

Summary
Dendritic cells (DCs) are the key professional antigen‐presenting cells which bridge innate and adaptive immune responses, inducing the priming and differentiation of naive to effector CD4+ T cells, the cross‐priming of CD8+ T cells and the promotion of B cell antibody responses. DCs also play a critical role in the maintenance of immune homeostasis and tolerance. DC–T cell interactions underpin the generation of an autoimmune response in rheumatoid arthritis (RA). Here we describe the function of DCs and review evidence for DC and T cell involvement in RA pathogenesis, in particular through the presentation of self‐peptide by DCs that triggers differentiation and activation of autoreactive T cells. Finally, we discuss the emerging field of targeting the DC–T cell interaction for antigen‐specific immunotherapy of RA.
Keywords: antigen presentation, autoantigen‐specific CD4+ T cells, autoimmunity, dendritic cells, immunotherapy, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) has a strong human leucocyte antigen (HLA) class II association: patients carrying HLA‐DR alleles that encode a ‘shared epitope’ (SE) five‐amino acid sequence motif (including HLA‐DRB1*04:01, *04:04, *04:05 and *01:01) exhibit a high risk of seropositive RA. In contrast, other HLA‐DR alleles, including HLA‐DRB1*03:01, are associated with seronegative RA and a milder disease course. The molecular mechanisms underpinning the association between particular HLA alleles and autoantibody positivity are increasingly appreciated, including the presentation of HLA class II‐restricted autoantigens to autoreactive CD4+ T cells. Dendritic cells (DCs) are the key professional antigen‐presenting cells (APCs) for T cell priming. They also play a major role in immune tolerance. DCs discriminate between self‐ and non‐self‐antigens on the basis of associated innate immune activating or suppressive signals, after which DC antigen uptake and presentation promotes T cell activation or regulation. DCs and T cells collaborate in the pathogenesis of RA, particularly through the presentation of antigen that triggers the differentiation of autoreactive T cells, as well as innate immune effector functions.
Based on human and rodent model evidence, we propose a working model of RA, where autoantigen‐specific CD4+ T cells, including follicular helper T cells (Tfh), are primed by major histocompatibility complex (MHC) class II+ DCs exposed to environmental inflammatory factors that enhance their maturation, as well as generation and presentation of neoepitopes. Autoreactive and potentially cross‐reactive Tfh propagate autoimmune arthritis through activation of B cells with genetic tolerance defects, followed by germinal centre formation and affinity maturation and glycosylation of autoantibodies, which contribute to the effector phase of the disease through innate mechanisms. Although partially regulated, the autoimmune response persists due to ongoing stimulation of autoreactive T cell clones by a variety of synovial MHC class II+ APCs and draining lymph node (dLN) DCs (Fig. 1 ). This paper reviews the evidence for the contribution of DCs and T cells to this model.
Figure 1.
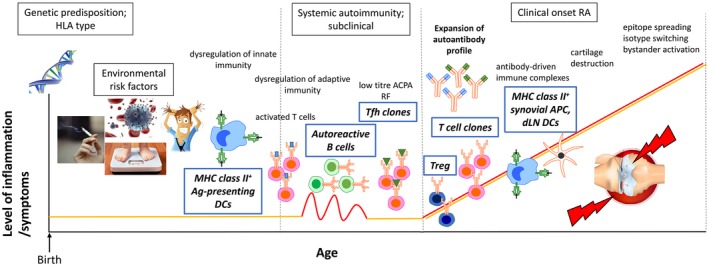
A working model for the development of rheumatoid arthritis (RA). Both genetic and multiple non‐genetic risk factors predispose to the development of RA. In this context, disease initiation probably involves dysregulation of innate and adaptive immunity. Mature major histocompatibility complex (MHC) class II+ dendritic cells (DCs) probably induced by the environmal inflammatory milieu, prime autoantigen‐specific CD4+ T cells, including follicular helper T cells (Tfh) cells. Subsequent germinal centre formation, affinity maturation of B cells and expansion of the autoantibody profile may lead to antibody‐driven immune complex formation and subsequent cartilage destruction coinciding with overt RA expression. Although partially regulated, the autoimmune response persists due to ongoing stimulation of autoreactive T cell clones by a variety of synovial MHC class II+ antigen‐presenting cells (APC) and draining lymph node (dLN) DCs. Downstream processes, including neoepitope formation, isotype switching and bystander activation, may further propagate autoimmunity.
DC antigen presentation
DCs are specialized APCs which link the innate and adaptive immune responses, activating and priming effector CD4+ T cells, cross‐presenting antigen to CD8+ T cells and promoting B cell antibody production 1. DCs also play important roles in the maintenance of immune tolerance. In RA, DCs are thought to drive the activation of self‐peptide‐reactive inflammatory T cells, Tfh and consequently B cells for stimulating autoantibodies 2, 3, 4.
DC subsets for antigen presentation
DCs are heterogeneous and can be divided into two major functionally distinct subsets: conventional myeloid DCs (cDCs) and plasmacytoid DCs (pDCs) 5. Human cDCs express typical myeloid markers and may be subdivided into CD1c+ and CD141+ fractions, while pDCs typically lack expression of these cell surface markers. Functionally, CD1c+ DCs prime CD4+ T cells, while CD141+ DCs cross‐present antigen to prime CD8+ and CD4+ T cells, thus stimulating CD4+ T cell help for cytotoxic T lymphocyte (CTL) generation and B cell activation 6. In contrast to cDCs, pDCs have lower levels of MHC class II expression and reduced APC capacity; however, they are specialized to secrete type I interferons (IFNs), and are therefore important players in viral immunity 1, 7. A further monocyte‐derived inflammatory DC population identified in ascites and synovial fluid co‐expresses CD1c and CD14 8, 9.
DC antigen presentation in immune homeostasis
DCs constitutively phagocytose self‐antigen derived from apoptotic cells during immune homeostasis in the absence of any overt inflammatory stimuli, and migrate from the periphery to draining lymph nodes via tissue lymphatics 10, 11. These apoptotic bodies actively promote regulation by stimulating transforming growth factor (TGF)‐β production, suppressing DC maturation, enhancing self‐antigen‐specific CD4+ peripheral regulatory T cell (Treg) induction and CD8+ T cell deletion. Immune tolerance is maintained through active suppression of inflammatory mediator production and DC activation 12, 13, 14, 15, 16.
DC antigen presentation in response to inflammatory signals
DC recognition of inflammatory stimuli – including pathogen‐associated molecular patterns (PAMPs), endogenous cytokines or damage‐associated molecular pattern molecules (DAMPs) – initiates a signalling cascade leading to activation of the nuclear factor kappa B (NF‐κB) transcription factor pathway (Fig. 2). The NF‐κB family consists of five binding proteins: p50, p52, RelA (p65), cRel and RelB, which can form homo‐ and heterodimers 17. NF‐κB activation comprises classical and alternative pathways, which are stimulated by different signals. The classical pathway (RelA‐p50, cRel‐p50) transcriptionally activates survival and inflammatory genes, and the alternative pathway plays additional roles in immune tolerance and activation of DCs and B cells for adaptive immunity 18, 19. RelB heterodimerizes with p50 for classical pathway or p52 for alternate pathway activation, while RelB‐RelA heterodimers are suppressive.
Figure 2.
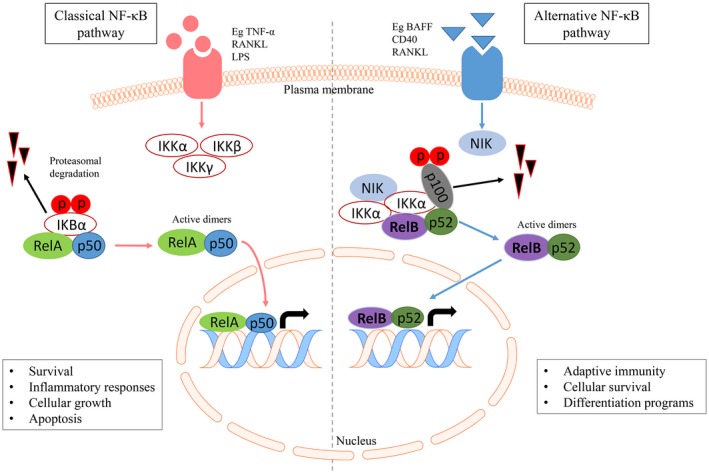
The classical and alternative nuclear factor kappa B (NF‐κB) pathways. The classical NF‐κB pathway (left): activation is induced by many extracellular signals including cytokines such as tumour necrosis factor (TNF). Activation leads to phosphorylation of inhibitory I‐κBα (I‐κB kinase) by the IKK complex, resulting in its degradation. The RelA/p50 complex is freed from the inhibitory interaction with IκBα and is an active dimer, which translocates to the nucleus to activate the transcription of target genes. c‐Rel and RelB also heterodimerize with p50. The alternative NF‐κB pathway (right): activation of this pathway occurs after stimulation by a more restricted set of ligands, including CD40, B cell activating factor (BAFF) and receptor activator of NF‐κB (RANK). Subsequently, NF‐κB‐inducing kinase (NIK) activation phosphorylates IKKα. pIKKα is crucial for phosphorylation of p100, leading to proteasome‐dependent processing of p100 to p52, ultimately liberating active RelB‐p52 dimers, which transactivate NF‐κB responsive genes. This figure was created using the Motfolio PPT Drawing Toolkits (www.motfolio.com), and adapted from 147, 148, 149.
