

ABSTRACT
HepaRG is a proliferative human hepatoma-derived cell line that can be differentiated into hepatocyte-like and biliary-like cells. Differentiated HepaRG cultures maintain key hepatic functions including drug transporters and xenobiotic-metabolizing enzymes. To gain insight into proliferative and differentiated HepaRG metabolism we profiled various bioenergetic parameters and investigated cell culture levels of adenosine triphosphate (ATP), lactate, and lactate dehydrogenase (LDH) activity. Compared to differentiated-derived HepaRG, cells from proliferative cultures had increased basal and ATP-linked respiration and decreased maximal and spare respiratory capacities. Basal ATP levels but not lactate or LDH activity were increased in samples from proliferative-derived compared to differentiated-derived HepaRG. Further extracellular acidification rate (ECAR) experiments revealed parameters associated with glycolysis and oxidative phosphorylation. Under basal conditions, cells derived from both cultures had similar ECARs; however, under stressed conditions, proliferative-derived HepaRG had increases in ECAR capacity and apparent glycolytic reserve. The biguanide metformin has been reported to protect differentiated HepaRG against acetaminophen (APAP)-induced cell injury, as well as offer protection against bioenergetic deficiencies; therefore, we studied the outcome of exposure to these drugs in both culture conditions. Proliferative- and differentiated-derived cells were found to have distinct mitochondrial bioenergetic alterations when exposed to the hepatotoxic drug APAP. Metformin offered protection against loss of APAP-induced cellular viability and prevented APAP-induced decreases in bioenergetics in differentiated- but not proliferative-derived HepaRG. Distinguishingly, treatment with metformin alone reduced ATP-linked respiration, maximal respiratory capacity, and basal respiration in proliferative-derived HepaRG. Our results support that HepaRG represents an appropriate model to study drug-induced bioenergetic dysfunction.
KEYWORDS: Extracellular flux analysis and bioenergetics, pharmacological stressors, proliferative and differentiated HepaRG
Introduction
Extracellular flux analysis is an important tool to quantitate cellular bioenergetic diversity in a wide variety of primary, stem, cancer, and immortalized cell lines [1–3]. Measuring bioenergetic parameters by extracellular flux analysis has been utilized to assess metabolic inhibition by mitochondrial-targeted anticancer compounds [3–5] and by compounds with off-target effects on mitochondrial function [6–8]. Off-target effects of drugs and environmental toxicants causing mitochondrial dysfunction are important factors to consider in toxicity studies and human cell lines have served as useful models for these experiments [6–8]. The use of in vitro human cell culture models in toxicity testing is becoming increasingly attractive due to the small quantities of compounds needed for testing, shortened experimental timelines, increased throughput to evaluate toxicants, and reduced number and suffering of animals [9,10]. Primary human hepatocytes isolated from liver and liver-derived immortalized cell lines are widely used as models for toxicological studies as the liver is the primary source of drug metabolism and biotransformation [9]. In hepatotoxicity cases, primary human hepatocytes are a desirable pertinent model; however, organ donors are scarce, the interdonor function is variable, and primary hepatocytes undergo early phenotypic changes in vitro [11]. Additionally, in culture, many human hepatocyte cell lines lack liver-specific functions including cytochrome P450-related enzyme activities [12]. The HepaRG cell line was originally derived from a liver tumor obtained from a patient suffering from hepatitis C infection and hepatocarcinoma [13]. Following the establishment of the cell line, the presence of the hepatitis C virus genome was no longer detectable but HepaRG supports hepatitis B virus (HBV) infection and is a useful tool to study mechanisms of HBV infectivity [13]. HepaRG is a proliferative human hepatoma-derived cell line that can be differentiated into hepatocyte-like and biliary-like cells [11,12]. Differentiated HepaRG cultures have been demonstrated to display toxicity towards compounds metabolized via cytochrome P450s [12]. In addition to cytochrome P450s (CYP1A1, 1A2, 2A2, 3A4, CYP4A11, 7A1, 2B6, 2C8, 2C9, 2C19, 2E1, 4F3), differentiated HepaRG cultures express phase II drug metabolizing genes (UGT1A1, GSTA1, GSTA4, GSTM1), membrane transporters (e.g. bile salt export pump), and transcription factors, PXR, CAR, PPARα, and AhR [11,12,14,15]. In terms of mitochondrial bioenergetic studies, differentiated HepaRG has been validated to mimic primary human hepatocyte bioenergetics utilizing the OROBOROS® Oxygraph 2K [16].
Acetaminophen (APAP) and aflatoxin B1 have been demonstrated to be cytotoxic to differentiated HepaRG and toxicity of these two compounds is mediated via the formation of toxic metabolites generated by cytochrome P450s [12]. An overdose of APAP leads to an excess of the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI), which depletes glutathione and binds to proteins [17]. Inhibition of mitochondrial respiration following APAP overdose is hypothesized to be caused in part by the formation of NAPQI adducts on oxidative phosphorylation (OXPHOS) proteins and peak levels of adducts have been detected in differentiated HepaRG at 6 hours after exposure to 20 mM APAP [17]. The biguanide metformin was previously demonstrated to protect differentiated HepaRG against APAP-induced cell injury and to attenuate APAP-induced mitochondrial bioenergetic deficiencies when cells were treated with 0.5 or 1 mM metformin 6 hours after exposure to 20 mM APAP [18]. Furthermore, metformin attenuated APAP-induced mitochondrial oxidant stress and dysfunction in mice [18]. Metformin is a drug widely used to treat diabetes and fertility and has been reported to decrease mitochondrial respiration in proliferative cell types such as normal immortalized fallopian tube secretory epithelial cells (FTSECs) and in high-grade serous ovarian cancer (HGSC) cells [19–21].
To gain insight into both proliferative and differentiated HepaRG metabolism we profiled various bioenergetic parameters utilizing the Seahorse XFp and investigated cell culture levels of adenosine triphosphate (ATP), lactate, and lactate dehydrogenase (LDH) activity. Proliferative and differentiated HepaRG cultures were also separately exposed to APAP, APAP + metformin, or metformin to determine effects on cellular viability and mitochondrial bioenergetics. To our knowledge, this is the first bioenergetic comparison examining HepaRG cells derived from both proliferative and differentiated cultures.
Materials and methods
Cell culture
The proliferating HepaRGTM hepatoma-derived cell line was purchased from Biopredic International (Saint-Grégoire, France). Cells were cultured and differentiated according to Biopredic International standard operating procedure and as previously described [12,13]. When proliferating HepaRGTM (proliferative HepaRG) are initially seeded at low density and then subjected to the differentiation process, differentiated cultures harbor both hepatocyte-like and biliary-like cells [11,12]. In this study, proliferative HepaRG were initially seeded at a low density of 2 × 104 cells/cm2 prior to the differentiation process; therefore, differentiated HepaRG cultures harbor both hepatocyte- and biliary-like cells as opposed to selectively detaching and re-seeding hepatocyte-like cells as has been described [22].
Briefly, proliferative HepaRG were grown in Working Growth Medium (WGM) consisting of William’s E Medium (Thermo Fisher Scientific), 2 mM GlutaMAXTM (Thermo Fisher Scientific) with the addition of HepaRGTM Growth Medium Supplement (Lonza). Cells were grown at 37°C, 5% CO2 in a humidified incubator and media was refreshed every three days. For day-to-day examination of cell culture, a Leica DMi1 inverted microscope with 10 and 20x phase contrast objectives was used. Images were collected on a Leica DMi8 with a 20x phase contrast objective. Proliferative HepaRG cells were passaged between days 12 and 15 post seeding by washing with pre-warmed Dulbecco’s phosphate-buffered saline (DPBS) followed by gentle trypsinization and neutralization with pre-warmed WGM. Tissue culture dishes were seeded with 2 × 104 proliferative cells/cm2 and cells were not passaged more than 18 times, P18. William’s Differentiation Medium (WDM) consisted of William’s E Medium (Thermo Fisher Scientific), 2 mM GlutaMAXTM (Thermo Fisher Scientific) with the addition of HepaRGTM Differentiation Medium Supplement (Lonza). The differentiation process was started two weeks after passaging proliferative HepaRG cells. WGM was replaced with Combination Medium (CM), consisting of a 1:1 mixture of WGM to WDM, and three days later CM was replaced with WDM. The medium was renewed every three days for two weeks and after two weeks cells attained differentiated hepatocyte-like morphology. Following treatment with pre-warmed trypsin and neutralization with pre-warmed medium, HepaRG viable cell counts were determined utilizing the TC20 Automated Cell Counter (Bio-Rad) and the trypan blue exclusion method.
Seahorse XFp extracellular flux analysis
Seahorse (SH) XFp cell culture miniplates, sensor cartridges with utility plates, and all reagents for Mito Stress and ECAR Stress tests (Glycolysis Stress tests), with the exception of oligomycin (Alfa AesarTM), were obtained from Agilent Technologies Cell Analysis Division (Lexington, MA, USA). The SH XFp Extracellular Flux Analyzer was used to simultaneously measure real-time oxygen (O2) consumption rates (OCR) and extracellular acidification rates (ECAR) of adherent proliferative- and differentiated-derived HepaRG cells in 8-well cell culture miniplates. The eight wells on SH XFp cell culture miniplates (referred to here as miniplates) are designated wells A through H. Each XFp disposable sensor cartridge contains eight probe sleeves with embedded pairs of fluorescence biosensors for measuring the extracellular flux changes of oxygen (532 nm excitation/650 nm emission) and pH (470 nm excitation/530 nm emission) in the medium surrounding cells seeded in each well of the miniplate. OCR is reported in the unit of picomoles (pmol) O2 per minute and ECAR in milli-pH (mpH) units per minute [23]. Additionally, four injector ports (ports A, B, C, & D) are adjacent to each sensor/probe sleeve such that miniplate wells A – H are equipped to receive four compounds during the course of the experiment. The applicable assay template file (*.asyt) was initiated during the 1-hour miniplate incubation period (see Replacement of miniplate well WGM or WDM with assay medium below) and the utility plate and sensor cartridge were loaded into the XFp. Following calibration of the sensor cartridge, the utility plate was replaced with the appropriate miniplate. Unless otherwise indicated, the XFp Cell Mito Stress Test protocol was run and consisted of equilibration, basal OCR/ECAR measurement (3 cycles), injection of oligomycin port A (3 cycles), injection of FCCP port B (3 cycles), and injection of a mixture of rotenone/antimycin A port C (3 cycles). In some experiments, such as optimizing cell-seeding density, the assay medium was loaded into the sensor cartridge compound injection ports instead of stressors. Miniplate wells A and H never received cells (background correction) but were treated identically to wells with cells during XFp analyses and protein normalization (see below) receiving growth media, assay media, and buffers during growth incubations, washes, and cell lysis.
Day 1 – To maintain cell adherence, miniplate wells were treated with a sterile solution of 0.1% gelatin (EMD MilliporeTM), parafilm was wrapped around the edges of the miniplates to prevent evaporation, and plates were stored at room temperature overnight.
Day 2, Part I – Hydrate sensor cartridge and seeding miniplates. First, 200 μl of tissue culture grade water (TC H2O) was added to each well of the XFp sensor cartridge utility plate and 400 μl of TC H2O was added to each moat chamber. Second, the sensor cartridge was placed on top of the utility plate and incubated overnight in a humidified 37°C incubator without CO2 along with a 20 ml aliquot of XF Calibrant. Finally, the gelatin solution was removed from the miniplates. The appropriate number of proliferative- or differentiated-derived HepaRG cells was resuspended in 80 μl per well of fresh WGM or WDM respectively. Cells were seeded in duplicate or triplicate into miniplate wells (0.106 cm2) for each cell type, see Day 2 Part II below. Miniplates were examined using a DMi1 microscope to ensure cells were evenly distributed and approximately 50–70% confluent and then incubated overnight in a 37°C, 5% CO2 incubator. Cellular confluency between 50–70% yielded OCR readings that were within the range recommended by the manufacturer, 20 to 150 pmol O2/minute. Lastly, the XFp was turned on to allow the chamber to stabilize at 37°C overnight.
