

Abstract
Efficacy trials, designed to gain regulatory marketing approval, evaluate drugs in optimally selected patients under advantageous conditions for relatively short time periods. Effectiveness trials, designed to evaluate use in usual practice, assess treatments among more typical patients in real‐world conditions with longer follow‐up periods. In “efficacy‐to‐effectiveness (E2E) trials,” if the initial efficacy trial component is positive, the trial seamlessly transitions to an effectiveness trial component to efficiently yield both types of evidence. Yet more time could be saved by simultaneously addressing efficacy and effectiveness in an “efficacy and effectiveness too (EE2) trial.” Additionally, hybrids of the E2E and EE2 approaches with differing degrees of overlap of the two components could allow flexibility for specific drug development needs. In planning EE2 trials, each stakeholder's current and future needs, incentives, and perspective must be considered. Although challenging, the ultimate benefits to stakeholders, the health system, and the public should justify this effort.
Clinical trials provide the foundational evidence for market approval and use of drugs. However, clinical trials executed to meet regulatory requirements to gain market approval do not always provide evidence needed by other stakeholders, such as patients, clinicians, payers, and the general public. Nor do they provide generalizable evidence of effectiveness of a new drug in real‐world practice conditions. There is a need for trial designs that could provide useful evidence to all stakeholders with minimal additional costs and delays. Ideally, these designs would enable evaluations of new treatments in the broad range of patients and settings that are representative of real‐world use.
Key contributors to the lack of evidence relevant to all stakeholders are differences between study subjects and investigators included in clinical trials designed for regulatory approval vs. patients and clinical practice settings in which treatments will be used once approved for marketing. For regulatory approval, the efficacy and safety of a drug is generally demonstrated in groups of patients with relatively homogenous characteristics, selected to maximize the study's chances of rejecting the null hypothesis and to minimize the risk of spurious safety signals being imputed to the drug. It would be very helpful if this initial evaluation took place in the broad range of patients and settings representative of its ultimate real‐world use. The risks of broader criteria for patients in these studies are the potential for confounding factors that could make it more difficult to demonstrate efficacy and safety, and thereby reducing the probability of gaining market approval.
In broad terms, respectively, these two approaches are referred to as “efficacy trials” and “effectiveness trials.” Ideally, efficacy trials, which assess a new drug in optimally selected patients and conditions typically for relatively short periods, should be supplemented by effectiveness trials that include the spectrum of patients and conditions in which the drug ultimately will be used.1, 2, 3 However, follow‐up effectiveness trials are infrequently done. Therefore, a more complete understanding of relative benefits and risks of use based on a clinical trial in a broader span of patients and settings is rarely achieved.
To address the need for timely high‐quality broader evidence of treatment benefit, we previously proposed a study design that integrates these two approaches, the “efficacy‐to‐effectiveness (E2E) trial.”4 With the E2E approach, a positive effect seen in the initial “efficacy” trial (e.g., by prespecified analyses and/or by the Data Safety Monitoring Board) can be seamlessly transitioned to an effectiveness trial portion, even while a regulatory decision is pending. Rather than disassembling the efficacy trial infrastructure and operations, the effectiveness portion builds on the original study to assess effectiveness in more usual care. For example, the trial enrollment criteria can be broadened, more real‐world study sites added, and plans made for longer treatment and follow‐up periods. The idea is that the effectiveness trial proceeds without delay, potentially being complete near the time of regulatory approval for marketing.
Besides providing a more complete understanding of a treatment's benefit, the E2E approach collects data to better understand heterogeneity of treatment effects across a wider group of potential patients and treatment circumstances. This heterogeneity can be captured in multivariable predictive models to aid the treatment's optimal use in those patients most likely to benefit,5, 6, 7, 8, 9, 10 thereby promoting the treatment's use in the most appropriate patients and its impact on public health.
Although the seamless sequential E2E design can be seen as a significant improvement, it still essentially requires the time, logistics, and costs of two clinical trials. As a further efficiency, we believe that in many cases, the need for two trials could be eliminated without loss in evidence generation. This would be achieved through simultaneous efficacy and effectiveness trials, termed “efficacy and effectiveness too (EE2)” trials.11 Such trials can provide both the efficacy evidence for regulatory requirements and more generalizable effectiveness evidence for a broader set of stakeholders, while integrated into a single trial.
