the G2/M phase of the cell cycle. Although CDK11p58 shares the same C-terminal sequence as CDK11p110, the two isoforms possess different functions. CDK11p58 has been identified as a negative regulator of oncogenesis, while CDK11p110 has been identified as a positive regulator [16,18,19]. Previous studies have shown that CDK11p11 s overexpressed in several kinds of human malignancies and is highly associated with poor outcomes in cancer patients. There is no effective inhibitor for this target; however, CDK11p110 knockdown has been demonstrated to decrease cell viability and increase apoptosis in breast cancer, ovarian cancer, osteosarcoma and liposarcoma [20–23]. These findings collectively suggest that CDK11p110 might be a novel and promising therapeutic target in cancer.
Previous studies found that knockdown of CDK11p110 can decrease cell viability and induce apoptosis in several kinds of cancers; however, the functional roles and molecular mechanisms of CDK11p110 in ESCC tumorigenicity remain unknown. The present study aimed to explore the significance of CDK11p110 in ESCC. We first analyzed the CDK11 expression level in esophageal cancer tissues and normal tissues through analysis of TCGA datasets, and we then detected CDK11p110 expression via ESCC tissue microarray analysis of clinical specimens. Subsequently, we investigated the changes in the proliferation, clonogenicity, migration, apoptosis and cell cycle of ESCC cells by RNAi-mediated CDK11p110 knockdown. Additionally, we constructed stable ESCC cell lines with CDK11p110 overexpression or knockdown to assess the function of CDK11p110 in ESCC subcutaneous xenograft models. The study provides evidence that CDK11p110 plays a critical role in the formation and progression of ESCC and that it might serve as a promising target for further ESCC treatment options.
Materials and methods
Cell lines and culture conditions
Human ESCC cell lines KYSE150 and TE-1 were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Other four ESCC cell lines, EC109, EC9706, KYSE70, TE-7 and human esophageal epithelial cell lines HET-1A were provided by doctor Lifeng Li (Biotherapy Center, Cancer Center, The First Affiliated Hospital of Zhengzhou University). All cells lines were cultured in RPMI-1640 (Hyclone; Thermo Fisher Scientific) medium supplemented with 10% (v/v) of fetal bovine serum (FBS, Gibco; Life Technologies), penicillin G (100 U/mL), and streptomycin (100 μg/mL) in an incubator with 5% CO2 at 37°C.
Tissue microarray analysis
The microarray chip with 15 samples of ESCC tissues and matched adjacent tissues (HEsoS030PG04) was purchased from Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). The detection and analysis were performed by Wuhan Servicebio Technology Co., Ltd. (Wuhan, China). Sections were semiquantitatively scored for the CDK11p110 staining patterns as follows: the staining intensity was quantified as 0 (negative), 1+ (weak), 2+ (intermediate), or 3+ (strong). The percentage of immunoreactive tumor cells was scored as follows: 0 (0%), 1 (1–20%), 2 (21–40%), 3 (41–60%), 4 (61–80%) or 5 (81–100%). The final immunoreactive score was obtained by multiplying the percentage and the intensity values (range 0–15), and the samples were grouped by immunoreactive score as follows: 1+ (score 0–2), 2+ (score 3–5), 3+ (score 6–8), 4+ (score 9–11) or 5+ (score 12–15).
Expression data collection
The mRNA expression data that included esophageal cancer tissues from 162 patients and 11 normal esophageal tissues was downloaded from TCGA database; expression levels of CDK11 (CDK11A and CDK11B) were retrieved from this data set.
Esophageal squamous cell carcinoma specimens
Nine pairs of esophageal cancer tissues and their matching adjacent normal tissues were collected in liquid nitrogen from surgical resections performed at the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China). No patients in this study received any therapeutic intervention, and all diagnoses were confirmed by pathological analysis. The use of clinical materials in this study for research purposes was approved by the ethics committee of Zhengzhou University, Henan, China; and we obtained prior consent from all patients. CDK11p110 expression levels were detected by western blotting. All experiments were performed in accordance with relevant guidelines and regulations.
Synthetic CDK11P110 siRNA and transfection
CDK11p110 knockdown in human ECSS cells was performed by the transfection of CDK11p110 siRNA synthesized by Guangzhou RiboBio Co., Ltd. The siRNA sequence targeting CDK11p110 corresponded to the coding regions (5ʹ-AGAUCUACAUCGUGAUGAAtt-3ʹ, antisense 5ʹ-UUCAUCACGAUGUAGAUCUtg-3ʹ) of the CDK11p110 gene, as previously reported [22]. Nonspecific siRNA oligonucleotides from RiboBio were used as negative controls. Various concentrations (0, 10, 20, and 40 nM) of CDK11p110 siRNA (siCDK11p110) or nonspecific siRNA (siNC) were transfected into EC109 or KYSE150 cells with Lipofectamine™ RNAiMAX reagent (Thermo Fisher Scientific, #13778075) according to the manufacturer’s instructions.
Immunofluorescence assay
The ESCC cell lines EC109 and KYSE150 were separately cultured on an eight-well chamber slide (Lab-Tek Chamber Slide System, Nunc). After cell attachment, cells were transfected with siCDK11p110 or siNC and incubated for an additional 48 hours. Then, cells were washed three times in PBS and immediately fixed in 4% paraformaldehyde for 15 minutes at room temperature. After fixation, cells were blocked in 1% BSA in PBST for 60 min. Following incubation with CDK11p110 primary antibody (1:50 dilution, Santa Cruz Biotechnology, sc-928) at 4°C overnight, the cells were washed three times in PBS and incubated with DyLight 488-conjugated goat anti-rabbit IgG secondary antibody (1:50 dilution, Abbkine, #A23220) for 30 minutes at 37°C. At the end of the incubation period, cells were washed 3 times with PBS, and DAPI (Zhongshan Golden Bridge Biotechnology) was used to stain the nuclei. Images were visualized by a ZEISS fluorescence microscope (Oberkochen, Germany) equipped with Zen Imaging software.
Western blot analysis
EC109 and KYSE150 cells were separately cultured on 6-well plates. After attachment, cells were transfected with siCDK11p110 or siNC, and incubated for an additional 72 hours. CDK11p110, β-actin and cleaved PARP protein expression levels were analyzed by western blotting. The experimental procedure for western blotting was performed according to the previously described protocol [24,25]. The following antibodies were used: the primary antibodies included anti-human CDK11p110 (1:500 dilution, Santa Cruz Biotechnology, #sc-928), anti-β-actin (1:5000 dilution, Proteintech, #66009-l-lg), and anti-cleaved PARP (1:1000 dilution, Cell Signaling Technology, #5625); the secondary antibodies included goat anti-mouse IgG (1:7500 dilution, Jackson, #115-035-003) and goat anti-rabbit IgG (1:7500 dilution, Jackson, #111-035-003). Bands were detected using a Bio-Rad ChemiDoc Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Cell proliferation assay
The growth and proliferation of EC109 and KYSE150 cells after CDK11p110 knockdown were assessed using the CCK8 assay. For each transfection, 3000 cells were cultured in 96-well plates and transfected with different concentrations of CDK11p110 siRNA. After 3–6 days of culture, Cell Counting Kit-8 reagent (CCK8) was added to each well, and the detection was performed as previously describe [26,27]. The optical density (OD) at 450 nm was determined by a microplate reader (Thermo Fisher Scientific, MULTISKAN FC). Untreated cells served as the control. The OD value represented the relative cell viability.
