

Summary
There is a close cross‐talk between complement, Toll‐like receptors (TLRs) and coagulation. The role of the central complement component 5 (C5) in physiological and pathophysiological hemostasis has not, however, been fully elucidated. This study examined the effects of C5 in normal hemostasis and in Escherichia coli‐induced coagulation and tissue factor (TF) up‐regulation. Fresh whole blood obtained from six healthy donors and one C5‐deficient individual (C5D) was anti‐coagulated with the thrombin inhibitor lepirudin. Blood was incubated with or without E. coli in the presence of the C5 inhibitor eculizumab, a blocking anti‐CD14 monoclonal antibody (anti‐CD14) or the TLR‐4 inhibitor eritoran. C5D blood was reconstituted with purified human C5. TF mRNA was measured by quantitative polymerase chain reaction (qPCR) and monocyte TF and CD11b surface expression by flow cytometry. Prothrombin fragment 1+2 (PTF1·2) in plasma and microparticles exposing TF (TF‐MP) was measured by enzyme‐linked immunosorbent assay (ELISA). Coagulation kinetics were analyzed by rotational thromboelastometry and platelet function by PFA‐200. Normal blood with eculizumab as well as C5D blood with or without reconstitution with C5 displayed completely normal biochemical hemostatic patterns. In contrast, E. coli‐induced TF mRNA and TF‐MP were significantly reduced by C5 inhibition. C5 inhibition combined with anti‐CD14 or eritoran completely inhibited the E. coli‐induced monocyte TF, TF‐MP and plasma PTF1·2. Addition of C5a alone did not induce TF expression on monocytes. In conclusion, C5 showed no impact on physiological hemostasis, but substantially contributed to E. coli‐induced procoagulant events, which were abolished by the combined inhibition of C5 and CD14 or TLR‐4.
Keywords: CD14, complement C5, Escherichia coli, tissue factor, Toll‐like receptor 4
Introduction
Sepsis is still a major threat to human health, and the incidence is rising 1. It is commonly associated with coagulation disturbances ranging from coagulation activation to disseminated intravascular coagulation 2. Tissue factor (TF) is suggested to be a major initiator of bacteria‐induced coagulation in human sepsis 3.
TF initiates the extrinsic coagulation pathway and results in thrombin activation and clot formation. We have previously demonstrated that the Escherichia coli‐induced coagulation in human whole blood measured by prothrombin fragment 1+2 (PTF1·2) is TF‐dependent, using a blocking antibody against TF 4. The E. coli‐induced monocyte TF surface expression was efficiently inhibited by the combined inhibition of complement C3 and CD14 4. Furthermore, the Neisseria meningitidis‐induced monocyte TF surface expression was largely complement component 5 (C5)‐dependent 5.
The complement system is a complex plasma cascade and an important component of innate immunity. The system is activated via three routes, the classical, alternative and lectin pathways, all leading to the activation of C5 and the terminal pathway 6. The terminal complement C5b‐9 complex (TCC) can either be deposited on a surface as the membrane attack complex (MAC) or remain in the fluid phase as soluble TCC (sC5b‐9) 6. Complement opsonizes bacteria, enhances phagocytosis and may induce MAC‐mediated lysis of some complement‐sensitive bacteria, in particular Neisseria species 7. Activation of C5 also induces formation of C5a, which is an important proinflammatory mediator with a wide range of effects on host cells, particularly leukocytes 8.
There are many links between the complement and coagulation systems 9, but the role of C5 in normal coagulation kinetics and hemostasis is only partially investigated. One reason for this is that genetic deficiency of C5 is extremely rare 10. However, clinical use of the anti‐C5 antibody eculizumab (Soliris®) is considerably increasing the patient population with a ‘functional’ C5 deficiency and, thus, increasing the relevance of studies on complement in hemostasis. Muhlfelder et al. showed that C5 increased the TF activity on leukocytes 11, due most probably to activation of C5a receptor type 1 (C5aR1) 5, 12.
Earlier studies have suggested that C6‐deficient rabbits had an increased tendency to bleed, as prolonged clotting time and reduced prothrombin consumption were observed, and reconstitution with purified C6 corrected this condition 13. Furthermore, C6‐deficient mice and rats also had prolonged bleeding 14. In contrast, humans with complement C6‐deficiency 15, 16 and a C7‐deficient individual 17 were shown to have normal hemostasis. Zymosan failed to aggregate platelets in both C3, C5, C6 and C7‐deficient plasmas, suggesting a role of C5 in platelet aggregation 18.
Combined inhibition of C3 and CD14 significantly inhibited the E. coli‐induced cytokine release, phagocytosis and oxidative burst in human whole blood 19. CD14 is the co‐receptor of several Toll‐like receptors (TLRs), including TLR‐4 and TLR‐2 20, indicating that TLRs may be involved in the E. coli‐induced TF up‐regulation.
The aim of this study was to investigate how C5 affects normal hemostasis and coagulation kinetics, and the roles of C5 as well as CD14 and TLR‐4 in bacteria‐induced coagulation in fresh human whole blood from healthy controls or a C5‐deficient individual (C5D).
Materials and methods
Blood sampling for hemostasis investigations
Venous blood from the male C5D individual, who was diagnosed due to repeated meningococcal sepsis 10, and healthy controls (one male and five females) for hemostasis investigation were collected in tubes with 3·2% sodium citrate (Vacuette; Greiner Bio‐One GmbH, Kremensmünster, Austria). The tubes were centrifuged at 2500 g for 10 min at room temperature. The study was approved by the regional ethics committee of the Northern Norway Regional Health Authority. The donors provided written informed consent.
Whole blood experiments and inhibitors
Whole blood experiments were performed according to the previously described whole blood model 21. Blood was collected in 4·5 ml Nunc polypropylene tubes (Roskilde, Denmark) containing a 50 mg/l final concentration of lepirudin (Refludan®; Celgene, Uxbridge, UK). All equipment and reagents were endotoxin‐free. Blood (975 µl) was preincubated for 5 min with 195 µl of phosphate‐buffered saline (PBS; Sigma‐Aldrich, St Louis, MO, USA) or inhibitor. Anti‐human C5 IgG2/4 eculizumab (Alexion Pharma GmbH, New Haven, CT, USA) was used at a final concentration of 77 mg/l, an immunoglobulin (Ig)G2/4 isotype control antibody (NHDL, produced in our laboratory) at 77 mg/l, anti‐human CD14 IgG2/4 r18D11 (anti‐CD14 22) at 15 mg/l and the TLR‐4 inhibitor eritoran (E5564, kindly provided from Eisai, Andover, MA, USA) at 1 µM. PMX53 [AcF‐(OPdChaWR)] was synthesized as described previously 23, purified by reverse‐phase high‐performance liquid chromatography, and used at a final concentration of 10 µM. In experiments with blood from the C5D, purified human C5 at a final concentration of 50 mg/l (Complement Technology, Tyler, TX, USA) or 17·5 mg/l human serum albumin (HSA) (Octapharma, Lachen, Switzerland) were added. After preincubation, 195 µl of PBS, heat‐inactivated E. coli (final concentration 1 × 107/ml; strain LE392, ATCC 33572; American Type Culture Collection, Manassas, VA, USA) 21 or ultrapure lipopolysaccharide (LPS) (final concentration 100 ng/ml; LPS‐EB Ultrapure from E. coli strain 0111; InvivoGen, Eugene, OR, USA) were added. The time zero sample (T0) was processed immediately after blood sampling. Samples for flow cytometric analysis of CD11b surface expression were incubated at 37°C for 10 min. All other samples were incubated at 37°C and tilted up and down 10 times per min on a Rock‐n‐Roller (Labinco, Breda, the Netherlands) for 120 min before further processing. For flow cytometric analysis of TF, 45 µl of whole blood was added to 5 µl citrate. For analysis of PTF1·2, soluble TCC, cytokines and TF mRNA, ethylenediamine‐tetraacetic acid (EDTA, 10 mM final concentration) was added to stop further coagulation and complement activation. All tubes were centrifuged at 3000 g at 4°C for 20 min, and plasma was stored at –80°C until analysis, except for quantitative PCR (qPCR) analysis. Here, the volume of plasma removed above the blood cells was replaced with PBS without CaCl2 and MgCl2 and TempusTM blood RNA solution (Applied Biosystems, Foster City, CA, USA) was added at twice the blood volume. The tubes were stored at –20°C until analysis. For analysis of TF function in plasma micro‐particles (TF‐MP), 25 µl citrate solution was added to 225 µl blood. The tubes were centrifuged twice, first at 1500 g at room temperature for 15 min to remove cells. The plasma was centrifuged a second time at 13 000 g at room temperature for 2 min and the supernatant was stored at –80°C until analysis.
