

Summary
Epigenetic effects of anti‐psychotic medications are poorly understood. We have appropriated a model whereby heterochromatin is established through 24‐ or 48‐h lipopolysaccharide (LPS) treatment, and tested the epigenetic effects of risperidone along the adenylyl cyclase/protein kinase A (AC/PKA) pathway in human liposarcoma cells that express the LPS‐sensitive Toll‐like receptor (TLR)‐4. Human SW872 cells were cultured with LPS and mRNA expression levels and epigenetic modifications of dimethylated lysine 9 of histone 2 (H3K9me2), geterochromatin protein 1γ (HP1γ) and phospho‐H3S10 at promoters of interleukin (IL)‐6, tumor necrosis factor (TNF)‐α and IL1β were measured. Pharmacological manipulation of the AC/PKA pathway was achieved through treatment with a PKA inhibitor (H89), mitogen‐ and stress‐activated kinase 1 (MSK1) inhibitor (SB‐747651A) or forskolin. Twenty‐four and 48‐h LPS treatment establishes heterochromatin at selected promoters, corresponding to decreased mRNA expression. Concurrent risperidone treatment with LPS treatment can both ‘block’ and ‘reverse’ heterochromatin formation. Forskolin treatment resulted in a similar disassembling effect on heterochromatin. Conversely, inhibition of PKA by H89 or MSK1 both blocked ‘normalizing’ effects of risperidone on LPS‐induced heterochromatin. Our results demonstrate that risperidone can disassemble heterochromatin, exerting this effect along the G‐protein/AC/PKA pathway. This approach can also be utilized to investigate functional outcomes of single or combined pharmacological treatments on chromatin assemblies in human cells.
Keywords: anti‐psychotics, epigenetics, heterochromatin, phosphorylation, risperidone, schizophrenia
Introduction
Many long‐term illnesses can be characterized by dysregulation of gene expression due to effects of predisposition and the impact of the environment. Epigenetic promoter assemblies are prime candidates for long‐term gene regulation. It can be hypothesized that co‐ordinated regulation of disease‐relevant gene networks can be catalyzed over time by the accumulation of a transcriptionally restrictive form of chromatin known as heterochromatin 1. Heterochromatin can be characterized by the presence of heterochromatin protein 1γ (HP1γ), dimethylated lysine 9 of histone 2 (H3K9me2) and a decrease of phosphorylated histones, such as the serine on the 10th residue of histone 3 (phospho‐H3S10). As HP1γ and H3S10 are both targets of kinase activity, we examined the effects that well‐established kinase signaling pathways may have on chromatin assemblies. Phosphorylation of H3S10 is catalyzed by multiple kinases, including G‐protein coupled protein kinases A and C 2, 3, 4. A terminal step in this kinase cascade is the activity of mitogen‐ and stress‐activated kinases 1 and 2 (MSK1 and 2). H3S10 phosphorylation, a transcriptionally permissive epigenetic modification, is an important component of chromatin decondensation, which allows transcription factors access to DNA and increases the expression of early response genes, such as c‐fos and c‐jun 5, 6. H3S10 is phosphorylated by MSK1 following activation of the extracellular signal‐regulated kinases (ERK1/2), which is an important step in the transmission of a signal from the cell surface to the nucleus (H3S10 phosphorylation by MSK1) 7, 8, 9. While associated with transcriptional activity, H3S10 phosphorylation also has the capability to ‘disassemble’ heterochromatin by encouraging the demethylation and acetylation of the neighboring H3K9 residue 10. Additionally, phosphorylation of HP1γ causes it to dissociate from its moored ligand, H3K9me2/3 4, 11, 12. A combination of these changes renders the H3K9me2 methyl groups accessible to removal by histone demethylases (KDMs) 9, 13, 14, 15, 16, 17. Considering that all anti‐psychotic medications act upstream from these chromatin phosphorylation events, the effect of anti‐psychotics on heterochromatin assemblies becomes an attractive target for study.
Heterochromatin formations are increased in both peripheral blood mononuclear cells (PBMCs) and postmortem brain from patients with schizophrenia 1, 18, 19, 20, 21, 22. Curiously, we have also found increased phospho‐H3S10 in PBMC nuclear extracts from subjects with schizophrenia treated with anti‐psychotics 23. This work prompted the hypothesis that anti‐psychotics may be acting in the process of heterochromatin removal and the establishment of transcriptionally active chromatin as one mechanism for their therapeutic efficacy. It has been shown previously that treatment with the anti‐psychotics haloperidol or raclopride will induce a rapid and sustained increase in histone H3 phosphorylation in striatopallidal medium spiny neurons in mice 24, an effect that is attenuated through inhibition of downstream kinases, most notably protein kinase A (PKA) 3.