Day 2, Part II – Optimizing cell-seeding density. Miniplates were seeded the day before an experiment. To optimize differentiated-derived HepaRG cell-seeding density cells were seeded in duplicate on miniplates at 8.9 × 104, 1.3 × 105, and 1.9 × 105 viable cells/cm2 (9.4 × 103, 1.34 × 104, and 2 × 104 cells per well respectively). On separate miniplates proliferative cells were seeded in duplicate at 4.4 × 104, 6.3 × 104, and 9.4 × 104 viable cells/cm2 (4.7 × 103, 6.7 × 103, and 1 × 104 cells per well respectively). For these cell-seeding tests, basal OCR values ranged from 37 to 115 pmol O2/minute while basal ECAR values ranged from 2 to 20 mpH/minute. For each experiment, two miniplates were run and the well with the highest basal rate (pmol O2/minute for OCR and mpH/minute for ECAR) was set to 100% baseline. The other wells were normalized relative to 100% baseline.
Unless otherwise described, an optimal seeding density of 9.4 × 104 viable proliferative-derived cells/cm2 (1 × 104 cells/well) was used and seeding densities of 9.4 × 104 or 1.9 × 105 viable differentiated-derived cells/cm2 (1 × 104 or 2 × 104 cells/well respectively) were used to obtain OCR readings within the recommended range of the XFp. Following normalization of data obtained from miniplates seeded with either 9.4 × 104 or 1.9 × 105 differentiated-derived cells/cm2, the calculated bioenergetic parameters were comparable (Table 1, the inter-experimental coefficients of variation for parameters were at most ~16%). Note, 1.9 × 105 viable differentiated-derived cells/cm2 resulted in high basal OCR readings, 120 ± 12 pmol O2/minute (mean value ± SD; n = 3); therefore, differentiated cells were not plated at the high-density of 4.5 × 105 cells/cm2 described elsewhere [11,12] as OCR values exceeded the XFp recommended upper limit of detection, 150 pmol O2/minute. Thus, we refer to the cells studied here as differentiated-derived cells. Furthermore, as we studied differentiated cells at a seeding density greater than 1.5 × 105 cells/cm,2 a density that has been demonstrated to support unipotent hepatocyte-like behavior, it is unlikely the cells underwent transdifferentiation [24]. In addition, we did not observe reversion to a homogenous population of epithelial-like cells during visual inspection of miniplates following seeding of differentiated cells. However, it is important to note that transdifferentiation has been observed when differentiated HepaRG are seeded at low densities such as 1 × 103 cells/cm2 and that hepatocyte-like cells proliferate when seeded in WGM at 5–7 × 104 cells/cm2 [22,24]. Finally, the total cellular protein content for each well was determined for all experiments (see Protein normalization below).
Table 1.
Proliferative- and differentiated-derived HepaRG mitochondrial parameters.
| Bioenergetic parameter | Proliferative OCRsa (pmol O2/min/μg cellular protein) |
Differentiated OCRsa (pmol O2/min/μg cellular protein) |
Fold-Change | P-valueb (Fold-change) |
|---|---|---|---|---|
| Basal respiration | 22.00 ± 1.42 | 18.39 ± 2.41 | 1.2 | < 0.02 |
| ATP-linked respiration | 16.19 ± 0.61 | 11.38 ± 1.86 | 1.4 | < 0.001 |
| Maximal respiratory capacity | 97.46 ± 5.47 | 116.75 ± 8.98 | 0.8 | < 0.002 |
| Spare respiratory capacity | 75.46 ± 4.60 | 98.37 ± 6.89 | 0.8 | < 0.0001 |
| Proton leak-linked respiration | 5.81 ± 1.51 | 7.01 ± 0.65 | 0.8 | 0.2 |
| Non-mitochondrial respiration | 8.91 ± 1.33 | 7.62 ± 0.70 | 1.2 | 0.06 |
| Spare respiratory capacity as a %c | 443.70% ± 21.28% | 639.43% ± 45.56% | 0.7 | < 0.0001 |
| Coupling efficiencyc | 0.74 ± 0.05 | 0.62 ± 0.02 | 1.2 | < 0.003 |
| Ratio of ATP-linked respiration to maximal respirationc | 0.17 ± 0.01 | 0.10 ± 0.01 | 1.7 | < 0.003 |
aData are presented as mean ± SD, n = 6 (triplicate from two independent experiments performed on different days with different preparations of cells).
bP-values < 0.05 were accepted as significantly different.
cInternally normalized parameters that are independent of cell number. These parameters are useful when difficulties arise controlling cell number or total protein [25].
Day 3 – The TC H2O in the XFp sensor cartridge utility plate wells/moats was removed and replaced with equal volumes of prewarmed XF Calibrant then returned to humidified 37°C incubator without CO2 for at least one hour. Next, the appropriate assay medium was prepared and pre-warmed to 37°C. Mito Stress test assay medium consisted of XF Base Medium (0 mM glucose, Agilent Technologies) supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose. ECAR Stress test assay medium consisted of XF Base Medium (Agilent Technologies) supplemented with 2 mM glutamine, 0.5 μM hydrocortisone (Sigma-Aldrich), and 5 μg/ml recombinant human insulin (Sigma-Aldrich). Assay medium pH was adjusted to 7.4 ± 0.1 using 0.1 N NaOH then the medium was sterilized by filtering through a 0.2 μm filter. Assay medium was stored at 4°C if not used immediately otherwise it was pre-warmed at 37°C for 15 minutes before use.
Replacement of miniplate well WGM or WDM with assay medium – Prior to replacing the 80 μl of WGM or WDM per miniplate well, assay medium was pre-warmed to 37°C. Sixty microliters of WGM or WDM was removed from each well. Subsequently, wells were gently washed twice by adding and removing 200 μl of assay medium from each well position. Following the second wash, 160 μl of assay medium was added to each well to give a final well volume of 180 μl. Miniplates were observed utilizing an inverted phase contrast microscope to ensure cells were not dislodged from the well bottom and to confirm cellular confluency was about 50–70%. The miniplate was placed in a humidified 37°C incubator without CO2 for 1 hour prior to running a bioenergetic assay.
Optimization of OXPHOS stressors. Oligomycin – A 25.3 mM stock solution of oligomycin was prepared in dimethyl sulfoxide (DMSO), aliquoted, and stored at −80°C. Differentiated- and proliferative-derived HepaRG cells were tested in separate miniplates, i.e. one cell type was seeded at equal density into wells B – G of a miniplate, and wells A and H were used for background correction. The day of XFp experiments during the 1-hour miniplate incubation period, a vial of oligomycin was thawed on ice and diluted in Mito Stress test assay medium to 160 μM. The 160 μM stock solution was diluted to 40 μM then subsequent 2-fold serial dilutions were made to 20 and 10 μM in Mito Stress test assay medium. To determine the effect of oligomycin on HepaRG OCRs, various concentrations of oligomycin were loaded into the sensor cartridge and a Mito Stress test was performed. A hydrated sensor cartridge was removed from the non-CO2 incubator then loaded with 20 μl of 10, 20, and 40 μM oligomycin into port A of wells B & C, D & E, and F & G respectively to obtain final well concentrations of 1, 2, and 4 μM. For each experiment, two miniplates were run successively and the well with the highest rate (pmol O2/minute/μg cellular protein) measured just prior to oligomycin injection was set to 100% baseline and the other wells were normalized relative to it. For both proliferative- and differentiated-derived HepaRG maximal decreases in OCRs were observed with 2 μM oligomycin; therefore, this concentration was used for subsequent experiments.
Titration of carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP) – FCCP titrations were carried out according to the manufacturer’s protocol using Agilent Technologies’ sourced FCCP. Briefly, differentiated- and proliferative-derived HepaRG cells were tested in separate miniplates and cells were seeded at equal densities in wells B – G leaving wells A and H for background correction. Stock solutions of oligomycin (160 μM) and FCCP (50 μM) were made in Mito Stress test assay medium. A hydrated sensor cartridge was removed from the non-CO2 incubator. Oligomycin was diluted to 20 μM as described above and 20 μl was loaded into all A ports of the sensor cartridge. Sensor cartridges were divided into two groups to assess low range and high range concentrations of FCCP via 5-point titration curves. The low range final well concentrations tested were 0.125, 0.25, and 0.5 μM (Ports B, C, and D of wells A – D respectively) while the high range final well concentrations tested were: 0.5, 1.0, and 2.0 μM (Ports B, C, and D of E – H respectively). FCCP was diluted 5-fold in assay medium to 10 μM then 2-fold serial dilutions were made to 5, 2.5, and 1.25 μM. Ports B, C, and D of wells A to D were loaded with 22, 28, and 30 μl of 1.25, 1.25, and 2.5 μM FCCP. Ports B, C, and D of wells E to H were loaded with 22, 28, and 30 μl of 5, 5, and 10 μM FCCP. The XFp Cell Characterization (FCCP Titration) protocol was run and consisted of equilibration, basal OCR/ECAR measurement (3 cycles), injection of oligomycin port A (3 cycles), injection of FCCP-1 port B (3 cycles), injection of FCCP-2 port C (3 cycles), and injection of FCCP-3 port D (3 cycles). The well with the highest rate (pmol O2/minute/μg cellular protein) measured prior to 2 μM oligomycin injection was set to 100% baseline and the other wells were normalized relative to it. Peak increases in proliferative- and differentiated-derived OCRs were observed at 1 μM FCCP; therefore, this concentration was used for subsequent experiments.
Mito Stress tests and sensor cartridge loading – Stock solutions of stressors were made and diluted in Mito Stress test assay medium. A 160 μM oligomycin stock solution was made as described above then diluted 8-fold to 20 μM. A hydrated sensor cartridge was removed from the non-CO2 incubator and 20 μl of 20 μM oligomycin was loaded into all A ports. A 50 μM FCCP stock solution was made then diluted 1 in 5 to 10 μM and 22 μl was loaded into all B ports. A 25 μM rotenone + 25 μM antimycin A stock solution was made then diluted 1 in 5 to 5 μM of both rotenone and antimycin A then 25 μl was loaded into all C ports. For side-by-side comparisons of differentiated- and proliferative-derived HepaRG during a Mito Stress test each cell type was seeded/technically replicated in triplicate in miniplate wells. Replacement of WGM and WDM with Mito Stress test assay medium and the XFp Cell Mito Stress Test protocol were carried out as described above. Independent experiments were run on different days using different passages/preparations of cells.
Equations used to calculate mitochondrial respiration parameters – Mitochondrial respiration parameters were calculated as follows: I. Basal respiration = (Last rate before the first injection) − (Non-mitochondrial respiration rate), II. Proton leak = (Minimum rate after oligomycin injection) − (Non-mitochondrial respiration rate), III. Maximal respiratory capacity = (Maximum rate after FCCP injection) − (Non-mitochondrial respiration rate), IV. Spare respiratory capacity or reserve respiratory capacity = (Maximal respiratory capacity) − (Basal respiration), V. Non-mitochondrial respiration = Minimum rate after rotenone + antimycin A injection, VI. ATP-linked respiration = (Last rate before oligomycin injection) − (Minimum rate after oligomycin injection), VII. Spare respiratory capacity as a percent = (Maximal respiratory capacity)/(Basal respiration) * 100, VIII. Coupling efficiency = (ATP-linked respiration)/(Basal respiration), IX. Ratio of ATP-linked respiration to maximal respiration = (ATP-linked respiration)/(Maximal respiratory capacity) [25].
ECAR Stress tests and sensor cartridge loading – Stock solutions of glucose and stressors were made and diluted in ECAR Stress test assay medium. Twenty microliters of a 100 mM glucose stock solution was loaded into all A ports. Next, a 160 μM oligomycin stock solution was made as described above then diluted 1 in 8 to 20 μM and 22 μl was loaded into all B ports. A 500 mM 2-deoxyglucose (2-DG) stock solution was made then 25 μl was loaded into all C ports. For side-by-side comparisons of differentiated- and proliferative-derived HepaRG during an ECAR Stress test each cell type was seeded/technically replicated in duplicate or triplicate in miniplate wells. WGM or WDM were replaced with ECAR Stress test assay medium as described above. The ECAR Stress test protocol was run and consisted of equilibration, basal OCR/ECAR measurement (3 cycles), injection of glucose port A (3 cycles), injection of oligomycin port B (3 cycles), and injection of 2-DG port C (3 cycles). Independent experiments were run on different days using different passages/preparations of cells.
Equations used to calculate ECAR bioenergetic parameters – ECAR bioenergetic parameters were calculated as follows: I. Glucose-stimulated ECAR = (Maximum rate before oligomycin injection) − (Last rate before glucose injection), II. ECAR capacity = (Maximum rate after oligomycin injection) − (Last rate before glucose injection), III. Apparent glycolytic reserve = (ECAR capacity) − (Glucose-stimulated ECAR), IV. Basal ECAR = Last rate measurement before glucose injection.