Coagulation analyses
Routine coagulation analyses were performed in citrated plasma using a STA‐R Evolution® instrument and reagents from Diagnostica Stago (Asniéres, France). STA® SPA+ reagent was used for prothrombin time‐international normalized ratio (PT‐INR), and STA®‐PPT for activated partial thromboplastin time (APTT; Diagnostica Stago) and clot detection methods. The colorimetric kits STACHROM® protein C and STACHROM® ATIII (Diagnostica Stago) were used for analysis of protein C and anti‐thrombin III, respectively. The clot‐based test kit STACLOT® protein S (Diagnostica Stago) and the COATEST® APCTM Resistance V kit from Chromogenix (Bedford, MA, USA) were used for analysis of protein S and activated protein C (APC) resistance, respectively. FVIII, von Willebrand factor antigen and activity, lupus anti‐coagulant, anti‐cardiolipin IgG and IgM and anti‐beta2‐glycoprotein I IgG and IgM were estimated in blood samples from the C5D individual by routine analyses at the Department of Medical Biochemistry, Oslo University Hospital, Oslo, Norway.
Thromboelastometry on whole blood
Clot development in fresh citrated blood was analyzed using ROTEM® delta (Tem Innovations GmbH, Munich, Germany) following the manufacturer's instructions. The non‐activated thromboelastometry (NATEM) analyses were performed as previously described 24, 25. Star‐tem reagent (Tem Innovations GmbH) containing CaCl2 was used to recalcify the samples.
Platelet function analysis in whole blood
Platelet function in whole blood was analyzed using the INNOVANCE® PFA200 instrument from Siemens Healthcare (Marburg, Germany) and Dade PFA collagen/ADP test cartridges (Siemens Healthcare). The test mimics venous damage and platelet adhesion and aggregation repair of a vessel injury. Closure time is the time before the hole in the cuvette is closed by platelets, and the results are given in seconds.
Real‐time quantitative polymerase chain reaction (qPCR) of TF mRNA levels
The MagMaxTM for stabilized Blood Tubes RNA Isolation kit from Thermo Fisher Scientific (Vilnius, Lithuania) was used to isolate the RNA. The concentration of RNA was measured using a Nanodrop 2000c (Thermo Fisher Scientific, Wilmington, DE, USA). The TaqMan® RNA–CTTM 1‐step kit (Applied Biosystems) was used to analyze the TF and control mRNA levels from 2 ng of total RNA per sample. Hs00175225_m1 (Applied Biosystems) was used to amplify the TF mRNA. Human beta‐2‐microglobulin (Hu B2 M; Applied Biosystems) was used as an endogenous control, as it is stably expressed in the whole blood model 26. The samples were analyzed in triplicate using QuantStudio 6 Flex from Applied Biosystems. The delta‐delta Ct‐method was used to calculate the relative concentration of TF mRNA. The results are given as relative quantitation (RQ), using the PBS control sample incubated for 120 min at 37°C as a calibrator and set to 1.
Flow cytometric analysis of TF surface expression
The monocyte TF surface expression in whole blood (12·5 µl) was analyzed by adding fluorescein isothiocyanate (FITC)‐labeled anti‐human TF antibody (2·5 µl) (product no. 4508CJ, clone VD8; American Diagnostica, Inc., Stamford, CT, USA) or a FITC‐labeled mouse IgG1 isotype control antibody (2·5 µl) (Becton Dickinson, San Jose, CA, USA). To gate the monocytes and leukocytes, phycoerythrin (PE) (Becton Dickinson)‐labeled anti‐CD14 antibody (2·5 µl) and peridinin‐chlorophyll (PerCP)‐labeled anti‐CD45 antibody (2·5 µl) (Becton Dickinson) were added to the blood samples in a 96‐well microtiter plate (Nunc) and incubated in the dark for 15 min at room temperature. The samples were lysed by adding EasyLyse™ (250 µl) (S2364; Dako Cytomation, Glostrup, Denmark) and incubated for 15 min at room temperature protected from light. The samples were centrifuged at 300 g for 5 min and washed with PBS containing 0·1% (w/v) bovine serum albumin (BSA), and the cells were resuspended in PBS. The samples were analyzed using a BD LSR II flow cytometer (Becton Dickinson) and results expressed as median fluorescence intensity (MFI). The MFI results are given as the differences between the MFI with anti‐TF antibody and the isotype control antibody.
Flow cytometric analysis of CD11b surface expression
Increasing amounts of human recombinant C5a (Hycult Biotechnology, Uden, the Netherlands) were added to blood from healthy donors. After a 10‐min incubation at 37°C, 25 µl whole blood was added to 25 µl 0·5% (v/v) paraformaldehyde (PFA; Sigma Aldrich, Oslo, Norway), and cells were fixed at 37°C for 4 min. The fixed blood cells were stained with PE‐labeled anti‐CD11b (Becton Dickinson) or PE‐labeled mouse IgG2a (Becton Dickinson) in the dark for 15 min at room temperature. In addition, PerCP‐labeled CD45 (Becton Dickinson) and FITC‐labeled anti‐CD14 (Becton Dickinson; clone MP9) were used to gate leukocytes and monocytes. The samples were mixed with 1 ml PBS and analyzed using NovoCyte flow cytometer (ACEA Biosciences, Inc., San Diego, CA, USA) and results recorded as the median fluorescence intensity (MFI).
Enzyme‐linked immunosorbent assays (ELISA)
Soluble TCC levels were measured in plasma using an antibody against a specific C9 neoepitope in the TCC complex, as described previously 27. PTF1·2 in plasma was measured using an Enzygnost®F1+2 (monoclonal) kit from Siemens Healthcare (Marburg, Germany). The PTF1·2 results in Fig. 1b,d were normalized to the sample incubated for 120 min at 37°C with E. coli, which was set to 100%. TF‐MP was measured in plasma using the Zymuphen MP‐TF kit (Aniara Diagnostica, West Chester, OH, USA). Optical density was measured using an MRX microplate reader (Dynex Technologies, Denkendorf, Germany).