Endotoxin tolerance is a well‐established paradigm, whereby after initial exposure and acute immune response to lipopolysaccharide (LPS) immune cells will enter what is known as a ‘tolerized’ state, which includes the targeted assembly of heterochromatin modifications along gene promoters. These epigenetic changes result in a muted proinflammatory response to a potentially cell‐lethal second challenge of endotoxin. The primary signaling pathway in response to LPS stimulation is the Toll‐like receptor (TLR)‐mediated activation of the transcription nuclear factor kappa B (NF‐κB) 25. In the acute phase of LPS stimulation in naive macrophages, H3K9 dimethylation is lost at tumor necrosis factor (TNF)‐α and interleukin (IL)‐1β promoters, leading to a significant increase in gene expression 26, 27, 28. After lengthier treatment with LPS, this effect is reversed. Activation of NF‐κB subunits, such as RelB, recruit the H3K9 methyltransferase G9a, which prevents promoter demethylation and recruits heterochromatin protein 1 (HP1), resulting in suppression of the transcription of target genes 29, 30.
SW872 cells are a human liposarcoma cell line that expresses both TLR‐4, the receptor through which LPS acts to establish endotoxin tolerance to influence cytokine release, and the dopamine D2 receptor, one target of anti‐psychotics 31, 32, but not serotonin receptors 2A or B, additional targets of anti‐psychotics. SW872 cells also manifest a functional signaling AC/cAMP pathway, capable of driving target promoter activity 31. We can demonstrate that the adenylyl cyclase (AC)/PKA pathway is operative in these cells through the application of forskolin, a well‐known activator of AC, to mimic the increased adenylate cyclase (AC) activity caused by risperidone application. We hypothesized that the anti‐psychotic drug risperidone would block the increases in heterochromatin formation after 24–48 h of LPS treatment in SW872 cells through increased signaling along the G‐protein/AC/PKA pathway towards the epigenome. Furthermore, when this pathway is inhibited along its theoretical path, either early with the block of PKA or at its termination, with the block of MSK1 risperidone treatment would not generate these effects.
There are no studies, to our knowledge, that have examined the ability of anti‐psychotics to disassemble preformed heterochromatin structures. We have appropriated the endotoxin tolerance paradigm to first locate the induced heterochromatin by a physiological pathway, and to then disassemble it by engaging an alternate path to observe if the phosphorylation events caused through G protein‐coupled receptors (GPCR) signaling can be responsible for prevention or removal of heterochromatin formation.
Materials and methods
Cell culture, reagents and treatment
SW872 cells (ATCC HTP‐92) were grown in a media consisting of RPMI‐1640 (gibco #111875‐093; ThermoFisher Scientific, Fremont, CA, USA) supplemented with penicillin/streptomycin (100 μ/ml; gibco #15140122) and 10% fetal bovine serum (FBS) (gibco #10‐437‐028) and incubated at 37°C in 5% CO2. Cells were grown to a maximum confluence of 80% before passage through trypsin dissociation (gibco #25300‐054). Cells at 75% confluency were treated for periods from 24 to 48 h with the indicated chemicals. Dosage for each compound was as follows throughout the study: LPS (100 ng/ml; Sigma #L4391; Sigma, St Louis, MO, USA); risperidone (10 µM; Sigma #R3030); H89 (50 µM; Sigma #B1427); SB‐747651A (10 µM; Axon Medchem; axon 1897; Axon Medchem, Reston, VA, USA); and forskolin (10 µM; Sigma; #F6886). The dose of risperidone that was used was selected based on literature indicating that risperidone 10 μM may be neuroprotective when compared to higher doses, which may induce oxidative stress and reduced cell viability 33, 34. Amounts of other drugs were selected based on dose–response curves established in our laboratory. Cell death in response to treatment exposure was examined using Trypan blue, and showed no significant differences between treatment conditions 35.
Chromatin immunoprecipitation and real‐time reverse transcription–polymerase chain reaction (RT–PCR)
Cells were harvested, homogenized in 500 μl RPMI‐1640 media, and proteins were cross‐linked to DNA by adding methanol‐free formaldehyde (ThermoFisher #28908) and incubated at 37⁰C for 5 min. The sample was quenched with 1 M glycine (Sigma #G7126), spun down and washed with phosphate‐buffered saline (PBS) in the presence of 1 : 100 protease inhibitors (Calbiochem #539134; Calbiochem, La Jolla, CA, USA). The samples were spun down again, supernatant removed and resuspended in sodium dodecyl sulfate (SDS) lysis buffer [1% SDS, 10 mM ethylenediamine tetraacetic acid (EDTA), 50 mM Tris‐HCl, pH 8.1], again in the presence of 1 : 100 protease inhibitor. Samples were sonicated for 20 min at 10% df in a Covaris m220 sonicator. At this point, the Upstate Cell Signaling protocol for their ChIP Assay kit (#17‐295; Cell Signaling, Beverly, MA, USA) was closely followed and DNA was precipitated by ethanol precipitation. The antibodies used targeted H3K9me2 (Abcam #Ab1220; Abcam, Cambridge, MA, USA), phospho‐H3S10 (Active Motif #39636; Active Motif, Inc., Carlsbad, CA, USA) and HP1γ (Abcam #Ab10480). Table 1 shows the specific primers that were designed for the gene promoters of interest [IL‐6, TNF‐α, and IL‐1β, with Kruppel‐like factor 4 (KLF)‐4 as a negative control]. For detection and measurement of expression, Fermentas Maxima SYBR Green/ROX qPCR Master Mix (K0222; Fermentas, Waltham, MA, USA) was used. PCR mixtures were run on a ThermoScientific PikoReal real‐time PCR system. Cycle threshold (CT) values were used for percentage of input calculation 36. All experiments had an n = 3.