Protein normalization
Following each SH XFp experiment, OCR and ECAR values were normalized to total cellular protein as has been described [1,26–28]. Briefly, total cellular protein content in each miniplate well was measured using the PierceTM bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Assay medium was removed from all wells, including background correction wells A and H, and each well was gently washed with 200 μl DPBS pre-warmed to 37°C. Next, the miniplate was inspected under a phase contrast microscope to assure cells remained adherent to the wells and miniplates were incubated overnight at −80°C to assist with cell lysis. The next day miniplates were removed from the freezer, stored on ice, and cells were lysed in ice-cold RIPA lysis buffer supplemented with 1 in 101 dilution of HALTTM protease inhibitor cocktail (Thermo Fisher Scientific) by pipetting up and down ten times. The miniplate was inspected under a phase contrast microscope to assure that cells were removed from the bottom of the wells and that cell lysis was complete. A range of 0 to 20 μg bovine serum albumin (BSA) standards diluted in RIPA lysis buffer + HALT were run in parallel with 10 μl samples on flat bottom 96-well polystyrene plates (Fisher Scientific). The sample to working reagent ratio was 1 to 20. Plates were mixed on a microplate shaker for 1 minute, incubated at 37°C for 30 minutes, then cooled to room temperature prior to measuring the absorbance at 562nm on a Synergy H1 microplate reader (BioTek). R-squared values for linear regression of BSA standards were consistent between experiments and the average R-squared value for all experiments was 0.99. For 9.4 × 104 proliferative-derived cells/cm2 seeded, the average protein content measured by the BCA assay was 3.3 ± 0.4 μg per well (n = 12, triplicate wells from two independent Mito Stress tests and two independent ECAR Stress tests run on different days with different preparations of cells). For 9.4 × 104 and 1.9 × 105 differentiated-derived cells/cm2 seeded, average protein contents measured by BCA assay were 3.0 ± 0.4 and 5.5 ± 0.6 μg per well respectively (n ≥ 5 for each, duplicate or triplicate wells from independent Mito Stress and ECAR Stress tests run on different days with different preparations of cells).
Quantitation of cell culture L-lactate levels
Total cell culture lactate levels were estimated using the Lactate-GlowTM Assay (Promega) according to the manufacturer’s recommendations for detecting extracellular + intracellular lactate. Proliferative and differentiated HepaRG cells were seeded side-by-side on Corning 96-well flat clear-bottom white-walled polystyrene microplates in 100 μl WGM and WDM respectively and the plates were incubated overnight at 37°C, 5% CO2. Replicate 96-well plates were seeded with identical concentrations of cells in parallel to normalize lactate data to total cellular protein and plate wells were processed as described above for miniplates under Protein normalization. Each cell density was replicated in 7 wells of the 96-well plates and growth media without cells was loaded into empty wells to determine background relative light units, RLUs (negative controls).
The next day, lactate standards were prepared on ice in Glucose-supplemented ECAR Stress test assay medium (ECAR Stress test assay medium as described above supplemented with 10 mM glucose) and contained 0.244, 0.98, 3.906, or 15.625 μM lactate. Growth media was removed from all wells of a 96-well plate including the negative controls and the wells were gently washed with pre-warmed 100 μl Glucose-supplemented ECAR Stress test assay medium. Next, 40 μl of Glucose-supplemented ECAR Stress test assay medium was carefully added to each well and thereafter 40 μl of each lactate standard was loaded into empty wells on the plate in triplicate followed by incubation at 37°C without CO2 for 20 minutes. The lactate detection reagent was prepared according to the manufacturer’s instruction using the components in the kit. To block lactate production, 5 μl of inactivation solution (0.6 N HCl, 0.25% dodecyltrimethylammonium bromide, DTAB) was added to each well, including negative control and lactate standard wells, and then the plate was mixed for 5 minutes on a microplate shaker. Next, to each well 5 μl of neutralization solution (1 M Tris base) was added, the plate was mixed for 1 minute, and 50 μl of lactate detection reagent was added. The plate was mixed for 1 minute followed by a 1-hour incubation at room temperature in the dark, and RLUs were measured on a Synergy H1 microplate reader (BioTek). Linear regression of mean luminescent values (RLUs) versus lactate standard concentrations generated consistent R-squared values of 0.99. RLU values for the 0.244 μM lactate standard, (0.2 μM following addition of inactivation and neutralization solutions) produced the lowest sample signals on the plate and were at most 2-fold above background.
Within the range of lactate standard RLUs, a linear increase in RLUs was detected for each experiment utilizing differentiated-derived HepaRG seeded at 8.9 × 104, 1.3 x105, and 1.9 × 105 viable cells/cm2 when plotting RLUs vs. cell seeded (R-squared values of 0.97 and 0.98 for two independent experiments run on different days using different preparations of cells and 7 replicate wells of each cell density) and utilizing proliferative-derived HepaRG seeded at 4.4 × 104, 6.3 × 104, and 9.4 × 104 viable cells/cm2 (R-squared values of 0.99 and 0.98 for two independent experiments as described for differentiated HepaRG). Proliferative HepaRG seeded at 4.4 × 104 cells/cm2 produced the lowest RLUs, which were at least 4-fold above background luminescence. The concentration of lactate in wells containing cells (corrected for background luminescence) were estimated from standard curves generated using the background corrected lactate standard RLU values.
The values presented in Figure 4(a) are normalized to total cellular protein and are mean ± SD of n = 42 replicates for each of proliferative- and differentiated-derived HepaRG (3 cell densities × 7 replicate wells × 2 independent experiments). For both cell types, the RLUs/μg cellular protein inter-experimental coefficients of variation were at most 22%.
Figure 4.
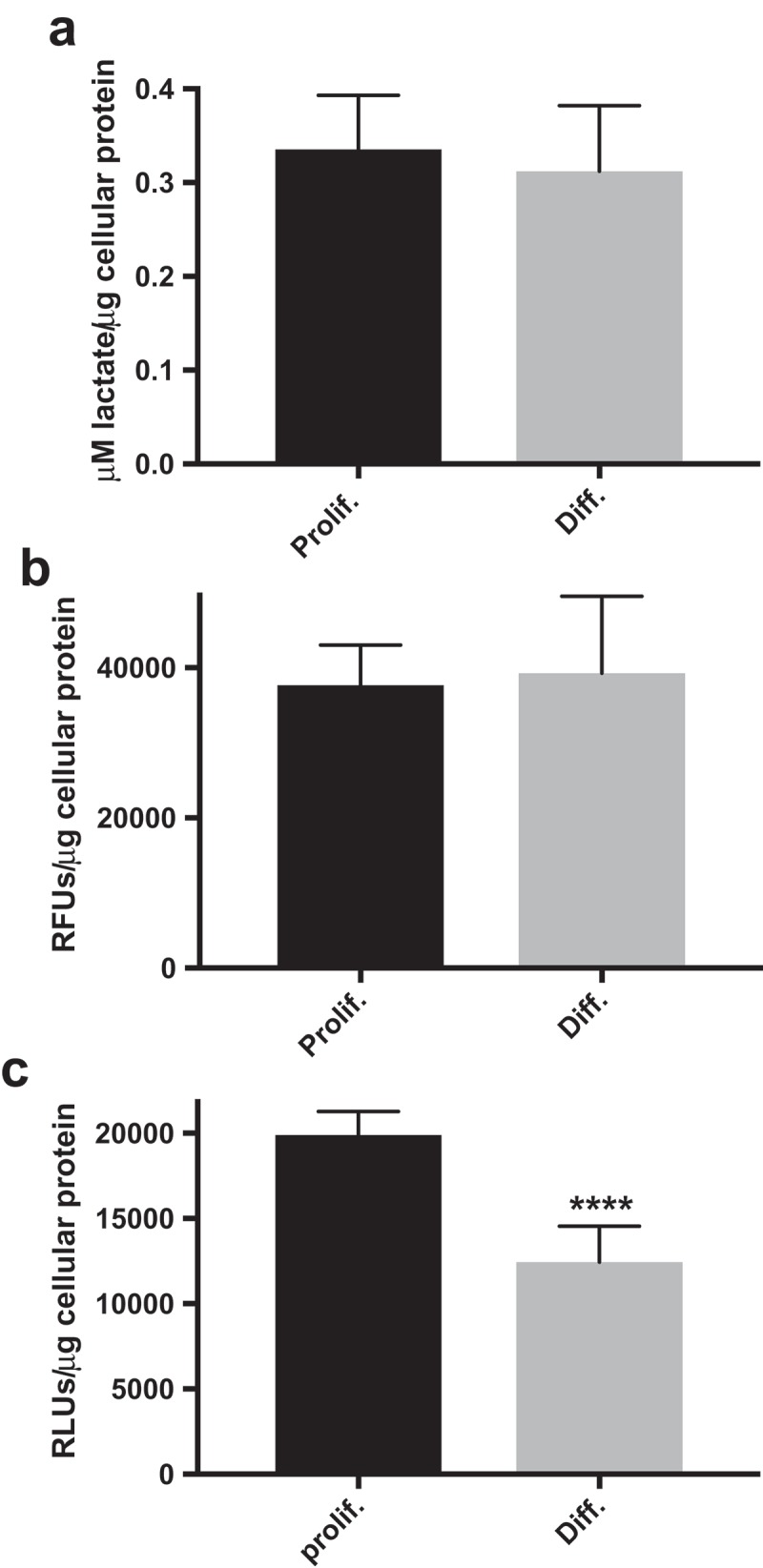
Proliferative-derived HepaRG cell cultures have increased ATP levels. Quantitation of cell culture (a) lactate, (b) LDH activity, and (c) ATP levels in proliferative (Prolif.)- and differentiated (Diff.)-derived HepaRG. Lactate, LDH activity, and ATP levels were determined with Lactate-Glow,TM CytoTox-ONE,TM and CellTiter-Glo® 2.0 assays respectively (****, P < 0.0001). Data are presented as mean ± SD, n ≥ 24 (≥12 from two independent experiments performed on different days with different preparations of cells).
Quantitation of cell culture LDH enzymatic activity
Relative amounts of proliferative- and differentiated-derived HepaRG cellular LDH activity were estimated by lysing cells in the presence of Triton X-100 followed by the detection of LDH activity in lysates utilizing the CytoTox-ONETM Homogeneous Membrane Integrity Assay (Promega). The day before the experiment proliferative- and differentiated-derived HepaRG cells were seeded side-by--side on Corning 96-well flat clear-bottom black-walled polystyrene microplates in 100 μl WGM and WDM respectively and plates were incubated overnight at 37°C, 5% CO2. Replicate 96-well plates were seeded with identical concentrations of cells in parallel to normalize relative fluorescence units (RFUs) to total cellular protein using the procedure described above for miniplates under Protein normalization. Differentiated-derived HepaRG cells were seeded at 8.9 × 104, 1.3 x105, and 1.9 × 105 viable cells/cm2 and proliferative-derived HepaRG cells were seeded at 4.4 × 104, 6.3 × 104, and 9.4 × 104 viable cells/cm.2 Each cell density was replicated in 8 wells of 96-well plates and for each density half of the wells were treated with Triton X-100 and the other half with vehicle. Growth media without cells was loaded into empty wells to serve as negative controls to determine background RFUs.
The day after seeding 96-well plates growth media was removed from all wells, including the negative controls, and wells were gently washed with 100 μl Glucose-supplemented ECAR Stress test assay medium. Next, 100 μl Glucose-supplemented ECAR Stress test assay medium was added to each well and the plate was incubated at 37°C without CO2 for 45 minutes. CytoTox-ONE Reagent was prepared and protected from light according to the manufacturer’s instruction using the components in the kit. The 96-well plate and the CytoTox-ONE Reagent were equilibrated to room temperature for 20 minutes. Next, 10 μl of 1.8% Triton X-100 (weight/volume) in water was added to four of the eight replicate wells for each cell density while 10 μl of vehicle was added to the remaining four wells for each density. The plate was mixed on a microplate shaker. Cell lysis due to Triton X-100 exposure and lack of cell lysis due to vehicle treatment was confirmed by visual inspection of wells using a Leica DMi1 inverted microscope. One volume (110 μl) of CytoTox-ONE Reagent was added to each well, the plate was mixed for 30 seconds, and then incubated in the dark for 10 minutes. To each well 55 μl of Stop solution was added, the plate was mixed for 10 seconds, and fluorescence was immediately recorded using 560nm excitation and 590nm emission on a Synergy H1 microplate reader (BioTek).