NF‐κB proteins are retained in an inactive form in the cytosol, bound to an I‐κB kinase (IKK) inhibitor protein. Activation of NF‐κB‐inducing kinase (NIK) and other kinases leads to phosphorylation of I‐κB and subsequent nuclear translocation of RelB/p50 and RelB/p52 transcription factor dimers 17, 20. RelB has a unique and critical role in DC maturation, as RelB‐deficient mice have normal numbers of undifferentiated immature DC precursors but are deficient in mature DCs 21, 22. After NF‐κB pathway activation, mature DCs exhibit enhanced capacity for antigen presentation for priming of naive CD4+ and/or CD8+ T cells by up‐regulating surface MHC class II expression as well as adhesion and co‐stimulatory molecules, including CD40, CD80 and CD86, and secreting cytokines to support T cell differentiation 23, 24, 25. RelB transcriptionally regulates CD40 and after CD40 ligation, RelB translocates to the nucleus of DCs for prolonged periods 26.
DC autoantigen presentation in RA
In RA patients, DCs are recruited in high concentrations to joint synovial fluid and tissues 27, 28, 29. These synovial DCs are generally mature and NF‐κB is over‐expressed. Nuclear RelB expression correlates with disease activity, indicating that DCs are responsive to local inflammatory PAMPs, DAMPs and/or cytokines, including tumour necrosis factor (TNF) 19. Multiple subsets of MHC class II+ APCs are present in RA synovial tissue, including Toll‐like receptor (TLR)‐activated myeloid‐derived cells, CD11c+CD20+ activated B cells and MHC class IIhi fibroblast‐like synoviocytes 30. Thus, after initiation of inflammatory arthritis, multiple APC types could perpetuate the presentation of synovial fibroblast or cartilage autoantigens such as vimentin, aggrecan, type II collagen (CII) and HCgp130 to synovial migratory memory T cells previously primed by DCs in lymph nodes draining the site of initial antigen presentation 4.
There are a number of sources for potentially immunogenic autoantigens presented by DCs in the context of RA‐risk HLA‐DR allomorphs, inducing the expansion of self‐peptide‐specific T cells prior to RA expression. These include infectious and self‐protein antigens that are post‐translationally modified by citrullination and carbamylation. The latter processes, enhanced under conditions of stress and damage, expand the production of neoepitopes. The peripheral adaptive immune system may be particularly reactive to neoepitopes if they were not presented by the thymus to negatively select autoreactive T cells during development. Furthermore, histones are citrullinated during the process of neutrophil extracellular trap (NET) formation. In this process, decondensed DNA is expelled from dying neutrophils after their recruitment to sites of infection, resulting in containment of bacteria by a web of extruded material. This process releases extracellular citrullinated autoantigens and possibly also infectious antigens in synovial fluid 31, 32. Citrullination of self‐epitopes derived from vimentin, fibrinogen, aggrecan, enolase, cartilage intermediate layer protein (CILP) and type II collagen (CII) enhances peptide binding to RA‐associated HLA‐DR alleles containing the shared epitope (SE) motif and promotes subsequent autoreactive T cell expansion. Thus, neoepitopes contribute to the molecular basis conferred by SE‐positive (SE+) HLA‐DR alleles to RA susceptibility 33, 34. Furthermore, cross‐presenting DCs taking up citrullinated necrotic antigen under conditions of stress or infection may be co‐stimulated by citrullinated antigen‐specific CD4+ T cells that could provide help to citrullinated peptide‐specific cytotoxic T lymphocytes (CTL) and anti‐citrullinated protein antibodies (ACPA+) B cell germinal centre (GC) differentiation.
DCs as master orchestrators of immune responses
CD1c+ DCs, pDCs and moDCs are all consistently enriched in rheumatoid synovial fluid and tissue 28, 35. RA synovial DCs secrete chemokines that attract proinflammatory immune cells including macrophages, neutrophils and monocytes 4, 36 (Fig. 3). DC secretion of chemokines chemokine (C‐C motif) ligand 3 (CCL3), CCL17, chemokine (C‐X‐C motif) ligand 19 (CXCL19) and CXCL10 also recruits T cells to the RA synovium 37. Enhanced expression of MHC class II and co‐stimulatory molecules (CD40, CD80, CD86) by mature CD1c+ DCs in RA patients is sufficient to trigger T cell activation in vitro 37. In comparison with DCs from healthy controls, RA CD1c+ DCs secrete increased amounts of proinflammatory cytokines interleukin (IL)‐1β, IL‐6, IL‐23 and IL‐12 8, 27, 37, 38. These cytokines are known to induce CD4+ T cell differentiation into activated T helper type 1 (Th1) (IL‐12, IL‐1) and Th17 (IL‐1β, IL‐6 and IL‐23) cells, which are crucial players in RA pathogenesis. Thus, cDCs are partially responsible for the elevated inflammatory Th1 and Th17 cells in RA synovial fluid and tissue 39, 40. DC‐mediated activation and differentiation of these inflammatory T cells potentiate further synovial immune responses; for example, Th17 cells mediate neutrophil recruitment, B cell activation and osteoclastogenesis leading to bone resorption and cartilage degradation 41 and Th1 cells promote macrophage activation 42. In addition, RA synovial DCs are capable of inducing Treg, at least in vitro 43, potentially through prolonged engagement of regulatory T cell receptor programmed cell death 1 (PD‐1) 43, 44. Briefly, PD‐1 is expressed by activated T and B cells 45. After engagement with its ligands programmed death ligand 1 (PD‐L1) and PD‐L2, which are widely expressed in non‐lymphoid tissue, PD‐1 signals a negative feedback loop that attenuates the T cell response and mediates peripheral tolerance 44, 46. Of interest, transcripts of the co‐inhibitory PD‐L1 ligand are highly expressed in RA synovium, but expression of PD‐L1 protein appears to be blocked by serum soluble (s)PD‐L1, which is present at high levels in seropositive RA, thereby down‐regulating the PD‐1/PD‐L1 inhibitory signalling axis 47.
Figure 3.
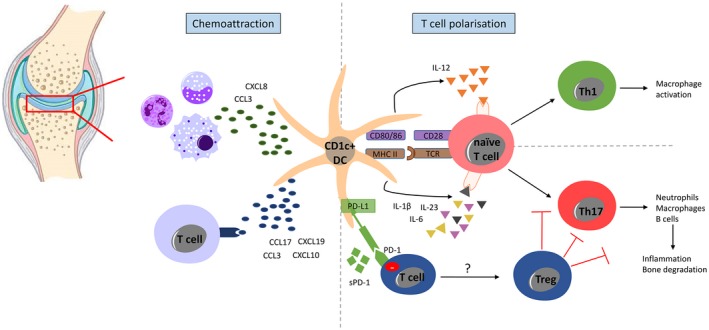
Synovial CD1c+ dendritic cell (DC)‐mediated chemoattraction, T cell activation and polarization. Intra‐articular DCs secrete multiple chemokines, including chemokine (C‐C motif) ligand 3 (CCL3), CCL17, C‐X‐C motif chemokine ligand19 (CXCL19) and CXCL10, to induce T cell migration to the rheumatoid arthritis (RA) synovium. RA synovial CD1c+ DCs also secrete chemokines (including CCL3 and CXCL8) that attract proinflammatory immune cells, including macrophages, neutrophils and monocytes. RA synovial CD1c+ DCs have enhanced expression of human leucocyte antigen D‐related (HLA‐DR) and co‐stimulatory molecules CD40, CD80 and CD86 required for T cell activation. Synovial CD1c+ DCs from RA patients produce cytokines required for T cell polarization towards T helper type 17 (Th17) [interleukin (IL)‐1β, IL‐6, and IL‐23] and Th1 (IL‐12) differentiation. Synovial fluid from seropositive RA patients show high levels of serum soluble (s) programmed cell death 1 (PD‐1), which may interfere with the PD‐1/programmed death ligand 1 (PD‐L1) inhibitory signalling loop. This figure was generated using the Motfolio PPT Drawing Toolkits (www.motfolio.com), and knee image was obtained from https://www.pearsonschoolsandfecolleges.co.uk/FEAndVocational/SportsStudies/ALevel/OCRALevelPE2008/Samples/SamplepagesfromOCRASPEStudentBook/chapter1_sample.pdf, page 7, figure 1.4.
Treg in RA
Treg play an indispensable role in regulating self‐peptide specific T cells. There are two classes of Treg identified in humans: constitutively generated thymic Treg (tTreg) and peripherally induced Treg (pTreg). The transcription factor forkhead box protein 3 (Foxp3) is a critical regulator for CD4+ Treg development and function 48, 49. tTreg can be further delineated in humans as resting Treg (Foxp3loCD45RA+CD25hi rTreg) and activated Treg (Foxp3hiCD45RA–CD25hi aTreg) 50. pTreg may be Foxp3+ antigen‐specific cells induced in response to antigen presented by tolerogenic DCs, or Foxp3– CD4+ T cells that acquire regulatory function in the periphery when exposed to inflammation. These pTreg secrete IFN‐γ and IL‐10 (Tr1 cells) or TGF‐β 51. Induction of pTreg represents an important mechanism for antigen‐specific control of autoreactive T cells escaping selection and entering the periphery in the face of deficient central tolerance or age‐related expansion of autoreactive effector‐memory T cell clones 52.
Treg may regulate in different ways, including deletion and anergy of effector T cells, and generation of new pTreg (‘infectious’ tolerance). For example, murine self‐reactive T cells acquired a folate receptor 4 (FR4)hiCD73hi anergic phenotype in the presence of Foxp3+ Treg 53. In RA patients, CD4+CD25+Foxp3+ Treg with high plasticity for IL‐17 production accumulated in inflamed synovium 54, 55. Moreover, a subset of Foxp3+CD39+ Treg in RA synovial tissue (ST) suppressed the production of IFN‐γ, TNF and IL‐17F but not IL‐17A 56. While peripheral blood (PB) Treg in RA patients were shown to retain suppressive capacity, this capacity was lost in synovial fluid Treg 57. Furthermore, Foxp3+ Treg activated by an inflammatory cytokine milieu produced IL‐17 58. Conversely, TNF inhibition induced a distinct population of Treg, which enhanced regulatory function in RA via TGF‐β 59, 60. Together these results suggest that the inflammatory cytokine milieu contributes to Treg dysfunction, preventing efficient control of autoreactive T cells recognizing self‐antigens.