Clonogenic assay
A clonogenic assay was performed to detect the effect of CDK11p110 knockdown on the colony formation capability of EC109 or KYSE150 cells. Cells were trypsinized and seeded in triplicate at a density of 200 cells/well in 12-well plates in 1 mL of medium containing 10% FBS. After incubation at 37°C with 5% CO2 for 24 h, cells were transfected with 10 nM siCDK11p110 or siNC and further grown for 10–15 days. When a macroscopic cell colony had formed, cells were fixed with methanol (4°C for 30 min) and stained with crystal violet staining solution, and colonies were counted on a ZEISS Primovert inverted microscope (Oberkochen, Germany) only if they contained more than 50 cells. Untreated cells served as the control. The clonogenicity value (%) represented the relative colony formation capability of the cells.
Cell migration assay
The migration of EC109 and KYSE150 cells was determined with a 24-well, 8.0 μm pore size Transwell plate (Corning, #3422). Before the experiment, Transwell inserts were rehydrated in 200 μL of serum-free RPMI-1640 medium and incubated at 37°C for 30 minutes. Then, after the rehydration medium was removed from the insert, 600 μL of fresh medium containing 20% FBS was added to the lower chamber, and cells transfected with 20 nM siCDK11p110 or siNC for 48 hours were trypsinized and plated in the upper chamber at a density of 1 × 105 cells/well in 100 μL serum-free medium. After incubation at 37°C for 24 hours, the upper chambers were fixed with methanol at RT for 30 minutes and stained with crystal violet staining solution for an additional 1 hour. Next, cotton swabs were used to remove nonmigrated cells from the upper chamber. Representative images were photographed with an inverted microscope (Oberkochen, Germany). For further objective evaluation, the cropped membrane was incised and dipped into 150 mL of 33% acetic acid to dissolve the crystal violet on a horizontal shaker at RT for 10 minutes. Then, the solution was transferred to 96-well plates. The absorbance was measured at 570 nm using a microplate reader (Thermo Fisher Scientific, MULTISKAN FC).
Cell apoptosis analyses
Flow cytometry was used to analyze the effect of CDK11p110 knockdown on EC109 and KYSE150 cells. Briefly, after cell attachment in 6-well culture plates, cells were transfected with 20 nM siCDK11p110 or siNC and incubated for an additional 72 hours. Following the instructions provided by the manufacturer of the Annexin V-FITC Apoptosis Kit (KeyGen Biotech, Nanjing, China), cells were resuspended in annexin V binding buffer; annexin V-FITC and propidium iodide (PI) were then added, and the cells were incubated for 30 minutes at 4°C. The fluorescently labeled cells were analyzed with a FACSCalibur system (FACSCanto ll, BD Biosciences, NJ, USA).
Analysis of cell-cycle arrest
Analysis of cell-cycle arrest was conducted by flow cytometry. After transfection with 20 nM of siCDK11p110 or siNC for 72 hours, cells were collected and washed with cold PBS, then fixed in 70% ethanol at −20°C overnight. Then cells were washed with PBS and resuspended in RNase A at 37°C for 30 minutes, followed by dyeing with propidium Iodide (KeyGEN BioTHCH, Nanjing, China) for 30 minutes. Cells were analyzed for DNA content by flow cytometry (FACSCantoll, BD, NJ, USA) and the population of cells in each cell cycle phase was analyzed by Modfit software.
Lentivirus production and establishment of cell lines with stable CDK11P110 overexpression or knockdown
To establish cell lines with stable CDK11p110 overexpression, the human gene encoding for CDK11p110 was amplified by PCR using the pUC-CDK11p110 plasmid synthesized by Applied Biological Materials Inc. as the template. Then, the CDK11p110 genes were cloned into pCDH-CMV-MCS-EF1-copGFP vectors (System Biosciences). Following the manufacturer’s instructions, lentiviruses were produced by cotransfection of the lentiviral packaging system by adding a mixture of pCDH-CMV-CDK11p110-EF1-copGFP and the packaging plasmids pSPAX2 and pMD2.0G into HEK293T packaging cells. An empty vector, namely, pCDH, was transfected into the cells to be used as controls. EC109 and KYSE150 cells in medium with 6 μg/mL polybrene were infected with lentivirus supernatant for 48 h, followed by puromycin selection for 7 days. To establish cell lines with stable CDK11p110 knockdown, shRNA were obtained using the pLKO.1-puro vector purchased from Sigma (Sigma, USA). The shRNA sequences were as follows: shCDK11p110-1 (TRCN0000314694): 5ʹ-CCGGCGATCAGATCAACAAGGTGTTCTCGAGAACACCTTGTTGATCTGATCGTTTTTG-3ʹ and shCDK11p110-2 (TRCN0000314617): 5ʹ-CCGGACGGCCTCAAGCATGAGTATTCTCGAGAATACTCATGCTTGAGGCCGTTTTTTG-3ʹ. The lentiviruses were produced in HEK293T cells as described above. Tumor cells were infected with viruses expressing shCDK11p110-1 and shCDK11p110-2 to knock down CDK11p110 expression. An empty vector, namely, pLKO.1, was transfected into the cells to be used as controls. Puromycin was added for selection for 7 days. The expression levels of CDK11p110 in the stable cell lines were confirmed by western blot assay. In addition, the growth and proliferation of these cell lines with stable CDK11p110 overexpression or knockdown were assessed using the CCK8 assay.
In vivo subcutaneous xenograft models
For evaluation of the effect of CDK11p110 on ESCC tumor growth, an animal experiment was performed with an EC109 xenograft model. The experimental procedures were in accordance with the National Guidelines for Housing and Care of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Institute of Medicine, University of Zhengzhou, Henan Province. Female athymic mice (BALB/c nude, 6–8 weeks old, 18–20 g) were purchased from Vital River Laboratories (Beijing, China). The mice were injected subcutaneously in the right flank with untransfected EC109 cells, pLKO.1 vector-expressing EC109 cells, shCDK11p110-1-expressing EC109 cells, shCDK11p110-2-expressing EC109 cells, pCDH-expressing EC109 cells, or pCDH-CDK11p110-expressing cells (2 × 106 cells in 100 μL PBS) to establish xenograft tumors (n = 7 per group). Tumor size was measured every 3–4 days, and tumor volume was calculated with the following formula: V (mm3) = 0.5 × a× b2, where a and b represent the long and the perpendicular short diameters of the tumor, respectively [28]. The mice were sacrificed at 20 d after inoculation, and solid tumors were dissected, measured and photographed. At the end of the experiment, the fresh tumor tissue specimens were collected for immunohistochemical analyses using antibodies specific for CDK11p110 and Ki67 (1:1000 dilution, Servicebio Technology Co., Ltd., #GB13030-2) as described previously [29].
