

Summary
Anti‐phospholipid syndrome (APS) is characterized by arterial and/or venous thrombosis and pregnancy morbidity. It is well known that in these patients thrombosis may be the result of a hypercoagulable state related to anti‐β2‐glycoprotein I (β2‐GPI) antibodies. Moreover, platelets may play a role in thrombotic manifestations by binding of anti‐β2‐GPI antibodies. Platelets express tissue factor (TF), the major initiator of the clotting cascade, after activation. We primarily analyzed whether anti‐β2‐GPI antibodies may trigger a signal transduction pathway leading to TF expression in human platelets. Platelets from healthy donors were incubated with affinity purified anti‐β2‐GPI antibodies for different times. Platelet lysates were analyzed for phospho‐interleukin‐1 receptor‐associated kinase 1 (IRAK), phospho‐p65 nuclear factor kappaB (NF‐κB) and TF by Western blot. IRAK phosphorylation was observed as early as 10 min of anti‐β2‐GPI treatment, with consequent NF‐κB activation, whereas TF expression, detectable at 45 min, was significantly increased after 4 h of anti‐β2‐GPI treatment. Virtually no activation was observed following treatment with control immunoglobulin IgG. We then analyzed TF expression in platelets from 20 APS patients and 20 healthy donors. We observed a significant increase of TF in APS patients versus control subjects (P < 0·0001). This work demonstrates that anti‐β2‐GPI antibodies may trigger in vitro a signal transduction pathway in human platelets, which involves IRAK phosphorylation and NF‐κB activation, followed by TF expression. Furthermore, ex vivo, platelets of APS patients showed a significantly increased expression of TF. These findings support the view that platelets may play a role in the pathogenesis of APS, with consequent release of different procoagulant mediators, including TF.
Keywords: anti‐β2‐GPI antibodies, anti‐phospholipid syndrome, platelets, tissue factor
Introduction
Anti‐phospholipid syndrome (APS) is characterized by arterial and/or venous thrombosis and pregnancy morbidity. In patients with APS, thrombosis may be the result of a hypercoagulable state related to the induction of a proinflammatory and procoagulant phenotype induced by a heterogeneous family of antibodies, among which anti‐β2‐glycoprotein I (anti‐β2‐GPI) antibodies are the best characterized 1, 2, 3, 4.
It is well known that platelets may play an active role in the pathogenesis of APS 5. In particular, a role of the platelets in APS‐related thrombosis has been demonstrated in experimental models, which revealed that platelets are activated following the infusion of anti‐β2‐GPI antibodies 6. Moreover, anti‐β2‐GPI–β2‐GPI complexes bind to the platelet thrombus, with consequent amplification of platelet activation 7. It has also demonstrated that inhibition of platelet activation prevents the activation of endothelial cells and fibrin generation 7. Thus, not only in vivo, platelets are a target for circulating anti‐β2‐GPI–β2‐GPI complexes, but also the enhancement of endothelium activation is platelet thrombus‐dependent.
The activation‐signaling pathways of platelets consequent to the presence of β2‐GPI and anti‐β2‐GPI antibodies are mediated by interaction with phospholipids on the cell surface (phosphatidylserine, phosphatidylethanolamine) or with platelet membrane receptors 5, 8. Their combination in complexes contributes to platelet activation in an apolipoprotein E receptor 2′ (apoER2′) and glycoprotein Ib‐α (GPIb‐α)‐dependent manner. ApoER2′ and GPIb‐α are two platelet receptors for β2‐GPI that, in turn, cross‐link anti‐β2‐GPI antibodies 8. This interaction induces the activation of the platelet signaling pathway, expressed by p38 mitogen‐activated protein kinase (MAPK) phosphorylation, and the prothrombotic phenotype, expressed by GP IIb/IIIa conformational change, P‐selectin expression and thromboxane B2 production 8.
Recent evidence showed that platelets can express on their surface tissue factor (TF), the major initiator of the clotting cascade. In particular, it was shown that resting unstimulated platelets express TF mRNA and this protein is enhanced or induced following cell activation 9.