Table 1.
Primer sequences
| Primer name | Forward primer | Reverse primer |
|---|---|---|
| TNF‐α promoter | GCTTGTGTGTCCCCAACTTT | TGTGCCAACAACTGCCTTTA |
| IL‐6 promoter | TGGAGATGTCTGAGGCTCATT | ACACACCCCTCCCTCACAC |
| IL‐1β promoter | CCCCTAAGAATTCCCATCAAGC | GAGCTGTGAAATTTTCCCTTGG |
| KLF‐4 promoter | CGAGATGGCTGGTTGAAAACTG | AGGCACGAATGGGGAGTTATG |
| TNF‐α mRNA | AAGCCTGTAGCCCATGTTGT | TGAGGTACAGGCCCTCTGAT |
| IL‐6 mRNA | AAAGAGGCACTGGCAGAAAA | AGCTCTGGCTTGTTCCTCAC |
| IL‐1β mRNA | TGGCGAGCTCAGGTACTTCT | AAACCTCTTCGAGGCACAAG |
| KLF‐4 mRNA | CCCCGTGTGTTTACGGTAGT | GTTCCCATCTCAAGGCACAC |
mRNA extraction and real‐time RT–PCR
Total RNA was pre‐treated with DNAse, quantified, and then converted to cDNA using the Applied Biosystems (Life Technologies, Carlsbad, CA, USA) High Capacity Archive Kit (4368813). For detection and measurement of expression, Fermentas Maxima SYBR Green/ROX qPCR Master Mix (#K0222) was used. Primers for mRNA were designed to cross over one intron to amplify only cDNA and yielding an amplicon of between 75 and 200 base pairs. Dissociation curves were conducted to establish the presence of a single amplicon at the predicted melting temperature and a lack of primer–dimer formation. A comparative CT validation experiment was performed to determine target and reference primer efficiency. CT values were used for relative quantification of target gene expression and normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), as GAPDH mRNA levels did not significantly alter in experimental conditions (data not shown). All experiments had an n = 3.
Results
Risperidone co‐treatment with LPS ‘blocks’ the effects of LPS on mRNA expression levels and chromatin assemblies
After initial exposure and resultant immune response to LPS immune cells will enter what is known as a ‘tolerized’ state, which is characterized by the assembly of heterochromatin modifications along gene promoters of targets in the LPS/NF‐κB signaling pathway. This heterochromatin assembly then results in a decrease of gene expression of these pathway genes. As expected, LPS treatment for 24 h reduces mRNA expression of the selected target genes IL‐6, TNF‐α and IL‐1β, compared to vehicle‐treated cells, serving as a validation of well‐established paradigm. Treatment with the anti‐psychotic risperidone concurrently with LPS for 24 h abolishes the reduction in mRNA levels of IL‐6, TNF‐α and IL‐1β seen with 24‐h LPS treatment, while the negative control gene KLF‐4 remains unaffected, suggesting that the effect is specific to LPS‐targeted immune promoters (Fig. 1a).
Figure 1.
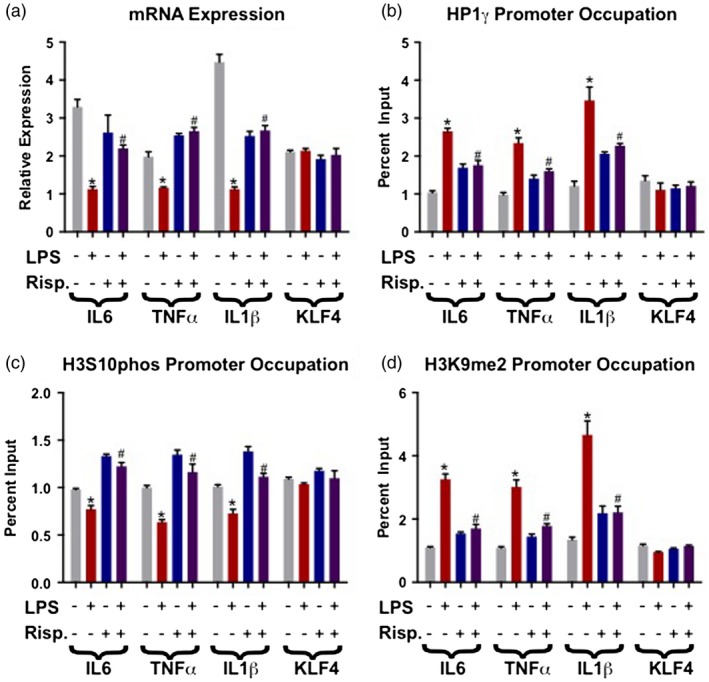
Risperidone (Risp) reverses the effects of lipopolysaccharide (LPS) on heterochromatin assemblies and mRNA expression at select promoters. In these experiments all cells were exposed to the differing drugs for 24 h, and treatment conditions remained the same between all graphs. (a) LPS significantly reduces mRNA expression of all three genes of interest [interleukin (IL)‐6, tumor necrosis factor (TNF)‐α, IL‐1β], but does not alter the negative control Kruppel‐like factor 4 (KLF)‐4. LPS + Risp co‐treatment reverses this effect, resulting in significant mRNA increases compared to the 24 h LPS‐only condition. (b) LPS treatment increases levels of the repressive epigenetic modification heterochromatin protein 1γ (HP1γ) along IL‐6, TNF‐α and IL‐1β promoters. LPS + Risp co‐treatment decreases HP1γ promoter occupancy when compared to LPS‐only. (c) LPS significantly decreases phospho‐H3S10 at the IL‐6, TNF‐α and IL‐1β promoters, an effect reversed by LPS + Risp co‐treatment. (d) LPS‐only significantly increased levels of the repressive epigenetic modification dimethylated lysine 9 of histone 2 (H3K9me2) at the IL‐6, TNF‐α and IL‐1β promoters, again with LPS + Risp significantly decreasing occupancy when compared to LPS alone. The following symbols were used to draw attention to specific Tukey post‐hoc analyses: *P < 0·05 vehicle versus LPS‐only; #P < 0·05, LPS‐only versus LPS + Risp.