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.0 software (GraphPad Software, La Jolla, CA, USA). Continuous variables were presented as mean ± standard deviation (SD). The Kolmogorov-Smirnov test was used to check for normal distribution of continuous data. F test was used to test homogeneity of variance. One-way ANOVA or student’s t test were used to analyze the data which was normal distribution and homogeneity of variances. In addition, the data from two groups were analyzed by student’s t test and one-way ANOVA for more than two groups. Mann-Whitney U test was performed for nonparametric data. P-values less than 0.05 were considered to indicate statistically significant differences.
Results
CDK11 expression analysis by TCGA database and tissue microarray
To explore the potential role of CDK11p110 in human ESCC formation and development, we first analyzed the mRNA expression level of CDK11 in human esophageal cancer in TCGA datasets. Compared with that in normal tissues, the mRNA expression of both CDK11A and CDK11B was significantly elevated in esophageal cancer tissues (Figure 1(a)). In addition, we then detected the CDK11p110 protein expression level via human ESCC tissue microarray analysis and analyzed the correlation of CDK11p110 expression with the ESCC patients. As shown in Figure 1(b), compared with that in the adjacent nontumorous tissues, the CDK11p110 expression level was significantly increased in the ESCC tissues (Mann-Whitney U test, P = 0.0003). Representative images of different immunohistochemical staining intensities representing different CDK11p110 protein expression levels in ESCC tissues are shown in Figure 1(c). In addition, CDK11p110 immunoreactivity was found to be mostly localized to the nucleus of tumor cells.
Figure 1.
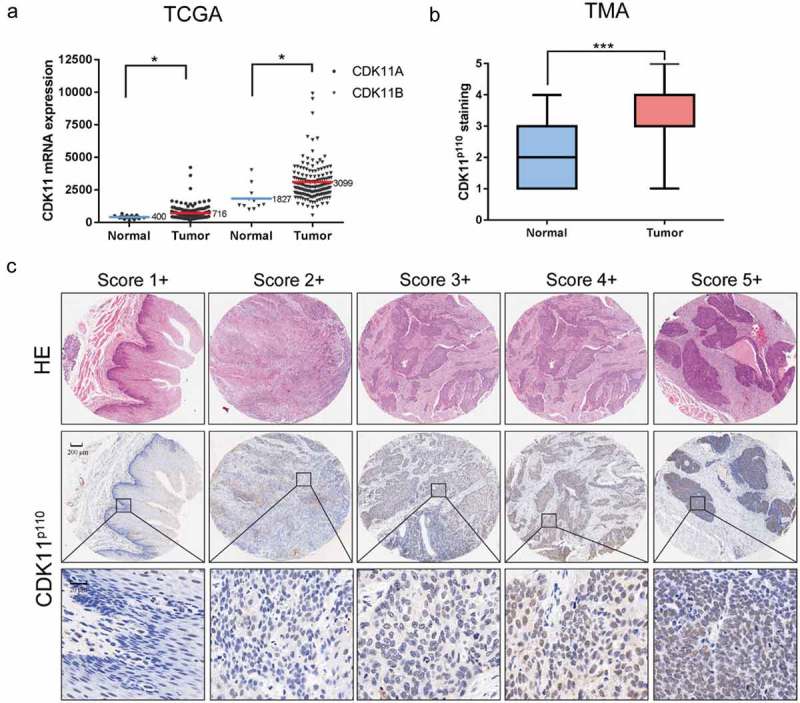
Analysis of CDK11 expression in human esophageal cancer via TCGA database and tissue microarray analysis. (a) Expression of CDK11A and CDK11B in normal tissue (n = 11) and esophageal cancer tissue (n = 162) from TCGA datasets. *P < 0.05, showing a significant difference. (b) Distribution of CDK11p110 staining scores among ESCC tissues and their adjacent normal tissues in tissue microarray (TMA) analysis. Statistical analysis was performed with the Mann-Whitney U test. ***P < 0.001, showing a significant difference. CDK11p110 expression was classified into categories 1–5 according to the CDK11p110 staining intensity and percentage values, as described in the Materials and Methods section. (c) Representative images showing the different CDK11p110 protein immunohistochemical staining categories in ESCC and adjacent normal tissues on TMA sections. The HE staining was performed by Shanghai Outdo Biotech Co., Ltd. (Shanghai, China) and was used to show the tumor area.
CDK11P110 is highly expressed in human ESCC tissues and cell lines
To further validate the clinical significance of CDK11p110 expression in patients with ESCC, western blotting was performed to evaluate the protein levels of CDK11p110 in nine paired clinical specimens and in seven cell lines. As shown in Figure 2(a), compared with those in the adjacent normal tissues, the relative levels of the CDK11p110 protein were obviously elevated in the human ESCC tissues. Additionally, all six human ESCC cell lines, especially EC109, EC9706, KYSE150 and TE-7, exhibited high levels of CDK11p110 expression, whereas the expression of CDK11p110 was lower in the human esophageal epithelial cell line HET-1A than in most of the ESCC cell lines (Figure 2(b)). To further confirm the expression of CDK11p110 and to determine its subcellular localization in the ESCC cell lines, immunofluorescence was performed in the EC109 and KYSE150 cell lines, which showed high expression of CDK11p110. As shown in Figure 2(d), the CDK11p110 protein was mainly localized in the nucleus of ESCC cells.
Figure 2.
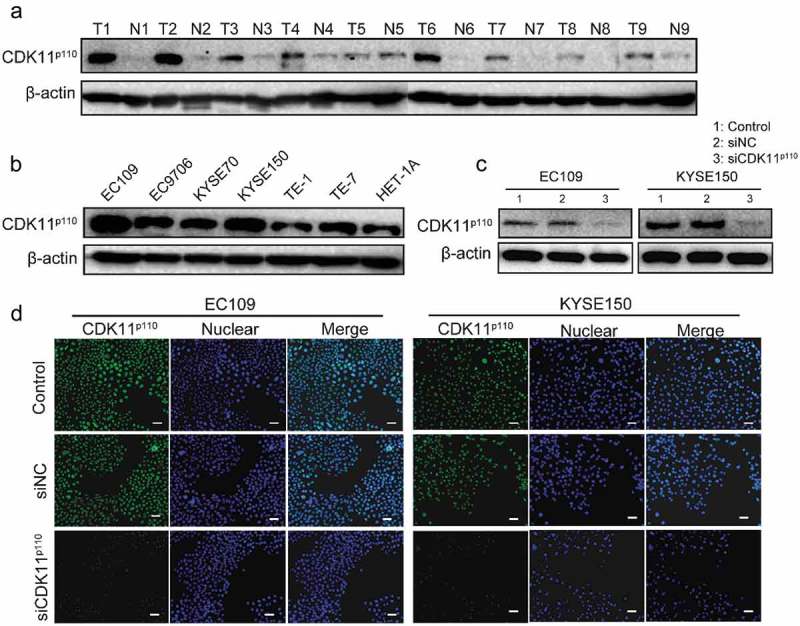
CDK11p110 is highly expressed in human ESCC tissues and cell lines, and CDK11p110 siRNA decreases the expression of CDK11p110 in cells. (a) Expression levels of CDK11p110 in nine pairs of ESCC tissues and their matching adjacent normal tissues were confirmed by western blotting. All the gels were run under the same experimental conditions. ESCC tissues (T1-T9) and adjacent normal tissues (N1-N9) are shown. (b) Levels of CDK11p110 expression in the human esophageal epithelial cell line HET-1A and in six ESCC cell lines, namely, EC109, EC9706, KYSE70, KYSE150, TE-1 and TE-7, were detected using western blotting. (c) Evaluation of CDK11p110 protein knockdown by 20 nM siRNA in EC109 and KYSE150 cells by western blot analysis. (d) Evaluation of CDK11p110 expression and confirmation of CDK11p110 protein knockdown by siRNA in EC109 and KYSE150 cells by immunofluorescence analysis. Cells were visualized under a fluorescence microscope after incubation with primary antibodies to CDK11p110 and DyLight 488-conjugated goat anti-rabbit IgG secondary antibody. Green signals indicate CDK11p110. Blue signals indicate DAPI staining, which identifies nuclei. Scale bar: 100 μm.