An example of a clinical trial designed to provide simultaneous evidence of efficacy and effectiveness is the Immediate Myocardial Metabolic Enhancement During Initial Assessment and Treatment in Emergency Care (IMMEDIATE) trial of intravenous glucose‐insulin‐potassium for acute coronary syndrome.12, 13 This was a trial executed to the high standards required to show efficacy but in a broad community‐based patient population in diverse settings. Its analyses were all done in the broad group of participants, without separation into efficacy and effectiveness analyses—and it demonstrated a positive response across the entire group. However, its study design and analysis are probably unlikely to be frequently possible as most sponsors will not want to have the efficacy analysis include the heterogeneity of patients and care settings included in the entire effectiveness cohort. Therefore, although it illustrates the efficiency of the simultaneous efficacy and effectiveness trials, for reasons noted, this is not the model of EE2 study that we propose in this paper. Our proposed EE2 model will have two categories of analyses: (i) efficacy and (ii) effectiveness. In distinction, the IMMEDIATE trial used generalizable community‐based effectiveness enrollment criteria, as in the effectiveness component, but there was only one analytic group—as the entire cohort was analyzed for efficacy. Thus, the IMMEDIATE trial can be seen as a special type of EE2 trial, with scientific and efficiency advantages, but the focus of the framework presented here is the two‐analysis approach.
The two‐cohort approach is likely more responsive to the needs of sponsors who focus on traditional efficacy testing and prefer narrower and/or enriched trial samples that will maximize the probability of a positive outcome. The E2E/EE2 designs provide an alternative way of addressing a sponsor's and regulator's need for efficacy study designs while also generating data on populations as would be expected in real‐world clinical practice.
EE2 Study designs
Enrollment criteria
Conceptually, the efficacy and effectiveness cohorts in an EE2 trial can be depicted as in Figure 1. Participants in the efficacy portion of the trial are a subset of the overall effectiveness cohort, as determined by the respective inclusion/exclusion criteria for the efficacy and effectiveness components of the study. The breadth of the effectiveness cohort will depend on the objectives of the study. The expansion of criteria will aim toward broad generalizability and may also include sufficient numbers of patients representing special populations that reflect those found in real‐world treatment settings (i.e., minorities that are frequently under‐represented in traditional efficacy clinical trials). Such populations might also include those with comorbidities, those not receiving their first treatment for the disease, and those of certain ages, clinical, socioeconomic, and/or other characteristics.
Figure 1.

Depiction of efficacy and effectiveness cohorts in an efficacy and effectiveness too trial.
Timing of efficacy and effectiveness components
The model of an EE2 trial is to have simultaneous efficacy and effectiveness components. This stands in contrast to the E2E model, previously described, in which the efficacy and effectiveness components are in sequence. Both designs address the need for greater efficiency and certainty in generating both kinds of evidence. More generally, EE2 designs could be considered as including trials with various timing of the two components, ranging from completely simultaneous to completely sequential (i.e., E2E). Such flexibility would allow for efficiency advantages while aligning to the model that best suits specific drug development needs. Three models of combined efficacy/effectiveness trials are illustrated below. In these models, timing and other variants not shown may be warranted in specific circumstances. In addition, it should be noted that the effectiveness component may be longer than the efficacy component, because it may reflect extended periods of treatment and longer term outcomes measures that map to real‐world practice considerations. Broader inclusion criteria may provide easier and more timely recruitment, although this would be study‐specific. It is also possible that the less restrictive recruitment of an effectiveness component might mitigate delays due to the more restrictive efficacy enrollment criteria, although, again, this would be study‐specific.
One option, diagramed below (Figure 2), is the completely sequential E2E trial that begins with the efficacy component and transitions to the effectiveness component upon completion of the efficacy trial's enrollment. Evaluation of the results of the first component for regulatory approval can start while the effectiveness portion is ongoing. In some cases, marketing might start, for example, in an adaptive or accelerated approval pathway, even prior to the conclusion of the full E2E trial and/or full approval.
