

Abstract
Legislative initiatives have been successful in increasing the availability of approved therapies for paediatric patients. However, additional measures to ensure the timely completion of paediatric studies are necessary to further increase the number of medicines available to children. Over the last 3 years, international experts convened to revise the ICH E11 guideline on clinical investigations of medicinal products in paediatric populations to harmonize approaches to paediatric extrapolation, striving to reduce substantial differences between regions in the acceptance of data for global paediatric medicine development programmes. Several areas of therapeutics development in children, such as human immunodeficiency virus and partial‐onset seizures, have been streamlined and require fewer children enrolled in clinical trials because of the appropriate application of paediatric extrapolation. Based on this experience, it is clear that for paediatric extrapolation strategies to reach their full potential there is the need to understand the quality and quantity of data, often collected in adult patients, that will inform the appropriateness of the use of paediatric extrapolation, as well as to identify gaps in knowledge with respect to disease pathophysiology, organ maturation or drug target ontogeny. The generation of information that enhances our current understanding of these gaps in knowledge can further decrease the need for larger, paediatric clinical trials and can increase the efficiency of paediatric therapeutics development as well as protect children from participation in unnecessary studies. We hope that this publication will increase awareness, input and support from all the stakeholders involved in paediatric therapeutics development.
Keywords: drug regulation, clinical pharmacology, evidence‐based medicine, clinical pharmacology, optimal design pharmacodynamics, paediatrics
Over the last 20 years, paediatric drug development has advanced from routine exclusion of paediatric patients from clinical trials to early consideration of paediatric studies during adult development when use in paediatric patients is anticipated. This shift has been driven by legislation both in the USA and EU and has led to a change in paradigm—that paediatric patients can be treated with drugs for which effectiveness and safety have been demonstrated, rather than reliance on often misleading assumptions of effectiveness and/or safety from adult data. Progress in paediatric drug development brings changes to the current paradigm, and is teaching us a few lessons about drug development in general while, out of necessity, paving the way for more efficient clinical trials.
Regulatory standards for approval of drugs, vaccines and biological products (medicines) are the same for adult and paediatric patients. Approval must be based on evidence obtained, in general, from adequate and well‐controlled investigations. Often, drug development proceeds in adults first and, once approved in adults, medicines are prescribed as off‐label to children, out of need, long before establishment of effectiveness and safety and appropriate dosing of a medicine in the paediatric populations. Off‐label use and lack of well‐tested medicines for children have caused several major catastrophes in the past, and on‐label paediatric use has been the exception rather than the rule 1.
To address the lack of medicines approved for paediatric use, transforming legislations were passed first in the USA with the Food and Drug Administration (FDA) Modernization Act of 1997, Best Pharmaceuticals for Children Act of 2002, and Pediatric Research Equity Act of 2003, and then in the EU with the Paediatric Regulation of 2006 2. The EU Paediatric Regulation requires prior approval of a paediatric investigation plan (PIP), a development plan aimed at ensuring that the necessary data are obtained to support authorization in children. Applications for authorization of all new medicines must include study results defined in PIPs agreed by the European Medicines Agency (EMA) Paediatric Committee, unless a waiver is granted. Paediatric trials can be deferred until after adult development for specific reasons. Similarly, in the USA, a Pediatric Study Plan is required under certain circumstances, usually by End of Phase 2, and must be reviewed by the FDA Pediatric Review Committee. The PIP and Pediatric Study Plan are similar and usually compatible with a single global approach. Based on data collected since the implementation of the legal framework in the USA and from the first 10 years of the Paediatric Regulation, these initiatives have been successful in increasing the availability of approved therapies for paediatric patients 3.
Despite these successes, challenges still remain. A recent cohort study 4 demonstrated that after a median follow‐up of 7 years from approval of PIPs, only 17% of medicines authorised for adults had completed required paediatric trials, and 38% of all required paediatric studies were completed. In addition, studies to be completed after initial authorization were unlikely to be completed as compared to those to be completed before authorization. To date, the conditions with the highest number of completed PIPs are immunology/rheumatology (14%) and infectious diseases (14%) 5. There is a need for new ways to ensure timely completion of paediatric studies to further increase the medicines available to children. In the USA, there continues to be a similar lag (approximately 8 years) between approval for use in adults and time to paediatric‐specific labelling 6, 7.