In addition, the knockdown efficacy of CDK11p110 in the ESCC cell lines EC109 and KYSE150 was assessed by western blot and immunofluorescence analysis. The results showed that the CDK11p110 expression level was significantly reduced in EC109 or KYSE150 cells after transfection with CDK11p110 siRNA (siCDK11p110) (Figure 2(c,d)).
Knockdown of CDK11P110 decreases ESCC cell viability, proliferation, clonogenicity and migration
Since CDK11p110 was obviously highly expressed in human ESCC tissues and cells, a proliferation assay was performed to determine whether CDK11p110 silencing could inhibit ESCC cell growth. As shown in Figure 3(a), when CDK11p110 siRNA was transfected into ESCC cell lines EC109 or KYSE150 for 72 h, cell viability was significantly reduced in a dose-dependent manner as determined by a CCK8 assay, while cells transfected with nonspecific siRNA (siNC) revealed no significant inhibition of growth compared with the control cells. Moreover, RNAi-mediated CDK11p110 knockdown decreased cell proliferation and inhibited cell growth in a time-dependent manner in both the EC109 and the KYSE150 ESCC cell lines (Figure 3(b)). These results confirmed that CDK11p110 was critical for ESCC cell growth.
Figure 3.
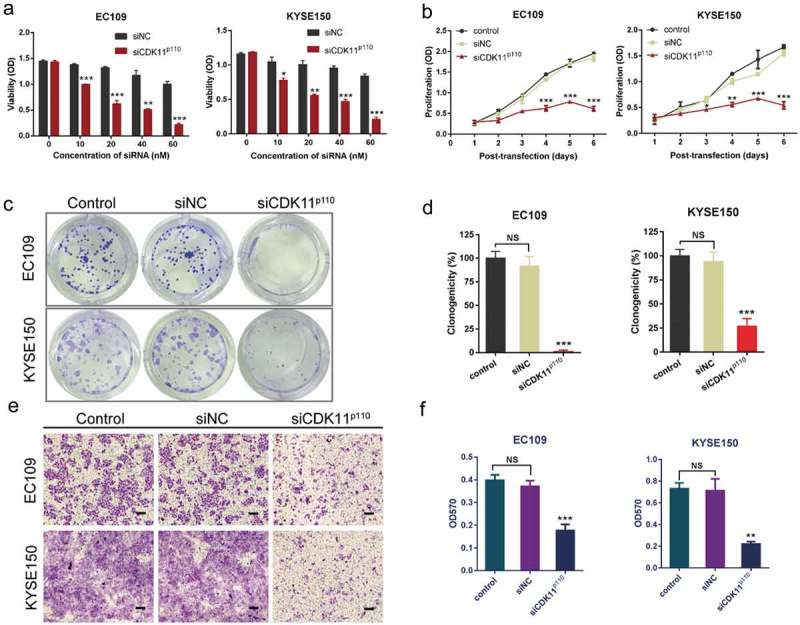
Effects of CDK11p110 silencing on ESCC cell growth and migration. (a) After EC109 and KYSE150 cells were transfected with CDK11p110 siRNA in a dose-dependent manner for 72 hours, cell viability was determined by a CCK8 assay. *P < 0.05, **P < 0.01, ***P < 0.001, compared with siNC groups. (b) EC109 and KYSE150 cells were transfected with siCDK11p110. Proliferation of cancer cells was assessed by a CCK8 assay at different time points post-transfection. *P < 0.05, **P < 0.01, ***P < 0.001, compared with siNC groups. (c) Macroscopic images showing alterations in the colony formation capability of EC109 and KYSE150 cells after CDK11p110 knockdown with siRNA. A clonogenic assay was used. (d) Statistical analysis shows that knockdown of CDK11p110 in EC109 and KYSE150 cells by siRNA caused a significant decrease in colony formation capability. ***P < 0.001, compared with siNC groups. NS, not significant. (e) Representative images showing migration of EC109 and KYSE150 cells after CDK11p110 knockdown. After transfection with siCDK11p110 or siNC for 72 hours, the cell migration activity of EC109 and KYSE150 cells was assessed by Transwell assays. Scale bar: 200 μm. (f) Statistical analysis shows that the knockdown of CDK11p110 in EC109 and KYSE150 cells caused a significant decrease in cell migration capability. **P < 0.01, ***P < 0.001, compared with siNC groups. NS, not significant. All the data are presented as the mean ± SD (n = 3) and comparisons among three groups were analyzed by one-way ANOVA and comparisons between two groups were performed by student’s t test.
Subsequently, a cell colony formation assay was conducted to investigate the effect of CDK11p110 knockdown on cell clonogenicity. As demonstrated in Figure 3(c,d), the colony formation capability of both EC109 and KYSE150 cells was noticeably inhibited after transfection with siCDK11p110 compared with that in the siNC and control groups (both P < 0.001); this effect was not observed in cells transfected with the same dose of siNC.
For further determination of the effect of CDK11p110 knockdown on the migration ability of human ESCC cells, a Transwell assay was performed after transfection with siCDK11p110 for 72 h. As shown in Figure 3(e,f), the migration of both EC109 and KYSE150 cells was significantly repressed in the siCDK11p110-transfected groups (both P < 0.01) compared with that in the siNC and control groups. These phenotypes were not observed with the transfection of nonspecific siRNA.
CDK11P110 knockdown induces apoptosis and cell cycle arrest in the G2/M phase in ESCC cells
To investigate the underlying mechanisms by which CDK11p110 knockdown inhibits ESCC cell growth and proliferation, we detected apoptosis in CDK11p110 knockdown cells with two independent experimental approaches, including flow cytometry analysis and apoptosis-associated protein detection. As shown in Figure 4(a), the flow cytometry analysis indicated that significantly increased cell apoptosis rates were observed in CDK11p110-silenced EC109 and KYSE150 cells compared with those in the control and siNC groups (P < 0.05), while Figure 4(b) shows the statistical analysis of repeated experiments. In addition, as presented in Figure 4(c), substantial expression of the apoptosis-associated cleaved PARP protein was detected by western blot analysis in EC109 and KYSE150 cells transfected with siCDK11p110 for 72 h but not in control and siNC-transfected cells. These results suggested that the reduction in ESCC cell proliferation by CDK11p110 silencing is associated with the induction of apoptosis.
Figure 4.