Figure 2.

Sequential efficacy‐to‐effectiveness trial beginning with efficacy component, transitioning to effectiveness component after efficacy trial's enrollment.
A second option (Figure 3), at the opposite end of the spectrum, is the completely simultaneous conduct of the efficacy and effectiveness components; both efficacy and effectiveness cohorts are enrolled from the beginning of the study. This approach could pose a risk for the pharmaceutical sponsor in the form of upfront investment in the effectiveness component before efficacy has been demonstrated; however, it would likely be more efficient from an operational study execution perspective. An incentive for this approach could be the possibility of earlier satisfaction of other stakeholders, such as payers.
Figure 3.

Simultaneous conduct of efficacy and effectiveness components; both cohorts are enrolled from beginning of study.
A third option (Figure 4) is a staggered EE2 trial. The trial would begin with only the efficacy component, and then at a prespecified point, where an interim assessment of results would be done. The study would need to be powered to accommodate the interim analysis, with an alpha penalty associated with the decision. If a positive signal is observed in the interim, then the effectiveness component would begin. This design allows for some assurance of success for the pharmaceutical sponsor before expanding the trial but also some time efficiency by obtaining effectiveness evidence earlier. Although not an intrinsic requirement of the EE2 design, this could be seen as including some adaptive trial features at the time of the expansion that might further enhance the efficiency of the overall trial.
Figure 4.

Staggered efficacy and effectiveness too trial; efficacy cohort starts before full effectiveness cohort.
As indicated above, the IMMEDIATE trial is a special case of an EE2 trial. The efficacy analyses for regulatory approval were performed on the effectiveness cohort. Although not the focus of this article, for contrast to the above designs it is depicted in Figure 5.
Figure 5.

Single enrollment that satisfies both efficacy and effectiveness criteria, with efficacy analysis on the entire effectiveness cohort.
Analytic framework
Figure 6 shows the relationships between the efficacy and effectiveness cohorts. It starts with a determination of whether patients meet the broad effectiveness inclusion/exclusion criteria, represented by the larger circle in Figure 1. If a patient is so qualified, then there is an additional determination of whether the efficacy inclusion/exclusion criteria are also met. If so, the patient would be in the central circle, representing inclusion in both effectiveness and efficacy cohorts. If the efficacy criteria are not met, then such a patient would be in the area of the larger circle not included in the central circle.
Figure 6.
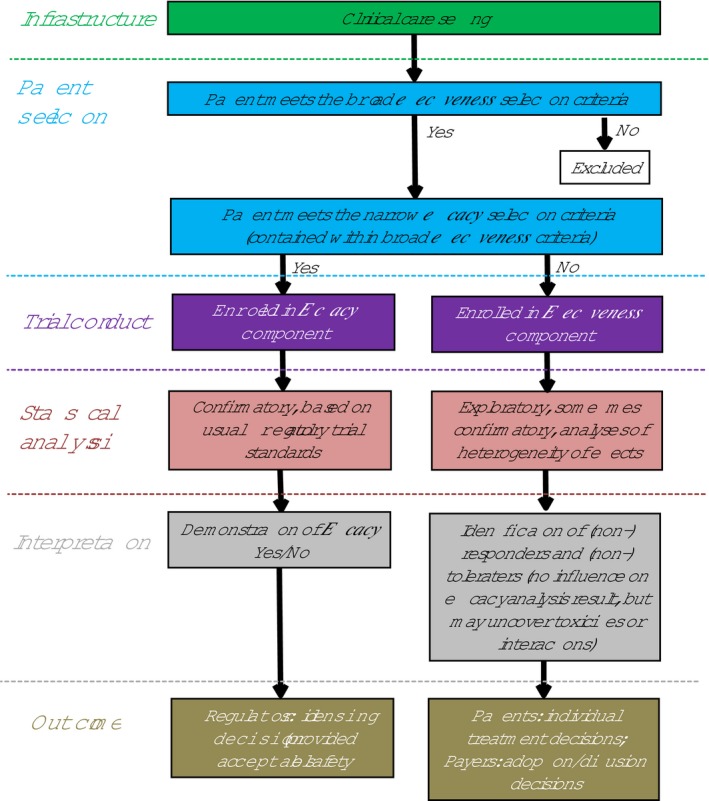
Efficacy and effectiveness too trial flowchart.