In previous studies we demonstrated that anti‐β2‐GPI antibodies may activate a signaling pathway in human monocytes, involving Toll‐like receptor (TLR)‐4 10. In human platelets the signal transduction pathway involving TLR‐4 is triggered after interaction with the soluble receptor CD14. The intracellular signaling pathway, triggered by TLR‐4, generally involves myeloid differentiation primary response 8 (MyD88) and interleukin (IL)‐1 receptor‐associated kinase (IRAK), leading to nuclear factor kappaB (NF‐κB) activation and consequent cytokine and TF release. In platelets, the intracellular signaling pathway that controls TF pre‐mRNA splicing involves CDC2‐like kinase (Clk)1, an enzyme which was identified in platelets 11, 12.
In this investigation, we analyzed whether anti‐β2‐GPI antibodies may trigger a signal transduction pathway leading to TF expression in platelets from healthy donors and whether platelets of APS patients may constitutively express high levels of TF.
Materials and methods
Platelets preparation
Platelets were prepared from blood samples of healthy donors, with the addition of acid citrate dextrose (ACD) as anti‐coagulant. Platelet‐rich plasma (PRP) was preliminary obtained from the whole blood by centrifugation at 150 g for 15 min at 20°C.
Two‐thirds of the PRP, adding anti‐coagulant to prevent platelet activation, were transferred into a another new sterile tube, without disturbing the buffy coat layer, in order to avoid contamination. PRP was centrifuged at 900 g for 10 min at 20°C (with no brake applied). Platelet‐poor plasma (PPP) was removed and platelet pellets were resuspended in calcium‐free Tyrode's buffer, containing 10% (v : v) ACD. Then, after washing as above, platelets pellets were resuspended in calcium‐free Tyrode's buffer containing bovine serum albumin (BSA, 3 mg/ml), which was tested as β2‐GPI‐free by high‐performance liquid chromatography (HPLC).
Platelet counts were performed by a hemocytometer (Coulter, Beckman Coulter, Brea, CA, USA), which revealed that leukocyte contamination was <1 leukocyte/107 platelets. The purity of the isolated platelets was confirmed by staining with a fluorescein isothiocyanate (FITC)‐conjugated anti‐CD41 antibody (Beckman Coulter) and analyzing by flow cytometry (Coulter Epics, Beckman Coulter, Hialeah, FL, USA).
Purification of anti‐β2‐GPI antibodies
Human anti‐β2‐GPI antibodies were purified by affinity chromatography, as previously reported 10 from three patients [positive for anti‐β2‐GPI antibodies by enzyme‐linked immunosorbent assay (ELISA)] who had been diagnosed as affected by APS according to the Sidney Classification Criteria 13 and, as a control, from three healthy donors. The antibodies displayed lupus anti‐coagulant activity, as revealed by at least two clotting tests with abnormal values that returned to normal values on confirmatory testing. In all tests, the stimulatory effect of the three antibodies was virtually the same (data not shown).
In‐vitro incubation of human platelets with anti‐β2‐GPI antibodies
For in‐vitro studies, platelets were resuspended in calcium‐free Tyrode's buffer containing BSA (3 mg/ml). Purified normal platelets (300 × 106/ml) were incubated at 37°C for different times (10 min, 45 min and 4 h) with human affinity‐purified anti‐β2‐GPI immunoglobulin IgG (200 μg/ml), according to the method of Raschi et al. 14, with normal human serum IgG (200 μg/ml) or lipopolysaccharide (LPS) (100 ng/ml). All materials contained <0·00025 ng of endotoxin/μg of protein, as determined by the Limulus amebocyte lysate test (Associates of Cape Cod, Falmouth, MA, USA).
Interleukin (IL)‐1 receptor‐associated kinase (IRAK) phosphorylation and phospho‐nuclear factor‐kappaB (NF‐kB) assays
Human platelets were left untreated or treated for 10 min, 45 min and 4 h at 37°C with affinity‐purified human anti‐β2‐GPI with normal human serum IgG or with LPS and then resuspended in lysis buffer containing 20 mM HEPES, pH 7.2, 1% Nonidet P‐40, 10% glycerol, 50 mM NaF and 1 mM Na3VO4, including protease inhibitors. For immunoblot, the protein lysates were separated by 7·5% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred electrophoretically to nitrocellulose membrane (Bio‐Rad Laboratories, Richmond, CA, USA). The membrane was blocked at room temperature for 1 h with 25 mM Tris‐HCl, 150 mM NaCl, pH 7.4 and 0·05% Tween‐20 (TBS‐T) containing 3% BSA, and subsequently incubated with polyclonal rabbit anti‐phospho‐IRAK (Cell Signaling, Inc., Danvers, MA, USA) or polyclonal rabbit anti‐phospho‐NF‐κB p65 (Cell Signaling, Inc.).