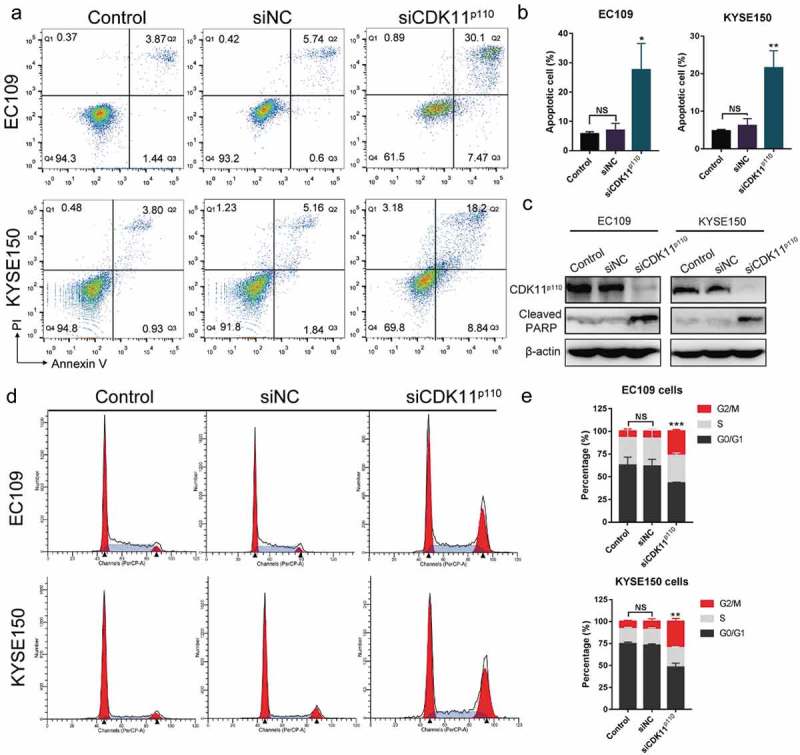
Silencing of CDK11p110 induces apoptosis and cell cycle arrest at the G2/M phase in ESCC cell lines. (a) Apoptosis was determined by a flow cytometry analysis. EC109 and KYSE150 cells were transfected with siNC or siCDK11p110 for 72 hours. (b) Apoptosis rate of EC109 and KYSE150 cells after CDK11p110 knockdown. *P < 0.05, **P < 0.01, compared with siNC groups. NS, not significant. (c) The levels of the apoptosis-associated protein cleaved PARP were detected by western blot analysis in EC109 and KYSE150 cells after transfection with siNC or siCDK11p110 for 72 hours. (d) The alterations in cell cycle distribution of EC109 and KYSE150 cells were determined by flow cytometry after transfection with siNC or siCDK11p110 for 72 hours. (e) Ratios of cells in different cell cycle phases in EC109 and KYSE150 cells. Statistical analysis shows that the knockdown of CDK11p110 in EC109 and KYSE150 cells caused a significant G2/M arrest. **P < 0.01, ***P < 0.001 compared with siNC group. NS, not significant. All the data are presented as the mean ± SD (n = 3) and comparisons among three groups were analyzed by one-way ANOVA and comparisons between two groups were performed by student’s t test.
To further explore the potential mechanism underlying the induction of ESCC cell apoptosis by CDK11p110 silencing, FACS-based DNA content analysis was used to determine the cell cycle phase distributions in human ESCC cells after CDK11p110 knockdown for 72 hours. As shown in Figure 4(d), after transfection with siCDK11p110, notable G2/M cell cycle arrest accompanied by a reduction in the fraction of cells in the G0/G1 phase was observed in both EC109 and KYSE150 cells (both P < 0.01) compared with those parameters in the control or siNC-transfected groups. Figure 4(e) shows the statistical analysis of three independent experiments. This results indicated that CDK11p110 silencing could induce cell cycle arrest in the G2/M phase in human ESCC cells and that this cell cycle arrest is probably one of the causes of apoptosis.
The effect of stable CDK11P110 overexpression or knockdown in ESCC cell lines
To explore the functional role of CDK11p110 in vivo, we first established ESCC cell lines with stable overexpression and knockdown of CDK11p110 by a lentiviral system. In these analyses, proliferation and CDK11p110 protein levels in the stable cell lines were detected by CCK8 and western blot assays, respectively. As shown in Figure 5(a), for both EC109 and KYSE150 cells, the viability of the stable CDK11p110 knockdown cells infected with lentiviruses carrying pLKO.1-shCDK11p110-1 and pLKO.1-shCDK11p110-2 was markedly inhibited compared with that of the group expressing pLKO.1 (P < 0.001), and this result validated the CDK11p110 siRNA results. Additionally, the western blot assay showed that the CDK11p110 protein level was significantly decreased in the two stable knockdown cell lines compared with that in the control cell line (Figure 5(b)). Moreover, Figure 5(c) showed that for both EC109 and KYSE150 cells, the proliferation of stable CDK11p110-overexpressing cells infected with pCDH-CDK11p110 lentiviruses was slightly increased compared with that of the pCDH-vector group (P < 0.05). In addition, the western blot assay showed that the CDK11p110 protein level was highly increased in the two stable overexpression cell lines (Figure 5(d)). These results not only confirmed the successful construction of the stable cell lines but also further supported the idea that CDK11p110 plays a critical role in the growth and proliferation of ESCC cells.
Figure 5.
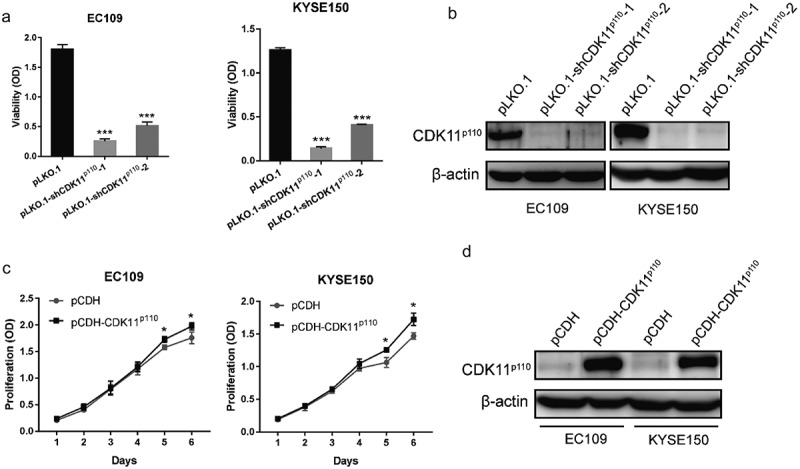
Effect of stable overexpression and knockdown CDK11p110 in ESCC cell lines. (a) EC109 and KYSE150 cell viability was evaluated for 72 h using a CCK8 assay after infection with lentiviruses expressing the pLKO.1 vector, pLKO.1-shCDK11p110-1 or pLKO.1-shCDK11p110-2 and puromycin selection for 7 days. ***P < 0.001 compared with the pLKO.1 group. (b) CDK11p110 expression was detected by western blot assay in stable CDK11p110 knockdown EC109 and KYSE150 cell lines. (c) EC109 and KYSE150 cell proliferation was evaluated at the indicated time points using a CCK8 assay after infection with lentiviruses expressing the pCDH vector or pCDH-CDK11p110 and puromycin selection for 7 days. *P < 0.05 compared with the pCDH-vector group. (d) CDK11p110 expression was detected by western blot assay in stable CDK11p110-overexpressing EC109 and KYSE150 cell lines. All the data are presented as the mean ± SD (n = 3) and comparisons among three groups were analyzed by one-way ANOVA and comparisons between two groups were performed by student’s t test.