Figure 1.

Effect of the anti‐C5 antibody eculizumab and C5 deficiency on Escherichia coli (E. coli)‐induced tissue factor function in plasma microparticles (TF‐MP) and on coagulation (PTF1·2). Whole blood from healthy donors was preincubated with phosphate‐buffered saline (PBS), eculizumab (Eculiz.) or control antibody (Ctrl.Ab.) prior to incubation with 1 × 107/ml E. coli (a,b). Whole blood from the C5‐deficient individual was incubated with PBS, purified C5 or human serum albumin (HSA) prior to the incubation (c,d). E. coli‐induced coagulation was measured by prothrombin fragment 1+2 (PTF1·2) in plasma using enzyme‐linked immunosorbent assay (ELISA) (b,d), and expressed as a percentage of the positive control sample incubated with E. coli (set to 100%). TF‐MP (a,c) are given in pg/ml. The results are expressed as means ± standard deviation (s.d.) for normal blood experiments (n = 6), and as means (line) and scatterplot of two experiments performed on two different days with C5‐deficient blood. * P < 0·05.
Statistical analysis
Analysis of normality distribution was performed using the Shapiro–Wilk test in IBM spss Statistics for Windows version 22·0 (IBM Corp., Armonk, NY, USA). If the normality test failed, the results were log‐transformed. The results that were still skewed were analyzed using non‐parametric tests. Data were analyzed using GraphPad Prism version 6·0 (GraphPad Software, San Diego, CA, USA). The results from experiments with stimuli were analyzed with a paired Student's t‐test comparing the results with and without E. coli or LPS and the exact P‐value is reported. Wilcoxon's matched‐pairs signed‐rank test was used to analyze non‐parametric data. Furthermore, a one‐way repeated‐measures analysis of variance (anova) and Dunnett's multiple comparisons test using PBS as a control were used to compare the effects of various additives. Non‐parametric data were analyzed using Friedman's test and Dunn's multiple comparisons test. The Pearson correlation in GraphPad Prism was used to analyze the correlation between coagulation measured as PTF1·2 and the monocyte TF surface expression or TF‐MP in plasma. Two‐way repeated‐measures anova and Sidak's multiple comparison tests were used to compare results in the presence or absence of the C5a receptor 1 (C5aR1) antagonist PMX53 at different C5a concentrations. A P‐value below 0·05 was considered statistically significant.
Results
No impact of C5 on hemostasis or coagulation kinetics under physiological conditions
Samples from the healthy blood donors were within the reference ranges for the selected routine coagulation analysis (Table 1). Plasma from the C5D individual displayed normal coagulation measured by PT‐INR, APTT, protein C, protein S and anti‐thrombin levels, APC resistance, von Willebrand factor activity, lupus anti‐coagulant and D‐dimer (Table 1).
Table 1.
Results from routine coagulation analysis of the C5D individual and healthy donors
| Analyses | Results | Reference ranges | |
|---|---|---|---|
| Healthy donors | C5Da | ||
| P‐PT‐INRb | 1·0 ± 0·1 | 1·1 | <1·2 |
| P‐APTTc (s) | 34 ± 1·6 | 38 | 30–42 |
| P‐D‐dimer (mg/l) | n.d.d | 0·3 | <0·5 |
| P‐fibrinogen (g/l) | n.d. | 2·0 | 1·5‐4·0 |
| P‐anti‐thrombin (%) | 102 ± 5·0 | 92 | 80–120 |
| P‐protein C (%) | 99 ± 15 | 76 | >70 |
| P‐protein S (%) | 88 ± 12 | 97 | >65 |
| P‐n‐APCe‐sensitivity ratio | 1·0 ± 0·02 | 1·03 | >0·80 |
| P‐von Willebrand antigen (%) | n.d. | 68 | 50–200 |
| P‐von Willebrand activity (%) | n.d. | 66 | 50–200 |
| P‐factor VIII (%) | n.d. | 68 | 50–150 |
| P‐lupus anti‐coagulant SCT | n.d. | 1·07 | <1·25 |
| P‐anti‐cardiolipin‐antibody IgG (GPL)f | n.d. | 3·9 | <40 |
| P‐anti‐cardiolipin‐antibody IgM (MPL)g | n.d. | 6·1 | <40 |
| P‐anti‐beta2‐GPI IgG (SGU)h | n.d. | 5·0 | <20 |
| P‐anti‐beta2‐GPI IgM (SMU)i | n.d. | 5·5 | <20 |
Results from C5‐deficient individual (n = 1) and healthy donors given in means ± standard deviation (s.d.) (n = 6). C5Da = complement component 5‐deficient individual; PT‐INRb = prothrombin international normalized ratio; APTTc = activated partial thromboplastin time; n.d.d = not done; APCe = activated protein C; (GPL)f = immunoglobulin (Ig)G phospholipid units corresponding to 1 µg/ml of cardiolipin antibody; (MPL)g = IgM phospholipid units corresponding to 1 µg/ml of cardiolipin antibody; (SGU)h = standard IgG anti‐beta‐2‐GPI unit; (SMU)i = standard IgM anti‐beta‐2‐GPI unit.
Analyses of coagulation kinetics in fresh blood from both the healthy donors and the C5D using thromboelastometry and platelet function analyzer showed that C5 had no apparent impact on clot formation and platelet aggregation under physiological conditions (Table 2). A statistical comparison of the ROTEM results in the C5D and the six healthy donors after adding eculizumab was not possible due to data from only one C5D individual.
Table 2.
Thromboelastometry and platelet function analyses of blood from healthy donors and the C5D individual
| Test | Additions | Healthy donors | C5Da |
|---|---|---|---|
| CTb (s)c | Undiluted | 824 ± 159 | 968 ± 109 |
| PBSd | 1073 ± 143 | 1059 ± 370 | |
| Eculizumab | 1059 ± 83 | n.d.e | |
| Control antibodyf | 934 ± 94 | n.d. | |
| Alpha‐angle (°)g | Undiluted | 47 ± 7·0 | 28 ± 1·0 |
| PBS | 41 ± 6·9 | 32 ± 8·5 | |
| Eculizumab | 39 ± 4·6 | n.d. | |
| Control antibody | 39 ± 5·2 | n.d. | |
| CFTh (s) | Undiluted | 259 ± 63 | 514 ± 22 |
| PBS | 333 ± 76 | 481 ± 187 | |
| Eculizumab | 366 ± 72 | n.d. | |
| Control antibody | 351 ± 61 | n.d. | |
| MCFi (smm)j | Undiluted | 48 ± 5·1 | 43 ± 2·3 |
| PBS | 46 ± 5·0 | 43 ± 1·5 | |
| Eculizumab | 46 ± 3·7 | n.d. | |
| Control antibody | 46 ± 4·3 | n.d. | |
| PFA200k (s) | Undiluted | 80 ± 21 | 106 ± 4·9 |
| PBS | 86 ± 24 | 121 ± 12 | |
| Eculizumab | 94 ± 20 | n.d. | |
| Control antibody | 103 ± 20 | n.d. |
Thromboelatometry was performed using non‐activated thromboelastometry (NATEM) reagents. Results are expressed as seconds, degrees or millimeter and are given as mean ± standard deviations (s.d.) for healthy donors (n = 6). Results for the C5D are from two analyses performed at two different days and are given as means ± s.d.