Antibody binding was visualized by means of horseradish peroxidase (HRP)‐conjugated anti‐rabbit IgG, followed by enhanced chemiluminescence (ECL) reaction using the ECL Western blotting system. In order to adjust for total protein content, the nitrocellulose membranes were stripped and reprobed with polyclonal anti‐IRAK antibody (MBL, Woburn, MA, USA) with anti‐NF‐κB (Cell Signaling, Inc.) or with anti‐actin mAb (Sigma‐Aldrich, St Louis, MO, USA).
TF assay
Platelets were incubated for 10 min, 45 min and 4 h at 37°C with normal human serum IgG, with human affinity‐purified anti‐β2‐GPI or with LPS. Platelets were then lysed as reported above and then subjected to SDS‐PAGE on 10% gels. The proteins were electrophoretically transferred to nitrocellulose membranes, and after blocking with Tris‐buffered saline Tween 20 (TBS‐T) containing 3% BSA were probed with anti‐TF monoclonal antibody (mAb) (NF‐κB Millipore, Darmstadt, Germany). Bound antibodies were visualized with HRP‐conjugated anti‐mouse IgG, and immunoreactivity was assessed by ECL reaction using the ECL Western blotting system.
In parallel experiments, platelets incubated for different times (10 min, 45 min and 4 h) with human affinity‐purified anti‐β2‐GPI, as above, were stained with FITC‐conjugated anti‐TF mAb (American Diagnostica, Greenwich, CT, USA). Fluorescence intensity was analyzed with flow cytometry (Coulter Epics, Beckman Coulter).
Patients
Twenty adult patients classified as affected by APS according to the Sydney Classification Criteria 13, attending the Lupus Clinic, Sapienza University of Rome, were consecutively enrolled.
In addition, 20 healthy subjects (normal blood donors) matched for age were included as controls. This study was approved by the local ethics committees and participants gave written informed consent.
Blood from peripheral veins were collected and platelets were prepared as described above. After washing in phosphate‐buffered saline (PBS), platelets were lysed and then subjected to SDS‐PAGE on 10% gels. TF expression was investigated, as reported above, by Western blotting analysis using anti‐TF mAb (Merck Millipore).
Statistical analysis
Statistical analysis, as shown in Fig. 1 was performed by paired Student's t‐test. All cultures were performed in triplicate. Differences in densitometric values measured on samples from healthy subjects and APS patients were evaluated by means of the Mann–Whitney non‐parametric test. All the statistical analyses of Fig. 3 were performed by GraphPad Prism Software Inc. (San Diego, CA, USA). P‐values ≤ 0·05 were considered significant.
Figure 1.
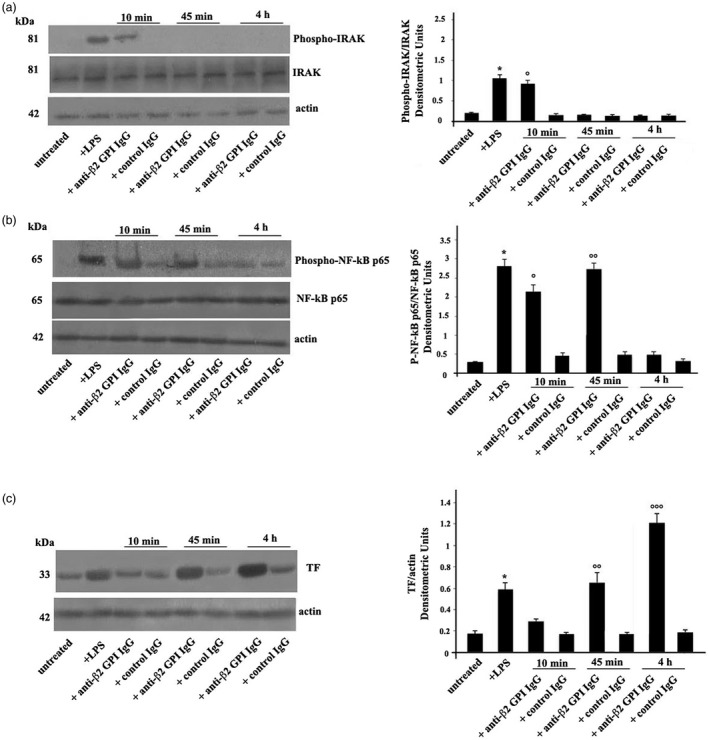