Why does such a lag exist? Paediatric drug development raises recognized specific challenges: e.g. developmental changes may require specific investigations in specific age or weight groups, different safety considerations, the need for paediatric‐specific endpoints and formulations, and the need to incorporate specific research protection. Paediatric drug development is therefore expensive, with a small market that may limit the return on investment.
To reduce the requirement for data where they already exist while addressing the challenges faced by all stakeholders involved in paediatric development (children and their families, healthcare professionals, academia, regulators and pharmaceutical companies), one area advanced by both EU and USA is the use of paediatric extrapolation. This allows maximizing the use of existing data across products life cycle to increase efficiency of paediatric drug development. Extrapolation of efficacy findings from adults to the paediatric population was first proposed by the FDA in the 1994 Pediatric Labeling Rule as a potential strategy to consider the use of adult data when designing paediatric drug development programmes. Paediatric extrapolation has been further described in FDA and international guidance to industry 7, 8. The EMA has developed a more specific framework for the use of paediatric extrapolation, based on the current understanding of quality and quantity of data, often collected in adult patients, informing the appropriateness of use of paediatric extrapolation. The framework acknowledges, for example, that chronological age alone may not be the most appropriate categorical determinant to define developmental subgroups in paediatric studies. Rather than the traditional development groups based on age (e.g. infants, adolescents), physiological development and organ maturation, pathophysiology and natural history of the disease or condition, and pharmacology of the medicine are factors to be considered in determining appropriate paediatric subpopulations for study (i.e. the source and target populations for extrapolation). Accordingly, the inclusion of paediatric subpopulations in adult trials or even adult subpopulations in paediatric trials may be justifiable 7. Conversely, many diseases of the preterm and term neonate are unique or have unique manifestations precluding extrapolation of efficacy from older patients and may require novel methods of outcome assessment to optimize development. Where there is sufficient understanding of the pathophysiology and the pharmacology of the medicine to support extrapolation across a range of paediatric subsets, trials should focus on those paediatric subsets or disease subsets where gaps in knowledge are greatest (generally infants and neonates). This will avoid unnecessary delays to obtain the needed evidence in these populations. To support this approach, a concept paper was published in 2013, followed by a reflection paper in 2018 9.
Paediatric extrapolation is an approach that may be used when, based on the course of the disease and expected response to a medicine in the paediatric and reference (adult or other paediatric) populations, to provide evidence in support of effective and safe use of this medicine in the paediatric population. Paediatric extrapolation requires evidence‐based sets of assumptions from existing knowledge, using, for example, systematic reviews and synthesis of relevant available evidence, while any uncertainties, including gaps in knowledge, need be quantified with respect to disease pathophysiology, organ maturation or drug target ontogeny. For example, identification of a clear exposure–response relationship in a reference population can be used to predict an effective dose and regimen for the intended population. Extrapolation exercises can take different forms although they share a common goal. The long experience with antibacterial agents illustrates well that the requirements for evidence generation to support licensing in the paediatric population are a continuum. Requirements range from the identification of an appropriate dose for the target population based on pharmacokinetic characterization only, where there is evidence to support that achieving similar exposure is sufficient to expect similar efficacy in the target population, to full clinical development where no extrapolation is possible. For the majority of infectious diseases occurring both in adults and in paediatric subgroups with same pathogens and similar distribution, extrapolation of efficacy against the infectious agent is possible, based on similar pathophysiology of the infectious disease and spectrum of activity of the antibacterial agent. However, extrapolation of efficacy from adults to paediatric patients may not be possible when the infectious disease occurs only or predominantly in paediatric patients (e.g. neonatal sepsis) or there are differences between these populations in the nature or precipitating factors for the infectious process and known different responses to treatment 10.