LPS applied for 24 h increases heterochromatin modifications on the target promoters IL‐6, TNF‐α and IL‐1β. Relative to the LPS‐only condition, 24‐h co‐treatment with LPS and risperidone significantly reduces the heterochromatin modifications HP1γ and H3K9me2 (Fig. 1b,c) when compared to the LPS‐only condition, and increases the open chromatin mark phospho‐H3S10 (Fig. 1d) on IL‐6, TNF‐α and IL‐1β promoters, but not on the negative control KLF‐4 promoter.
Risperidone ‘reversal’ of LPS induced heterochromatin through the AC/PKA pathway
To further characterize the mechanism of this effect, we examined whether risperidone can ‘reverse’ heterochromatin formation once it has been established. Cells were treated with LPS for 24 h to establish the formation of heterochromatin, which was followed by an additional 24‐h treatment with risperidone or risperidone plus H89, a PKA inhibitor meant to counteract the downstream function of risperidone and to exclude non‐specific effects. We found that 24 h of risperidone treatment reverses the effects of LPS‐established H3K9me2. The addition of the kinase inhibitor resulted in abrogation of the ‘reversing’ effects of risperidone by significantly increasing H3K9me2 assemblies and decreasing H3S10 phosphorylation on the promoters of IL‐6, TNF‐α and IL‐1β when compared to the LPS + risperidone condition (Fig. 2a,b). Again, no effect was seen at the KLF‐4 promoter.
Figure 2.
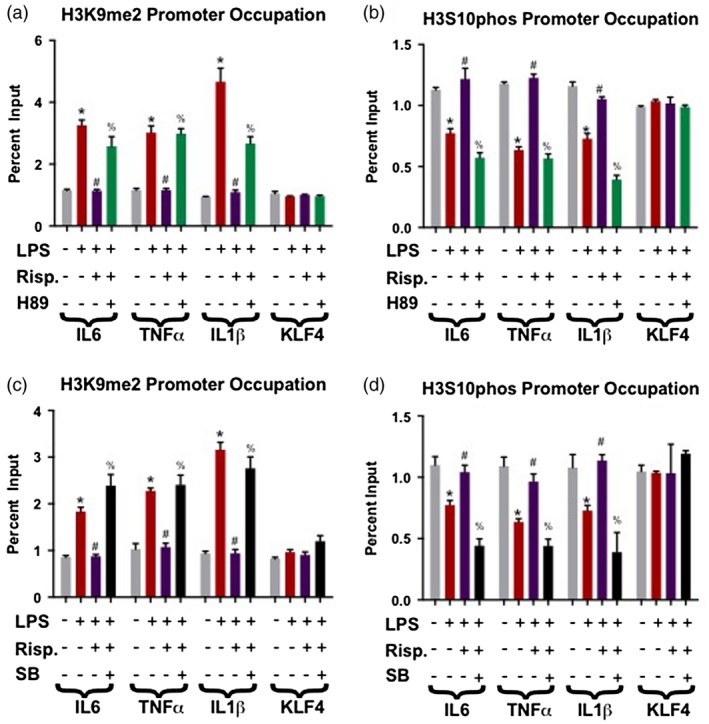
Inhibition of either G protein‐coupled receptors (GPCR) protein kinase A (PKA) or nuclear mitogen‐ and stress‐activated kinase 1 (MSK1) abrogates the ‘reversal’ effect of risperidone (Risp) on lipopolysaccharide (LPS)‐induced heterochromatin. In these experiments, cells were exposed to LPS for 48 h and supplemented with either Risp, the PKA inhibitor‐H89 or the MSK1 inhibitor SB‐70746501A for the latter 24 h before harvest. (a) Increased dimethylated lysine 9 of histone 2 (H3K9me2) promoter occupancy by LPS‐only was blocked by Risp, which was in turn blocked by co‐treatment with the PKA inhibitor‐H89 (LPS + Risp + H89). There were no changes for the negative control Kruppel‐like factor 4 (KLF)‐4. (b) LPS‐only significantly decreased phospho‐H3S10 levels at interleukin (IL0‐6, tumor necrosis factor (TNF)‐α and IL‐1β promoters. LPS + Risp co‐treatment ablated the LPS‐only decreases seen, with additional co‐treatment with H89 returning promoter occupation back to LPS‐only levels. (c) The LPS‐only induced increased promoter occupancy of H3K9me2 was abrogated by LPS + Risp co‐treatment. This Risp effect was blocked in turn by the MSK1 inhibitor SB‐70746501A (LPS + Risp + SB). (d) LPS‐only significantly reduced phospho‐H3S10 levels at the IL‐6, TNF‐α and IL‐1β promoters, again with Risp co‐treatment ablating this effect. However, when SB‐70746501A and Risp were added together (LPS + Risp + SB), the phospho‐H3S10 promoter occupancy of IL‐6, TNF‐α and IL‐1β was significantly decreased from vehicle and LPS + Risp treatment levels. The following symbols were used to draw attention to specific Tukey post‐hoc analyses: *P < 0·05 vehicle versus LPS‐only; #P < 0·05, LPS‐only versus LPS + Risp; %P < 0·05, LPS + Risp versus LPS + Risp + H98 or SP.