CDK11P110 plays a critical role in the formation and progression of ESCC cell subcutaneous xenograft tumors
To further support the biological significance of the in vitro findings regarding the function of CDK11p110 in ESCC, we subsequently established xenograft tumor models by subcutaneously injecting EC109 cells with stable CDK11p110 overexpression or knockdown into nude mice to examine the effect of CDK11p110 on ESCC tumor growth in vivo (Figure 6(a)). As shown in Figure 6(b,c), the increase in the volume of tumors formed by CDK11p110-overexpressing EC109 cells was significantly enhanced compared with that of tumors formed by control cells (untransfected) or by pCDH vector-transfected cells (p < 0.05). At the termination of the experiment, the net weight of tumors formed by CDK11p110-overexpressing EC109 cells was also significantly increased compared with that of tumors formed by the control and pCDH-expressing cells (p < 0.01; Figure 6(d)). Moreover, immunohistochemical staining indicated a significant increase in the expression of CDK11p110 and the Ki67 cell proliferation index in tumors formed by EC109 cells stably overexpressing CDK11p110 (Figure 6(e)). Conversely, the increase in the volume of tumors formed from control or pLKO.1-transfected EC109 cells was strongly suppressed in EC109 cells with stable shRNA-mediated knockdown of CDK11p110 (p < 0.001; Figure 6(f,g)). Similarly, at the termination of the experiment, the net weight of tumors formed by CDK11p110-knockdown cells was also dramatically decreased compared with that of tumors formed by control or pLKO.1-transfected cells (p < 0.001; Figure 6(h)). Additionally, immunohistochemical staining showed a significant decrease in the expression of CDK11p110 and Ki67 in tumors formed by stable shRNA-mediated knockdown cells (Figure 6(e)), indicating that the tumor suppressive effect was exerted by CDK11p110 depletion. Overall, these data support an important role of CDK11p110 in controlling ESCC cell formation and progression in vivo.
Figure 6.
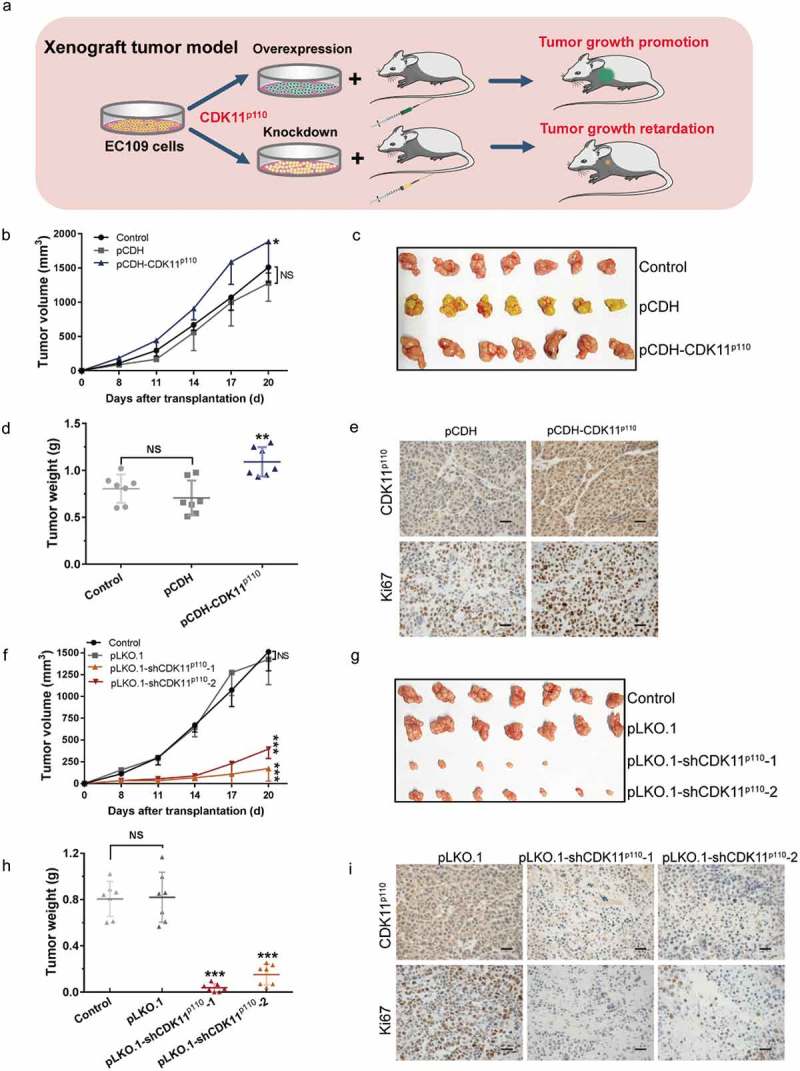
In vivo EC109 xenograft model confirmed the critical role of CDK11p110 in ESCC cell tumorigenicity. (a) Stable CDK11p110 overexpression or knockdown EC109 xenograft model in athymic nude mice. (b) Subcutaneous tumor growth of CDK11p110-overexpressing EC109 cells, vector pCDH-transfected cells and control (untransfected) cells in nude mice. *P < 0.05, against pCDH group. NS, not significant. The data are the means ± SD (n = 7/group). (c) Photographs of the excised EC109 tumors from all groups of mice, including those inoculated with control, pCDH- or pCDH-CDK11p110-transfected EC109 cells. (d) Tumor weights in different groups of mice inoculated with control, pCDH- and pCDH-CDK11p110-transfected EC109 cells. **P < 0.01 compared with the pCDH EC109 cell groups. NS, not significant. The data are the means ± SD (n = 7/group). (e) The expression of CDK11p110 and of the tumor proliferation marker Ki67 in stable CDK11p110-overexpressing EC109 subcutaneous xenografts were examined by immunohistochemistry. Scale bar: 20 μm. (f) Subcutaneous tumor growth of CDK11p110-knockdown EC109 cells, pLKO.1-transfected cells and control cells in nude mice. ***P < 0.001, versus the pLKO.1 group. NS, not significant. The data are the means ± SD (n = 7/group). (g) Photographs of the excised EC109 tumors from all groups of mice including those inoculated with control, pLKO.1-, pLKO.1-shCDK11p110-1- and pLKO.1-shCDK11p110-2-transfected EC109 cells. (h) Tumor weights in different groups inoculated with control, pLKO.1- and pLKO.1-shCDK11p110-1- and pLKO.1-shCDK11p110-2-transfected EC109 cells. ***P < 0.001, versus the pCDH-EC109 group. NS, not significant. The data are the means ± SD (n = 7/group). (i) Confirmation of CDK11p110 expression and Ki67 staining in stable CDK11p110-knockdown EC109 subcutaneous xenografts by immunohistochemistry. Scale bar: 20 μm. For all data, comparisons between three groups were analyzed by one-way ANOVA and comparisons between two groups were performed by student’s t test.