(a) Anti‐β2‐glycoprotein I (GPI) antibodies induce interleukin receptor‐associated kinase (IRAK) phosphorylation. Human platelets from healthy donors, untreated or treated with lipopolysaccharide (LPS) (100 ng/ml), with control human serum immunoglobulin IgG (200 μg/ml), or with human affinity‐purified anti‐β2‐GPI IgG (200 μg/ml) for 10 min, 45 min and 4 h at 37°C, were analyzed by Western blotting using rabbit anti‐phospho‐IRAK. The membrane was stripped and reprobed with polyclonal anti‐IRAK antibody. Loading control was evaluated using anti‐actin monoclonal antibody (mAb). Right panel, densitometric phospho‐IRAK/IRAK ratios are shown. Results represent the mean ± standard deviation (s.d.) from three independent experiments. *P + LPS <0·001 versus untreated, °P + anti‐β2‐GPI IgG (10 min) <0·001 versus untreated. (b) Anti‐β2‐GPI antibodies induce nuclear factor kappaB (NF‐κB) activation. Human platelets, untreated or treated with LPS, with control human serum IgG, or with human affinity‐purified anti‐β2‐GPI IgG for 10 min, 45 min and 4 h, were analyzed by rabbit anti‐phospho‐NF‐κB p65. Loading control was evaluated using anti‐actin mAb. Right panel, densitometric phospho‐NF‐κB p65/NF‐κB p65 ratios are shown. Results represent the mean ± s.d. from three independent experiments. *P + LPS <0·001 versus untreated, °P + anti‐β2‐GPI IgG (10 min) <0·001 versus untreated. °°P + anti‐β2‐GPI IgG (45 min) <0·001 versus untreated. (c) Anti‐β2‐GPI antibodies induce tissue factor (TF) expression. Human platelets, untreated or treated with LPS, with control IgG, or with anti‐β2‐GPI IgG for 10 min, 45 min and 4 h, were analyzed by anti‐TF mAb. Loading control was evaluated using anti‐actin mAb. Densitometric analysis on the right panel. Results represent the mean ± s.d. from three independent experiments. *P + LPS <0·001 versus untreated, °°P + anti‐β2‐GPI IgG (45 min) <0·001 versus untreated. °°°P + anti‐β2‐GPI IgG (4 h) <0·001 versus untreated.
Figure 3.
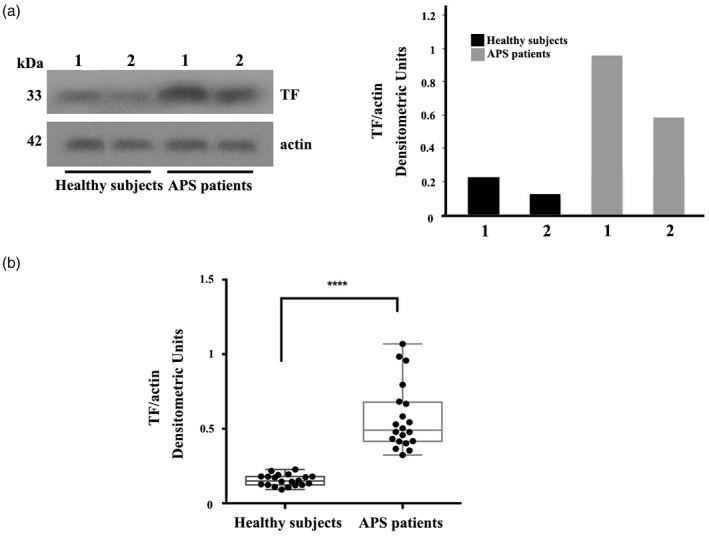
Increase of tissue factor (TF) expression in platelets from anti‐phospholipid syndrome (APS) patients. (a) TF expression in platelets was analysed in 20 samples from APS patients and 20 samples from healthy donors by Western blot using anti‐TF. Western blot and densitometric analysis from two representative samples are shown. (b) Densitometric values of TF/actin ratio calculated in healthy subjects and APS patients are represented and summarized by box‐plot statistics. A statistically significant difference between expression of TF in APS patients versus control subjects was found (P < 0·0001).
Results
Characteristics of patients
The 20 APS patients enrolled into this study were all Caucasian; 13 were primary APS and seven APS associated with systemic lupus erythematosus (SLE). The clinical and demographic characteristics of the enrolled patients are reported in Table 1.