T cell help for autoantibody production
In the inflamed RA synovium, follicular structures resembling germinal centres (termed ectopic lymphoid‐like structures; ELS) show evidence of sustained activation‐induced cytidine deaminase (AID) expression 61, 62 – an enzyme critical for regulating somatic hypermutation and class‐switch recombination of the immunoglobulin (Ig) genes 62. The detection of class‐switched ACPA, produced by mature B cells resident in ELS in RA synovium, implies cognate antigen‐specific CD4+ T cell help 61. However, antigen‐specific CD4+ T helper cells in ELS have not yet been demonstrated. Moreover, RA ELS showing a synovial lymphoid “pathotype”, characterized by high levels of T and B cell gene expression, were significantly associated with ACPA seropositivity and high disease activity 63, 64.
As seropositive RA is characterized by prominent autoantibody production and T cell–B cell aggregates in the RA synovium, Rao et al. 65 hypothesized that synovial PD‐1+C‐X‐C motif chemokine receptor (CXCR5)– CD4+ T cells observed at increased frequency in seropositive RA patients may be Tfh cells 65. Typically, Tfh cells are identified by the co‐expression of CXCR5, PD‐1 and inducible T cell co‐stimulator (ICOS), and may contribute to autoimmune diseases by facilitating the aberrant generation of autoantibodies and aiding the formation or maintenance of pathogenic ELS 66, 67. After Tfh activation, up‐regulation of CXCR5 in concert with CCR7 down‐regulation are crucial chemokine responsiveness changes that guide activated T cells toward the T cell–B cell border in the GC and to enter the B cell follicle 68. PD‐1, although inhibitory, controls GC positioning and helper functions of Tfh cells 69. PD‐1+CXCR5– RA synovial cells, termed ‘peripheral helper’ T (Tph) cells, were non‐exhausted, activated cells specialized to promote B cell help, plasma cell differentiation and to enhance antibody production. Tph were transcriptomically distinct from Tfh cells, in that they displayed a unique chemokine receptor expression profile, including CCR2, that direct migration to inflamed peripheral sites and thus endow Tph with a unique capacity to interact with B cells within non‐lymphoid structures of the inflamed) synovium.
In RA sera and synovial fluid, ACPA of the IgG isotype have been shown to harbour N‐linked glycans in the variable antigen‐binding domains in both heavy and light chains, and to a much greater extent than other IgG molecules 70. Interestingly, Fab glycosylation of ACPA‐IgG was also observed in patient samples collected at the time of RA diagnosis (disease duration < 1 year). However, none of the ACPA‐IgG N‐glycosylation sites were germline‐encoded, thus suggesting that Fab glycosylation was introduced after extensive somatic hypermutation 70. Furthermore, in contrast to ACPA‐IgG, ACPA‐IgM had no additional glycosylation in the variable region 71. These results strongly suggest that glycosylation of the Fab region of ACPA‐IgG results from somatic mutation during ACPA maturation, could be T cell‐dependent, and may already develop before onset of RA 72.
T cell receptor (TCR) diversity for recognition of antigen
CD4+ and CD8+ surface TCRs recognize antigen processed by APCs and presented by MHC class II and class I molecules, respectively. The TCR is a heterodimer comprising an α and β chain, both of which are formed by somatic rearrangements of variable (V), diversity (D; β chain only) and junctional (J) gene segments 73, 74 (Fig. 4a). Recombined gene segments are spliced together with the constant region (C) to form a functional TCRαβ complex of a single specificity 74.
Figure 4.
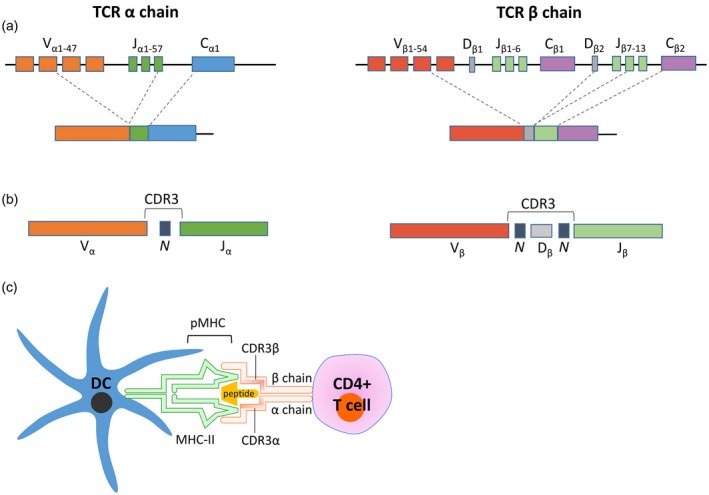
Schematic demonstrating the generation of the T cell receptor, the resultant complementarity‐determining region 3 (CDR3) of both α and β chains and T cell receptor–peptide–major histocompatibility complex (MHC) interaction. (a) Functional T cell receptors (TCRs) comprise an α and a β chain formed by somatic recombination of variable (V), diversity (D; β chain only) and junctional (J) gene segments. Recombined gene segments are then spliced together with the constant region (C) to form the functional TCR‐αβ. (b) CDR3 is formed at the V, (D), J junction, where imprecise V, (D), J joining and random addition of non‐template encoded nucleotides (N) makes this region the most diverse component of the TCR. (c) The heterodimeric TCR on a CD4+ T cell engages peptide displayed in the major histocompatibility complex class II (MHC‐II) on dendritic cells (DC). The CDR3 region is the main TCR domain in contact with the antigenic peptide. Figure (c) was created with the help of Motfolio PPT Drawing Toolkits (www.motfolio.com), and figures adapted from 74, 150.
These somatic gene segment rearrangements result in combinatorial diversity 73, 74. Further random insertion/deletion of nucleotides at the V, (D), J junction during recombination creates junctional diversity of the hypervariable complementarity‐determining region 3 (CDR3) 74, 75 (Fig. 4b). As a result, the CDR3 region of the TCR is unique and can serve as a molecular fingerprint of each T cell clone. The current dogma proposes that the CDR3 loops make the greatest contact with antigenic peptides presented by HLA 76, 77, 78 (Fig. 4c). It is likely that the TCR evolved to position the highly diverse CDR3 residues to where they have maximal contact with HLA‐presented antigen in order to recognize the enormous diversity of antigenic peptides, which is essential for avoiding blind spots during pathogen recognition and for distinguishing between foreign and self‐antigens 74, 79.
TCR repertoire analysis: evidence for autoantigen‐specific CD4+ T cell involvement
While several lines of evidence suggest that self‐antigen‐specific CD4+ T cells are involved in RA pathogenesis, their identification in inflamed RA synovial tissue has been a challenge. Characterization of CD4+ T cells and determining their antigen‐specificity is crucial to understanding their role in autoimmune inflammation, and to develop T cell‐directed immunotherapies.
Elucidation of the TCR repertoire makes it possible to infer which T cells, if any, are expanding in response to potentially autoantigenic stimuli in RA. TCR repertoire biases may already manifest in the naive pool. Proposed mechanisms include convergent recombination, whereby certain TCR sequences are produced at high frequency 80, 81. Furthermore, TCRs with a selective advantage, such as structural features that are optimal for peptide recognition within the context of particular MHC molecules, may provoke emergence of a biased repertoire 82. Preferential (clonal) expansion of particular precursor T cells from the naive repertoire in response to antigen priming can further skew the usage of particular TCRs. This is because expanding daughter T cell clonotypes use an identical nucleotide sequence in the CDR3 region, in addition to identical V, (D) and J genes in TCRα and β chains 74, 83, 84, 85, 86.
TCR bias, oligoclonal T cell expansion in RA patients
In an attempt to unravel the role of antigen‐specific T cells in RA, numerous studies have examined the TCR repertoire in the synovial compartment and PB of RA patients, aiming to identify biased TCRα and/or β usage that would suggest oligoclonal expansions. However, no consensus has emerged (Table 1). There are many possible reasons why these studies failed to elucidate a common TCR clonal signature, including non‐homogeneous patient cohorts comprising individuals of different HLA haplotypes, patients treated with various immunosuppressive therapies, differences in disease stage of recruited patients and dilution of clonal bias by multiple autoantigen specificities in RA.
Table 1.
Summary of studies of TCR αβ V gene usage in rheumatoid arthritis (RA) patients
| Authors | Source | TCRα/β V gene usage |
|---|---|---|
| 137 | PB, ST and SF | all Vβ genes in SF and ST relative to PB |
| 138 | PB and SM | Vβ3, Vβ17, Vβ22 and Vβ4 in SM relative to PB |
| 139 | PB, ST and SF | Vβ6, Vβ15 in ST; Vβ14 in SF; Vβ1, Vβ4, Vβ5.1, Vβ10, Vβ16, Vβ19 in ST, relative to PB |
| 140 | PB and SF | Vβ14 in SF compared to PB |
| 141 | PB and SF | Vβ17 in PB and SF of RA patients compared to healthy controls |
| 142 | PB and SF | Vβ2, Vβ6; Vβ13.1 and Vβ13.2 in SF compared to PB |
| 143 | PB and SF | Vβ7 in SF compared to PB |
| 144 | PB and SF | Vα10, Vα15, Vα18; Vβ4, Vβ5, Vβ13 in SF compared to PB |
| 145 | PB, ST and SF | Vβ14 and Vβ16 in ST; Vβ16 in SF, relative to PB |
| 146 | SF | Vα17 |
PB = peripheral blood; ST = synovial tissue; SF = synovial fluid.