Inhibition of MSK1 at the end of the AC/PKA pathway
MSK1 is a nuclear H3S10 kinase, and the ultimate step in the AC/PKA signaling pathway. Cells underwent similar treatment paradigms as in the experiment above (24‐h LPS, 24‐h risperidone, 48‐h LPS + 24‐h risperidone, 48‐h LPS + 24‐h risperidone + 24‐h SB‐70746501A) to determine if the functional end‐point of the kinase cascade (phosphorylation of H3S10) would be affected. As seen previously, 24‐h treatment with LPS resulted in significantly increased H3K9me2 promoter occupation and decreased phospho‐H3S10 in nearly all cases, while risperidone alone did not have a significant effect (Fig. 2c,d). However, when risperidone was added to the LPS treatment after 24 h, chromatin modification levels ‘normalize’ back to vehicle standards. The most interesting result, however, comes with the inclusion of the MSK1 inhibitor SB‐70746501A (SB) to the LPS + risperidone condition. Treatment with this inhibitor and blocking of the putative action of the anti‐psychotic resulted in chromatin modification levels indistinguishable from the LPS‐only condition (i.e. significantly elevated H3K9me2 and reduced phospho‐H3S10).
Forskolin activation of the AC/PKA pathway simulates the effects of risperidone ‘reversal’
Cells were treated in three conditions: 48‐h vehicle, 48 h of LPS and a 48‐h treatment with LPS with forskolin added for the final 24 h of treatment. Forty‐eight‐hour treatment with LPS, and resultant induction of heterochromatin formation, significantly decreased mRNA expression of IL‐6, TNF‐α and IL‐1β. When forskolin was added for the final 24 h of the 48‐h LPS treatment, mRNA expression levels of IL‐6, TNF‐α and IL‐1β were significantly increased from LPS‐only treatment levels. KLF‐4 mRNA expression did not significantly change in either treatment (Fig. 3a).
Figure 3.
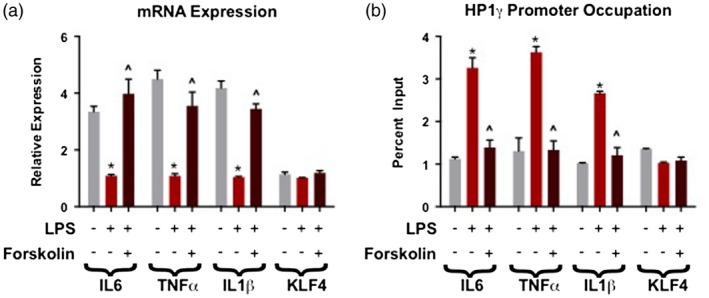
Induction of adenylyl cyclase/protein kinase A (PKA) pathway with forskolin activity mimics the ‘reversal’ effect of risperidone on lipopolysaccharide (LPS)‐induced heterochromatin. In these experiments, cells were exposed to LPS for 48 h and supplemented with forskolin for 24 h before harvest. (a) Forskolin treatment significantly increased mRNA expression levels of interleukin (IL)‐6, tumor necrosis factor (TNF)‐α and IL‐1β from LPS‐only treatment levels, but does not alter the negative control Kruppel‐like factor 4 (KLF)‐4. (b) Forskolin resulted in significantly decreased HP1γ occupancy on IL‐6, TNF‐α and IL‐1β promoters from the LPS‐only condition. The following symbols were used to draw attention to specific Tukey post‐hoc analyses: *P < 0·05 vehicle versus LPS‐only; ^P < 0·05, LPS‐only versus LPS + forskolin.
Cells were treated for 48 h with vehicle, LPS or LPS where forskolin was added for the final 24 h of treatment and chromatin immunoprecipitated for HP1γ. Forskolin treatment had the same effect as risperidone in significantly reducing or ‘reversing’ the formation of heterochromatin protein assemblies in response to LPS on the IL‐6, TNF‐α and IL‐1β promoters when compared to the LPS‐only condition (Fig. 3b).