Table 1.
Clinical characteristics of patients studied
| Characteristics n (%) | APS patients |
|---|---|
| (n = 20) | |
| Females/males | 16/4 |
| Age (years) mean ± s.d. | 45·9 ± 11·86 |
| Disease duration (months) mean ± s.d. | 7·14 ± 7·92 |
| Pregnancy morbidity | 3 (18·7) |
| Spontaneous abortions* | 0 |
| Normal fetus deaths** | 3 (100) |
| Premature births*** | 1 (33·3) |
| Vascular thrombosis# | 16 (80) |
| Arterial thrombosis | 10 (50) |
| Venous thrombosis | 8 (40) |
| Recurrent thrombosis | 4 (20) |
| Non‐criteria APS features | |
| Livedo reticularis | 2 (10) |
| Thrombocytopenia | 8 (40) |
| Migraine | 1 (5) |
| Seizures | 3 (15) |
3 or + losses <10 weeks of gestation;
1 or + losses ≥10 weeks of gestation;
preterm birth <34 weeks due to eclampsia, pre‐eclampsia or placental insufficiency.
#Thrombosis (arterial, venous or in small vessels) in any tissue, confirmed by imaging or histopathology (thrombosis without significant inflammation).
Anti‐β2‐GPI antibodies induce IRAK phosphorylation
Western blot analysis using anti‐phospho‐IRAK antibodies showed that affinity‐purified anti‐β2‐GPI IgG induced IRAK phosphorylation as early as 10 min in purified human platelets obtained from healthy donors. As expected by activation control, similar results were obtained upon LPS triggering. Conversely, platelets stimulated with control human IgG virtually did not show anti‐phospho‐IRAK reactivity. Loading control was evaluated using anti‐actin mAb (Fig. 1). Densitometric analysis is reported in the right panel.
Anti‐β2‐GPI antibodies induce NF‐kB activation
Then, we investigated the effects of affinity purified anti‐β2‐GPI IgG on p65 NF‐κB activation. Western blot analysis of platelet lysates revealed that anti‐β2‐GPI IgG, as well as LPS, induced NF‐κB phosphorylation after 10 or 45 min, as revealed by anti‐phospho‐NF‐κB p65 antibody reactivity. Conversely, platelets stimulated with control human IgG showed virtually no anti‐phospho‐NF‐κB p65 reactivity. Loading control was evaluated using anti‐actin mAb (Fig. 1b). Densitometric analysis is reported in the right panel.
Anti‐β2‐GPI antibodies induce TF expression
TF expression was significantly increased in anti‐β2‐GPI‐stimulated platelets compared to untreated platelets as well as platelets stimulated with control human IgG (Fig. 1c). Western blot analysis of TF on platelet lysates obtained from anti‐β2‐GPI antibody‐treated platelets revealed that TF levels were clearly detectable at 45 min with a significant increase after 4 h of anti‐β2‐GPI treatment. Virtually no activation was observed following treatment with control IgG. Loading control was evaluated using anti‐actin mAb. Densitometric analysis is reported in the right panel.
These findings were verified by flow cytometry analysis, which revealed a similar rate of TF expression (Fig. 2a), confirming a significant increase after 45 min or 4 h of anti‐β2‐GPI treatment compared to control IgG (Fig. 2b).
Figure 2.