Following exposure to Triton X-100, linear increases in RFUs were detected by plotting RFUs versus differentiated- or proliferative-derived HepaRG at the aforementioned seeding densities. Utilizing linear regression analysis, R-squared values of 0.99 were consistently obtained between experiments for both cell types; quadruplicates of each cell density for two independent experiments run on different days using different preparations of cells. For Triton X-100 treated samples, proliferative cells seeded at 4.4 × 104 cells/cm2 produced the lowest RFUs, which were a least 3-fold above background fluorescence. Both cell types treated with vehicle produced even lower RFUs, which were at most 2-fold above background fluorescence when seeded at the highest densities for each cell type described above.
The experiment was performed twice on different days using different preparations of both proliferative- and differentiated-derived cells. Following subtraction of background fluorescence (negative control wells), RFUs were normalized to total cellular protein in the well; thus, the values presented for total LDH activity obtained from lysates (Figure 4(b)) were normalized to total cellular protein and are the mean ± SD of n = 24 replicates for each cell type (3 cell densities × 4 replicate wells × 2 independent experiments). The inter-experimental coefficients of variation were 20% and 30% for proliferative- and differentiated-derived HepaRG respectively.
Quantitation of cell culture ATP levels
Relative levels of cellular adenosine triphosphate (ATP) were estimated using the CellTiter-Glo® 2.0 Assay (Promega) as per the manufacturer’s recommendations. The day before the experiment, proliferative- and differentiated-derived HepaRG were seeded into Corning 96-well flat clear-bottom white-walled polystyrene microplates for protein normalization and for quantitation of ATP levels. Differentiated-derived cells were seeded at 8.9 × 104, 1.3 x105, and 1.9 × 105 viable cells/cm2 and proliferative-derived HepaRG were seeded at 4.4 × 104, 6.3 × 104, and 9.4 × 104 viable cells/cm.2 Each cell density was replicated in 8 wells. Growth media without cells was loaded into empty wells to serve as negative controls to determine background RLUs.
The day after seeding the 96-well plates, growth media was removed from all wells, including the negative controls. Wells were gently washed with 100 μl Mito Stress test assay medium then 100 μl Mito Stress test assay medium was added to each well. The plate was incubated at 37°C without CO2 for 45 minutes and thereafter the plate was equilibrated to room temperature for 20 minutes. Next, 100 μl of CellTiter-Glo 2.0 reagent was added to each well, the plate was mixed for 4 minutes, and then incubated at room temperature for 8 minutes. During this incubation time, cell lysis was quickly confirmed by visual inspection of wells under the microscope. Luminescence was measured on the Synergy H1 microplate reader (BioTek). Linear increases in luminescence were detected by plotting RLUs versus differentiated- or proliferative-derived HepaRG at the aforementioned seeding densities. Utilizing linear regression analysis, R-squared values of 0.99 were consistently obtained between experiments for both cell types; 8 replicates of each cell density for two independent experiments run on different days using different preparations of cells. Proliferative cells seeded at 4.4 × 104 cells/cm2 produced the lowest RLUs, which were a least 40-fold above background luminescence.
The values presented for cellular ATP in Figure 4(c) are normalized to total cellular protein in the well and are mean RLU/μg values ± SD of n = 48 replicates (3 cell densities × 8 replicate wells × 2 experiments). The RLUs/μg cellular protein inter-experimental coefficients of variation for both proliferative- and differentiated-derived HepaRG were at most 17%.
Acetaminophen (APAP) and metformin treatment of HepaRG
APAP (Alfa Aesar) was dissolved in William’s E Medium, 2 mM GlutaMAX and filter sterilized with a 0.22 μm filter into a sterile bottle then Growth Medium Supplement was added as described above in Cell Culture to give a final concentration of 20 mM APAP in WGM as previously described [17]. To expose proliferative HepaRG cultures to drugs, tissue culture dishes were separately treated with WGM containing 20 mM APAP, 1 mM metformin (EMD Millipore), 20 mM APAP + 1 mM metformin, or vehicle control 7 days post-seeding. Briefly, growth medium was removed from tissue culture dishes then prewarmed WGM + APAP was initially added to half of the dishes while WGM vehicle was added to the other half. All samples were incubated at 37°C, 5% CO2, time point 0 hours. After a 6 hour incubation period, half of the WGM + APAP and half of the WGM vehicle treated dishes were removed from the incubator and media was replaced with prewarmed WGM + APAP + metformin and WGM + metformin respectively. The rationale for adding metformin 6 hours later has been described [18]. The dishes were returned to the 37°C, 5% CO2 incubator and incubated overnight. Twenty-four hours after the initial APAP treatment (time point 0 hours), media was aspirated from all of the culture dishes, the dishes were washed with DPBS, trypsinized, and neutralized with pre-warmed WGM and then the numbers of viable cells were determined via the trypan blue exclusion method as described above. Viable cell counts were used to seed XFp miniplates. Miniplate wells were seeded with 9.4 × 104 or 1.9 × 105 proliferative-derived cells/cm,2 Mito Stress tests were run, and respiration parameters (OCR values) were normalized to total cellular protein as described above.
Confluent HepaRG were differentiated in tissue culture dishes as described above in Cell Culture. Differentiated cultures were separately treated with vehicle and drugs (APAP, metformin, and APAP + metformin) in WGM as DMSO has been demonstrated to prevent APAP-induced hepatotoxicity [12,29]. Drug treatments, harvesting, and counting of viable differentiated-derived cells were conducted as described above for proliferative-derived cells. Miniplate wells were seeded with 1.9 × 105 differentiated-derived cells/cm,2 Mito Stress tests were run, and respiration parameters (OCR values) were normalized to total cellular protein as described above.
The drug exposure experiments were performed at least twice on different days using different preparations of cells. For each experiment at least two SH miniplate were run for each treatment. Following normalization of bioenergetic OCR parameter values to μg cellular protein (pmol O2/min/μg cellular protein), vehicle controls were set to 100% and data are presented relative to vehicle control (%OCR).
Statistical analyses
All data presented are mean values ± standard deviations (SDs). Statistical significance between two parametric groups was determined using a Student’s or a Welch’s t-test while significance between two non-parametric groups was determined using a Mann-Whitney U test. Comparisons of more than two groups of parametric data were assessed by one-way analysis of variance (ANOVA) followed by Tukey’s test or Welch’s ANOVA with Games-Howell post hoc. Comparisons of greater than two groups of non-parametric data were assessed by Kruskal-Wallis tests followed by Dunn’s post hoc test. P-values less than 0.05 were considered significant.
Results
Optimization of HepaRG cell seeding density for extracellular flux analysis
To better understand the usefulness of HepaRG to study alterations in bioenergetics we cultivated HepaRG cells in proliferative and differentiated cultures then harvested cells for bioenergetic analyses. To differentiate HepaRG, tissue culture vessels were initially seeded with cells in the proliferative phase of growth. Proliferative HepaRG initially appeared homogenously epithelial-like in morphology and were maintained for two weeks in growth medium followed by additional maintenance in medium supplemented with 2% DMSO to induce differentiation [13,24], Figure 1(a). Following the differentiation process HepaRG cells morphologically resemble two distinct populations of hepatocyte-like and biliary- (epithelial-) like cells [12,15,24], Figure 1(b). HepaRG cells were observed daily using a phase contrast microscope and recognizable hepatocyte-like cells, epithelial-like cells, and bile canaliculus-like structures were noted as previously described [13]. Cells derived from both proliferative and differentiated HepaRG cultures were seeded onto specialized 8-well cell culture miniplates (miniplates) for XFp Extracellular Flux analysis. Oxygen (O2) consumption rates (OCRs) and extracellular acidification rates (ECARs) were initially monitored by testing various cell densities. Proliferative cells were seeded at 47% (4.7 × 103 cells), 67% (6.7 × 103 cells), and 100% (1 × 104 cells per well) and differentiated cells were seeded at 47% (9.4 × 103 cells), 67%, (1.34 × 104 cells), and 100% (2 × 104 cells per well). Linear correlations between OCRs and ECARs versus cell density were observed for both cell types (Figure 1). Following these tests, miniplate wells were washed with DPBS, examined under a phase contrast microscope to ensure cells were not washed away, and total cellular protein per well was determined by bicinchoninic acid (BCA) assay. In agreement with the number of cells seeded per well as described above, the protein concentration measured in wells seeded with proliferative cells was 51% ± 4%, 61% ± 2%, and 93% ± 10% and for differentiated cells was 39% ± 9%, 62% ± 1%, and 94% ± 8%, mean values with errors as standard deviations (SDs) are reported (n = 4, duplicate densities from two miniplates). These tests confirmed that HepaRG cells remain adherent to miniplates during measurement of basal extracellular OCRs and ECARs.
Figure 1.
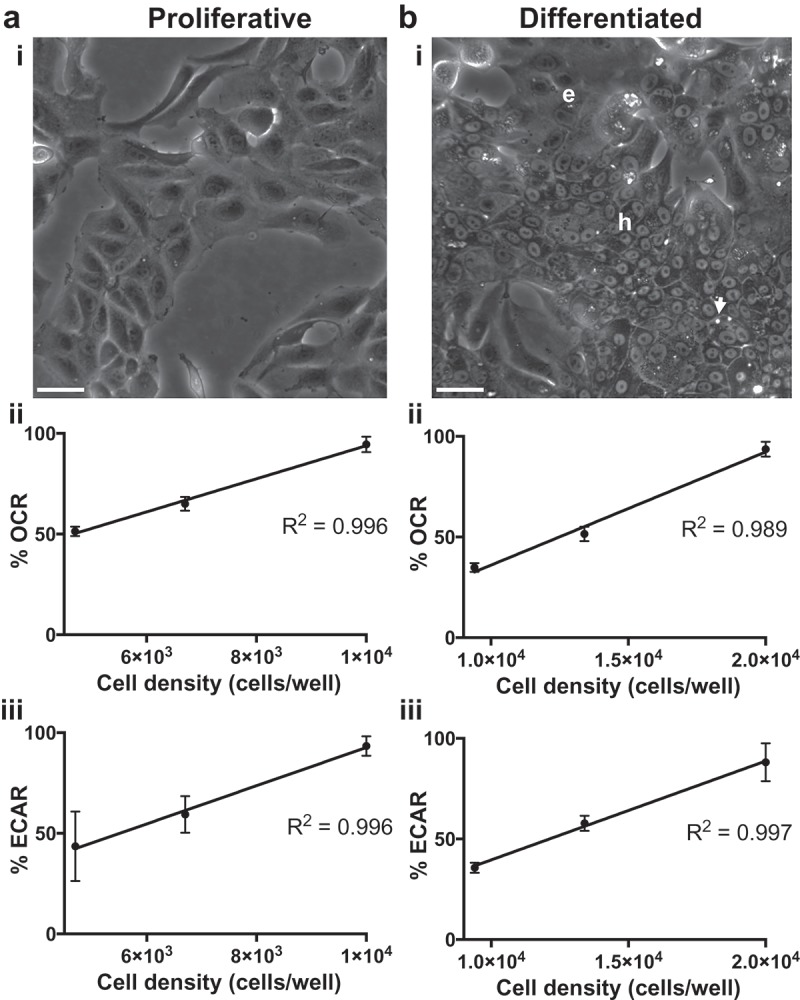
HepaRG morphology and optimization of cell seeding density for XFp analysis. (a) i. Proliferative & (b) i. Differentiated HepaRG cell cultures. Hepatocyte-like and epithelial-like cells are indicated by “h” and “e” respectively. The arrow emphasizes a bile canaliculus-like structure. Scale bars, 40 μm. Various densities of viable cells per well were seeded into miniplate wells to determine basal oxygen consumption rates (OCRs) for (a) ii. Proliferative-derived and (b) ii. Differentiated-derived cells and basal extracellular acidification rates (ECARs) for (a) iii. Proliferative-derived and (b) iii. Differentiated-derived cells. Data are presented as mean ± SD, n = 4 (duplicates from two miniplates). R-squared values were ~0.99 as determined by linear regression analysis.