Discussion
More than 50% of ESCC patients present with unresectable or metastatic disease at the time of diagnosis [4]. Unfortunately, there is a serious deficiency of effective clinical therapy strategies targeting ESCC, and most of the current research in esophageal cancer focuses on esophageal adenocarcinoma and gastroesophageal junction cancer [4,30–32]. Accumulating studies have demonstrated that CDKs are overexpressed or activated in cancer, and targeting CDKs in tumor cells has become a promising therapeutic strategy against cancer [8,9]. As a member of CDK family, CDK11p110 in particular is reported to be critical for the growth and proliferation of some types of cancer cells [33]. However, the biological functions of CDK11p110 in ESCC remain unclear. In this study, we have shown that the expression of the kinase CDK11p110 is commonly increased in ESCC cell lines and tissues. Knockdown of CDK11p110 with CDK11p110-specific siRNA significantly decreased the proliferation, inhibited the migration, and induced the apoptosis of ESCC cells and induced G2/M arrest in these cells. Additionally, CDK11p110 overexpression increased the growth of subcutaneous xenograft tumors in nude mice, and the opposite phenomenon occurred in CDK11p110-depleted xenograft tumors. Moreover, the Ki67 proliferation index was closely associated with the elevation or depletion, respectively, of CDK11p110 in vivo. Our results suggest that CDK11p110 not only plays a vital role in the growth of ESCC but also serves as a novel promising therapeutic target for the treatment of human ESCC.
CDK11 is functionally relevant to many biological processes, such as RNA transcription and splicing, mitosis, autophagy, and apoptosis. This distinct and unique biological roles of CDK11 have been discovered in human cancers; CDK11 may play critical regulatory roles in human tumorigenesis and in cancer cell growth and proliferation. Particularly, CDK11p110, the larger and major isoform of CDK11, functions in the processes of transcription and RNA splicing by binding to its regulatory partner cyclin L [33,34]. Recent studies have reported that the expression of CDK11p110 is increased and that CDK11p110 plays a pivotal part in survival in various cancers, including osteosarcoma, liposarcoma, breast cancer and ovarian cancer [20–23]. CDK11p110 has been proposed as a potential therapeutic target in these cancers. In this study, we focused on the critical role of CDK11p110 in ESCC cell tumorigenicity. First, we analyzed the mRNA expression profiles of esophageal cancer patients from TCGA datasets and found that CDK11 mRNA of was highly expressed in esophageal cancer tissues. Then, the level of the CDK11p110 protein was detected in an ESCC tissue microarray. An immunohistochemical assay showed that CDK11p110 is highly expressed in ESCC tissues. To further confirm the expression of CDK11p110 in human ESCC, we determined CDK11p110 expression in ESCC tissues and adjacent tissues from clinical patients, as well as in a normal esophageal epithelial cell line and six ESCC cell lines, by western blotting. These results indicated that CDK11p110 was highly expressed in human ESCC tissues and cell lines and were consistent with previous results demonstrating the overexpression of CDK11p110 in other types of cancer [33,35,36]. For further study, a wider analysis of CDK11p110 expression in a large cohort of patients with ESCC will be needed to firmly establish whether the expression of CDK11 is correlated with the progression and prognosis of ESCC, as it appears to be for breast cancer. Nonetheless, these results provide a rationale for the further evaluation of the role of CDK11p110 in ESCC.
To this end, we investigated the function of CDK11p110 in ESCC both in vivo and in vitro. Compared with siNC transfection, CDK11p110 knockdown in EC109 and KYSE150 cells by siRNA significantly suppressed cell viability and colony formation ability. In addition, this important function of CDK11p110 in ESCC cells was further validated in stable knockdown ESCC cell lines established through CDK11p110 gene-specific lentiviral shRNA transfection. Conversely, overexpression of CDK11p110 in EC109 and KYSE150 cells produced the effect of promoting cell proliferation. Furthermore, ESCC xenograft tumor models established by subcutaneous injection of EC109 cells with stable CDK11p110 overexpression and knockdown into nude mice confirmed that CDK11p110 overexpression enhances the growth of subcutaneous xenograft tumors in nude mice, whereas CDK11p110 depletion inhibits tumor growth in this model. These results are congruent with those in osteosarcoma, liposarcoma, breast cancer and ovarian cancer, in which CDK11p110 knockdown reduces cell or tumor growth [20–23]. As is well known, tumor cell migration is important not only for cancer development but also for cancer metastasis [28]. CDKs and their cyclin family are important for cell migration in several types of tumors. Besides, it was reported that knockdown or knockout of CDK11p110 significantly inhibited cell migration in breast cancer and osteosarcoma [22,35]. Therefore, we assessed the function of CDK11p110 in migration of esophageal squamous cell carcinoma cells using migration assay. In this study, knockdown of CDK11p110 in EC109 and KYSE150 cells resulted in the inhibition of migration activity in these ESCC cell lines. As reported, several invasion-associated cellular factors, such as integrin β3, MT1-MMPs, and VEGF were significantly reduced in CRISPR-Cas9 CDK11 knockout osteosarcoma cell lines [35]. It is of interest to study the mechanism of CDK11p110 modulating cell migration in esophageal squamous cell carcinoma in the future studies. Overall, those results highlighted the importance of CDK11p110 in supporting the growth and proliferation of ESCC cells in vitro and in vivo.
To establish the mechanisms of CDK11p110 knockdown-induced cell growth inhibition in ESCC, the cell apoptosis rate was measured using flow cytometry, and the expression of the apoptosis-related cleaved PARP protein was examined after the transfection of siCDK11p110 into EC109 and KYSE150 cells. As expected, the proportion of apoptotic cells and expression of cleaved PARP increased significantly, suggesting that CDK11p110 was involved in ESCC cell apoptosis signaling. Considering that CDK11p110 plays a critical role in regulating the cell cycle, we further explored the potential mechanism of cell apoptosis by determining cell cycle alterations. The results showed that cell cycle progression was blocked in the G2/M phase in both EC109 and KYSE150 cells after CDK11p110 knockdown, which was different from the result reported in breast cancer that the cell cycle was blocked in the G1 phase. The postulated explanation for the difference in cell cycle arrest in ESCC and breast cancer was that cancer cell lines have different genetic backgrounds. The further in vivo study found that the Ki67 proliferation index was markedly decreased in xenograft tumors formed by subcutaneous injection of stable CDK11p110 knockdown EC109 cells and vice versa, which indicated that the expression of Ki67 correlates well with the differential expression of CDK11p110. Collectively, these results support the idea that CDK11p110 sustains tumor cell survival and proliferation through controlling aspects of apoptosis, G2/M cell cycle progression and Ki67 expression in ESCC. However, further studies will be needed to understand the precise mechanism of action.
In summary, this study provided evidence that CDK11p110 is highly expressed in human ESCC tissues and demonstrated that CDK11p110 is essential for the growth and proliferation of ESCC cells in vitro and in vivo. CDK11p110 knockdown exerts suppressive effects in ESCC cells by inducing apoptosis and G2/M cell cycle arrest and by reducing the Ki67 index. These findings provide a theoretical basis for the exploration of CDK11p110 as a novel therapeutic target in ESCC.
Funding Statement
This work was supported by grants from the Major Science and Technology Project of Henan Province (No. 161100310100) and the National Natural Science Foundation of China (No. 31670895, U1504831, and 81703417).