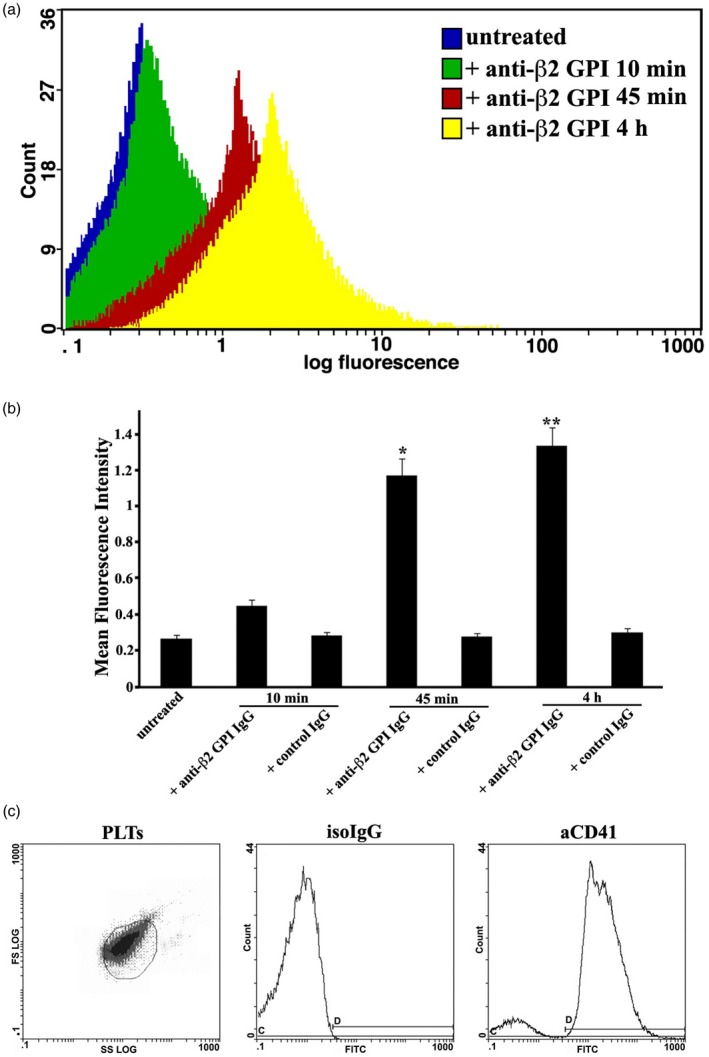
Cytofluorimetric analysis of tissue factor (TF) expression on platelets. (a) Human platelets, untreated or treated with control immunoglobulin IgG, or with anti‐β2‐ glycoprotein I (GPI) IgG for 10 min, 45 min and 4 h, were stained with fluorescein isothiocyanate (FITC)‐conjugated anti‐TF monoclonal antibody (mAb) and analyzed by cytofluorimetric analysis. Platelets were gated on the basis of forward‐ (FSC) and side‐scatter (SSC) parameters. Histograms show the log fluorescence versus the cell number. Cell number is indicated on the y‐axis and fluorescence intensity is represented in three logarithmic units on the x‐axis. Fluorescence intensity was analyzed with a Coulter Epics Beckman cytometer. (b) Results represent the mean ± standard deviation (s.d.) from three independent experiments. *P + anti‐β2‐GPI IgG (45 min) <0·001 versus untreated. **P + anti‐β2‐GPI IgG (4 h) <0·001 versus untreated. (c) Purity of platelets (PLTs) was checked by flow cytometry using fluorescein isothiocyanate (FITC)‐conjugated anti‐CD41 IgG (aCD41) and FITC‐conjugated IgG as isotypic control (isoIgG).
The purity of platelets preparations was analyzed by flow cytometry and shown in Fig. 2c.
Increase of TF expression in platelets from APS patients
Because we demonstrated in vitro that in normal platelets TF expression was significantly increased upon anti‐β2‐GPI stimulation compared with untreated normal platelets, we also decided to analyze TF expression in platelets from 20 patients with APS (Fig. 3). We observed that platelets from patients with APS (both primary and associated to SLE), despite variability among the different patients (Fig. 3a), showed a significantly increased expression of TF compared to healthy donors as assessed by Western blot and confirmed by densitometric analyses. In addition, as represented by box‐plot statistics calculated in healthy subjects and APS patients, Western blot analysis of TF and densitometric analysis revealed a significant difference in protein detection (P < 0·0001, Fig. 3b). No significant difference was found between patients with primary and secondary APS (data not shown).
Discussion
This study demonstrates that human anti‐β2‐GPI antibodies induce TF expression in platelets in which these antibodies are able to trigger a signal transduction pathway(s). Moreover, our results show a significant increase of TF in platelets from APS patients in comparison with control subjects.
Following the demonstration that in endothelial and monocytic cells anti‐β2‐GPI antibodies trigger a signaling cascade comparable to that used by TLR‐4 10, with consequent IRAK phosphorylation and NF‐κB activation, we analyzed whether the same signaling pattern may be activated in human platelets. Using a previously described methodological approach 10, we demonstrated that anti‐β2‐GPI antibodies from APS patients induce IRAK phosphorylation.