HepaRG from proliferative and differentiated cultures are sensitive to OXPHOS stressors
To determine mitochondrial bioenergetic profiles, cells are exposed to known pharmacological stressors of the OXPHOS machinery. Mitochondrial OXPHOS is the O2-dependent process of coupling substrate oxidation to the production of the energy-rich molecule adenosine triphosphate (ATP). During OXPHOS molecular O2 is reduced to water (H2O). The XFp O2 biosensor measures the real-time rate at which cells convert O2 to H2O, the O2 consumption rate (OCR). Empirical determination of the optimal concentrations for two stressors, oligomycin and FCCP, is important in order to achieve the greatest effect on OCR [30–32]. Excess FCCP may cause an abrupt reduction in OCRs [25]; therefore, we investigated the optimal concentrations of these stressors utilizing HepaRG obtained from proliferative and differentiated cultures (Figure 2). Oligomycin is an inhibitor of the mitochondrial ATP synthase (complex V) and is used to estimate the proportion of basal respiration that is used to drive ATP synthesis [25], Figure 2(a). Upon treatment with oligomycin, a backup of protons builds in the intermembrane space causing a decrease in electron flow through the OXPHOS machinery and subsequent decrease in O2 consumption [31]. By injecting 1, 2, or 4 μM oligomycin into miniplate wells seeded with proliferative or differentiated HepaRG cells, we tested extracellular OCR dose responses. For both cell types measurable decreases in OCRs were observed at concentrations of 2 μM oligomycin and higher; therefore, both cell types are sensitive to the complex V inhibitor and 2 μM oligomycin was used for subsequent experiments (Figure 2(b)).
Figure 2.
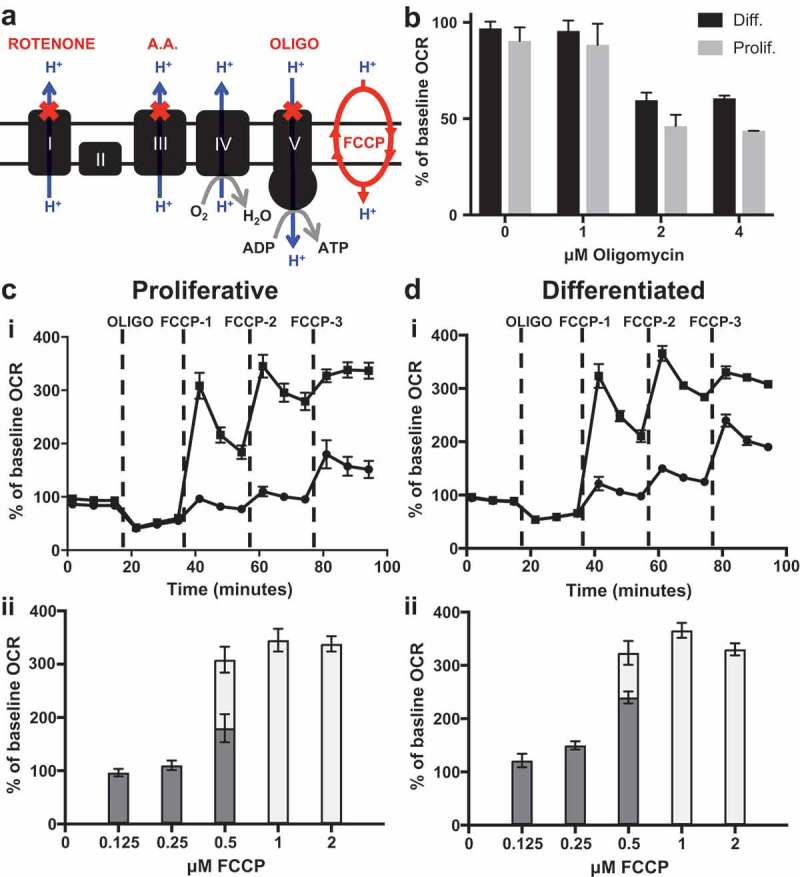
HepaRG cells are sensitive to OXPHOS metabolic stressors. (a) Cartoon of the mitochondrial inner membrane OXPHOS machinery targeted by key stressors of the Agilent Seahorse XFp Cell Mito Stress Test. (b) Optimization of the oligomycin concentration required to inhibit cellular OCRs. Data are presented as mean ± SD, n = 4 (duplicates from two miniplates). (c) & (d) FCCP dose-response tests to stimulate oxygen consumption are shown for (c) proliferative-derived and (d) differentiated-derived HepaRG. The three sequential injections of FCCP are represented by FCCP-1, −2, and −3 (c) & (d) i. OCR responses to low range FCCP concentrations are represented by black circles (i) and dark grey bars (ii) while responses to high range FCCP concentrations are represented by black squares (i) and light grey bars (ii). Mean % of baseline OCR values measured immediately post FCCP injections are graphed in (c) & (d) ii to emphasize stimulation of oxygen consumption. Data in (c) & (d) are presented as mean ± SD, n = 6 (triplicate from two miniplates). For several data points, error bars are not shown as the error bars are shorter than the height of the symbol. A.A., antimycin A; OLIGO, oligomycin.
Taking advantage of the four-injection ports per well located on the XFp sensor cartridge, 5-point FCCP titration curves were conducted (Figure 2(c,d)). FCCP is a protonophore that can shuttle protons across the inner mitochondrial membrane and dissipate the proton-motive force (PMF), Figure 2(a). Following oligomycin injection, FCCP was added allowing protons that have been built up in the intermembrane space to dissipate back into the matrix and allowing electrons to resume their flow through the electron transport chain (ETC) to O2. Because mitochondrial respiration is coupled to ATP synthesis the process of moving protons back into the matrix independent of ATP synthesis is referred to as uncoupling and FCCP is typically referred to as an uncoupler [25]. Sequential injection of 2 μM oligomycin followed by 0.125, 0.25, and 0.5 μM FCCP final well concentrations (low range analysis) or 0.5, 1.0 and 2.0 μM FCCP final well concentrations (high range analysis) were performed and the effect of the stressors on OCRs were determined (Figure 2(c,d)). Proliferative- and differentiated-derived HepaRG cells had peak OCRs when treated with 1 μM FCCP, thus both cell types are sensitive to the protonophore and 1 μM was chosen for further study. These results emphasize the importance of optimizing FCCP concentration to maximally activate OCRs following oligomycin injection, which is necessary to determine respiratory parameters (see below).
Cells derived from proliferative and differentiated cultures have distinct bioenergetics
Seahorse XFp Mito Stress tests and ECAR Stress tests were carried out to determine bioenergetic parameters associated with OCRs and ECARs respectively. OCRs and ECARs are extracellular indicators of the chief energy-producing conduits in the cell, OXPHOS and glycolysis. To determine bioenergetic profiles, proliferative- and differentiated-derived HepaRG were analyzed side-by-side on XFp miniplates and exposed to stressors of the OXPHOS machinery. The compilation of mitochondrial parameters from Mito Stress tests are summarized in Table 1 and those from ECAR Stress tests are summarized in Table 2.
Table 2.
Proliferative- and differentiated-derived HepaRG ECAR parameters.
| Bioenergetic parameter | Proliferative ECARsa (mpH/min/μg cellular protein) |
Differentiated ECARsa (mpH/min/μg cellular protein) |
Fold-Change | P-valueb (Fold-change) |
|---|---|---|---|---|
| Glucose-stimulated ECAR | 0.84 ± 0.34 | 1.14 ± 0.56 | 0.7 | 0.3 |
| ECAR capacity | 7.32 ± 1.24 | 3.94 ± 0.78 | 1.9 | < 0.0001 |
| Apparent glycolytic reserve | 6.48 ± 1.4 | 2.80 ± 0.70 | 2.3 | < 0.001 |
| Basal ECAR | 3.97 ± 0.93 | 2.85 ± 0.93 | 1.4 | 0.08 |
aData are presented as mean ± SD, n ≥ 5 (duplicate or triplicate from two independent experiments performed on different days with different preparations of cells).
bP-values < 0.05 were accepted as significantly different.
HepaRG from proliferative cultures have increased basal and ATP-linked respiration and decreased maximal and spare respiratory capacities
Mito Stress tests were conducted by sequentially injecting oligomycin, FCCP, and rotenone plus antimycin A. Rotenone and antimycin A are inhibitors of OXPHOS complexes I and III respectively and were the last stressors added during the experiment to terminate electron flow through the ETC and to enable calculation of oxygen consumption from non-mitochondrial oxidases, Figures 2(a) and 3(a i) [25,33]. Both proliferative- and differentiated-derived HepaRG cells were sensitive to rotenone and antimycin A as indicated by the acute inhibition of cellular OCRs following their injection (Figure 3(a)). The non-mitochondrial respiration parameter is representative of the OCR remaining following rotenone plus antimycin A treatment. Non-mitochondrial respiration was not significantly different between the two cell types (Table 1). For each cell type, the basal respiration parameter is the difference between the OCR measured just prior to oligomycin injection and the non-mitochondrial respiration parameter. Basal respiration represents the sum of the respiration used to power ATP production and respiration associated with proton leakage across the inner membrane. Basal respiration was slightly higher in cells derived from proliferative cultures in comparison to those derived from differentiated cultures (Table 1). Following injection of oligomycin to inhibit complex V, ATP production slows and respiration utilized to power ATP production decreases. The amount of basal OCR sensitive to oligomycin represents an estimation of ATP-linked respiration [25]. A 1.4-fold increase in ATP-linked respiration was observed in HepaRG cells derived from proliferative cultures. The estimation of the fraction of basal respiration used to drive ATP synthesis [25,28] also known as coupling efficiency was slightly increased 1.2-fold in proliferative-derived HepaRG. After subtracting the non-mitochondrial respiration rate, FCCP-stimulation of OCR provides an estimate of the maximal respiratory capacity of cells and by extension the maximal capacity to oxidize substrates via the OXPHOS machinery. Next, the difference between maximal respiratory capacity and basal respiration was used to calculate spare respiratory capacity. Spare respiratory capacity has been defined as the extra mitochondrial capacity available to produce ATP during stress or increased work. Spare respiratory capacity has been suggested to be important for cellular function and long-term survival [34]. In proliferative-derived HepaRG, both maximal and spare respiratory capacities were decreased ~20% relative to differentiated-derived cells. Also, an ~ 30% relative decrease in proliferative- to differentiated-derived HepaRG spare respiratory capacity as a percent was observed and this decrease is similar to that observed for spare respiratory capacity (Table 1). During substrate oxidation, a portion of the PMF is not utilized to drive ATP synthesis via complex V and some protons “leak” back into the matrix via inducible uncoupling proteins and through the presence (not the activity) of the inner membrane adenine nucleotide translocase [35]. With regards to proton leak-linked respiration, or the remaining mitochondrial respiration in the presence of oligomycin, no significant difference between the two cell types was detected (Table 1).
Figure 3.