C5Da = complement component 5‐deficient individual; CTb = clotting time; (s)c = second; PBSd; phosphate‐buffered saline; n.d.e = not done; (°)g = degrees; CFTh = clot formation time; MCFi = maximum clot firmness; (mm)j = millimeter; PFA200k, Siemens Healthcare platelet function analyzer 200 closure time.
C5 inhibition and absence of C5 (C5D) reduced the E. coli‐induced TF whole blood mRNA levels and monocyte TF surface expression
Incubation for 120 min with E. coli significantly (P = 0·0056) increased the TF mRNA level in blood from healthy donors compared with control samples incubated with PBS (Fig. 2a). Eculizumab significantly (P < 0·05) reduced the E. coli‐induced TF mRNA level from 6·4 to 1·9 RQ compared to PBS, whereas the control antibody had no effect. In blood from healthy donors, monocyte TF surface expression was significantly (P = 0·0002) increased from MFI = 88 in the PBS control to MFI = 851 in the presence of E. coli (Fig. 2b). Eculizumab significantly (P < 0·05) reduced the E. coli‐induced TF surface expression to MFI = 327, whereas the control antibody had no effect. Eculizumab completely blocked the E. coli‐induced complement activation measured as formation of sC5b‐9 in plasma (data not shown).
Figure 2.

Effect of the anti‐human C5 antibody eculizumab and C5 deficiency on Escherichia coli (E. coli)‐induced tissue factor (TF) mRNA levels and monocyte TF surface expression on whole blood monocytes. Whole blood from healthy donors was preincubated with phosphate‐buffered saline (PBS), eculizumab (Eculiz.) or control antibody (Ctrl.Ab.). The blood samples were either processed immediately (time zero samples) or incubated for 120 min at 37°C with PBS (control) or 1 × 107/ml E. coli. TF mRNA expression (a,c) was examined using real‐time quantitative PCR (qPCR) and expressed as the relative quantity (RQ) compared to the PBS control (set to 1). Monocyte TF surface expression (b,d) was examined using flow cytometry and given as median flow intensity (MFI). Whole blood from the C5‐deficient individual was incubated with PBS, purified complement component C5 or human serum albumin (HSA) prior to incubation with E. coli (c,d). The results are given as means ± standard deviation (s.d.) for normal blood experiments (n = 6), and as mean (line) and scatterplot of two experiments performed on two different days with C5D blood. * P < 0·05.
In blood from the C5D individual, E. coli increased the TF mRNA level from 1·0 to 7·5 RQ (Fig. 2c). After reconstitution with purified C5, the E. coli‐induced TF mRNA level further increased to 23·4 RQ. Monocyte TF surface expression increased in the presence of E. coli to MFI = 232 compared to MFI = 59 in the PBS control (Fig. 2d). Reconstitution with purified C5 further increased the E. coli‐induced TF surface expression to MFI = 470. The HSA control had no effect on either E. coli‐induced TF mRNA or monocyte TF surface expression.
C5 inhibition and absence of C5 (C5D) reduced the E. coli‐induced TF‐MP and PTF1·2 levels
Incubation of whole blood from healthy donors with E. coli for 120 min significantly (P = 0·0002) increased the levels of TF‐MP from 0·84 to 8·8 pg/ml (Fig. 1a). Eculizumab significantly (P < 0·05) reduced the E. coli‐induced TF‐MP levels to 4·9 pg/ml, whereas the control antibody had no effect. The impact of C5 inhibition on coagulation was evaluated by analyzing the PTF1·2 levels in plasma (Fig. 1b). E. coli significantly (P < 0·05) increased the PTF1·2 levels from 4·4% in the PBS control to 100% after 120 min incubation with E. coli (set to 100%). Eculizumab significantly (P < 0·05) reduced the E. coli‐induced PTF1·2 level to 44% of the PBS control. The control antibody had no effect on the E. coli‐induced PTF1·2 level; however, the results displayed high biological variation.
In blood from the C5D, E. coli increased the TF‐MP levels from 0·36 to 2·9 pg/ml (Fig. 1c). After reconstitution with purified C5, E. coli increased the TF‐MP levels from 2·9 to 5·7 pg/ml and the PTF1·2 level from 100% in the absence of recombinant C5 to 471% in C5 reconstituted blood (Fig. 1d). In the absence of E. coli and C5, PTF1·2 were 12% of the sample incubated with E. coli. The HSA control had no effect on either the E. coli‐induced TF‐MP or E. coli‐induced PTF1·2 levels.
E. coli‐induced PTF1·2 levels correlated with procoagulant activity of TF‐MP
We then tested if the functional measure of coagulation activation, reflected by plasma PTF1·2, correlated significantly with TF surface expression on monocytes and TF‐MP. Data for PTF1·2, TF‐MP and monocyte TF surface expression from one experiment with each blood donor were included in the correlation analysis (n = 6). PTF1·2 did not correlate with TF surface expression on monocytes (r = 0·49, P > 0·05, Fig. 3a), but with TF‐MP levels (r = 0·85, P = 0·0319, Fig. 3b).
Figure 3.

Correlations between coagulation activation measured as prothrombin fragment 1+2 (PTF1·2) in plasma and monocyte tissue factor (TF) surface expression (a) or tissue factor function in plasma microparticles (TF‐MP) (b). The PTF1·2 level was analyzed by enzyme‐linked immunosorbent assay (ELISA) and expressed in nmol/l. The TF surface expression on monocytes was analyzed by flow cytometry and expressed as median fluorescence intensity (MFI). The TF‐MP levels were expressed in pg/ml. The Pearson's correlation analyses were performed using GraphPad Prism. Results from separate experiments with each blood donor are included (n = 6).
Recombinant human C5a increased the CD11b surface expression but not monocyte TF surface expression in whole blood
We next examined if recombinant human C5a alone enhanced the TF surface expression on monocytes. The effect of increasing doses of C5a on TF and CD11b surface expression on monocytes in human whole blood was examined using flow cytometry. CD11b surface expression was used as a control readout, as C5a was reported to enhance CD11b surface expression 28. Human C5a increased CD11b surface expression from MFI = 3187 at baseline to MFI = 9161 after a 10‐min incubation with the highest C5a concentration in a dose‐dependent manner (Fig. 4). The C5aR1 antagonist PMX53 significantly (P < 0·05) reduced C5a‐induced CD11b expression on monocytes. In contrast, TF surface expression was unchanged after a 120‐min incubation with C5a and PMX53 had no effect (Fig. 4).
Figure 4.