Increasing experience of extrapolation approaches over time has led to agreed development programmes that do not require adequate and well‐controlled trials in children to establish efficacy. Human immunodeficiency virus (HIV) infection is one example where the pharmacokinetic/pharmacodynamic‐response relationship is assumed to be independent of age, and similar activity can be obtained from similar exposure in children and adults, based on efficacy data from controlled trials 11. On this basis, in 2007, the EU guideline on clinical development of antivirals for HIV infection accepted extrapolation of efficacy from the adult to the paediatric populations. To assess the impact of extrapolating efficacy, we reviewed all antiretrovirals for HIV infection and analysed the EMA data for all those authorized before and after the EU Regulation (i.e. comparing 1997–2006 and 2007–2016). We observed that extrapolating efficacy from adult data paralleled a dramatic reduction in the number of enrolled children. Up to 2016, 18 single‐substance medicines and six fixed‐dose combinations had been authorized in at least one paediatric subset. Before 2007, the 11 paediatric marketing authorizations were based on 48 clinical trials enrolling 4971 paediatric participants. Twelve of 48 clinical trials (25%) were powered for efficacy as primary endpoint and required enrolment of 2979 paediatric participants (59%). Between 2007 and 2016, the 13 paediatric marketing authorizations were based on 12 clinical trials enrolling 621 patients; all the clinical trials had pharmacokinetics as primary objective to determine paediatric doses, and provided systemic drug exposures similar to that known to be effective in adults and safety data (Figure 1). Not requiring studies powered for efficacy for a paediatric indication led to a smaller number of enrolled subjects. The use of extrapolation of efficacy in paediatric HIV avoided unnecessary studies, increased efficiency, reduced testing burden to patients, and allowed better allocation of resources, and for most of them leading to faster availability of approved medicines for paediatric HIV patients.
Figure 1.
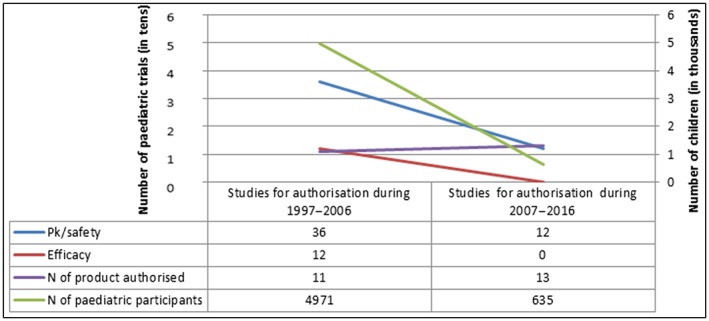
Paediatric population in clinical trials for authorization of HIV medicines in 1997–2006 and 2007–2016
Epilepsy is another area where FDA and EMA have used increased disease and treatment knowledge in children to review the acceptability of paediatric extrapolation. Reviews were conducted to determine whether the efficacy of antiepileptic drugs (AEDs) in adults could be used to predict efficacy in children with partial‐onset seizure (POS). A first study by Pellock et al. in 2008 12, reviewed 30 adjunctive therapy trials of both partial‐onset seizures and primary generalized tonic–clonic seizures in adults and children aged 2–18 years. Following the Pellock review, the FDA conducted systematic and quantitative analyses of AEDs approved for use in paediatric patients; these AEDs spanned a variety of putative mechanisms of action. Data from 26 placebo‐controlled trials in over 4600 adults and 1400 children aged 4 years and older were analysed with the goal to compare responses and exposures at the approved doses between adults and various paediatric age subsets. In 2018, the FDA concluded that the relationship between exposure and response (reduction in seizure frequency) in adults and children was sufficiently similar to allow for extrapolation of efficacy of drugs approved for the treatment of POS in adults to paediatric patients 4 years of age and older 13. However, clinical studies to identify a dose and characterize adequately the safety in children still need to be conducted.
The methods and acceptability of paediatric extrapolation for specific paediatric drug development programmes are rapidly evolving and present both challenges and opportunities. For example, innovative strategies such as modelling and simulation, or alternative statistical approaches to support the acceptability and strengthen the appropriate use of extrapolation, are actively considered by medicine developers and regulatory authorities globally.
Modelling and simulation help use all available data optimally to make predictions in various areas of drug development, including clinical trials and are essential tools to increase efficiency and accuracy of paediatric drug development, especially for the purpose of extrapolation. Modelling and simulation use mathematical techniques to quantify the current state of knowledge and to make predictions, most often on pharmacokinetics and pharmacodynamics, efficacy, and, potentially, on safety of new medicines to improve decision‐making. Use of modelling and simulation may help decrease the amount of data required for a paediatric drug development programme, regardless of whether paediatric extrapolation is considered acceptable. Importantly, the assumptions made initially need to be revisited (iterations) and potentially revised based on newly generated data.