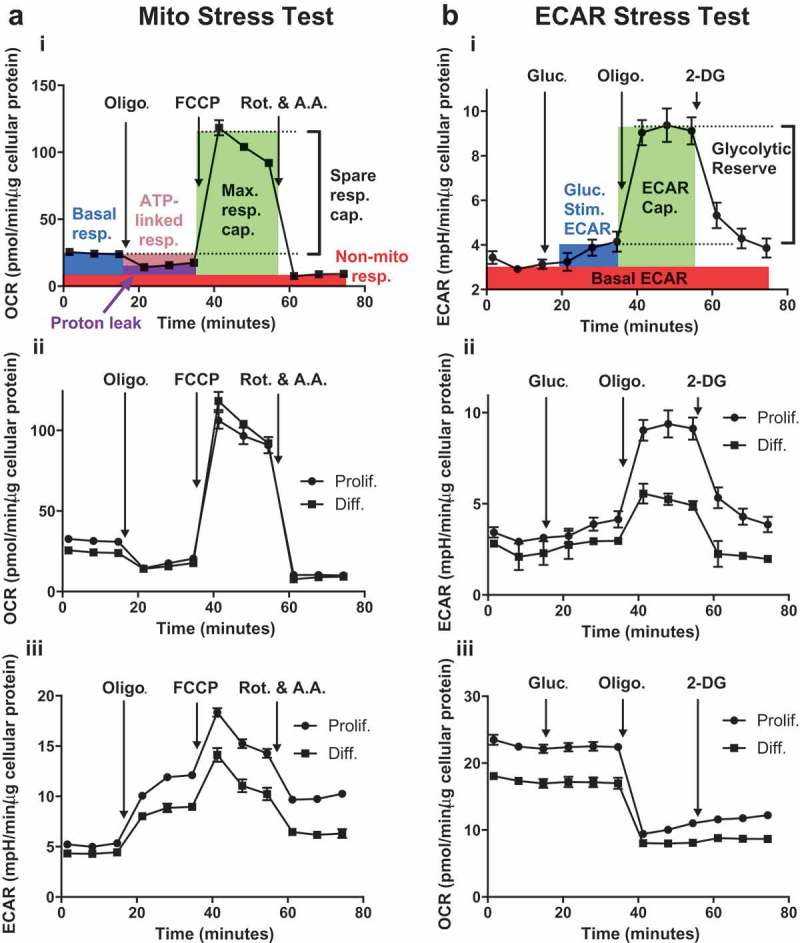
HepaRG cells derived from proliferative and differentiated cultures have distinctive bioenergetic parameters. (a) Results from a representative Mito Stress test comparing proliferative- (Prolif.) and differentiated- (Diff.) derived HepaRG side-by-side on a cell culture miniplate. i. Description of mitochondrial bioenergetic parameters: basal respiration (Basal resp.), ATP-linked respiration (ATP-linked resp.), maximal respiratory capacity (Max. resp. cap.), spare respiratory capacity (Spare resp. cap.), proton leak, and non-mitochondrial respiration (Non-mito resp.). Metabolic stressors are injected sequentially from Ports A (2 μM oligomycin final well concentration), B (1 μM FCCP final well concentration), and C (0.5 μM antimycin A + 0.5 μM rotenone, final well concentrations). ii. OCR and iii. ECAR results for both cell types. (b) Results from a representative ECAR Stress test comparing Prolif. and Diff.-derived HepaRG side-by-side on a cell culture miniplate. i. Description of ECAR bioenergetic parameters: basal ECAR, Glucose-stimulated ECAR (Gluc.-Stim.), ECAR capacity (ECAR Cap.), and apparent glycolytic reserve (Glycolytic Reserve). First, final well concentrations of 10 mM glucose were injected from A ports, followed by 2 μM oligomycin (B ports), and ~51 mM 2-DG (C ports). ii. ECAR and iii. OCR results for both cell types. Data are presented as mean ± SD, n = 3. For several data points errors are not shown as the error bars are shorter than the height of the symbol. Oligo., oligomycin; Rot., rotenone; A.A., antimycin A; Gluc., glucose; 2-DG, 2-deoxyglucose.
As discussed earlier, the cell seeding density experiments demonstrated that the relative amount of total cellular protein measured in the wells was as would be expected based on the number of cells seeded. In agreement, when bioenergetic parameters are normalized to cells seeded, the relative changes between the two cell types remain similar to those observed when data is normalized to μg cellular protein per well (compare fold-changes in Supplementary Table 1 and Table 1). Furthermore, we can assess mitochondrial function utilizing internally normalized parameters that are calculated independently of cell number or total protein. Two of these parameters were discussed above, coupling efficiency and spare respiratory capacity as a percent. Additionally, the ratio of ATP-linked respiration to maximal respiration can be used to estimate OXPHOS’ ability to generate ATP relative to its maximum capacity. This parameter was increased nearly 2-fold in proliferative derived HepaRG (Table 1 and Supplementary Table 1).
HepaRG TCA cycle activity likely contributes to ECAR and 2-deoxyglucose inhibits extracellular acidification
A second XFp biosensor measures the extracellular acidification rate (ECAR) resulting from the cytoplasmic breakdown of glucose-derived pyruvate to lactate and the respiratory evolution of carbon dioxide (CO2). Glycolysis is the major cytosolic O2-independent metabolic pathway that converts glucose into two molecules of each of the following: pyruvate, ATP, and NADH. When pyruvate is shunted through the mitochondria to the pyruvate dehydrogenase complex (PDC) and subsequently through the tricarboxylic acid cycle (TCA), CO2 is generated. In solution, a molecule of CO2 can combine with a molecule of H2O forming carbonic acid that dissociates at physiological pH into the bicarbonate anion and a proton that contributes to medium acidification [25]. ECAR Stress tests were conducted by sequentially injecting glucose, oligomycin, and 2-deoxyglucose (2-DG) into a glucose-free medium bathing either differentiated- or proliferative-derived HepaRG cells, Figure 3. Following sugar injection, oligomycin inhibits the OXPHOS machinery and allows estimation of ECAR parameters. As a competitive inhibitor of hexokinase, 2-DG inhibits the cell’s ability to utilize free glucose to generate pyruvate via glycolysis and ECAR drastically decreases [31]. When analyzed side-by-side, proliferative- and differentiated-derived HepaRG cells were both sensitive to the hexokinase inhibitor as indicated by a reduction of ECARs following the last injections of 2-DG (Figure 3(b ii)). Parameters such as basal ECAR, glucose-stimulated ECAR, ECAR capacity, and apparent glycolytic reserve were determined (Table 2). Basal ECAR is the last of three ECARs measured immediately before sugar injection (Figure 3(b)). Glucose-stimulated ECAR is the maximal rate following glucose injection (but before oligomycin injection) minus basal ECAR. Glucose-stimulated ECAR represents the total extracellular acidification from cellular pathways metabolizing free glucose including the production of cytoplasmic lactate and mitochondrial protons produced via CO2 hydration and dissociation.
In support of the presence of mitochondrial proton production in HepaRG cells, Mito Stress tests revealed decreases in ECAR following injection of the OXPHOS inhibitors rotenone and antimycin A. When ECARs are measured following rotenone and antimycin A injection (the twelfth time point at ~74 minutes) as compared to the rates obtained following oligomycin injection (the sixth time point at ~35 minutes), a difference of 10.2 ± 0.3 vs. 12.3 ± 0.4 mpH/minute/μg cellular protein respectively for proliferative- and 6.4 ± 0.5 vs. 9.5 ± 0.7 mpH/minute/μg cellular protein respectively for differentiated-derived cells were observed (n = 6, mean values ± SD; triplicate determinations from two independent experiments, P-values < 0.0001). Figure 3(a iii) highlights ECAR data from a representative Mito Stress test. The further decrease in ECAR following blockage of complex I and III (relative to the increase caused by the upstream blockage of complex V) suggests that TCA activity is contributing modestly to ECAR in both cell types. However, as ECAR rates remain higher than initial basal rates for the two cell types following blockage of the ETC by rotenone and antimycin A, we hypothesize mitochondrial CO2 evolution is not the only contributing factor to ECAR (compare the twelfth time point at ~74 minutes to the third at ~15 minutes for both proliferative- and differentiated-derived HepaRG, Figure 3(a iii)). Therefore, glycolysis is likely active in both cell types. In contrast, if ECAR remained constant or increased after exposure to rotenone and antimycin A then this would suggest medium acidification results primarily from glycolysis as previously described [25].
HepaRG from proliferative cultures have increased ECAR capacity and apparent glycolytic reserve
ECAR capacity is a measurement of the extracellular acidification including glucose-stimulated ECAR and oligomycin-stimulated ECAR production, Figure 3(b). The apparent glycolytic reserve parameter is obtained by subtracting glucose-stimulated ECAR from ECAR capacity. Apparent glycolytic reserve is the estimated amount of unused glycolytic capability of the cell that could be utilized if cellular ATP demand was increased. Cells derived from proliferative and differentiated cultures did not have significant differences between their basal ECAR and glucose-stimulated ECAR parameters (Table 2). In contrast, when cells derived from both cultures are stressed with oligomycin, ECAR capacity and apparent glycolytic reserve both increased ~2-fold in proliferative- relative to differentiated-derived HepaRG. As part of the ECAR likely arises from mitochondrial protons as discussed above, the apparent glycolytic reserve parameter is an approximation of HepaRG glycolysis.
Basal ATP levels but not lactate or LDH activity are increased in proliferative-derived compared to differentiated-derived cell cultures
To better understand the metabolic differences observed between proliferative- and differentiated-derived HepaRG via XFp analyses, we conducted follow up experiments to quantitate cell culture lactate, ATP, and lactate dehydrogenase (LDH) activity. Basal levels of proliferative- and differentiated-derived HepaRG cell culture lactate were determined utilizing the Lactate-GlowTM Assay (Promega), Figure 4(a). The lactate detection reagent consists of LDH, NAD+, reductase, pro-luciferin reductase substrate, and luciferase. This assay utilizes an L-lactate selective LDH to oxidize lactate in biological samples to pyruvate and to generate NADH. In the presence of NADH, the reductase substrate is converted by reductase to luciferin, which in the presence of luciferase produces light. The luminescent signal that is produced is proportional to the amount of lactate in a sample. Luminescence was measured using a microplate reader and the concentration of lactate was estimated in each sample well of 96-well plates containing both cell types. Both proliferative- and differentiated-derived HepaRG produced ~0.3 μM lactate/μg cellular protein, Figure 4(a).
To determine proliferative- and differentiated-derived HepaRG LDH activity levels, we separately lysed both cell types in Triton X-100 then measured LDH activity in the whole-cell lysates using the CytoTox-ONETM Homogeneous Membrane Integrity Assay [36]. The CytoTox-ONETM assay measures LDH with a coupled enzymatic reaction that results in the conversion of resazurin into the fluorescent resorufin, which is proportional to the amount of LDH. The fluorescent signals were then measured using a microplate reader. Cell lysates from both cell types harbored indistinguishable LDH activities, ~40,000 RFUs/μg cellular protein.
Next, we examined proliferative- and differentiated-derived HepaRG cellular ATP levels utilizing the CellTiter-Glo® 2.0 Assay (Promega). The luciferase reaction for this assay generates a luminescent signal proportional to the amount of ATP present in the sample [37]. A significant 1.6-fold increase in proliferative-derived cellular ATP levels relative to differentiated-derived HepaRG levels was detected (Figure 4(c)).
Metformin prevents APAP-induced loss of cellular viability in differentiated HepaRG cultures
Both APAP and metformin have been reported to impair mitochondrial functions [17,18,21]. In mouse hepatocytes, it is generally accepted that APAP is hepatotoxic causing necrosis, and APAP overdose causing necrotic cell death has been proposed for differentiated HepaRG [17]. Metformin has traditionally been used to treat diabetes but more recently metformin has been associated with improved survival rates in cancer [21]. To gain an understanding of drug effects on proliferative versus differentiated HepaRG hepatocarcinoma-derived cells we explored the effects of APAP, metformin, or both drugs on cellular viability and mitochondrial bioenergetics. Both types of cultures were separately exposed to 20 mM APAP, 1 mM metformin, or 20 mM APAP + 1 mM metformin. As formation of APAP-protein adducts peak at 6 hours post-treatment, and to avoid potential metabolic activation of APAP, metformin was added 6 hours after APAP [17,18]. Proliferative HepaRG cultures displayed extreme sensitivity to APAP as indicated by ~50% decreases in cellular viability on treatment with APAP or APAP + metformin (Figure 5(a)). Relative to vehicle control treated cultures differentiated HepaRG treated with APAP had a 21% reduction in cellular viability (Figure 5(b)). Exposure to metformin only did not have an effect on proliferative or differentiated cellular viability; however, the addition of metformin 6 hours post-APAP to differentiated HepaRG prevented APAP-induced loss of cellular viability (Figure 5(b)).
Figure 5.

Metformin treatment of differentiated HepaRG blocks APAP-induced loss of cellular viability. Cells were treated with 20 mM APAP and 1 mM metformin (MET) was added 6 hours later. Cells were harvested 24 hours after the addition of APAP and cellular viability was determined by the trypan blue exclusion method. (a) Proliferative HepaRG cells are sensitive to APAP-induced loss of cellular viability in the absence or presence of MET (****, P < 0.0001 compared with control; $$$$, P < 0.0001 compared with MET). (b) Differentiated HepaRG cells are sensitive to APAP-induced loss of cellular viability. In the presence of MET the APAP-induced loss of viability is prevented (*, P < 0.05 compared with control; $, P < 0.05 compared with MET; #, P < 0.05 compared with APAP + MET). In (a) and (b) the number of viable cells treated with vehicle (control) was set to 100%. Data are presented as mean ± SD, n ≥ 8 (≥quadruplicate from two independent experiments performed on different days with different preparations of cells) relative to control.