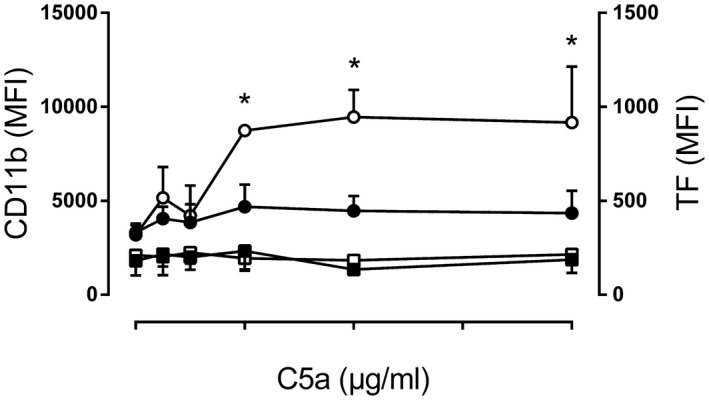
Effect of recombinant C5a on monocyte CD11b and tissue factor (TF) surface expression. Whole blood from healthy donors was incubated with phosphate‐buffered saline (PBS) or increasing concentrations of purified human C5a for 10 min at 37°C (CD11b analysis) or 120 min (TF analysis) followed by flow cytometry. The results from separate experiments using blood from healthy blood donors (n = 4) are expressed as median fluorescence intensity (MFI) ± standard deviation (s.d.). The left y‐axis indicates monocyte CD11b surface expression; samples added C5a (open circles) and samples added C5a and PMX53 (closed circle). The right y‐axis indicates monocyte TF surface expression samples added C5a (open square) and samples added C5a and PMX53 (closed square).
The combination of TLR‐4‐ and C5‐inhibition reduced E. coli‐induced monocyte TF surface expression, TF‐MP and plasma PTF1·2
We then examined the possible additional role for C5 and TLR‐4 and its co‐receptor CD14 on the E. coli‐induced monocyte TF surface expression using the TLR‐4/MD2‐specific inhibitor eritoran and a blocking anti‐CD14 mAb (Fig. 5). As a positive control, the effect of TLR‐4 inhibition on ultrapure E. coli LPS‐induced monocyte TF surface expression, TF‐MP levels and PTF1·2 was also examined.
Figure 5.

Effect of eculizumab, anti‐CD14 or eritoran on Escherichia coli (E. coli)‐induced monocyte tissue factor (TF) surface expression (a), tissue factor function in plasma microparticles (TF‐MP) (b) and prothrombin fragment 1+2 (PTF1·2) levels in plasma (c). The blood samples from healthy donors were preincubated with eritoran, eculizumab (Eculiz.), anti‐CD14 blocking monoclonal antibody (mAb) (aCD14), control antibody (Ctrl.Ab.) or a combination, as indicated, before they were either processed immediately (time zero samples) or incubated for 120 min at 37°C with phosphate‐buffered saline (PBS) (control), 100 ng/ml ultrapure E. coli lipopolysaccharide (LPS) (closed circles) or 1 × 107/ml E. coli (open circles). Monocyte TF surface expression was measured using flow cytometry and given as median fluorescence intensity (MFI). TF‐MP and PTF1·2 in plasma were analyzed using enzyme‐linked immunosorbent assay (ELISA) and expressed as pg/ml and nmol/l, respectively. In the PTF1·2 analysis, one data point of the control antibody data set was an outlier and replaced by the mean of this data set, both in the figure and the statistical analysis. All results are given as means ± standard deviation (s.d.) (n = 6). * P < 0·05.
Eculizumab alone and anti‐CD14 alone did not significantly reduce the E. coli‐induced monocyte TF surface expression, whereas the combination significantly (P < 0·05) reduced the monocyte TF surface expression from MFI = 1066 to MFI = 176 (Fig. 5a). Eritoran alone did not significantly reduce the E. coli‐induced monocyte TF surface expression, whereas combined with eculizumab the reduction was significant (P < 0·05) to MFI = 227.
Eculizumab alone significantly (P < 0·05) reduced the E. coli‐induced TF‐MP from 9·0 to 6·4 pg/ml (Fig. 5b). Anti‐CD14 alone did not significantly reduce TF‐MP, whereas combined with eculizumab the reduction from 9·0 to 3·4 pg/ml was significant (P < 0·05). Eritoran alone did not significantly reduce TF‐MP, whereas combined with eculizumab the reduction from 9·0 to 3·8 pg/ml was significant (P < 0·05). The control antibody unexpectedly and significantly increased (P < 0·05) the E. coli‐induced TF‐MP.
Eculizumab alone and anti‐CD14 alone did not significantly reduce the E. coli‐induced PTF1·2, whereas the combination significantly (P < 0·05) reduced the PTF1·2 from 18·8 to 2·7 nmol/ml analyzed by anova (Fig. 5c). However, a paired Student's t‐test showed that eculizumab alone significantly (P = 0·0356) reduced the E. coli‐induced PTF1·2 (Fig. 5c). Eritoran alone did not significantly reduce the PTF1·2, whereas combined with eculizumab the reduction from 18·8 to 4·0 nmol/ml was significant (P < 0·05).
Eritoran significantly (P < 0·05) reduced LPS‐induced monocyte TF surface expression (Fig. 5a), LPS‐induced TF‐MP (Fig. 5b) and LPS‐induced PTF1·2 (Fig. 5c). Data on LPS and E. coli‐induced monocyte TF surface expression were displayed in representative histograms for one donor (Fig. 6).
Figure 6.