Where extrapolation is not acceptable, adequate and well‐controlled paediatric studies are necessary to establish effectiveness. New approaches are welcome to maximize generation of data while reducing the number of children exposed, but innovative clinical trial designs can be challenging. Until recently, fully powered trials were required to demonstrate a statistically significant and clinically meaningful treatment effect. In many paediatric programmes, such studies were not feasible, or not designed to answer questions that the adult trials also failed to address, such as the investigation of the impact of covariates (e.g. body size, organ maturation) in children across the developmental spectrum. Therefore, alternative clinical trial designs should be set up to establish effectiveness, while meeting regulatory standards, and should be designed to answer questions that cannot be addressed by the adult trials in a timely manner. Additionally, while it is acknowledged that safety information in adults may be used to predict some adverse effects in children, data are still required to identify those age‐specific adverse effects, in particular related to growth and maturation.
Unlike medicines development for paediatric anti‐HIV or POS medicines, some paediatric conditions highlight the challenges to acceptability and use of paediatric extrapolation. For example, in Gaucher disease, when planning paediatric studies, one must carefully consider the impact of different mechanisms of action and disease‐modifying factors such as types of mutation, residual enzyme activity, age and epigenetic factors, which result in presentations of the paediatric disease that differ from those seen in adults. Nonetheless, extrapolation can still be considered to increase efficiency and reduce testing burden to patients, once the characteristics of different patient populations are taken into consideration 14. For diseases such as pulmonary arterial hypertension (PAH) where medicines are not directly targeting the known pathways that contribute to the disease, it would not be safe to assume that children will see a similar benefit from similar pharmacodynamic effect. There are significant remaining gaps in our knowledge of the natural history of PAH that need addressing. The use of extrapolation in paediatric PAH is not straightforward; the predictions that can be generated from existing knowledge will carry a high level of uncertainty related to pharmacology, disease progression and clinical response.
Undeniably, the use and understanding of paediatric extrapolation are rapidly evolving. Collection of data to support its use must be planned very early in product development, and requires careful planning designed to better understand the medicine's pharmacological behaviour and its clinical value for children. For example, there may be a need to include specific elements in the adult trials, such as additional time points or dose‐levels, to inform and strengthen the data supporting the evidence for effective and safe use of the medicine in adults and consequently in the paediatric population. Biomarkers, clinical and surrogate endpoints should ideally be appropriate to both adults and paediatric subsets; high‐quality longitudinal data on growing children should be collected to allow characterization of longer‐term benefits or harms, especially in neurodevelopmental and behavioural disorders, as well as characterization of ontogeny of drug targets, effect of developmental changes on medicine metabolizing enzymes across the developmental spectrum. Certain data generation (e.g. longer‐term safety) may not be needed before approval in paediatric subpopulations but collected postapproval. There is also growing interest in the potential use of real‐world evidence in paediatric medicines development programmes. While there may be a role for high quality real‐world evidence to maximize the efficiency of development programmes, this should not be considered an encouragement to rely on information from off‐label use as a primary source of paediatric data in the future.
Finally, there are other challenges in paediatric drug development that deserve mention. Children of all ages require special measures to protect their rights as study participants and to shield them from undue harm. With the aim to protect children from unexpected adverse effects, in most paediatric programmes, investigators and regulators opted for sequential developments, starting with the oldest and ending with the youngest children. After >10 years of experience, any protective effect of default use of sequential approaches has not been proven, while there is evidence of harm from prolonged use of unstudied medicines in the most fragile subset, i.e. the neonate, due to long delays in completing paediatric studies and prolonged off‐label use 15, 16, 17.
Based on our experience, paediatric extrapolation strategies can reduce the number of children enrolled in trials and facilitate earlier and timely access to appropriately‐studied medicines for children. Significant work remains to be done to increase our knowledge of paediatric therapeutics and move away from the unnecessary age‐based sequential development paradigm. Most importantly, regulators should continue looking outside the box for innovative drug development approaches for the benefit of the whole paediatric population. Children deserve to participate in and benefit from high quality and ethical clinical research, and to have access to medicines authorized appropriately, without being subject to unnecessary trials. To achieve this, all stakeholders involved in paediatric medicine development have a responsibility to design and conduct rational and ethically sound studies of medicines, based on all knowledge available through previous research. Indeed, the use of paediatric extrapolation is not only a strategy to increase the efficiency of medicines development; it is part of a new paradigm in paediatric medicine development and an ethical imperative.