Distinct mitochondrial bioenergetic alterations in proliferative- and differentiated-derived HepaRG are associated with exposure to APAP
As incubation with APAP induced loss of cellular viability in both types of culture, and metformin offered protection against the loss of cellular viability in differentiated cultures, we examined the effects of APAP or APAP + metformin treatment on mitochondrial bioenergetics using the Seahorse XFp. Compared to vehicle control, proliferative-derived HepaRG separately treated with APAP and APAP + metformin had 1.7- and 1.6-fold increases in proton leak-linked respiration respectively, Figure 6(a). Additionally, both treatments resulted in 1.2-fold increases in the ratio of ATP-linked respiration to maximal respiration respectively and slight decreases in percent spare respiratory capacity (~30% less for APAP and APAP + metformin) and coupling efficiency (~17% less for APAP and 14% less for APAP + metformin), Figure 6(a). Differentiated-derived cells exposed to APAP had an ~13% decrease in basal respiration and an ~15% decrease in ATP-linked respiration, Figure 6(b). When metformin was added 6 hours post-APAP treatment decreases in differentiated-derived HepaRG basal respiration and ATP-linked respiration were prevented, Figure 6(b).
Figure 6.
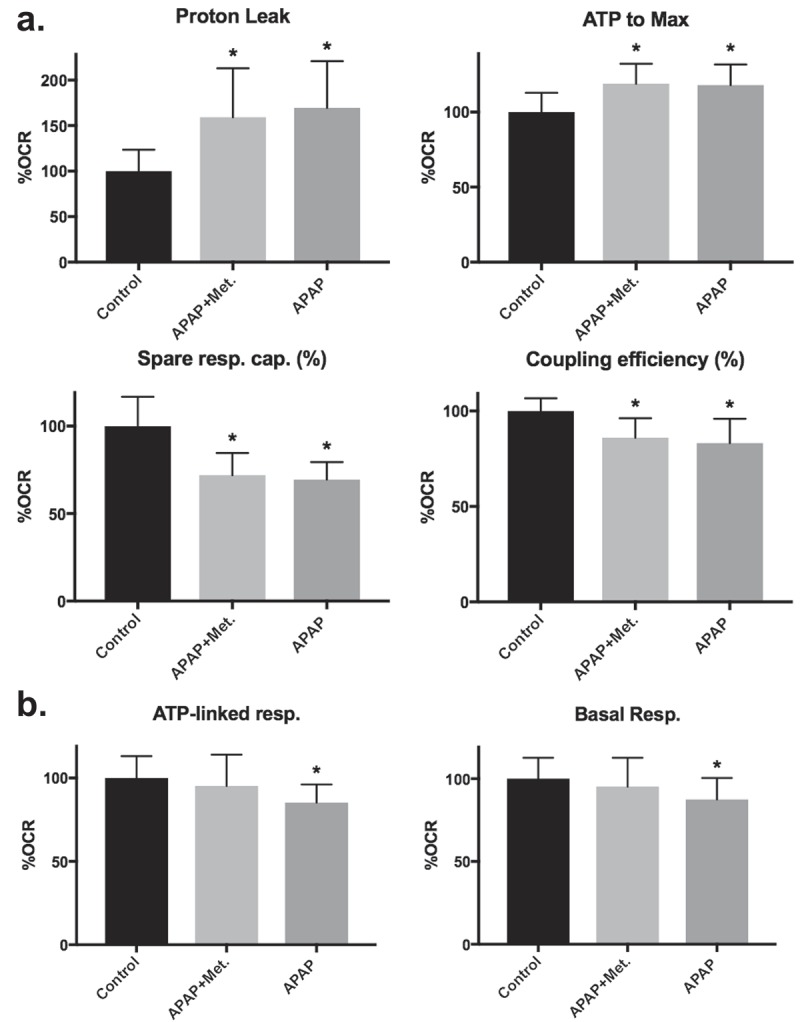
Metformin treatment prevents APAP-induced alterations in differentiated-derived HepaRG mitochondrial bioenergetics but not in proliferative-derived cultures. (a) Proliferative-derived HepaRG were separately treated with media containing vehicle control, 20 mM APAP + 1 mM metformin, and 20 mM APAP then seeded into miniplate wells for comparative analysis of bioenergetic parameters. For each experiment, two consecutive Mito Stress tests were run. Data are presented as mean ± SD, n ≥ 7 (triplicate or quadruplicate from two independent experiments using different preparations of cells) relative to vehicle control (set to 100% OCR). (b) Differentiated-derived cells were treated with drugs and seeded into miniplate wells as described in (a). For each experiment, at least two consecutive Mito Stress tests were run. Data are presented as mean ± SD, n ≥ 14 (≥triplicate from three independent experiments using different preparations of cells) relative to vehicle control (set to 100% OCR); *, P < 0.05 compared with control.
Metformin reduces ATP-linked respiration, maximal respiratory capacity, and basal respiration in proliferative-derived HepaRG
The effects of metformin on HepaRG mitochondrial bioenergetics were assessed by incubating cells with 1 mM metformin for 18 hours prior to seeding viable cells on SH miniplates. With the exception of coupling efficiency, significant differences on exposure to metformin were not detected for differentiated-derived HepaRG Mito Stress test parameters. In comparison to vehicle control treated cells, differentiated-derived HepaRG treated with 1 mM metformin had a slight decrease in coupling efficiency, 100 ± 4 to 94 ± 5% (n ≥ 9, ≥quadruplicate from two independent experiments with different preparations of cells; P < 0.05). In proliferative-derived HepaRG, maximal respiratory capacity, ATP-linked respiration, and basal respiration were all reduced ~30% compared to vehicle control treated cells (Figure 7).
Figure 7.
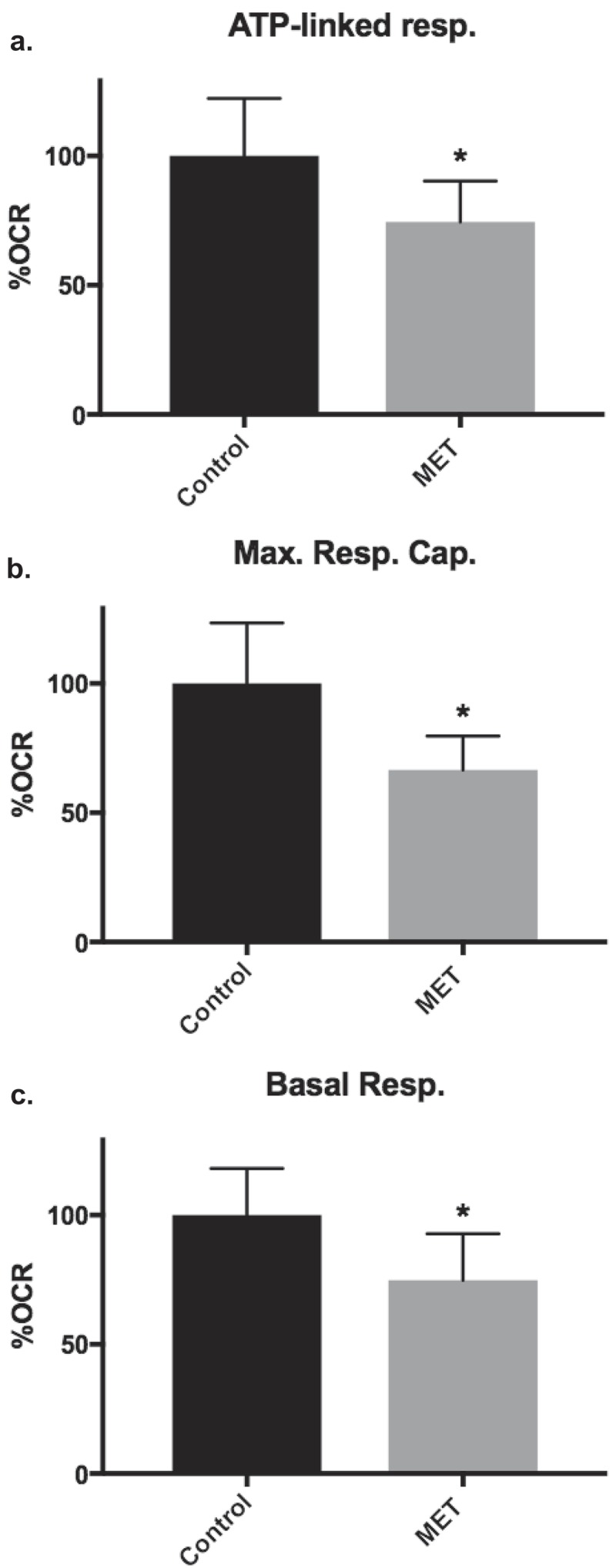
Metformin reduced proliferative-derived HepaRG mitochondrial bioenergetics. Proliferative-derived cells were separately treated with media containing vehicle control or 1 mM metformin then seeded into miniplate wells and Mito Stress tests were run. (a) ATP-linked respiration (ATP-linked resp.), (b) maximal respiratory capacity (Max. Resp. Cap.), and (c) basal respiration (Basal Resp.). Data are presented as mean ± SD, n = 7 (triplicate and quadruplicate from two independent experiments using different preparations of cells) relative to vehicle control (set to 100% OCR); *, P < 0.05 compared with control.
Discussion
Use of animal models has been controversial due to the lack of efficacy and unexpected toxicity in some model systems including rodents [38]. In one large study comparing human and animal drug toxicities, only 43% of rodent model cases correctly predicted human toxicity [39]. The hepatoma-derived HepaRG cell line represents a useful alternative to animal models to study the differentiation process, carcinogenesis, hepatocyte toxicity, and drug metabolism [12,15]. To better understand the practicality of utilizing HepaRG to analyze the pharmacological inhibition of metabolic energy-producing pathways, the XFp Extracellular Flux Analyzer was employed. Evidence suggests that carbon source choice in tissue culture media affects cellular physiology and replacing glucose with galactose has been reported to increase the susceptibility of cells to mitochondrial toxicants [40–42]. On the other hand, low glucose but not galactose has been reported to enhance oxidative mitochondrial metabolism [43]. To ensure the preservation of the HepaRG phenotype that was carefully selected for by Gripon et al., and has been used in many studies thereafter, we strictly adhered to the published culture conditions that were also reiterated by the vendor, Biopredic International [11–13,15]. Following supplement additions to Williams’ E Medium to make either WGM or WDM the final glucose concentrations were ~10 mM, which is similar to the human postprandial plasma level of ~9 mM [44].
Side-by-side comparisons of proliferative- and differentiated-derived HepaRG cells revealed that both cell types are sensitive to stressors of glycolysis (2-DG) and OXPHOS (oligomycin, FCCP, and rotenone plus antimycin A). Furthermore, distinct bioenergetic profiles were revealed by ECAR and mitochondrial OCR analyses. In comparison to differentiated-derived cells, proliferative-derived cells had increases in the proportions of basal respiration and ATP-linked parameters and slight decreases in maximal and spare respiratory capacities (Table 1). These results are in agreement with observations by Divakaruni et al., which suggest that other proliferative cells such as cortical astrocytes, C2C12 myoblasts, and A549 lung epithelial cells have lower spare capacities and proportionally higher ATP-linked respiration parameters in comparison to terminally differentiated cortical neurons, C2C12 myotubes, and white adipocytes respectively [25]. In agreement with the 1.4-fold increase in proliferative-derived HepaRG ATP-linked respiration (Table 1), we found 1.6-fold higher levels of ATP in proliferative-derived cell cultures in comparison to differentiated-derived cultures (Figure 4(c)). We hypothesize that an increase in proliferative cell ATP is likely due to higher biosynthetic demands required for cell division in comparison to differentiated cells. Differentiated cells exist in a quiescent stationary or contact inhibited phase [15,45]. Similar to other post-mitotic cells [25] differentiated-derived HepaRG had increased spare respiratory capacity suggesting differentiated cells maintain the ability to execute energy-demanding functions when the need arises.