Discussion
In this study, we demonstrate the ability of an anti‐psychotic to disassemble preformed heterochromatin along a targeted promoter. Human SW872 cells respond to LPS treatment in a manner similar to immune cells, whereby heterochromatin is induced along specific promoters 37, 38. We show that, once established, heterochromatinized promoters can be ‘re‐modeled’ through treatment with risperidone, which can activate the GPCR kinase cascade, from PKA to the terminal phosphorylation of H3S10 by MSK1 39, 40. The addition of risperidone appears to not only ‘block’ the heterochromatinizing effects of LPS treatment when applied concurrently (Fig. 2), but will also ‘reverse’ the heterochromatin state of promoters previously heterochromatinized by LPS treatment (Fig. 3). These blocking and reversing effects could be comparable to distinctions such as ‘prophylactic’ or ‘curative’ functions in the clinical setting. These effects of risperidone can be abrogated through addition of an inhibitor of PKA, the putative kinase through which risperidone is working, to shift the chromatin state at these promoters back to the heterochromatic LPS treatment state. Similarly, and most convincingly, the same effect is seen when the end‐point kinase MSK1 of the PKA cascade is inhibited (Fig. 2). In the opposite direction, treating LPS‐tolerized cells with forskolin, an AC activator, had a similar effect to risperidone by decreasing chromatin modifications characteristic of heterochromatin in this study. These results suggest that activity along the AC/PKA/MSK1‐initiated kinase signaling pathway is one possible mechanism explaining an additional therapeutic effect of anti‐psychotics.
Phosphorylation of H3S10 is a multi‐functional modification that has a role in both the cell cycle and regulation of gene transcription 4. In post‐mitotic cells, including neurons, this modification is associated with euchromatic regions and increased gene transcription in response to sensory stimulation and cognitive functioning 7, 9, 15. Chwang et al. demonstrate the role of the ERK/mitogen‐activated protein kinase (MAPK) and p38/MAPK pathways working downstream through MSK1, which leads to both H3S10 and CREB phosphorylation in the nucleus in behavioral conditioning and spatial learning 8, 9. Treatment with dopaminergic, muscarinic or glutamatergic agonists influences H3S10 phosphorylation in hippocampal neurons 15. At the cellular level, Tiwari et al., have recently established a link between phospho‐H3S10 chromatin modification in terminally differentiated pyramidal glutamatergic neurons and c‐Jun N‐terminal kinases (JNKs) of the MAPK cascades. When JNK/MAPK signaling is inhibited, a substantial reduction in H3S10 phosphorylation occurs at JNK target promoters, reducing expression of these genes. JNKs carry out several functions, including response to stress stimuli, T cell differentiation and apoptosis, but had not previously been linked to chromatin modification in neurons 17.
Facultative heterochromatin, characterized by the H3K9me2 modification, tends to occupy euchromatic regions and gene clusters, and represses genomewide tissue‐specific ontologies and networks. The co‐ordination of this genomewide repression is not well studied in chronic illness, but is a probable outcome of poorly co‐ordinated promoter activity. Over time, heterochromatin has the tendency to spread over kilobase distances, compounding untargeted gene repression 41.
HP1 attachment to H3K9me2/3 is one mechanism for propagating the spread of heterochromatin. The N‐terminus chromodomain of the HP1 protein has a high‐affinity attraction to the methylated lysine residue of H3K9me2/3 42 and the C‐terminus of the HP1 protein contains the chromo‐shadow domain, which is capable of homodimerization with a neighboring HP1 43. The concurrent attachment of an HP1 protein with H3K9me2 and dimerization with another HP1 protein from a neighboring nucleosome allows for higher‐order heterochromatin assembly. When a specific region is thus occupied, gene silencing tends to occur within a functional and co‐ordinated gene cluster, such as the expression of lipase genes in the liver but not in the brain 41, 44. The binding of HP1 to H3K9me2/3 is not always guaranteed, as while heterochromatin is an expression‐resistant assembly it is capable of disassembly and remains dynamic with unbound HP1 protein 45. The genomewide spread and segregation of restrictive chromatin has functional implications if it is considered as a constraint on plasticity or reactivity. In this framework, it is noteworthy that in the liver, 46% of the genome is occupied by H3K9me2/3 modifications, while in comparison only 4% of the embryonic stem cell and 10% of the adult neuron genome is similarly occupied, suggesting that the neuron is more similar to the embryonic stem cell by maintaining a relatively unrestricted genome 44. Enhanced genomic plasticity due to reduced H3K9me2/3 occupancy has been demonstrated by nuclear transfer experiments into recipient single cell embryos, where gene expression in the subsequent fetus is increased if the transplanted nuclei contained low levels of methylated H3K9 46.
A limitation of this study is the SW872 cell type, which includes the receptor/signaling machinery necessary for transmission towards epigenetic effects in the nucleus, but given the non‐neuronal lineage, they cannot immediately be applied to psychiatric disorders. Characterizing endotoxin tolerance and understanding its ability to be manipulated through anti‐psychotic use in cells of neuronal origin, such as a gliablastoma or neuroblastoma cell line, would be an interesting set of future experiments. However, risperidone has been shown to modulate cytokine production in antigen‐presenting dendritic cells, increasing IL‐10, IL‐6, IL‐8 and TNF‐α, but reducing IFN‐γ‐inducible protein‐10 (IP‐10) and IL‐12 47. Risperidone has also been shown to lower IFN‐γ production by T cells 48. Interestingly, a decrease in serum levels of IL‐6 and TNF‐α in first‐episode schizophrenia patients after treatment with risperidone has been shown 49. However, a similar study by Chen et al. demonstrated increased TNF‐α but decreased IL‐1β in plasma after risperidone treatment 47. Patient‐derived samples are recipients of multiple chromatin‐modifying stimuli, including additional medications and environmental stressors, suggesting additional in‐vivo inputs that were not modeled in our in‐vitro system. Lastly, measuring other psychotropic medications would be instructive to identify the specificity of this mechanism. Even given these limitations, there are significant theoretical and clinical implications for the findings of this study. Our ability to demonstrate anti‐psychotic effects along kinase pathways downstream of neurotransmitter receptors suggests that the current inadequate therapeutic response from anti‐psychotic medication can perhaps be supplemented with pharmacology capable of altering kinase activity, warranting future studies.