The presence of TLR‐4 in platelets is currently well ascertained, and has been shown to play a role in thrombocytopenia induced by systemic LPS 15. Thus, activation of the TLR‐4 signaling pathway in platelets by anti‐β2‐GPI may have specific functional implications, playing a role in some pathological manifestations of the syndrome. Indeed, TLR‐4 is able to signal through both MyD88‐IRAK‐dependent and ‐independent signaling pathways leading to the activation of transcription factors, with consequent functional effects on platelet activity, hemostasis and thrombosis 16.
In addition, we show that, following anti‐β2‐GPI antibody triggering, NF‐κB was activated. Although the role of NF‐κB in platelets is still not completely understood, recent evidence strongly supports the view that NF‐κB activation represents a key signaling pathway in the regulation of platelets physiology 16.
Regarding the molecular mechanisms by which anti‐β2‐GPI antibodies may trigger the signaling transduction pathway in human platelets we can hypothesize that, similarly to monocytes 10, anti‐β2‐GPI antibodies react with their target antigen strictly in association with TLR‐4. Because of the molecular mimicry among β2‐GPI and bacterial antigens or microbial products, adhered β2‐GPI might interact with TLR‐4 10 or, alternatively, anti‐β2‐GPI may cross‐link the molecule and TLR‐4. Indeed, we previously demonstrated that antibodies specific to a peptide of β2‐GPI cross‐react with TLR‐4 17. As a consequence, a proinflammatory and procoagulant phenotype is induced in endothelial cells 14, 18 and monocytes 10. Speculatively, other authors have shown that reduced TF expression in endothelial cells significantly decreased the clinical manifestation of APS in naive mice infused with anti‐β2GPI antibodies 19. Here, we show that platelets may also be involved, as we detected TF expression on their surface, following anti‐β2‐GPI antibody incubation. Taking into account that megakaryocytes are able to confer TF mRNA to platelets 20, TF expression on platelets may be achieved by different mechanisms: (i) fusion of TF containing microparticles with the plasma membrane of activated platelets, (ii) storage in α‐granules with translocation to the cell surface following platelets activation and (iii) neosynthesis of surface TF by activated platelets 9. Thus, we cannot exclude the possibility that platelets can take up TF from other cells, such as activated monocytes. In any case, our present study supports the view that an external stimulus, i.e. anti‐β2‐GPI antibodies, may trigger a signal transduction pathway leading to TF expression on platelet surface. Although the functional role of platelets‐associated TF is still controversial, the possibility arises that it may play a relatively minor role in hemostasis but a major role in pathological thrombosis.
Moreover, we demonstrate that platelets of both primary and secondary APS patients showed a significantly increased expression of TF. To our knowledge, this is the first demonstration of an increase of TF in platelets of APS patients in vivo. In previous studies 21, 22, 23, a similar increase in monocytes of patients with APS was observed.
We now suggest that, on the basis of both in‐vitro and ex‐vivo data, TF on platelets may also contribute to the prothrombotic state detectable in these patients. The TF procoagulant effect may depend upon antibody engagement of PL binding proteins on platelets and the consequent induction of intracellular signals that will result in the up‐regulation of TF. Although we did not demonstrate an association between TF expression on platelets and a specific clinical manifestation of the syndrome, only a follow‐up study, which also analyzes TF mRNA in a wide cohort of patients, including subjects with active thrombosis, could disclose the predictive meaning of this finding. In conclusion, the demonstration of TF expression in human platelets confirms and extends the possibility that different cell types are able to express this protein. Moreover, our findings support the view that platelets play an important role in the pathogenesis of APS, with consequent release of different procoagulant mediators, including TF.
Disclosures
None of the authors have conflicts of interest with regard to the work.
Acknowledgements
This study was supported by grants from Sapienza University, 2015 and 2016, to M. S.