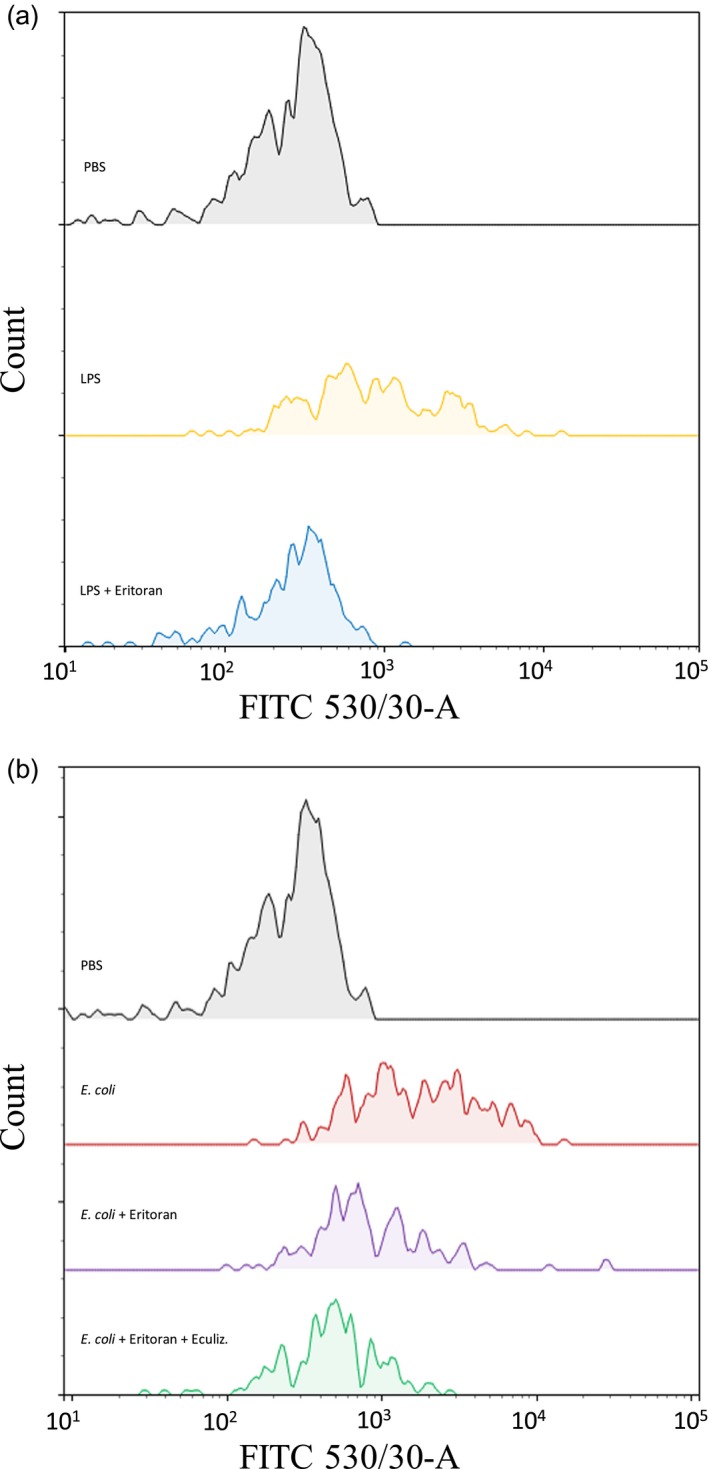
Representative histograms from the flow cytometric analysis of the monocyte tissue factor (TF) surface expression in blood from one of six healthy blood donors. The median fluorescence (MFI) signal derived from the fluorescein isothiocyanate (FITC)‐labeled anti‐TF antibody is depicted at the x‐axis and the monocyte count at the y‐axis. The isotype control antibody data were nearly identical for all samples and not included in this illustration. (a) Whole blood was incubated for 120 min at 37°C with phosphate‐buffered saline (PBS) (gray) or ultrapure E. coli lipopolysaccharide (LPS) (100 ng/ml) in the absence (yellow) or presence of eritoran (blue). (b) Whole blood was incubated for 120 min with PBS (gray) or E. coli (1 × 107/ml) in the absence (red) or presence of eritoran (purple) or a combination of eritoran and eculizumab (Eculiz.) (green).
Discussion
The present data demonstrate that blood from a C5‐deficient (C5D) individual displayed completely normal concentrations of hemostatic proteins and functional hemostatic analyses as normal individuals. Consistently, C5 had no impact on the physiological coagulation kinetics in vitro using whole blood samples from healthy controls inhibited with eculizumab. In contrast, C5 was crucially important in the E. coli‐induced hemostatic disturbance, reflected by increased TF mRNA generation, monocyte TF surface expression, plasma TF‐MP and PTF1·2 formation. The effect of C5 was most pronounced at the mRNA level. Finally, combined inhibition of C5 and CD14 or TLR‐4 most efficiently blocked the E. coli‐induced coagulation, with only minor effects of each inhibitor alone.
Inhibition of complement with eculizumab emerges as an important strategy for treatment of an increasing number of diseases involving complement activation, and is Food and Drug Administration (FDA)‐approved for clinical use with paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome and myasthenia gravis 29, 30. Here, we show that the presence of eculizumab in normal human blood did not affect platelet closure times or the coagulation kinetic readouts, implying that eculizumab does not interfere with normal hemostasis, and that C5 most probably does not play a role in hemostasis under physiological conditions. This is in agreement with the observation that C5‐deficient individuals do not have a general bleeding tendency 18.
Keshari et al. recently observed that coagulation and hemostasis can be activated independently of significant complement activation, as infusion of FXa and phosphatidylcholine–phosphatidylserine in baboons did not activate complement 31. Notably, in rodents and rabbits complement C6 and C7 deficiencies were accompanied by increased bleeding tendency or other coagulation disorders 13, 14. However, previous studies with human complement deficiencies, including C6 and C7, could not find such effects 15, 16, 17. In the present study, the results from thromboelastometry, platelet function and coagulation tests imply that human C5 deficiency neither causes hemostatic disorders nor pathological coagulation kinetics.
The relatively high biological variation of PTF1·2 levels is due most probably to different levels of TF in high and low TF responders 32. Eculizumab significantly reduced the E. coli‐induced PTF1·2 when three, but not seven, groups were compared by one‐way repeated‐measures anova. This is due probably to weaker statistical power when many groups are compared. When E. coli with and without eclizumab were compared using a paired Student's t‐test, the effect on PTF1·2 was statistically significant in all experiments. However, when the results after incubation with E. coli plus eculizumab were compared with those of E. coli plus the control antibody, the results were significantly different in six of seven readouts. The significant inhibitory effect of eculizumab on the E. coli‐induced coagulation in human whole blood indicated, however, an important role of C5 in bacteria‐induced coagulation. C5 blockade by eculizumab may reduce the complement‐mediated lysis of live E. coli bacteria and reduce the free LPS level in plasma 33. It was not possible to examine the effect of eculizumab on bacterial survival in this study, as we used heat‐inactivated E. coli bacteria. Patients treated with eculizumab may possibly have a reduced risk of E. coli‐induced coagulation. As bacteria‐induced thromboembolic events are significantly increased in PNH and catastrophic anti‐phospholipid syndrome, two diseases indicative for eculizumab treatment, the putative protective effect of eculizumab on bacteria‐induced thrombosis should be further examined 34.
This report indicates a significant role of C5 in the bacteria‐induced TF up‐regulation. We have previously reported that the E. coli‐induced coagulation is predominantly TF‐dependent 4. In this study, we reveal that the E. coli‐induced increase in TF mRNA levels in blood from healthy donors was virtually abolished by the addition of eculizumab, and was significantly enhanced in the C5D individual after reconstitution with purified C5. In reconstituted C5D blood, C5 reached a final concentration of 50 mg/l, which is within the normal physiological range 35.
TF gene transcription can be induced rapidly by various stimuli. In fact, Gregory et al. showed that monocyte TF mRNA expression is mainly regulated on the transcriptional level, and that the TF mRNA half‐life of approximately 1·5 h is somewhat short 36. In accordance, monocytes were previously reported to have nearly undetectable amounts of TF stored in their cytoplasm in resting state 32. Like N. meningitidis 5, E. coli induced TF surface expression on monocytes in a highly C5‐dependent manner. We assumed that nearly all monocyte TF surface expression originated from newly produced TF mRNA due to the significant role of C5 in the E. coli‐induced TF mRNA up‐regulation. However, the E. coli‐induced monocyte TF surface expression was only partially blocked by C5 inhibition using eculizumab. Both TF mRNA and monocyte surface TF were slightly increased in the absence of C5, indicating that other molecules and receptors are involved.