Disclosures
The authors declare there are no conflicts of interest in this study.
Acknowledgements
This work was supported in part by PHS grant (NIH) R01MH094358 (RPS).
References
- 1. Sharma RP, Gavin DP, Chase KA. Heterochromatin as an incubator for pathology and treatment non‐response: Implication for neuropsychiatric illness. Pharmacogenomics J 2012;12:361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang W, Mishra V, Batra S, Dillon I, Mehta KD. Phorbol ester promotes histone H3‐Ser10 phosphorylation at the LDL receptor promoter in a protein kinase C‐dependent manner. J Lipid Res 2004; 45:1519–27. [DOI] [PubMed] [Google Scholar]
- 3. Li J, Yin G, Schroeder FA et al Dopamine D2‐like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP‐protein kinase A and NMDA receptor signaling. J Neurochem 2004; 90:1117–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sawicka A, Seiser C. Sensing core histone phosphorylation – a matter of perfect timing. Biochim Biophys Acta 2014; 1839:711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davie JR. MSK1 and MSK2 mediate mitogen‐ and stress‐induced phosphorylation of histone H3: A controversy resolved. Sci STKE 2003; 2003:PE33. [DOI] [PubMed] [Google Scholar]
- 6. Soloaga A, Thomson S, Wiggin GR et al MSK2 and MSK1 mediate the mitogen‐ and stress‐induced phosphorylation of histone H3 and HMG‐14. EMBO J 2003; 22:2788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brami‐Cherrier K, Valjent E, Herve D et al Parsing molecular and behavioral effects of cocaine in mitogen‐ and stress‐activated protein kinase‐1‐deficient mice. J Neurosci 2005; 25:11444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chwang WB, O'Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem 2006; 13:322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chwang WB, Arthur JS, Schumacher A, Sweatt JD. The nuclear kinase mitogen‐ and stress‐activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci 2007; 27:12732–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 2005; 438:1176–80. [DOI] [PubMed] [Google Scholar]
- 11. Sabbattini P, Sjoberg M, Nikic S et al An H3K9/S10 methyl‐phospho switch modulates Polycomb and Pol II binding at repressed genes during differentiation. Mol Biol Cell 2014; 25:904–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fischle W, Tseng BS, Dormann HL et al Regulation of HP1‐chromatin binding by histone H3 methylation and phosphorylation. Nature 2005; 438:1116–22. [DOI] [PubMed] [Google Scholar]
- 13. Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 2002; 111:381–92. [DOI] [PubMed] [Google Scholar]
- 14. Cheung P, Tanner KG, Cheung WL et al Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol Cell 2000; 5:905–15. [DOI] [PubMed] [Google Scholar]
- 15. Crosio C, Heitz E, Allis CD, Borrelli E, Sassone‐Corsi P. Chromatin remodeling and neuronal response: Multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci 2003; 116:4905–14. [DOI] [PubMed] [Google Scholar]
- 16. Kovarik P, Stoiber D, Eyers PA et al Stress‐induced phosphorylation of STAT1 at Ser727 requires p38 mitogen‐activated protein kinase whereas IFN‐gamma uses a different signaling pathway. Proc Natl Acad Sci U S A 1999; 96:13956–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tiwari VK, Stadler MB, Wirbelauer C et al A chromatin‐modifying function of JNK during stem cell differentiation. Nat Genet 2011; 44:94–100. [DOI] [PubMed] [Google Scholar]
- 18. Sharma RP, Rosen C, Kartan C et al Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: Preliminary results from a clinical population. Schizophr Res 2006;88:227–31. [DOI] [PubMed] [Google Scholar]
- 19. Sharma RP, Grayson DR, Gavin DP. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: Analysis of the National Brain Databank microarray collection. Schizophr Res 2008;98:111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guidotti A, Auta J, Davis JM et al Toward the identification of peripheral epigenetic biomarkers of schizophrenia. J Neurogenet 2014; 28:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gavin DP, Sharma RP. Chromatin from peripheral blood mononuclear cells as biomarkers for epigenetic abnormalities in schizophrenia. Cardiovasc Psychiatry Neurol. 2009; 2009:409562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chase KA, Gavin DP, Guidotti A, Sharma RP. Histone methylation at H3K9: Evidence for a restrictive epigenome in schizophrenia. Schizophr Res 2013; 149:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharma RP, Feiner B, Chase KA. Histone H3 phosphorylation is upregulated in PBMCs of schizophrenia patients in comparison to healthy controls. Schizophr Res 2015; 169:498–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bertran‐Gonzalez J, Håkansson K, Anders B et al Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology 2009; 34:1710–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Venkatasubramanian G, Debnath M. The TRIPS (Toll‐like receptors in immuno‐inflammatory pathogenesis) Hypothesis: A novel postulate to understand schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2013; 44:301–11. [DOI] [PubMed] [Google Scholar]