References
- 1. Hughes GR. The anticardiolipin syndrome. Clin Exp Rheumatol 1985; 3:285–86. [PubMed] [Google Scholar]
- 2. Abrahams VM, Chamley LW, Salmon JE. Emerging treatment models in rheumatology: antiphospholipid syndrome and pregnancy pathogenesis to translation. Arthritis Rheum 2017; 69:1710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schreiber K, Sciascia S, de Groot PG et al Antiphospholipid syndrome. Nat Rev Dis Primers 2018; 4:17103. [DOI] [PubMed] [Google Scholar]
- 4. Rand JH. Molecular pathogenesis of the antiphospholipid syndrome. Circ Res 2002; 90:29–37. [DOI] [PubMed] [Google Scholar]
- 5. Baroni G, Banzato A, Bison E, Denas G, Zoppellaro G, Pengo V. The role of platelets in antiphospholipid syndrome. Platelets 2017; 28:762–66. [DOI] [PubMed] [Google Scholar]
- 6. Romay‐Penabad Z, Aguilar‐Valenzuela R, Urbanus RT et al Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood 2011; 117:1408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Proulle V, Furie RA, Merrill‐Skoloff G, Furie BC, Furie B. Platelets are required for enhanced activation of the endothelium and fibrinogen in a mouse thrombosis model of APS. Blood 2014; 124:611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang W, Gao F, Lu D et al Anti‐β2 glycoprotein I antibodies in complex with β2 glycoprotein I induce platelet activation via two receptors: apolipoprotein E receptor 2′ and glycoprotein I bα. Front Med 2016; 10:76–84. [DOI] [PubMed] [Google Scholar]
- 9. Nigel S. Key platelet tissue factor: how did it get there and is it important? Semin Hematol 2008; 45:S16–S20. [DOI] [PubMed] [Google Scholar]
- 10. Sorice M, Longo A, Capozzi A et al Anti‐beta2‐glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum 2007; 56:2687–97. [DOI] [PubMed] [Google Scholar]
- 11. Rondina MT, Schwertz H, Harris ES et al The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost 2011; 9:748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwertz H, Tolley ND, Foulks JM et al Signal‐dependent splicing of tissue factor pre‐mRNA modulates the thrombogenicity of human platelets. J Exp Med 2006; 203:2433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miyakis S, Lockshin MD, Atsumi T et al International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–06. [DOI] [PubMed] [Google Scholar]
- 14. Raschi E, Testoni C, Bosisio D, Borghi MO, Koike T, Mantovani A. Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood 2003; 101:3495–500. [DOI] [PubMed] [Google Scholar]
- 15. Andonegui G, Kerfoot SM, McNagny K, Ebbert KVJ, Patel KD, Kubes P. Platelets express functional Toll‐like receptor‐4. Blood 2005; 106:2417–23. [DOI] [PubMed] [Google Scholar]
- 16. Vallance TM, Zeuner MT, Williams HF, Widera D, Vaiyapuri S. Toll‐like receptor 4 signalling and its impact on platelet function, thrombosis, and haemostasis. Mediators Inflamm 2017; 2017:9605894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colasanti T, Alessandri C, Capozzi A et al Autoantibodies specific to a peptide of β2‐glycoprotein I cross‐react with TLR4, inducing a proinflammatory phenotype in endothelial cells and monocytes. Blood 2012; 120:3360–70. [DOI] [PubMed] [Google Scholar]
- 18. Raschi E, Chighizola CB, Grossi C et al β2‐glycoprotein I, lipopolysaccharide and endothelial TLR4: three players in the two hit theory for anti‐phospholipid‐mediated thrombosis. J Autoimmun 2014; 55:42–50. [DOI] [PubMed] [Google Scholar]
- 19. Blank M, Baraam L, Eisenstein M et al β2‐Glycoprotein‐I based peptide regulate endothelial‐cells tissue‐factor expression via negative regulation of pGSK3β expression and reduces experimental‐antiphospholipid‐syndrome. J Autoimmun 2011; 37:8–17. [DOI] [PubMed] [Google Scholar]
- 20. Brambilla M, Facchinetti L, Canzano P et al Human megakaryocytes confer tissue factor to a subset of shed platelets to stimulate thrombin generation. Thromb Haemost 2015; 114:579–92. [DOI] [PubMed] [Google Scholar]
- 21. Conti F, Spinelli FR, Alessandri C et al Subclinical atherosclerosis in systemic lupus erythematosus and antiphospholipid syndrome. Arterioscler Thromb Vasc Biol 2014; 34:661–8. [DOI] [PubMed] [Google Scholar]
- 22. Conti F, Sorice M, Circella A et al Beta‐2‐glycoprotein I expression on monocytes is increased in anti‐phospholipid antibody syndrome and correlates with tissue factor expression. Clin Exp Immunol 2003; 132:509–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dobado‐Berrios PM, López‐Pedrera C, Velasco F, Aguirre MA, Torres A, Cuadrado MJ. Increased levels of tissue factor mRNA in mononuclear blood cells of patients with primary antiphospholipid syndrome. Thromb Haemost 1999; 82:1578–82. [PubMed] [Google Scholar]