By application of powerful technologies in more homogeneous groups of RA patients, the TCR repertoire diversity in PB, particularly within the memory CD4+ T cell compartment, was shown to be significantly reduced in SE+ RA compared with SE– RA patients and healthy controls 87. This negative correlation was dose‐dependent, where the lowest TCR diversity occurred in RA patients carrying two SE alleles. These findings complement those of Scally et al. 33, wherein the SE was shown to permit the binding and presentation of RA‐associated peptides, associated with enrichment in peripheral blood of memory CD4+ T cells recognizing autoantigenic peptides presented in the SE. Expansion of such clonotypes contributes to reduced TCR diversity observed in SE+ RA patients 34, 87.
At the target site of autoimmune inflammation, TCR repertoire profiling in the synovial tissue (ST) of recent‐onset RA patients revealed several highly expanded clones 88. Furthermore, the degree of clonal dominance (exclusive expansion of a few clones) observed in recently diagnosed RA patients was significantly greater than in established RA patients 88. These findings suggest the contribution of a limited number of expanded T cell clones to early RA pathogenesis, and highlight that clonotypical analysis of ST harvested from patients with preclinical RA may be fruitful. Of the oligoclonal expansions found in a recent study of RA ST, the same expanded TCRβ clones dominated at different locations within the single inflamed joint as well as in different joints when compared to the paired PB repertoire 89. These findings suggest that there is widespread TCR recognition of the same antigens presented by APCs in ST.
In this regard, a small population of ‘circulating pathogenic‐like lymphocytes’ (CPLs) has been identified in blood of patients affected by RA, juvenile idiopathic arthritis (JIA) and healthy individuals 90, 91. CPLs express an activated, antigen‐experienced T cell signature, are capable of recirculating through inflamed sites, retain proinflammatory capacity and correlate with disease activity in both JIA and adult RA. Notably, CPLs have reduced TCR diversity and share a high fraction of CDR3 sequences with synovial T cells, consistent with antigen‐specific expansion in situ. Furthermore, a subset of synovial inflammation‐associated (ia) suppressive Treg cells, which appear to recirculate through inflamed sites of patients with JIA and adult RA, expand during inflammation and exhibit lower TCR diversity than other Treg, demonstrating an antigen‐driven response 92. While iaTreg are ontogenically related to the Treg cell lineage, they share substantial TCR repertoire overlap with pathogenic synovial effector T cells (Teff) and blood CPLs 92. These findings imply that iaTreg selectively expand in active disease from Teff, consistent with the origin of Treg type 1 (Tr1) cells 93. iaTreg may be unsuccessful at restraining these effector responses in vivo due to Teff resistance to Treg‐mediated suppression – such as protein kinase B hyperactivation in JIA and secretion of TNF‐related apoptosis‐inducing ligand (TRAIL) in RA by effector cells – in combination with the proinflammatory milieu 94, 95, 96, 97.
The deep sequencing technologies used for TCR repertoire profiling in these studies are advantageous in that these methods do not rely on prior (auto)antigen knowledge, which poses a major hurdle in autoimmune diseases where multiple putative autoantigens exist or where autoantigens are still yet to be defined 98, 99. Furthermore, the low natural frequency of antigen‐specific T cells can add serious constraints on the number of cells analysed, limiting depth of repertoire analysis. However, as perturbations of the TCR repertoire often occur as a result of transient or chronic infection, a greater understanding of the fine epitope‐specificity of the oligoclonally expanded T cell clones in the aforementioned studies is needed to determine their contribution to autoimmune pathogenesis 100.
Autoantigen‐specific CD4+ T cells in RA
In this regard, MHC class II multimers have emerged as a valuable tool for identifying and characterizing CD4+ T cells of a particular specificity, albeit a smaller, predefined subset of the repertoire. In the RA context, tetramers comprise fluorescently labelled multivalent complexes of HLA‐DR susceptibility molecules loaded with putative autoantigenic peptides (pMHCII) which bind the appropriate antigen‐specific CD4+ T cell receptor (Fig. 4a). As the pMHCII multimers are fluorescently labelled, CD4+ T cells specific for the (auto)antigen can be detected and quantified by flow cytometry 101, 102 (Fig. 4b).
Tetramer staining identified vimentin, cit‐vimentin and cit‐aggrecan‐reactive CD4+ T cells in the PB of RA patients, ACPA– first‐degree relatives (FDR) and also healthy controls, all HLA‐matched with RA susceptibility alleles 33, 34, 103. While tetramer+ CD4+ T cells from RA patients and FDR displayed an effector‐memory phenotype, those from healthy controls were more likely to be naive or Foxp3+. This suggests that while self‐antigen‐specific CD4+ T cells circulate in healthy individuals not suffering from autoimmune disease, there is a T cell regulatory capacity to control these autoreactive CD4+ T cells in healthy controls that is deficient in RA patients 33. Single‐cell sorting and TCR sequencing of tetramer+ CD4+ T cells identified repeated CDR3α and CDR3β sequences, indicating antigen‐driven oligoclonal expansion. In Indigenous North American subjects at high risk of RA, oligoclonal expansion, including public clonotypes, were observed both in RA patients and ACPA– FDR, suggesting that self‐peptide‐specific T cell expansion occurs in individuals with high‐risk HLA during the preclinical period prior to the development of ACPA 34 (Fig. 5).
Figure 5.
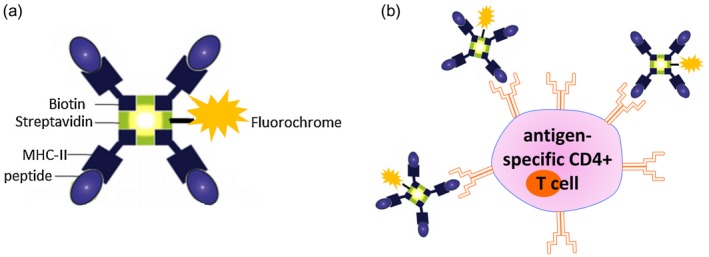
Class II tetramers as tools for antigen‐specific CD4+ T cell detection. (a) Schematic depicting the structure of a major histocompatibility complex (MHC) class II tetramer. Four identical biotinylated MHC class II molecules loaded with a candidate (auto)antigenic peptide are linked to a central streptavidin molecule. The central streptavidin is conjugated to a fluorochrome of interest. (b) Binding of fluorescently labelled class II tetramer molecules to the antigen‐specific CD4+ T cell via the T cell receptor (TCR) enables detection of antigen‐specific T cells by flow cytometry. Figure (b) was created with the help of Motfolio PPT Drawing Toolkits (www.motfolio.com), and figures adapted from 151 and https://www.intechopen.com/books/allergen/strategies-to-study-t-cells-and-t-cell-targets-in-allergic-disease, chapter 7, page 128, figure 1.
Antigen‐specific T cells in mouse models of RA
There is evidence in mouse models that an adaptive immune response commences prior to disease onset and then continues. Transgenic HLA‐DR1 humanized mouse models with collagen‐induced arthritis (CIA) indicate that, at disease onset, autoantigen‐specific effector CD4+ T cells preferentially use TCRβ chain variable genes (TRBV) 31 and 13, are highly clonal based on CDR3 length polymorphism analysis and migrate to arthritic joints 104. Selective use of TRBV genes results from clonal expansion due to recognition by the preferentially used TCRαβ of the dominant type II collagen257–274 epitope presented by HLA‐DR1, rather than predisposition to autoreactivity due to selection of a pathogenic naive repertoire by the DR1 transgene 104. However, it is unclear how autoantigen‐specific CD4+ T cells contribute to arthritis pathogenesis in the predisease period prior to the development of autoantibodies.
Conversely, the arthritogenic potential of antibodies in the effector phase has been demonstrated in animal models: transfer of autoantibodies present in serum are sufficient to induce arthritic disease 105, 106, 107. Downstream antibody‐driven innate mechanisms, including immune complexes which trigger vascular leak and Fc‐dependent mast cell activation, as well as macrophage and complement activation, together have been shown to provoke inflammatory arthritis in mouse models 108, 109, 110.
A novel murine transgenic (Tg) antigen‐induced arthritis model, in which ovalbumin (OVA)‐specific CD4+ TCR transgenic cells are adoptively transferred, provides insight into the contribution of antigen‐specific T cells to experimental arthritis. After immunization with OVA, heat‐aggregated ovalbumin (HAO) is injected into the footpad. Th1 OVA‐specific T cells recruited to the footpad are capable of breaking self‐tolerance to joint‐specific antigens and inducing mild arthritis, associated with the production of antibodies against CII, demonstrating bystander self‐antigen activation during an immune response to foreign antigen 111. HAO challenge, in the presence or absence of non‐specific inflammatory LPS, increased CII‐specific T cell proliferative autoreactivity and anti‐CII, anti‐ssDNA and rheumatoid factor (RF) autoantibody titres 112. Challenge with non‐specific inflammatory lipopolysaccharide (LPS) alone was insufficient to break self‐tolerance. The authors concluded that non‐self‐antigen‐specific T cell responses (such as might occur during infection) when recruited into the joint, and when boosted by pathogen‐associated molecules, may disrupt local immunoregulation 112. Bacterial DNA and peptidoglycans are detectable in synovial tissue of RA patients, which could stimulate both innate and adaptive immune responses 113. In an inflammatory setting, breakdown of tolerance to self‐antigen may occur when infectious antigen‐specific T cells provide help to DCs cross‐presenting both self‐ and foreign antigens. Autoreactive Tfh may in turn provide help to autoreactive B cell clones to stimulate their differentiation. In addition, inflammation driven by innate or T cell‐driven mechanisms may promote cartilage destruction, resulting in the uptake of neoepitopes by DCs, triggering epitope spreading to other T cell specificities 112. Interestingly, in this model, transferred OVA‐specific T cells could drive activation of autoreactive B cells and production of autoantibodies, thus contradicting the requirement for cognate T cell–B cell interactions to break tolerance. Autoreactive B cells may elicit help from activated OVA‐specific T cells in a bystander manner, or autoreactive B cells may co‐capture and present HAO and self‐antigen liberated in the inflamed joint to mount an autoantibody response dependent on OVA‐specific T cells, as has been recently reported in other mouse models of autoimmune disease 114.