As is depicted in Figure 6, these two cohorts have separate statistical analyses. The efficacy cohort will have prespecified clinical end points for which there is planned statistical power with typical analytic procedures, accepted by regulatory agencies. The effectiveness cohort may have prespecified statistically powered end points or may be exploratory without power for hypothesis testing, whereas yet other analyses may be directed at detecting and modeling heterogeneity of treatment effects. The effectiveness cohort, especially as it will likely have longer follow‐up, will also presumably be analyzed for safety signals. Again, in contrast, this two‐lane approach to analysis is distinct from the single‐lane analysis done in the IMMEDIATE trial, in which the efficacy analysis was done on the entire effectiveness cohort.
Although various arrangements of data analyses are possible, it is likely that the primary efficacy hypothesis/es will be tested first and according to a prespecified plan, even as the efficacy cohort is a subset of the broader effectiveness cohort. The effectiveness hypotheses will be considered as secondary hypotheses, and, in some cases, can be powered a priori for detecting statistical significance. In other cases, such as exploratory analyses, these secondary hypotheses may not be powered for significance. Thus, it is possible that the primary efficacy analysis would be positive and would qualify for regulatory purposes, whereas the secondary effectiveness analyses might not reach significance, albeit still informative to broader stakeholders. Alternatively, if the primary efficacy analysis is not positive, then although not likely qualifying for market approval, the broader array of data and analyses should inform future efforts.
When designing an EE2 study, the risks and benefits of the therapy on different patient populations for a number of factors will determine the optimal design with respect to the efficacy and effectiveness cohorts and the amount of overlap of the enrollment of the two cohorts and any use of an adaptive strategy. These will include the clinical development and marketing plans of the sponsor, the strength of the efficacy and effectiveness evidence already available, and other business and operational considerations. It is likely that simulations will need to be performed to identify the optimal design. From a statistical perspective, although this is not necessarily intrinsic to the EE2 approach, analyses related to the spectrum of overlap of the E2E/EE2 components could be thought of as an adaptive design.14 Based on this, appropriate looks and adaptations can take place at prespecified intervals throughout the initial E2E/EE2 design. This presumably would be analytically similar in a fully simultaneous EE2 trial, a fully sequential E2E trial, and in a staggered EE2 design, with the interim inspection with appropriate accommodations of alpha if warranted.
Discussion
Design considerations
In an EE2 trial, assessments of drug efficacy for regulatory approval and effectiveness for real‐world use are simultaneously addressed in a single trial. Thus, the overall cohort includes a cohort with well‐defined enrollment criteria to address the efficacy analyses, as well as a wider range of patients for whom the treatment might be used in real‐world practice. Additionally, the effectiveness portion of the EE2 may be conducted over longer periods of time than the efficacy portion of the study, as the long‐term outcomes may better reflect chronic usage of the treatment in clinical practice. Thus, ideally, both types of objectives are accomplished in one study.
In our previously published E2E study design,4 continuation on to the effectiveness portion of the study is predicated on the initial efficacy trial meeting its end point with significance. That the effectiveness trial component follows immediately and seamlessly, the positive efficacy study better ensures the generation of evidence on effectiveness, and sooner, than otherwise would be the case. However, we believe that total study duration could be even further shortened by having the efficacy and effectiveness components performed simultaneously, in an EE2 design. (In this context, the fully sequential E2E design is considered a specific type of EE2 trial.)
Additionally, specific trials could have designs that span the spectrum from having the two components completely sequential to have them completely simultaneous. Indeed, for partially overlapping components with staggered starts, while the efficacy component is ongoing, adaptive adjustments might be used in conjunction with the start of the effectiveness component. Recent efforts to reduce the duration and cost of therapy development programs have focused on umbrella and platform adaptive designs.15 These types of trial design innovations can be combined with EE2 design considerations and result in incremental improvement of the cost benefit ratio of additional information for patients and physicians.