Author contributions
Study Design: Quancheng Kan, Xin Tian
Study Conduct: Yue Du, Weiyan Cheng, Yongliang Yuan, Suhua Wang, Zhiheng Yang, Dan Yan
Data Analysis and Interpretation: Yue Du, Yongliang Yuan, Jian Xu, Dan Yan
Writing and Revision of the Manuscript: Xin Tian, Yue Du, Weiyan Cheng, Jian Xu
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Napier KJ, Scheerer M, Misra S.. Esophageal cancer: a review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol. 2014;6:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tian X, Sun B, Chen C, et al. Circulating tumor DNA 5-hydroxymethylcytosine as a novel diagnostic biomarker for esophageal cancer. Cell Res. 2018;28:597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Abnet CC, Arnold M, Wei WQ.. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. 2018;154:360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu Y, Xiong Z, Beasley A, et al. Personalized and targeted therapy of esophageal squamous cell carcinoma: an update. Ann N Y Acad Sci. 2016;1381:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vrana D, Hlavac V, Brynychova V, et al. ABC transporters and their role in the neoadjuvant treatment of esophageal cancer. Int J Mol Sci. 2018;19:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vijayaraghavan S, Moulder S, Keyomarsi K, et al. Inhibiting CDK in cancer therapy: current evidence and future directions. Target Oncol. 2018;13:21–38. [DOI] [PubMed] [Google Scholar]
- [7].Whittaker SR, Mallinger A, Workman P, et al. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017;173:83–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mills CC, Kolb EA, Sampson VB. Development of chemotherapy with cell-cycle inhibitors for adult and pediatric cancer therapy. Cancer Res. 2018;78:320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Santo L, Siu KT, Raje N. Targeting cyclin-dependent kinases and cell cycle progression in human cancers. Semin Oncol. 2015;42:788–800. [DOI] [PubMed] [Google Scholar]
- [10].Asghar U, Witkiewicz AK, Turner NC, et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].de Duenas EM, Gavila-Gregori J, Olmos-Anton S, et al. Preclinical and clinical development of palbociclib and future perspectives. Clin Transl Oncol. 2018:1–9. [DOI] [PubMed] [Google Scholar]
- [12].Finn RS, Martin M, Rugo HS, et al. Palbociclib and Letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925–1936. [DOI] [PubMed] [Google Scholar]
- [13].Shah A, Bloomquist E, Tang S, et al. FDA approval: ribociclib for the treatment of postmenopausal women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer. Clin Cancer Res. 2018;24:2999–3004. [DOI] [PubMed] [Google Scholar]
- [14].Shupp A, Casimiro MC, Pestell RG. Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget. 2017;8:17373–17382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17:353–377. [DOI] [PubMed] [Google Scholar]
- [16].Trembley JH, Loyer P, Hu D, et al. Cyclin dependent kinase 11 in RNA transcription and splicing. Prog Nucleic Acid Res Mol Biol. 2004;77:263–288. [DOI] [PubMed] [Google Scholar]
- [17].Wilkinson S, Croft DR, O’Prey J, et al. The cyclin-dependent kinase PITSLRE/CDK11 is required for successful autophagy. Autophagy. 2011;7:12951301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kren BT, Unger GM, Abedin MJ, et al. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as RNA interference targets for triple negative breast cancer therapy. Breast Cancer Res. 2015;17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chi Y, Huang S, Peng H, et al. Critical role of CDK11(p58) in human breast cancer growth and angiogenesis. BMC Cancer. 2015;15:701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Duan Z, Zhang J, Choy E, et al. Systematic kinome shRNA screening identifies CDK11 (PITSLRE) kinase expression is critical for osteosarcoma cell growth and proliferation. Clin Cancer Res. 2012;18:4580–4588. [DOI] [PubMed] [Google Scholar]
- [21].Jia B, Choy E, Cote G, et al. Cyclin-dependent kinase 11 (CDK11) is crucial in the growth of liposarcoma cells. Cancer Lett. 2014;342:104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou Y, Han C, Li D, et al. Cyclin-dependent kinase 11p110 (CDK11p110) is crucial for human breast cancer cell proliferation and growth. Sci Rep. 2015;5:10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu X, Gao Y, Shen J, et al. Cyclin-dependent kinase 11 (CDK11) is required for ovarian cancer cell growth in vitro and in vivo, and its inhibition causes apoptosis and sensitizes cells to paclitaxel. Mol Cancer Ther. 2016;15:1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xu J, Liu XJ, Li L, et al. An engineered TIMP2-based and enediyne-integrated fusion protein for targeting MMP-14 shows potent antitumor efficacy. Oncotarget. 2015;6:26322–26334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu WJ, Zhu KL, Xu J, et al. Enediyne-activated, EGFR-targeted human β-defensin 1 has therapeutic efficacy against non-small cell lung carcinoma. Lab Invest. 2018;98:1538–1548. [DOI] [PubMed] [Google Scholar]
- [26].Liu WJ, Liu XJ, Xu J, et al. EGFR-targeting, beta-defensin-tailored fusion protein exhibits high therapeutic efficacy against EGFR-expressed human carcinoma via mitochondria-mediated apoptosis. Acta Pharmacol Sin. 2018;39:1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xu J, Du Y, Liu XJ, et al. Recombinant EGFR/MMP-2 bi-targeted fusion protein markedly binding to non-small-cell lung carcinoma and exerting potent therapeutic efficacy. Pharmacol Res. 2017;126:66–76. [DOI] [PubMed] [Google Scholar]
- [28].Xu J, Du Y, Liu WJ, et al. Intensive fibrosarcoma-binding capability of the reconstituted analog and its antitumor activity. Drug Deliv. 2018;25:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Du Y, Shang BY, Sheng WJ, et al. A recombinantly tailored beta-defensin that displays intensive macropinocytosis-mediated uptake exerting potent efficacy against K-Ras mutant pancreatic cancer. Oncotarget. 2016;7:58418–58434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Maron SB, Catenacci DVT. Novel targeted therapies for esophagogastric cancer. Surg Oncol Clin N Am. 2017;26:293–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Janmaat VT, Steyerberg EW, van der Gaast A, et al. Palliative chemotherapy and targeted therapies for esophageal and gastroesophageal junction cancer. Cochrane Database Syst Rev. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gerson JN, Skariah S, Denlinger CS, et al. Perspectives of HER2-targeting in gastric and esophageal cancer. Expert Opin Investig Drugs. 2017;26:531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou Y, Shen JK, Hornicek FJ, et al. The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (CDK11) in human cancer. Oncotarget. 2016;7:40846–40859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dos Santos P, Nikolas F, Canduri F. The emerging picture of CDK11: genetic, functional and medicinal aspects. Curr Med Chem. 2018;25:880–888. [DOI] [PubMed] [Google Scholar]
- [35].Feng Y, Sassi S, Shen JK, et al. Targeting CDK11 in osteosarcoma cells using the CRISPR-Cas9 system. J Orthop Res. 2015;33:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liao Y, Sassi S, Halvorsen S, et al. Androgen receptor is a potential novel prognostic marker and oncogenic target in osteosarcoma with dependence on CDK11. Sci Rep. 2017;7:43941. [DOI] [PMC free article] [PubMed] [Google Scholar]