Abatacept is a soluble fusion protein comprising the extracellular domain of cytotoxic T lymphocyte 4 (CTLA‐4) linked to the Fc domain of human IgG (CTLA‐4‐Ig) used as a biological DMARD in RA. Abatacept binds to CD80/86 expressed by mature DCs with higher affinity than CD28, thereby interfering with co‐stimulation obligatory for full T cell activation 115, 116. CTLA‐4‐Ig prevents CIA, experimental autoimmune encephalomyelitis and murine lupus in vivo 117, 118, 119. Platt et al. 120 demonstrated that abatacept inhibits T cell activation, proliferation and inflammatory mediator production [IFN‐γ, IL‐17, macrophage inflammatory protein (MIP)‐1α, IL‐13] by cells in the joint and draining lymph nodes. Abatacept also suppressed the acquisition of a Tfh phenotype and reduced the proportion of antigen‐specific T cells migrating into B cell follicles: the few antigen‐specific T cells were restricted to the paracortical T cell zone, due probably to their inability to migrate to CXCL13 via CXCR5. Consistent with the expression of CD80/CD86 by activated B cells, abatacept also inhibited the endogenous breach of B cell self‐tolerance, with reduction in GC formation and autoantibody tires 120.
In this model, the majority of activated T cells infiltrating the inflamed joint were not OVA antigen‐specific 121. However, the adoptively transferred, OVA antigen‐specific population and a subpopulation of endogenous T cells interacted for prolonged periods with synovial DCs consistent with cognate antigen recognition. The articular T cells used a diverse range of TRBV genes and only one or two CDR3 clones were represented per TRBV, suggesting that T cell activation and clonal expansion occurred in secondary lymphoid organs prior to the migration and selective accumulation of particular clones in the joint. While abatacept seemed to interfere with priming of potentially autoreactive (Tfh) T cells in joint dLNs, it did not alter endogenous T cell–DC interactions in the inflamed joint.
Quality of DC–T cell interaction
As mentioned earlier, citrullination of self‐antigens modulates the quality of MHC binding and TCR signalling. For example, citrullinated aggrecan reduced T cell proliferation but enhanced Th17 differentiation, which was promoted by low‐strength TCR signalling 122. TCR signalling strength can also determine the severity of arthritis in murine models of RA 123, 124. In recent‐onset HLA‐DR SE+ RA patients, T cells were more likely to produce IL‐6 in response to cit‐aggrecan than cit‐vimentin or cit‐fibrinogen epitopes 101. Hyperactivity of the rat sarcoma extracellular signal‐regulated kinase (Ras‐Erk) pathway was shown to prevent negative regulation of TCR signalling by the phosphatase SHP‐1, leading to sustained TCR signalling in RA patients 125. Other genetic polymorphisms associated with RA may impact the quality of TCR signalling and immune synapse formation. RA‐associated polymorphisms in CD28, CTLA4, protein tyrosine phosphatase, non‐receptor type 22 (PTPN22), protein kinase C theta (PRKCQ) and CD247 (TCRζ) all impact TCR signalling 124, 126. Perturbations to the function of these molecules may impact TCR signalling thresholds and therefore the ensuing immune response. Conversely, RA‐associated polymorphisms in MHC class II, REL, CD40, peptidyl arginine deiminase type IV (PADI4), IL12B, TNF‐α‐induced protein 3 (TNFAIP3), CD83, CCL21, IL‐1 receptor‐associated kinase 1 (IRAK1) and NFKB Inhibitor Epsilon (NFKBIE) may impact DC activation and signal transmission to T cells. Modulating the quality of interactions between DCs and T cells is therefore an attractive therapeutic target for RA.
Antigen‐specific immunotherapy targeting the DC–T cell interaction
Despite considerable scientific and therapeutic advances, currently available treatment agents – although effective in reducing inflammation – do not address the upstream immune dysfunctions initiating the pathogenic processes. The collaborative interaction between specific autoantigen presentation by DCs to autoreactive T cells is a major untapped target in RA, which has been exploited in several recent and ongoing clinical trials to restore immune tolerance. If effective, tolerance‐inducing antigen‐specific immunotherapy would have lower toxicity and would represent a longer‐term solution to controlling or preventing autoimmune disease. Such strategies would specifically target the autoimmune disease process involving RA‐specific antigens presented by specific high‐risk HLA‐DR SE molecules and autoimmunity based on autoantibody positivity.
As discussed, DCs present RA autoantigens to T cells, and DCs with regulatory properties can promote the induction of pTreg. An important body of work demonstrated that certain subsets of DCs in vivo, such as CD103+ intestinal DCs, promote pTreg 127. Furthermore, after exposure to antigen, RelB‐deficient myeloid DCs or DCs generated in the presence of various NF‐κB suppressive drugs (‘tolerogenic DCs’) could restore antigen‐specific tolerance in primed mice or mice with antigen‐induced inflammatory arthritis 128, 129, 130. In the first Phase I proof‐of‐concept clinical trial of antigen‐specific DC immunotherapy, HLA‐DR SE+ ACPA+ RA patients were treated with autologous PB DCs modified with NF‐κB inhibitor, BAY11‐7082, and exposed to four citrullinated peptide autoantigens. Treatment was safe, and reduced both inflammation and CD4+ effector T cells and increased the Treg to effector T cell ratio. In treated subjects, ex‐vivo IL‐6 production to cit‐vimentin447–455 was suppressed 131. A second trial of tolerogenic DCs exposed to antigens in autologous synovial fluid also demonstrated safety and feasibility 132, and further DC trials are ongoing (see clinicaltrials.gov). These trials begin to build a mechanistic understanding around the potential for DC‐based antigen‐specific immunotherapies to rebalance antigen‐specific regulatory to effector T cells, and highlight the need for sensitive, standardized and clinically qualified immunological assays, such as tetramers, to determine the pharmacodynamic immunological effects of antigen‐specific therapies so that effects can be evaluated and compared in early‐stage trials 102.
What might the future hold for patients with and/or at risk of RA? Murine proof‐of‐concept studies show that DCs or T cells may be targeted in situ for induction of tolerance. In mice, liposomes encapsulating mBSA antigen and an NF‐κB inhibitor (curcumin, quercetin or BAY11‐7082) suppressed mBSA antigen‐induced inflammatory arthritis in an antigen‐specific manner. The liposomes suppressed antigen‐specific T cells and induced antigen‐specific pTreg 133. A Phase I clinical trial is in progress in patients with HLA‐DRB1*04:01 and 01:01+ RA to ascertain safety and immunomodulatory effects of liposomes encapsulating the collagen II259–273 epitope (restricted by these HLA‐DR allomorphs) and NF‐κB inhibitor 1α,25‐dihydroxyvitamin D3 (calcitriol) (see anzctr.org.au). Mouse models demonstrate other potential uses for immune tolerance in RA: PLGA nanoparticles encapsulating rapamycin delivered with adalimumab suppressed the development of anti‐drug antibodies and improved drug efficacy in inflammatory arthritis 134, 135. In an alternative approach, iron nanoparticles coated with peptide‐MHC molecules directly targeted autoreactive TCR to induce antigen‐specific Treg and suppress inflammatory arthritis 136. Of interest, these studies show that just as bystander activation can be sufficient to activate autoreactive T cells, Tr1 cells of a single autoantigen specificity are sufficient to regulate autoreactive T cells of multiple specificities.
Thus, basic and translational studies indicate that DC antigen presentation to T cells is a ripe area for future drug development in RA. The field is moving increasingly towards precise definition of target populations, more sophisticated immunophenotypical characterization of patients prior to treatment and more consistent application of immunomonitoring to clinical trials. If successful, immunotherapies targeting the DC‐antigen–T cell interaction should deliver improved safety, specificity and durability of disease control.
Disclosures
R. T. has filed provisional patents surrounding technology for targeting DCs for antigen‐specific tolerance, and is a director of the spin‐off company, Dendright, which is commercializing immunotherapy to target DCs to suppress rheumatoid arthritis in collaboration with Janssen Biotech Inc. R. T. has also received speaker fees and/or consulting fees from Janssen and Abbvie.
Acknowledgements
This study was supported by NHMRC grant no. 1083192 and is part of a project that has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 777357. This Joint Undertaking receives support from the European Union's Horizon 2020 research and innovation programme and EFPIA. R. T. is supported by Arthritis Queensland and a Fellowship from NHMRC.