The underlying assumption of the EE2 approach is that both efficacy and effectiveness trials are necessary. Efficacy trials are needed for regulatory approval and are the foundation for bringing new drugs to market. Effectiveness trials, which in the past have been somewhat discretionary, are increasingly wanted by patients and payers as evidence of applicability to real‐world settings and the range of patients seen in widespread usual practice. In addition, regulators, with their responsibilities to the public, also have become more interested in information from effectiveness trials.
To accrue these benefits, EE2 trials should have several key attributes:
The efficacy component design and end points must be sufficient for regulatory approval if the trial is positive, which will be evidenced by pretrial regulatory agency approval of the protocol.
The effectiveness component should test the treatment in patients and settings for which it is ultimately intended, including in participants often kept out of efficacy trials because of age, comorbidities, and diversity. In addition, the lengths of treatment should be more typical of real‐world use.
In addition to broadening entry criteria, the effectiveness component could have enrichment for certain subgroups to allow targeted analyses of special effects and of heterogeneity of treatment effects.
Enrollment into EE2 trials should be done in ways that are compatible with usual clinical care and with mechanisms for accurately assessing the denominator for reporting enrollment rates of eligible patients, which will facilitate understanding of study samples relative to their broad target populations.
The effectiveness component should not delay regulatory approval or, in the event that it is not statistically significant, prevent regulatory approval.
With such features, there should be substantial advantages in having the efficacy and effectiveness trial components executed simultaneously. It should reduce the time, effort, and costs that would be required for conducting two trials, as operational efficiencies in study setup and execution can be realized. It also could speed broad acceptance by patients, payers, and providers by allowing for incorporation of measures that align with each of these stakeholder's concerns. However, analytic plans will need to be suitable for pivotal trials for a variety of drugs and circumstances. Because the efficacy cohort inclusion criteria and clinical settings are a subset of the broader effectiveness cohort inclusion criteria and settings, the analysis plan will need to preserve the ability to test the efficacy cohort in alignment with regulatory requirements—and then be able to test prespecified hypotheses in the wider effectiveness cohort. Analytic adjustments will need to be made to accommodate hypothesis testing in fully simultaneous EE2 trials, staggered EE2 designs, and fully sequential E2E trials. In each case, the analytic approaches should first test the efficacy data for regulatory approval and then test effects in the wider use that will be of importance to patients, payers, and other stakeholders.
A set of circumstances that may seem challenging for EE2 approaches might be trials that use specific biological or genetic markers to indicate treatment, as are increasingly seen in cancer trials. Wider inclusion beyond those having the molecular marker presumably would be precluded. However, the broader effectiveness dimension of the trial could be based on allowing enrollment of patients with comorbidities, histories of tumors, or other features typical of patients who would receive this treatment in real‐world practice. Indeed, with the proliferation of such treatments, which often are expensive, all stakeholders have an interest in obtaining evidence about the optimal use and limits of such therapies.
Some types of treatments may be more or less suitable for EE2 trials. Experience will inform and alter approaches and processes, as will details of each case. At this early stage, we see some general features as playing roles in these considerations but with the understanding and handling of such issues will evolve. Some examples of potentially challenging areas are below.
A scenario unlikely to be suitable for an initial EE2 trial would be new molecular entities that do not yet have sufficient data to understand their safety profile when compared, ideally, with a placebo arm. At that early stage, it will be important to minimize comorbidities while learning about its safety profile without significant confounders.
An example where EE2 trials would work is the case of drugs that are graduates of successful phase II efficacy trials and have shown excellent potential for benefit. In this case, phase III confirmatory trials with a heterogeneous population would be helpful for market acceptance and dissemination.
Also potentially suitable might be new “me too” drugs, such as a new lipid‐lowering statin or angiotensin receptor blocker antihypertensives, for which the risks and benefits of the class are generally known and for which the demonstration of wide effectiveness and differentiating advantages in a broad population would be important.