Egorina et al. previously showed that LPS is necessary to induce exocytosis of TF 32. Also, there are several functional links between the LPS receptor CD14/TLR‐4 and CD11b or complement receptor CR3 (CD11b/CD18) 37. We found that the TLR‐4 inhibitor eritoran completely inhibited LPS‐induced, but did not significantly reduce the E. coli‐induced, TF surface expression. Together, these results indicate that E. coli‐induced monocyte TF surface expression is C5 and TLR‐4‐dependent. E. coli bacteria also activates TLR‐2 38. CD14 blockade has a broader effect on several TLRs than inhibition of TLR‐4 by eritoran as CD14 is a co‐receptor of several TLRs, including TLR‐2 and ‐4 39. This study indicates that combined inhibition of TLR‐4 with eritoran and eculizumab or combined inhibition of several TLRs by anti‐CD14 and eculizumab have similar effects on E. coli‐induced TF, suggesting that TLR‐4 is the main TLR involved.
In the present study, addition of human C5a alone to normal whole blood did not induce monocyte TF surface expression, despite its positive effect on CD11b. This is in contrast to a study by Janco et al., which showed that C5a induced procoagulant activity in isolated monocytes 40. We have no explanation for this discrepancy, but a possible LPS contamination in the previously used C5a preparation should be considered. Importantly, in blood, C5a is immediately cleaved by carboxypeptidase N to inactive C5a des‐Arg 41 which, however, has some affinity to C5aR and may induce CD11b up‐regulation 42. However, the selective C5aR1 antagonist PMX53 43 ablated the C5a‐induced CD11b expression on monocytes. This indicates that C5a‐mediated induction of CD11b in human monocytes is wholly dependent upon C5aR1 signaling, similar to that described for human neutrophils 44.
We have demonstrated earlier that E. coli‐induced TF expression on plasma microparticles is partly C5‐dependent 45, and have now verified this observation using C5‐deficient blood reconstituted with C5. Importantly, TF negative microparticles may also induce coagulation through a procoagulant negatively charged surface and FXI 46. In addition, LPS‐induced shedding of TF positive plasma microparticles has been observed in high responders 32. However, the high correlation between TF‐MP and PTF1·2 levels found in our model suggests that TF positive plasma microparticles play a significant role in the coagulation activation and PTF1·2 formation. The E. coli‐induced PTF1·2 was not significantly correlated with the TF surface expression on monocytes, due possibly to the limited number of blood donors. We demonstrated that LPS induces TF‐MP in a TLR‐4‐dependent manner in our model. E. coli‐induced TF‐MP levels, however, were significantly reduced only by the combined inhibition of CD14 or TLR‐4 and C5, indicating a role for both TLR‐4 and complement C5 for whole bacteria‐induced TF‐MP. We have no explanation for the unexpected increase in the E. coli‐induced TF‐MP and PTF1·2 levels by the control antibody. However, eculizumab did not have these effects, indicating that this is probably related to some unknown effects of the control antibody itself.
A limitation of this study is the inhibition of thrombin and the lack of endothelial cells as well as of blood flow in the whole blood model. This precludes us from studying the effects of these factors in the experimental system. However, the contribution of TF from endothelial cells in sepsis is unclear, as a study on the endothelial TF‐gene knock‐out mice did not affect coagulation in an endotoxin model 47. The TF‐positive microparticles seem to be an important contributor to TF's effect in sepsis 48. Nevertheless, so far the current whole blood model is the best available model to study the role of complement ex vivo in interaction with other biological systems. Another limitation is that only one C5D individual was included in our study, as this deficiency is extremely rare. It is therefore possible that our observations from this C5D individual do not reflect the theoretical entirety of C5D individuals in general.
Taken together, this study revealed that C5 plays a critical role together with TLR‐4 in E. coli‐induced coagulation without affecting physiological hemostasis. Consequently, C5‐deficient and patients treated with eculizumab may have a reduced risk of E. coli‐induced coagulation.
Disclosures
None.
Author contributions
A. L., C. L., S. N., K. T. L., T. E., T. E. M. and O. L. B. planned the experiments. A. L., H. F., D. C., G. B., M. M. and J. K. L. performed the experiments. T. M. W. contributed key reagents and assisted in experimental design and data interpretation. All authors participated in writing of the manuscript and approved the final version.
Acknowledgements
This study was supported financially by the research fund at Nordland Hospital Trust, the Research Council of Norway, the Norwegian Council on Cardiovascular Disease, the Northern Norway Regional Health Authority, the Southern and Eastern Norway Regional Health Authority, the Odd Fellow Foundation, and the European Community's Seventh Framework Programme under grant agreement no. 602699 (DIREKT).
References
- 1. Danai P, Martin GS. Epidemiology of sepsis: recent advances. Curr Infect Dis Rep 2005; 7:329–34. [DOI] [PubMed] [Google Scholar]
- 2. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med 2013; 369:840–51. [DOI] [PubMed] [Google Scholar]
- 3. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol 2017; 17:407–20. [DOI] [PubMed] [Google Scholar]
- 4. Landsem A, Fure H, Christiansen D et al The key roles of complement and tissue factor in Escherichia coli‐induced coagulation in human whole blood. Clin Exp Immunol 2015; 182:81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lappegard KT, Christiansen D, Pharo A et al Human genetic deficiencies reveal the roles of complement in the inflammatory network: lessons from nature. Proc Natl Acad Sci USA 2009; 106:15861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Merle NS, Church SE, Fremeaux‐Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol 2015; 6:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merle NS, Noe R, Halbwachs‐Mecarelli L, Fremeaux‐Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol 2015; 6:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol 2004; 4:133–42. [DOI] [PubMed] [Google Scholar]
- 9. Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol 2007; 28:184–92. [DOI] [PubMed] [Google Scholar]
- 10. Grimnes G, Beckman H, Lappegard KT, Mollnes TE, Skogen V. Recurrent meningococcal sepsis in a presumptive immunocompetent host shown to be complement C5 deficient – case report. APMIS 2011; 119:479–84. [DOI] [PubMed] [Google Scholar]
- 11. Muhlfelder TW, Niemetz J, Kreutzer D, Beebe D, Ward PA, Rosenfeld SI. C5 chemotactic fragment induces leukocyte production of tissue factor activity: a link between complement and coagulation. J Clin Invest 1979; 63:147–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ritis K, Doumas M, Mastellos D et al A novel C5a receptor–tissue factor cross‐talk in neutrophils links innate immunity to coagulation pathways. J Immunol 2006; 177:4794–802. [DOI] [PubMed] [Google Scholar]
- 13. Zimmerman TS, Arroyove CM, Muller‐Eberhard HJ. A blood coagulation abnormality in rabbits deficient in the sixth component of complement (C6) and its correction by purified C6. J Exp Med 1971; 134:1591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhole D, Stahl GL. Molecular basis for complement component 6 (C6) deficiency in rats and mice. Immunobiology 2004; 209:559–68. [DOI] [PubMed] [Google Scholar]