References
- 1. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 2013; 31:563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol 2013; 13:566–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lutzky V, Hannawi S, Thomas R. Cells of the synovium in rheumatoid arthritis. Dendritic cells. Arthritis Res Ther 2007; 9:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu MB, Langridge WHR. The function of myeloid dendritic cells in rheumatoid arthritis. Rheumatol Int 2017; 37:1043–51. [DOI] [PubMed] [Google Scholar]
- 5. Ziegler‐Heitbrock L, Ancuta P, Crowe S et al Nomenclature of monocytes and dendritic cells in blood. Blood 2010; 116:e74–80. [DOI] [PubMed] [Google Scholar]
- 6. Reynolds G, Haniffa M. Human and mouse mononuclear phagocyte networks: a tale of two species? Front Immunol 2015; 6:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siegal FP, Kadowaki N, Shodell M et al The nature of the principal type 1 interferon‐producing cells in human blood. Science 1999; 284:1835–7. [DOI] [PubMed] [Google Scholar]
- 8. Segura E, Touzot M, Bohineust A et al Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013; 38:336–48. [DOI] [PubMed] [Google Scholar]
- 9. Reynolds G, Milne P, Bigley V et al Use of the dendritic cell marker, B and T lymphocyte attenuator, to identify functionally distinct subsets of human CD1c+ dendritic cells. Lancet 2016; 387:S85. [Google Scholar]
- 10. Smith JB, McIntosh GH, Morris B. The traffic of cells through tissues: a study of peripheral lymph in sheep. J Anat 1970; 107(Pt 1):87–100. [PMC free article] [PubMed] [Google Scholar]
- 11. Pugh CW, MacPherson GG, Steer HW. Characterization of nonlymphoid cells derived from rat peripheral lymph. J Exp Med 1983; 157:1758–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang F‐P, Platt N, Wykes M et al A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to t cell areas of mesenteric lymph nodes. J Exp Med 2000; 191:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med 2000; 191:423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF‐beta, PGE2, and PAF. J Clin Invest 1998; 101:890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen‐specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med 2001; 193:233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10–producing, nonproliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med 2000; 192:1213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oeckinghaus A, Ghosh S. The NF‐κB family of transcription factors and its regulation. Cold Spring Harbor Perspec Biol 2009; 1:a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neumann M, Fries H‐W, Scheicher C et al Differential expression of Rel/NF‐κB and octamer factors is a hallmark of the generation and maturation of dendritic cells. Blood 2000; 95:277–85. [PubMed] [Google Scholar]
- 19. Pettit AR, Quinn C, MacDonald KP et al Nuclear localization of RelB is associated with effective antigen‐presenting cell function. J Immunol 1997; 159:3681–91. [PubMed] [Google Scholar]
- 20. Ghosh S, May MJ, Kopp EB. NF‐kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998; 16:225–60. [DOI] [PubMed] [Google Scholar]
- 21. Burkly L, Hession C, Ogata L et al Expression of relB is required for the development of thymic medulla and dendritic cells. Nature 1995; 373:531–6. [DOI] [PubMed] [Google Scholar]
- 22. DiMolfetto L, Reilly C, Wei Q, Lo D. Dendritic‐like cells from relB mutant mice. Adv Exp Med Biol 1997; 417:47–54. [DOI] [PubMed] [Google Scholar]
- 23. Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 1991; 9:271–96. [DOI] [PubMed] [Google Scholar]
- 24. Hartmann G, Weiner GJ, Krieg AM. CpG DNA: a potent signal for growth, activation, and maturation of human dendritic cells. Proc Natl Acad Sci 1999; 96:9305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cella M, Scheidegger D, Palmer‐Lehmann K et al Ligation of CD40 on dendritic cells triggers production of high levels of interleukin‐12 and enhances T cell stimulatory capacity: T‐T help via APC activation. J Exp Med 1996; 184:747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Sullivan BJ, Thomas R. CD40 ligation conditions dendritic cell antigen‐presenting function through sustained activation of NF‐κB. J Immunol 2002; 168:5491–8. [DOI] [PubMed] [Google Scholar]
- 27. Lebre MC, Jongbloed SL, Tas SW et al Rheumatoid arthritis synovium contains two subsets of CD83–DC–LAMP– dendritic cells with distinct cytokine profiles. Am J Pathol 2008; 172:940–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thomas R, Davis LS, Lipsky PE. Rheumatoid synovium is enriched in mature antigen‐presenting dendritic cells. J Immunol 1994; 152:2613–23. [PubMed] [Google Scholar]
- 29. Thomas R, MacDonald KPA, Pettit AR, Cavanagh LL, Padmanabha J, Zehntner S. Dendritic cells and the pathogenesis of rheumatoid arthritis. J Leukoc Biol 1999; 66:286–92. [DOI] [PubMed] [Google Scholar]
- 30. Zhang F, Wei K, Slowikowski K et al Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single‐cell transcriptomics and mass cytometry. bioRxiv 2018. 10.1101/351130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sorensen OE, Borregaard N. Neutrophil extracellular traps – the dark side of neutrophils. J Clin Invest 2016; 126:1612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spengler J, Lugonja B, Jimmy Ytterberg A et al Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol 2015; 67:3135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scally SW, Petersen J, Law SC et al A molecular basis for the association of the HLA‐DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med 2013; 210:2569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scally SW, Law S‐C, Ting YT et al Molecular basis for increased susceptibility of Indigenous North Americans to seropositive rheumatoid arthritis. Ann Rheum Dis 2017; 76:1915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jongbloed SL, Lebre MC, Fraser AR et al Enumeration and phenotypical analysis of distinct dendritic cell subsets in psoriatic arthritis and rheumatoid arthritis. Arthritis Res Ther 2006; 8:R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prevosto C, Goodall JC, Hill Gaston JS. Cytokine secretion by pathogen recognition receptor‐stimulated dendritic cells in rheumatoid arthritis and ankylosing spondylitis. J Rheumatol 2012; 39:1918–28. [DOI] [PubMed] [Google Scholar]
- 37. Moret FM, Hack CE, van der Wurff‐Jacobs KMG et al Intra‐articular CD1c‐expressing myeloid dendritic cells from rheumatoid arthritis patients express a unique set of T cell‐attracting chemokines and spontaneously induce Th1, Th17 and Th2 cell activity. Arthritis Res Ther 2013; 15:R155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Santiago B, Izquierdo E, Rueda P et al CXCL12gamma isoform is expressed on endothelial and dendritic cells in rheumatoid arthritis synovium and regulates T cell activation. Arthritis Rheum 2012; 64:409–17. [DOI] [PubMed] [Google Scholar]
- 39. Yamada H, Nakashima Y, Okazaki K et al Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis 2008; 67:1299–304. [DOI] [PubMed] [Google Scholar]
- 40. Arroyo‐Villa I, Bautista‐Caro MB, Balsa A et al Frequency of Th17 CD4+ T cells in early rheumatoid arthritis: a marker of anti‐CCP seropositivity. PLoS ONE 2012; 7:e42189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Azizi G, Jadidi‐Niaragh F, Mirshafiey A. Th17 cells in immunopathogenesis and treatment of rheumatoid arthritis. Int J Rheum Dis 2013; 16:243–53. [DOI] [PubMed] [Google Scholar]
- 42. Hume DA. The many alternative faces of macrophage activation. Front Immunol 2015; 6:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Estrada‐Capetillo L, Hernandez‐Castro B, Monsivais‐Urenda A et al Induction of Th17 lymphocytes and Treg cells by monocyte‐derived dendritic cells in patients with rheumatoid arthritis and systemic lupus erythematosus; Clin Dev Immunol 2013. doi:10.1155/2013/584303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moret FM, Hack CE, Vander Wurff‐Jacobs K et al Thymic stromal lymphopoietin, a novel proinflammatory mediator in rheumatoid arthritis that potently activates CD1c+ myeloid dendritic cells to attract and stimulate T cells. Arthritis Rheumatol 2014; 66:1176–84. [DOI] [PubMed] [Google Scholar]
- 45. Agata Y, Kawasaki A, Nishimura H et al Expression of the PD‐1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 1996; 8:765–72. [DOI] [PubMed] [Google Scholar]
- 46. Keir ME, Liang SC, Guleria I et al Tissue expression of PD‐L1 mediates peripheral T cell tolerance. J Exp Med 2006; 203:883–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo Y, Walsh AM, Canavan M et al Immune checkpoint inhibitor PD‐1 pathway is down‐regulated in synovium at various stages of rheumatoid arthritis disease progression. PLOS ONE 2018; 13:e0192704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299:1057–61. [DOI] [PubMed] [Google Scholar]
- 49. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4:330–6. [DOI] [PubMed] [Google Scholar]
- 50. Miyara M, Yoshioka Y, Kitoh A et al Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009; 30:899–911. [DOI] [PubMed] [Google Scholar]
- 51. Lan Q, Fan H, Quesniaux V et al Induced Foxp3(+) regulatory T cells: a potential new weapon to treat autoimmune and inflammatory diseases? J Mol Cell Biol 2012; 4:22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nguyen TL, Sullivan NL, Ebel M, Teague RM, DiPaolo RJ et al Antigen‐specific TGF‐beta‐induced regulatory T cells secrete chemokines, regulate T cell trafficking, and suppress ongoing autoimmunity. J Immunol (Balt 1950) 2011; 187:1745–53. [DOI] [PubMed] [Google Scholar]
- 53. Martinez RJ, Zhang N, Thomas SR, Nandiwada SL, Jenkins MK, Binstadt BA. Arthritogenic self‐reactive CD4+ T cells acquire an FR4hiCD73hi anergic state in the presence of Foxp3+ regulatory T cells. J Immunol 2012; 188:170–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mottonen M, Heikkinen J, Mustonen L, Isomaki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol 2005; 140:360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Komatsu N, Okamoto K, Sawa S et al Pathogenic conversion of Foxp3(+) T cells into TH17 cells in autoimmune arthritis. Nat Med 2014; 20:62–8. [DOI] [PubMed] [Google Scholar]