Proliferative- and differentiated-derived cell culture lactate and LDH levels were comparable as were ECAR bioenergetic parameters: glucose-stimulated ECAR and basal ECAR (Table 2 and Figure 4). These results suggest that under basal conditions both cell types utilize glycolysis to a similar extent. However, recognizable differences were revealed by ECAR bioenergetic analysis of the two cell types. ECAR capacity and apparent glycolytic reserve were significantly increased in proliferative-derived HepaRG cells (Table 2). Comparing HepaRG following proliferative and differentiated culture conditions revealed distinct bioenergetic signatures with the ratio of ATP-linked respiration to maximal respiration and apparent glycolytic reserve having the largest differences among the sets of Mito Stress test and ECAR Stress test parameters. Our bioenergetic results support that proliferating HepaRG require a large amount of energy and in addition to increased ATP-linked respiration maintain an unused reserve of glycolytic capacity to quickly generate energy if required. Furthermore, glycolysis intermediates are necessary for biosynthesis of amino acids, nucleotides, and lipids all of which are required by actively cycling cells. Intracellular glycogen metabolism, which converts glycogen into pyruvate, does not require hexokinase; therefore, glycogenolysis may account for some of the observed basal ECAR and for extracellular acidification remaining following 2-DG injection (Figure 3(b ii)). As mentioned previously, as pyruvate is metabolized through PDC and the TCA cycle, molecules of CO2 are yielded that contributes to medium acidification as CO2 hydrates and dissociates into bicarbonate anions and protons. Likewise, the TCA cycle intermediate α-ketoglutarate derived from glutamine anaplerosis can be utilized as a source of energy and may contribute to basal ECAR as Seahorse assay media contains 2 mM glutamine [26]. Beta-oxidation of endogenous fatty acids to produce acetyl CoA, which feeds into the TCA cycle, could also contribute to basal ECAR levels.
After 24 hours of exposure to the hepatotoxic drug APAP, both proliferative- and differentiated-derived HepaRG had reduced viability (Figure 5). The 21% loss in differentiated HepaRG viability is in agreement with another study indicating that after 24 hours of exposure to 20 mM APAP, differentiated HepaRG had a 29% increase in LDH release, which is an indicator of cell death [17]. In addition to increased LDH release, differentiated HepaRG mitochondrial membrane potential was decreased [17]. In another study, after exposing differentiated HepaRG cells for 24 hours to 10 mM APAP an ~20% LDH release has also been reported [46]. Treatment of differentiated HepaRG cultures with metformin 6 hours after APAP exposure has been demonstrated to protected cells from APAP-induced cell injury [18]. Similarly, when we examined differentiated HepaRG we observed metformin prevented the loss of APAP-induced cell viability. Distinguishingly, proliferative HepaRG were extremely sensitive to APAP treatment even in the presence of metformin (Figure 5).
Distinct mitochondrial bioenergetic profiles were observed for the two cell phases upon exposure to APAP. Metformin did not prevent APAP-induced alterations in proliferative-derived cell bioenergetics (Figure 6). Proliferative-derived HepaRG mitochondria responded to APAP or to APAP + metformin treatment by increasing proton leak and the ratio of ATP-linked respiration to maximal respiration. In addition, proliferative-derived HepaRG responded to APAP or to APAP + metformin treatment by slightly decreasing percent spare respiratory capacity and coupling efficiency. Treatment of proliferative-derived cells with only 1 mM metformin reduced ATP-linked respiration, maximal respiratory capacity, and basal respiration (Figure 7); however, at this concentration, there was no effect on cellular viability (Figure 5). The decrease in proliferative-derived HepaRG basal respiration upon exposure to metformin is consistent with decreased basal OCRs observed with FTSECs and in HGSC cells treated with metformin [21]. Interestingly, in 3 out of 5 HGSC cell lines 10 mM metformin completely inhibited proliferation [21]. Upon exposure to APAP differentiated-derived HepaRG had reduced ATP-linked respiration and basal respiration and the addition of metformin prevented these decreases (Figure 6(b)). Similarly, another study found that metformin prevented a decrease in APAP-induced coupled respiration in differentiated HepaRG [18]. In contrast, we did not observe an effect of APAP on differentiated-derived HepaRG proton leak and postulate this could be due to differences in cell seeding densities between the two studies.
Evidence supports that mitochondria are vulnerable to attack by certain drug classes and it has been argued that these important organelles could be vulnerable to environmental toxicants [47]. Drug classes such as antibiotics, thiazolidinediones, antivirals, statins, fibrates, and anticancer agents have been demonstrated to cause mitochondrial toxicity [6,42]. Recently, the HepG2 hepatocellular carcinoma cell line was utilized to screen unique environmental and drug-like compounds and 913 compounds that disrupted mitochondrial membrane potential were identified [48]. Potent compounds that were discovered include triethyltin bromide, carbocyanine, basic blue 7, and bryostatin. By selecting drugs or compounds known to disrupt mitochondrial functions, we suggest HepaRG could provide an important system to study mechanisms of mitochondrial function and dysfunction in proliferating and differentiated culture phases. In conclusion, our results support that HepaRG represents an appropriate model system to study drug-induced bioenergetic dysfunctions.
Funding Statement
This research was supported by start-up funds from Southern Illinois University School of Medicine and from a National Institute of Environmental Health Sciences Pathway to Independence Award to MJY [5R00ES022638-04].
Acknowledgments
We thank Dr. Kristin Delfino for assistance with statistical analyses of data and Dr. Christiane Guguen-Guillouzo and Mr. Joel Wheeler for critically reading this manuscript. Also, we are grateful for the recommendations that were provided by the anonymous reviewers.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- [1].Zhang J, Nuebel E, Wisidagama DRR, et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat Protoc. 2012;7:1068–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dar S, Chhina J, Mert I, et al. Bioenergetic adaptations in chemoresistant ovarian cancer cells. Sci Rep. 2017;7:8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kalyanaraman B, Cheng G, Hardy M, et al. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018;14:316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marrache S, Pathak RK, Dhar S.. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc Natl Acad Sci U S A. 2014;111:10444–10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liyanage SU, Hurren R, Voisin V, et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood. 2017;129:2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Young MJ. Off-target effects of drugs that disrupt human mitochondrial DNA maintenance. Front Mol Biosci. 2017;4:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nadanaciva S, Rana P, Beeson GC, et al. Assessment of drug-induced mitochondrial dysfunction via altered cellular respiration and acidification measured in a 96-well platform. J Bioenerg Biomembr. 2012;44:421–437. [DOI] [PubMed] [Google Scholar]
- [8].Attene-Ramos MS, Huang R, Sakamuru S, et al. Systematic study of mitochondrial toxicity of environmental chemicals using quantitative high throughput screening. Chem Res Toxicol. 2013;26:1323–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Soldatow VY, Lecluyse EL, Griffith LG, et al. Models for liver toxicity testing. Toxicol Res (Camb). 2013;2:23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang R, Xia M, Sakamuru S, et al. Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat Commun. 2016;7:10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Anthérieu S, Chesné C, Li R, et al. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab Dispos. 2010;38:516–525. [DOI] [PubMed] [Google Scholar]
- [12].Aninat C, Piton A, Glaise D, et al. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab Dispos. 2006;34:75–83. [DOI] [PubMed] [Google Scholar]
- [13].Gripon P, Rumin S, Urban S, et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A. 2002;99:15655–15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hart SN, Li Y, Nakamoto K, et al. A comparison of whole genome gene expression profiles of HepaRG cells and HepG2 cells to primary human hepatocytes and human liver tissues. Drug Metab Dispos. 2010;38:988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marion MJ, Hantz O, Durantel D. The HepaRG cell line: biological properties and relevance as a tool for cell biology, drug metabolism, and virology studies. Methods Mol Biol. 2010;640:261–272. [DOI] [PubMed] [Google Scholar]
- [16].Peyta L, Jarnouen K, Pinault M, et al. Reduced cardiolipin content decreases respiratory chain capacities and increases ATP synthesis yield in the human HepaRG cells. Biochim Biophys Acta. 2016;1857:443–453. [DOI] [PubMed] [Google Scholar]
- [17].McGill MR, Yan H-M, Ramachandran A, et al. HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology. 2011;53:974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Du K, Ramachandran A, Weemhoff JL, et al. Editor’s highlight: metformin protects against acetaminophen hepatotoxicity by attenuation of mitochondrial oxidant stress and dysfunction. Toxicol Sci. 2016;154:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nestler JE. Metformin in the treatment of infertility in polycystic ovarian syndrome: an alternative perspective. Fertil Steril. 2008;90:14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Evans JM, Donnelly LA, Emslie-Smith AM, et al. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hodeib M, Ogrodzinski MP, Vergnes L, et al. Metformin induces distinct bioenergetic and metabolic profiles in sensitive versus resistant high grade serous ovarian cancer and normal fallopian tube secretory epithelial cells. Oncotarget. 2018;9:4044–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Laurent V, Glaise D, Nübel T, et al. Highly efficient SiRNA and gene transfer into hepatocyte-like HepaRG cells and primary human hepatocytes: new means for drug metabolism and toxicity studies. Methods Mol Biol. 2013;987:295–314. [DOI] [PubMed] [Google Scholar]
- [23].Wu M, Neilson A, Swift AL, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–136. [DOI] [PubMed] [Google Scholar]
- [24].Cerec V, Glaise D, Garnier D, et al. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology. 2007;45:957–967. [DOI] [PubMed] [Google Scholar]
- [25].Divakaruni AS, Paradyse A, Ferrick DA, et al. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol. 2014;547:309–354. [DOI] [PubMed] [Google Scholar]
- [26].Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc. 2014;9:421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mookerjee SA, Goncalves RLS, Gerencser AA, et al. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta. 2015;1847:171–181. [DOI] [PubMed] [Google Scholar]
- [28].Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Park Y, Smith RD, Combs AB, et al. Prevention of acetaminophen-induced hepatotoxicity by dimethyl sulfoxide. Toxicology. 1988;52:165–175. [DOI] [PubMed] [Google Scholar]
- [30].Dranka BP, Benavides GA, Diers AR, et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011;51:1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Qian W, Van Houten B. Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods. 2010;51:452–457. [DOI] [PubMed] [Google Scholar]
- [32].Das KC. Hyperoxia decreases glycolytic capacity, glycolytic reserve and oxidative phosphorylation in MLE-12 cells and inhibits complex I and II function, but not complex IV in isolated mouse lung mitochondria. PLoS One. 2013;8:e73358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hill BG, Benavides GA, Lancaster JR, et al. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393:1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].van der Windt GJ, Everts B, Chang C-H, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brand MD. The efficiency and plasticity of mitochondrial energy transduction. Biochem Soc Trans. 2005;33:897–904. [DOI] [PubMed] [Google Scholar]
- [36].Kendig DM, Tarloff JB. Inactivation of lactate dehydrogenase by several chemicals: implications for in vitro toxicology studies. Toxicol In Vitro. 2007;21:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Swiss R, Will Y. Assessment of mitochondrial toxicity in HepG2 cells cultured in high-glucose- or galactose-containing media. Curr Protoc Toxicol. 2011;49:2–20. [DOI] [PubMed] [Google Scholar]
- [38].Uhl EW, Warner NJ. Mouse models as predictors of human responses: evolutionary medicine. Curr Pathobiol Rep. 2015;3:219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Olson H, Betton G, Robinson D, et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol. 2000;32:56–67. [DOI] [PubMed] [Google Scholar]
- [40].Young MJ, Humble MM, DeBalsi KL, et al. POLG2 disease variants: analyses reveal a dominant negative heterodimer, altered mitochondrial localization and impaired respiratory capacity. Hum Mol Genet. 2015;24:5184–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gohil VM, Sheth SA, Nilsson R, et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Marroquin LD, Hynes J, Dykens JA, et al. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97:539–547. [DOI] [PubMed] [Google Scholar]
- [43].Elkalaf M, Anděl M, Trnka J. Low glucose but not galactose enhances oxidative mitochondrial metabolism in C2C12 myoblasts and myotubes. PLoS One. 2013;8:e70772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].König M, Bulik S, Holzhütter HG. Quantifying the contribution of the liver to glucose homeostasis: a detailed kinetic model of human hepatic glucose metabolism. PLoS Comput Biol. 2012;8:e1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].van Wenum M, Adam AAA, Hakvoort TBM, et al. Selecting cells for bioartificial liver devices and the importance of a 3D culture environment: a functional comparison between the HepaRG and C3A cell lines. Int J Biol Sci. 2016;12:964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xie Y, McGill MR, Dorko K, et al. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 2014;279:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Meyer JN, Leung MCK, Rooney JP, et al. Mitochondria as a target of environmental toxicants. Toxicol Sci. 2013;134:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Attene-Ramos MS, Huang R, Michael S, et al. Profiling of the Tox21 chemical collection for mitochondrial function to identify compounds that acutely decrease mitochondrial membrane potential. Environ Health Perspect. 2015;123:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.