- 26. El Gazzar M, Yoza BK, Chen X et al G9a and HP1 couple histone and DNA methylation to TNFalpha transcription silencing during endotoxin tolerance. J Biol Chem 2008; 283:32198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan C, Li L, McCall CE, Yoza BK. Endotoxin tolerance disrupts chromatin remodeling and NF‐kappaB transactivation at the IL‐1beta promoter. J Immunol 2005; 175:461–8. [DOI] [PubMed] [Google Scholar]
- 28. Chen X, El Gazzar M, Yoza BK, McCall CE. The NF‐kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem 2009; 284:27857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+‐dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem 2011; 286:9856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoza BK, Hu JY, Cousart SL, Forrest LM, McCall CE. Induction of RelB participates in endotoxin tolerance. J Immunol 2006; 177:4080–5. [DOI] [PubMed] [Google Scholar]
- 31. Borcherding DC, Hugo ER, Idelman G et al Dopamine receptors in human adipocytes: Expression and functions. PLOS ONE 2011; 6:e25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nativel B, Marimoutou M, Thon‐Hon VG et al Soluble HMGB1 is a novel adipokine stimulating IL‐6 secretion through RAGE receptor in SW872 preadipocyte cell line: Contribution to chronic inflammation in fat tissue. PLOS ONE 2013; 8:e76039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt AJ, Krieg JC, Clement HW et al Effects of quetiapine, risperidone, 9‐hydroxyrisperidone and ziprasidone on the survival of human neuronal and immune cells in vitro . J Psychopharmacol 2010; 24:349–54. [DOI] [PubMed] [Google Scholar]
- 34. Yang MC, Chen KP, Lung FW. Generalized estimating equation model and long‐term exposure effect of antipsychotics on SH‐SY5Y cells against oxidative stressors. Eur J Pharmacol 2014; 740:697–702. [DOI] [PubMed] [Google Scholar]
- 35. Crowley LC, Marfell BJ, Christensen ME, Waterhouse NJ. Measuring cell death by trypan blue uptake and light microscopy. Cold Spring Harb Protoc 2016; 2016. doi: 10.1101/pdb.prot087155. [DOI] [PubMed] [Google Scholar]
- 36. Lacazette E. A laboratory practical illustrating the use of the ChIP‐qPCR method in a robust model: Estrogen receptor alpha immunoprecipitation using Mcf‐7 culture cells. Biochem Mol Biol Educ 2017; 45:152–60. [DOI] [PubMed] [Google Scholar]
- 37. Seeley JJ, Ghosh S. Molecular mechanisms of innate memory and tolerance to LPS. J Leukoc Biol 2017; 101:107–19. [DOI] [PubMed] [Google Scholar]
- 38. Chen J, Ivashkiv LB. IFN‐gamma abrogates endotoxin tolerance by facilitating Toll‐like receptor‐induced chromatin remodeling. Proc Natl Acad Sci USA 2010; 107:19438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Akam E, Strange PG. Inverse agonist properties of atypical antipsychotic drugs. Biochem Pharmacol 2004; 67:2039–45. [DOI] [PubMed] [Google Scholar]
- 40. Piao L, Park J, Li Y et al SOCS3 and SOCS6 are required for the risperidone‐mediated inhibition of insulin and leptin signaling in neuroblastoma cells. Int J Mol Med 2014; 33:1364–70. [DOI] [PubMed] [Google Scholar]
- 41. Kungulovski G, Nunna S, Thomas M et al Targeted epigenome editing of an endogenous locus with chromatin modifiers is not stably maintained. Epigenetics Chromatin 2015; 8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9‐methylated histone H3 tail. Science 2002; 295:2080–3. [DOI] [PubMed] [Google Scholar]
- 43. Brasher SV, Smith BO, Fogh RH et al The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J 2000; 19:1587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 2009; 41:246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Festenstein R, Pagakis SN, Hiragami K et al Modulation of heterochromatin protein 1 dynamics in primary mammalian cells. Science 2003; 299:719–21. [DOI] [PubMed] [Google Scholar]
- 46. Baxter J, Sauer S, Antoine P et al Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J 2004; 23:4462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen JJ, Chan HY, Chen CH, Gau SS, Hwu HG. Risperidone and olanzapine versus another first generation antipsychotic in patients with schizophrenia inadequately responsive to first generation antipsychotics. Pharmacopsychiatry 2012; 45:64–71. [DOI] [PubMed] [Google Scholar]
- 48. Dodd S, Maes M, Anderson G et al Putative neuroprotective agents in neuropsychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 2013;42:135–45. [DOI] [PubMed] [Google Scholar]
- 49. Lu LX, Guo SQ, Chen W et al Effect of clozapine and risperidone on serum cytokine levels in patients with first‐episode paranoid schizophrenia. Di Yi Jun Yi Da Xue Xue Bao 2004; 24:1251–4. [PubMed] [Google Scholar]