- 56. Herrath J, Chemin K, Albrecht I, Catrina AI, Malmström V. Surface expression of CD39 identifies an enriched Treg‐cell subset in the rheumatic joint, which does not suppress IL‐17A secretion. Eur J Immunol 2014; 44:2979–89. [DOI] [PubMed] [Google Scholar]
- 57. Wang T, Sun X, Zhao J et al Regulatory T cells in rheumatoid arthritis showed increased plasticity toward Th17 but retained suppressive function in peripheral blood. Ann Rheum Dis 2015; 74:1293–301. [DOI] [PubMed] [Google Scholar]
- 58. Beriou G, Costantino CM, Ashley CW et al IL‐17‐producing human peripheral regulatory T cells retain suppressive function. Blood 2009; 113:4240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nie H, Zheng Y, Li R et al Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF‐alpha in rheumatoid arthritis. Nat Med 2013; 19:322–8. [DOI] [PubMed] [Google Scholar]
- 60. Nadkarni S, Mauri C, Ehrenstein MR. Anti‐TNF‐alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF‐beta. J Exp Med 2007; 204:33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Humby F, Bombardieri M, Manzo A et al Ectopic lymphoid structures support ongoing production of class‐switched autoantibodies in rheumatoid synovium. PLOS Med 2009; 6:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation‐induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000; 102:553–63. [DOI] [PubMed] [Google Scholar]
- 63. Pitzalis C, Kelly S, Humby F. New learnings on the pathophysiology of RA from synovial biopsies. Curr Opin Rheumatol 2013; 25:334–44. [DOI] [PubMed] [Google Scholar]
- 64. DiCicco M, Humby F, Kelly S et al O49 synovial lymphocytic aggregates associate with highly active RA and predict erosive disease progression at 12 months: results from the Pathobiology of Early Arthritis Cohort. Rheumatology 2016; 55(Suppl 1):i59–i59. [Google Scholar]
- 65. Rao DA, Gurish MF, Marshall JL et al Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017; 542:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014; 41:529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol 2012; 8:337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haynes NM, Allen CDC, Lesley R et al Role of CXCR69 and CCR69 in follicular Th cell positioning and appearance of a programmed cell death gene‐1high germinal center‐associated subpopulation. J Immunol 2007; 179:5099–108. [DOI] [PubMed] [Google Scholar]
- 69. Shi J, Hou S, Fang Q et al PD‐1 controls follicular T helper cell positioning and function. Immunity 2018; 49:264–74.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rombouts Y, Willemze A, van Beers JJBC et al Extensive glycosylation of ACPA‐IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann Rheum Dis 2016; 75:578–85. [DOI] [PubMed] [Google Scholar]
- 71. Kempers AC, Hafkenscheid L, Dorjée AL et al The extensive glycosylation of the ACPA variable domain observed for ACPA‐IgG is absent from ACPA‐IgM. Ann Rheum Dis 2018; 77:1087–8. [DOI] [PubMed] [Google Scholar]
- 72. Kempers AC, Hafkenscheid L, Scherer HU, Toes REM. Variable domain glycosylation of ACPA‐IgG: a missing link in the maturation of the ACPA response? Clin Immunol 2018; 186:34–7. [DOI] [PubMed] [Google Scholar]
- 73. Lefranc MP, Giudicelli V, Ginestoux C et al IMGT, the international ImMunoGeneTics database. Nucleic Acids Res 1999; 27:209–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Turner SJ, Doherty PC, McCluskey J, Rossjohn J. Structural determinants of T‐cell receptor bias in immunity. Nat Rev Immunol 2006; 6:883–94. [DOI] [PubMed] [Google Scholar]
- 75. Davis MM. T cell receptor gene diversity and selection. Annu Rev Biochem 1990; 59:475–96. [DOI] [PubMed] [Google Scholar]
- 76. Davis MM, Bjorkman PJ. T‐cell antigen receptor genes and T‐cell recognition. Nature 1988;334:395–402. [DOI] [PubMed] [Google Scholar]
- 77. Pannetier C, Cochet M, Darche S et al The sizes of the CDR3 hypervariable regions of the murine T‐cell receptor beta chains vary as a function of the recombined germ‐line segments. Proc Natl Acad Sci USA 1993; 90:4319–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kjer‐Nielsen L, Clements CS, Purcell AW et al A structural basis for the selection of dominant alphabeta T cell receptors in antiviral immunity. Immunity 2003; 18:53–64. [DOI] [PubMed] [Google Scholar]
- 79. Arstila TP, Casrouge A, Baron V et al A direct estimate of the human alphabeta T cell receptor diversity. Science 1999; 286:958–61. [DOI] [PubMed] [Google Scholar]
- 80. Attaf M, Huseby E, Sewell AK. αβ T cell receptors as predictors of health and disease. Cell Mol Immunol 2015; 12:391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T‐cell responses? Nat Rev Immunol 2008; 8:231. [DOI] [PubMed] [Google Scholar]
- 82. Miles JJ, Bulek AM, Cole DK et al Genetic and structural basis for selection of a ubiquitous T cell receptor deployed in Epstein–Barr virus infection. PLOS Pathog 2010; 6:e1001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med 1999; 189:701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Day EK, Carmichael AJ, ten Berge IJM, Waller ECP, Sissons JGP, Wills MR. Rapid CD8+ T cell repertoire focusing and selection of high‐affinity clones into memory following primary infection with a persistent human virus: human cytomegalovirus. J Immunol 2007; 179:3203–13. [DOI] [PubMed] [Google Scholar]
- 85. La Gruta NL, Thomas PG. Interrogating the relationship between naive and immune antiviral T cell repertoires. Curr Opin Virol 2013; 3:447–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Goronzy JJ, Bartz‐Bazzanella P, Hu W, Jendro MC, Walser‐Kuntz DR, Weyand CM. Dominant clonotypes in the repertoire of peripheral CD4+ T cells in rheumatoid arthritis. J Clin Invest 1994; 94:2068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sakurai K, Ishigaki K, Shoda H et al HLA‐DRB1 shared epitope alleles and disease activity are correlated with reduced T cell receptor repertoire diversity in CD4+ T cells in rheumatoid arthritis. J Rheumatol 2018; 45:905–14. [DOI] [PubMed] [Google Scholar]
- 88. Klarenbeek PL, de Hair MJH, Doorenspleet ME et al Inflamed target tissue provides a specific niche for highly expanded T‐cell clones in early human autoimmune disease. Ann Rheum Dis 2012; 71:1088–93. [DOI] [PubMed] [Google Scholar]
- 89. Musters A, Klarenbeek PL, Doorenspleet ME et al In Rheumatoid arthritis, synovitis at different inflammatory sites is dominated by shared but patient‐specific T cell clones. J Immunol 2018; 201:417–422. [DOI] [PubMed] [Google Scholar]
- 90. Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol 2004; 172:5967–72. [DOI] [PubMed] [Google Scholar]
- 91. Spreafico R, Rossetti M, van Loosdregt J et al A circulating reservoir of pathogenic‐like CD4+ T cells shares a genetic and phenotypic signature with the inflamed synovial micro‐environment. Ann Rheum Dis 2016; 75:459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rossetti M, Spreafico R, Consolaro A et al TCR repertoire sequencing identifies synovial Treg cell clonotypes in the bloodstream during active inflammation in human arthritis. Ann Rheum Dis 2017; 76:435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cope A, Le Friec G, Cardone J, Kemper C. The Th1 life cycle: molecular control of IFN‐gamma to IL‐10 switching. Trends Immunol 2011; 32:278–86. [DOI] [PubMed] [Google Scholar]
- 94. Pesenacker AM, Wedderburn LR. T regulatory cells in childhood arthritis – novel insights. Exp Rev Mol Med 2013; 15:e13. [DOI] [PubMed] [Google Scholar]
- 95. Haufe S, Haug M, Schepp C et al Impaired suppression of synovial fluid CD4+CD25– T cells from patients with juvenile idiopathic arthritis by CD4+CD25+ Treg cells. Arthritis Rheum 2011; 63:3153–62. [DOI] [PubMed] [Google Scholar]
- 96. Wehrens EJ, Mijnheer G, Duurland CL et al Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c‐AKT hyperactivation in effector cells. Blood 2011; 118:3538–48. [DOI] [PubMed] [Google Scholar]
- 97. Xiao H, Wang S, Miao R, Kan W. TRAIL is associated with impaired regulation of CD4+CD25– T cells by regulatory T cells in patients with rheumatoid arthritis. J Clin Immunol 2011; 31:1112–9. [DOI] [PubMed] [Google Scholar]
- 98. Dornmair K, Meinl E, Hohlfeld R. Novel approaches for identifying target antigens of autoreactive human B and T cells. Semin Immunopathol 2009; 31:467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Vanderlugt CL, Miller SD. Epitope spreading in immune‐mediated diseases: implications for immunotherapy. Nat Rev Immunol 2002; 2:85–95. [DOI] [PubMed] [Google Scholar]
- 100. Fazou C, Yang H, McMichael AJ, Callan MF. Epitope specificity of clonally expanded populations of CD8+ T cells found within the joints of patients with inflammatory arthritis. Arthritis Rheum 2001; 44:2038–45. [DOI] [PubMed] [Google Scholar]
- 101. Law SC, Street S, Yu CH et al T‐cell autoreactivity to citrullinated autoantigenic peptides in rheumatoid arthritis patients carrying HLA‐DRB1 shared epitope alleles. Arthritis Res Ther 2012; 14:R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jansen D, Ramnoruth N, Loh KL et al Flow cytometric clinical immunomonitoring using peptide‐MHC class II tetramers: optimization of methods and protocol development. Front Immunol 2018; 9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Rims C, Uchtenhagen H, Kaplan MJ et al Citrullinated aggrecan epitopes as targets of auto‐reactive CD4+ T cells in patients with rheumatoid arthritis. Arthritis Rheum 2018. 10.1002/art.40768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Qian Z, Latham KA, Whittington KB, Miller DC, Brand DD, Rosloniec EF. An autoantigen‐specific, highly restricted T cell repertoire infiltrates the arthritic joints of mice in an HLA‐DR1 humanized mouse model of autoimmune arthritis. J Immunol 2010; 185:110–8. [DOI] [PubMed] [Google Scholar]