Competing Interests
There are no competing interests to declare.
The views expressed in this article are the personal views of the author(s) and may not be understood or quoted as being made on behalf of or reflecting the position of the EMA or one of its committees or working parties, or that of the US FDA.
Ollivier C., Mulugeta Y. (. L.)., Ruggieri L., Saint‐Raymond A., and Yao L. (2019) Paediatric extrapolation: A necessary paradigm shift, Br J Clin Pharmacol. 85, 675–679, 10.1111/bcp.13809.
References
- 1. Kimland E, Nydert P, Odlind V, Böttiger Y, Lindemalm S. Paediatric drug use with focus on off‐label prescriptions at Swedish hospitals – a nationwide study. Acta Paediatr 2012; 101: 772–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. European Union . Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. Available at https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2006_1901/reg_2006_1901_en.pdf (last accessed 1 June 2018).
- 3. Nordenmalm S, Tomasi P, Pallidis C. More medicines for children: impact of the EU paediatric regulation. Arch Dis Childhood 2018; 103: 557–564. [DOI] [PubMed] [Google Scholar]
- 4. Hwang TJ, Tomasi PA, Bourgeois FT. Delays in completion and results reporting of clinical trials under the Paediatric regulation in the European Union: a cohort study. PLoS Med 2018; 15: e1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. European Medicines Agency . 10‐year report to the European Commission. 2017. Available at https://ec.europa.eu/health/sites/health/files/files/paediatrics/docs/paediatrics_10_years_ema_technical_report.pdf (last accessed 1 June 2018).
- 6. Mehrotra N, Bhattaram A, Earp JC, Florian J, Krudys K, Lee JE, et al Role of quantitative clinical pharmacology in paediatric approval and labeling. Drug Metab Dispos 2016; 44: 924–933. [DOI] [PubMed] [Google Scholar]
- 7. Addendum to ICH E11: Clinical Investigation of Medicinal Products in the Paediatric population E11(R1). Available at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E11/E11‐R1EWG_Step4_Addendum_2017_0818.pdf (last accessed 1 June 2018).
- 8. US Food and Drug Administration . Draft guidance for industry: general clinical pharmacology considerations for pediatric studies for drugs and biological products. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425885.pdf (last accessed 1 June 2018).
- 9. EMA Reflection paper on extrapolation of efficacy and safety in paediatric medicine development. Available at (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/10/WC500236640.pdf (last accessed 29 October 2018).
- 10. CHMP Addendum to the guideline on the evaluation of medicinal products indicated for treatment of bacterial infections to address paediatric‐specific clinical data requirements. Available at https://www.ema.europa.eu/documents/scientific‐guideline/draft‐addendum‐guideline‐evaluation‐medicinal‐products‐indicated‐treatment‐bacterial‐infections_en.pdf (last accessed 30 April 2018).
- 11. CHMP Guideline on the clinical development of medicinal products for the treatment of HIV infection. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500209918.pdf (last accessed 1 June 2018).
- 12. Pellock JM, Carman WJ, Thyagarajan V, Daniels T, Morris DL, D'Cruz O. Efficacy of antiepileptic drugs in adults predicts efficacy in children: a systematic review. Neurology 2012; 79: 1482–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drugs for treatment of partial onset seizures: full extrapolation of efficacy from adults to pediatric patients 4 years of age and older. Guidance for industry. Available at https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM596731.pdf (last accessed 1 June 2018).
- 14. Paediatric Gaucher disease: A strategic collaborative approach from EMA and FDA. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/06/WC500230342.pdf (last accessed 1 June 2018).
- 15. Neville KA, Frattarelli DA, Galinkin JL, Johnson TD, Neville KA, Paul IM, et al Off‐label use of drugs in children. Paediatrics 2014; 133: 563–567. [DOI] [PubMed] [Google Scholar]
- 16. Czaja AS, Reiter PD, Schultz ML, Valuck RJ. Patterns of off‐label prescribing in the paediatric intensive care unit and prioritizing future research. J Paed Pharmacol Therap 2015; 20: 186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Allegaert K, Anker JN. Adverse drug reactions in neonates and infants: a population‐tailored approach is needed. Br J Clin Pharmacol 2015; 80: 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]