- 15. Frank MM, Leddy JP, Gaither T, Heusinkveld RS, Breckenridge RT, Klemperer MR. Hereditary C6 deficiency in man. Birth Defects Orig Artic Ser 1975; 11:318–21. [PubMed] [Google Scholar]
- 16. Heusinkveld RS, Leddy JP, Klemperer MR, Breckenridge RT. Hereditary deficiency of the sixth component of complement in man. II. Studies of hemostasis. J Clin Invest 1974; 53:554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boyer JT, Gall EP, Norman ME, Nilsson UR, Zimmerman TS. Hereditary deficiency of the seventh component of complement. J Clin Invest 1975; 56:905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Breckenridge RT, Rosenfeld SI, Graff KS, Leddy JP. Hereditary C5 deficiency in man. III. Studies of hemostasis and platelet responses to zymosan. J Immunol 1977; 118:12–6. [PubMed] [Google Scholar]
- 19. Brekke OL, Christiansen D, Fure H et al Combined inhibition of complement and CD14 abolish E. coli‐induced cytokine‐, chemokine‐ and growth factor‐synthesis in human whole blood. Mol Immunol 2008; 45:3804–13. [DOI] [PubMed] [Google Scholar]
- 20. Zanoni I, Granucci F. Role of CD14 in host protection against infections and in metabolism regulation. Front Cell Infect Microbiol 2013; 3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mollnes TE, Brekke OL, Fung M et al Essential role of the C5a receptor in E. coli‐induced oxidative burst and phagocytosis revealed by a novel lepirudin‐based human whole blood model of inflammation. Blood 2002; 100:1869–77. [PubMed] [Google Scholar]
- 22. Lau C, Gunnarsen KS, Hoydahl LS et al Chimeric anti‐CD14 IGG2/4 hybrid antibodies for therapeutic intervention in pig and human models of inflammation. J Immunol 2013; 191:4769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kumar V, Lee JD, Clark RJ, Woodruff TM. Development and validation of a LC‐MS/MS assay for pharmacokinetic studies of complement C5a receptor antagonists PMX53 and PMX205 in mice. Sci Rep 2018; 8:8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luddington RJ. Thrombelastography/thromboelastometry. Clin Lab Haematol 2005; 27:81–90. [DOI] [PubMed] [Google Scholar]
- 25. Abdelfattah K, Cripps MW. Thromboelastography and rotational thromboelastometry use in trauma. Int J Surg 2016; 33:196–201. [DOI] [PubMed] [Google Scholar]
- 26. Lau C, Nygard S, Fure H et al CD14 and complement crosstalk and largely mediate the transcriptional response to Escherichia coli in human whole blood as revealed by DNA microarray. PLOS ONE 2015; 10:e0117261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bergseth G, Ludviksen JK, Kirschfink M, Giclas PC, Nilsson B, Mollnes TE. An international serum standard for application in assays to detect human complement activation products. Mol Immunol 2013; 56:232–9. [DOI] [PubMed] [Google Scholar]
- 28. Brekke OL, Christiansen D, Fure H, Fung M, Mollnes TE. The role of complement C3 opsonization, C5a receptor, and CD14 in E. coli‐induced up‐regulation of granulocyte and monocyte CD11b/CD18 (CR3), phagocytosis, and oxidative burst in human whole blood. J Leukoc Biol 2007; 81:1404–13. [DOI] [PubMed] [Google Scholar]
- 29. Baines AC, Brodsky RA. Complementopathies. Blood Rev 2017; 31:213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dhillon S. Eculizumab: a review in generalized myasthenia gravis. Drugs 2018; 78:367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keshari RS, Silasi R, Lupu C, Taylor FB Jr, Lupu F. In vivo‐generated thrombin and plasmin do not activate the complement system in baboons. Blood 2017; 130:2678–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Egorina EM, Sovershaev MA, Bjorkoy G et al Intracellular and surface distribution of monocyte tissue factor: application to intersubject variability. ArteriosclerThromb Vasc Biol 2005; 25:1493–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keshari RS, Silasi R, Popescu NI et al Inhibition of complement C5 protects against organ failure and reduces mortality in a baboon model of Escherichia coli sepsis. Proc Natl Acad Sci USA 2017; 114:E6390–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grimnes G, Isaksen T, Tichelaar YIGV, Brækkan SK, Hansen J‐B. Acute infection as a trigger for incident venous thromboembolism: results from a population‐based case–crossover study. Res Pract Thromb Haemost 2018; 2:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kohler PF, Ten Bensel R. Serial complement component alterations in acute glomerulonephritis and systemic lupus erythematosus. Clin Exp Immunol 1969; 4:191–202. [PMC free article] [PubMed] [Google Scholar]
- 36. Gregory SA, Morrissey JH, Edgington TS. Regulation of tissue factor gene expression in the monocyte procoagulant response to endotoxin. Mol Cell Biol 1989; 9:2752–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hajishengallis G, Lambris JD. More than complementing Tolls: complement‐Toll‐like receptor synergy and crosstalk in innate immunity and inflammation. Immunol Rev 2016; 274:233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dziarski R, Wang Q, Miyake K, Kirschning CJ, Gupta D. MD‐2 enables Toll‐like receptor 2 (TLR2)‐mediated responses to lipopolysaccharide and enhances TLR2‐mediated responses to Gram‐positive and Gram‐negative bacteria and their cell wall components. J Immunol 2001; 166:1938–44. [DOI] [PubMed] [Google Scholar]
- 39. Barratt‐Due A, Pischke SE, Nilsson PH, Espevik T, Mollnes TE. Dual inhibition of complement and Toll‐like receptors as a novel approach to treat inflammatory diseases‐C3 or C5 emerge together with CD14 as promising targets. J Leukoc Biol 2017; 101:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Janco RL, Morris P. Serum augments the generation of monocyte procoagulant stimulated by bacterial lipopolysaccharide or chemotactic fragments of C5. Thromb Res 1983; 32:73–86. [DOI] [PubMed] [Google Scholar]
- 41. Mueller‐Ortiz SL, Wang D, Morales JE, Li L, Chang JY, Wetsel RA. Targeted disruption of the gene encoding the murine small subunit of carboxypeptidase N (CPN1) causes susceptibility to C5a anaphylatoxin‐mediated shock. J Immunol 2009; 182:6533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reis ES, Chen H, Sfyroera G et al C5a receptor‐dependent cell activation by physiological concentrations of desarginated C5a: insights from a novel label‐free cellular assay. J Immunol 2012; 189:4797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hawksworth OA, Li XX, Coulthard LG, Wolvetang EJ, Woodruff TM. New concepts on the therapeutic control of complement anaphylatoxin receptors. Mol Immunol 2017; 89:36–43. [DOI] [PubMed] [Google Scholar]
- 44. Bekker P, Dairaghi D, Seitz L et al Characterization of pharmacologic and pharmacokinetic properties of CCX168, a potent and selective orally administered complement 5a receptor inhibitor, based on preclinical evaluation and randomized Phase 1 clinical study. PLOS ONE 2016; 11:e0164646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brekke OL, Waage C, Christiansen D et al The effects of selective complement and CD14 inhibition on the E. coli‐induced tissue factor mRNA upregulation, monocyte tissue factor expression, and tissue factor functional activity in human whole blood. Adv Exp Med Biol 2013; 735:123–36. [DOI] [PubMed] [Google Scholar]
- 46. Mooberry MJ, Bradford R, Hobl EL, Lin FC, Jilma B, Key NS. Procoagulant microparticles promote coagulation in a factor XI‐dependent manner in human endotoxemia. J Thromb Haemost 2016; 14:1031–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pawlinski R, Mackman N. Cellular sources of tissue factor in endotoxemia and sepsis. Thromb Res 2010; 125(Suppl 1):S70–S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemost 2018; 16:231–41. [DOI] [PubMed] [Google Scholar]