- 105. Stuart JM, Dixon FJ. Serum transfer of collagen‐induced arthritis in mice. J Exp Med 1983; 158:378–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Holmdahl R, Jansson L, Larsson A, Jonsson R. Arthritis in DBA/1 mice induced with passively transferred type II collagen immune serum. Immunohistopathology and serum levels of anti‐type II collagen auto‐antibodies. Scand J Immunol 1990; 31:147–57. [DOI] [PubMed] [Google Scholar]
- 107. Korganow AS, Ji H, Mangialaio S et al From systemic T cell self‐reactivity to organ‐specific autoimmune disease via immunoglobulins. Immunity 1999; 10:451–61. [DOI] [PubMed] [Google Scholar]
- 108. Binstadt BA, Patel PR, Alencar H et al Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol 2006; 7:284–92. [DOI] [PubMed] [Google Scholar]
- 109. Nigrovic PA, Binstadt BA, Monach PA et al Mast cells contribute to initiation of autoantibody‐mediated arthritis via IL‐1. Proc Natl Acad Sci USA 2007; 104:2325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Matsumoto I, Maccioni M, Lee DM et al How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint‐specific autoimmune disease. Nat Immunol 2002; 3:360–5. [DOI] [PubMed] [Google Scholar]
- 111. Maffia P, Brewer JM, Gracie JA et al Inducing experimental arthritis and breaking self‐tolerance to joint‐specific antigens with trackable, ovalbumin‐specific T cells. J Immunol 2004; 173:151–6. [DOI] [PubMed] [Google Scholar]
- 112. Nickdel MB, Conigliaro P, Valesini G et al Dissecting the contribution of innate and antigen‐specific pathways to the breach of self‐tolerance observed in a murine model of arthritis. Ann Rheum Dis 2009; 68:1059–66. [DOI] [PubMed] [Google Scholar]
- 113. van Der Heijden IM, Wilbrink B, Tchetverikov I et al Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum 2000; 43:593–8. [DOI] [PubMed] [Google Scholar]
- 114. Sanderson NSR, Zimmermann M, Eilinger L et al Cocapture of cognate and bystander antigens can activate autoreactive B cells. Proc Natl Acad Sci USA 2017; 114:734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Walunas TL, Lenschow DJ, Bakker CY et al CTLA‐4 can function as a negative regulator of T cell activation. Immunity 1994; 1:405–13. [DOI] [PubMed] [Google Scholar]
- 116. Tan P, Anasetti C, Hansen JA et al Induction of alloantigen‐specific hyporesponsiveness in human T lymphocytes by blocking interaction of CD28 with its natural ligand B7/BB1. J Exp Med 1993; 177:165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Knoerzer DB, Karr RW, Schwartz BD, Mengle‐Gaw LJ. Collagen‐induced arthritis in the BB rat. Prevention of disease by treatment with CTLA‐4‐Ig. J Clin Invest 1995; 96:987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Arima T, Rehman A, Hickey WF, Flye MW. Inhibition by CTLA4Ig of experimental allergic encephalomyelitis. J Immunol 1996; 156:4916–24. [PubMed] [Google Scholar]
- 119. Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science 1994; 265:1225–7. [DOI] [PubMed] [Google Scholar]
- 120. Platt AM, Gibson VB, Patakas A et al Abatacept limits breach of self‐tolerance in a murine model of arthritis via effects on the generation of T follicular helper cells. J Immunol 2010; 185:1558–67. [DOI] [PubMed] [Google Scholar]
- 121. Prendergast CT, Patakas A, Al‐Khabouri S et al Visualising the interaction of CD4 T cells and DCs in the evolution of inflammatory arthritis. Ann Rheum Dis 2018; 77:579–88. [DOI] [PubMed] [Google Scholar]
- 122. Tibbitt C, Falconer J, Stoop J, van Eden W, Robinson JH, Hilkens CMU. Reduced TCR‐dependent activation through citrullination of a T‐cell epitope enhances Th17 development by disruption of the STAT3/5 balance. Eur J Immunol 2016; 46:1633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Thomas R, Turner M, Cope AP. High avidity autoreactive T cells with a low signalling capacity through the T‐cell receptor: central to rheumatoid arthritis pathogenesis? Arthritis Res Ther 2008; 10:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sakaguchi S, Benham H, Cope AP, Thomas R. T‐cell receptor signaling and the pathogenesis of autoimmune arthritis: insights from mouse and man. Immunol Cell Biol 2012; 90:277–87. [DOI] [PubMed] [Google Scholar]
- 125. Singh K, Deshpande P, Pryshchep S et al ERK‐dependent T cell receptor threshold calibration in rheumatoid arthritis. J Immunol 2009; 183:8258–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Purvis HA, Stoop JN, Mann J et al Low strength T‐cell activation promotes Th17 responses. Blood 2010; 116:4829‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Coombes JL, Siddiqui KR, Arancibia‐Carcamo CV et al A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF‐beta and retinoic acid‐dependent mechanism. J Exp Med 2007; 204:1757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Martin E, Capini C, Duggan E et al Antigen‐specific suppression of established arthritis in mice by dendritic cells deficient in NF‐kappaB. Arthritis Rheum 2007; 56:2255–66. [DOI] [PubMed] [Google Scholar]
- 129. Martin E, O'Sullivan B, Low P, Thomas R. Antigen‐specific suppression of a primed immune response by dendritic cells mediated by regulatory T cells secreting interleukin‐10. Immunity 2003; 18:155–67. [DOI] [PubMed] [Google Scholar]
- 130. Stoop JN, Harry RA, von Delwig A et al Therapeutic effect of tolerogenic dendritic cells in established collagen‐induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum 2010; 62:3656–65. [DOI] [PubMed] [Google Scholar]
- 131. Benham H, Nel HJ, Law SC et al Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype‐positive rheumatoid arthritis patients. Sci Transl Med 2015; 7:290ra87. [DOI] [PubMed] [Google Scholar]
- 132. Bell GM, Anderson AE, Diboll J et al Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis 2017; 76:227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Capini C, Jaturanpinyo M, Chang H‐I et al Antigen‐specific suppression of inflammatory arthritis using liposomes. J Immunol 2009; 182:3556–65. [DOI] [PubMed] [Google Scholar]
- 134. Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle‐mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2012; 109:11270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kishimoto TK, Ferrari JD, LaMothe RA et al Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat Nanotechnol 2016; 11:890–9. [DOI] [PubMed] [Google Scholar]
- 136. Clemente‐Casares X, Blanco J, Ambalavanan P et al Expanding antigen‐specific regulatory networks to treat autoimmunity. Nature 2016; 530:434–40. [DOI] [PubMed] [Google Scholar]
- 137. Yamamoto K, Sakoda H, Nakajima T et al Accumulation of multiple T cell clonotypes in the synovial lesions of patients with rheumatoid arthritis revealed by a novel clonality analysis. Int Immunol 1992; 4:1219–23. [DOI] [PubMed] [Google Scholar]
- 138. Alam A, Lule J, Coppin H et al T‐cell receptor variable region of the beta‐chain gene use in peripheral blood and multiple synovial membranes during rheumatoid arthritis. Hum Immunol 1995; 42:331–9. [DOI] [PubMed] [Google Scholar]
- 139. Jenkins RN, Nikaein A, Zimmermann A, Meek K, Lipsky PE. T cell receptor V beta gene bias in rheumatoid arthritis. J Clin Investigation 1993; 92:2688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Paliard X, West S, Lafferty J et al. Evidence for the effects of a superantigen in rheumatoid arthritis. Science 1991; 253:325–9. [DOI] [PubMed] [Google Scholar]
- 141. Zagon G, Tumang JR, Li Y, Friedman SM, Crow MK. Increased frequency of V beta 17‐positive T cells in patients with rheumatoid arthritis. Arthritis Rheum 1994; 37:1431–40. [DOI] [PubMed] [Google Scholar]
- 142. Cooper SM, Roessner KD, Naito‐Hoopes M et al Increased usage of V beta 2 and V beta 6 in rheumatoid synovial fluid T cells. Arthritis Rheum 1994; 37:1627–36. [DOI] [PubMed] [Google Scholar]
- 143. Sottini A, Imberti L, Gorla R, Cattaneo R, Primi D. Restricted expression of T cell receptor V beta but not V alpha genes in rheumatoid arthritis. Eur J Immunol 1991; 21:461–6. [DOI] [PubMed] [Google Scholar]
- 144. Lunardi C, Marguerie C, So AK. An altered repertoire of T cell receptor V gene expression by rheumatoid synovial fluid T lymphocytes. Clin Exp Immunol 1992; 90:440–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Sun W, Nie H, Li N et al Skewed T‐cell receptor BV14 and BV16 expression and shared CDR3 sequence and common sequence motifs in synovial T cells of rheumatoid arthritis. Genes Immun 2005; 6:248–61. [DOI] [PubMed] [Google Scholar]
- 146. Fischer D‐C, Opalka B, Hoffmann A et al Limited heterogeneity of rearranged T cell receptor Vα and Vβ transcripts in synovial fluid T cells in early stages of rheumatoid arthritis. Arthritis Rheum 1996; 39:454–62. [DOI] [PubMed] [Google Scholar]
- 147. Baud V, Collares D. Post‐translational modifications of RelB NF‐κB subunit and associated functions. Cells 2016;5; pii:E22. doi: 10.3390/cells5020022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Viennois E, Chen F, Merlin D. NF‐kappaB pathway in colitis‐associated cancers. Transl Gastrointest Cancer 2013; 2:21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Olivotto E, Otero M, Marcu KB, Goldring MB. Pathophysiology of osteoarthritis: canonical NF‐κB/IKKβ‐dependent and kinase‐independent effects of IKKα in cartilage degradation and chondrocyte differentiation. RMD Open 2015; 1(Suppl 1): e000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Woodsworth DJ, Castellarin M, Holt RA. Sequence analysis of T‐cell repertoires in health and disease. Genome Med 2013; 5:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Law S‐C, Benham H, Reid HH, Rossjohn J, Thomas R. Identification of self‐antigen–specific T cells reflecting loss of tolerance in autoimmune disease underpins preventative immunotherapeutic strategies in rheumatoid arthritis. Rheum Dis Clin N Am 2014; 40:735–52. [DOI] [PubMed] [Google Scholar]