Likely suitable as well could be already‐marketed drugs that have well‐established benefit‐risk profiles, for which a new therapeutic indication is wanted. An example of this might be a tumor necrosis factor (TNF) inhibitor marketed for several years for rheumatoid arthritis (RA), for which the sponsor seeks indication for one of the spondyloarthropathies. Combination drugs, such as a sodium‐glucose cotransporter type 2 inhibitor with metformin for diabetes, inhaled beta‐2 adrenergic bronchodilator, steroids, and/or anticholinergic bronchodilators for obstructive lung disease, are also examples of drugs that could benefit from an EE2 trial because of their extensive safety and efficacy profiles yet need for more data on their combined use. Another logical candidate for such a trial would be a targeted molecular agent for one type of cancer for which the sponsor is seeking approval for a different cancer harboring the same molecular target.
A different type of suitable case for an EE2 trial might be when a drug is tested in a definable population, indeed, perhaps identified in analyses of heterogeneity of effects uncovered in a prior EE2 trial. Such characteristics could be specific clinical or physiologic features or responses to a drug, biomarker, or genetic features. Another approach could be as has been suggested for the use of genetic markers that reflect coronary heart disease as a way to target anticholesterol statin treatment to the population for which treatment will be most advantageous.16, 17 This approach also could be applied to syndromes that we currently treat without adequate knowledge about the specific subgroups that have different pathophysiological processes and who, therefore, would benefit from different treatments. For example, it is understood that complex chronic disorders, such as “type 2 diabetes,” are very heterogeneous, and so it could be more productive to stratify patients who are more likely to respond to incretin‐pathway agents, sodium‐glucose cotransporter type 2 inhibitors, etc.18 This approach could allow current rather crude diagnoses of complex chronic disorders or syndromes to be more refined, perhaps by genetic‐based diagnostic tools, to allow for better stratification and targeted therapies. An EE2 trial could have an overall target population defined by such indicators, and still the efficacy and effectiveness components could be important. The latter could examine treatment effects in wider spectra of age, comorbidities, settings, and other features, which would inform wider practice.
Balancing stakeholders’ needs and interests
Although patients and payers will appreciate EE2 results relevant to real‐world practice, pharmaceutical companies may have reservations about the effectiveness component. They may have concerns that their substantial investments in drug development and clinical trials will be undermined by an increased noise‐to‐signal ratio. Heterogeneity of trial participants could dilute or overwhelm efficacy signals that would have been detected in more selective trial samples. In addition, even if the effectiveness trial component is positive, the results could imply a higher number needed to treat per person who benefits, which could have an adverse impact on use and reimbursement.
A countervailing aspect (besides prespecifying as the primary analyses of the efficacy cohort) could be prespecified analyses of targeted populations and/or exploratory analyses of the effectiveness data that could inform treatment decisions for certain patient groups or be used to create multivariable models to identify patients who would benefit from treatment. Additionally, these additional analyses of the wider effectiveness cohort, even if showing a less or no overall effect, will be of interest to patients, clinicians, and payers. Indeed, if broader enrollment criteria are not used, patients, clinicians, and payers will have concerns about the use of new treatments in far wider spectra of patients than were shown to have benefit in a pivotal clinical trial. Absent that, there will remain questions about for whom the treatment might not work, might be toxic, and/or will generate new costs, which could affect the ability to market the new drug. Thus, it will likely be in all stakeholders’ interests to have a clear plan for use of both efficacy and effectiveness results.
In creating such an EE2 plan, ideally each stakeholder's needs, incentives, and perspectives will be explicitly called out and addressed. The specifics of the inclusion and exclusion criteria and analytic plans will benefit by having key stakeholders—pharmaceutical manufacturers, patients, payers, and regulatory agencies, and others—involved in the design of the study. This will not be easy; hierarchies of research questions and hypotheses will need to be agreed upon, as will matches of study samples and clinical use to the ultimate target populations and wide practice. Compromises and trade‐offs will be necessary. However, we believe that the benefits to all stakeholders, and to the health system and the public, will justify this work. Moreover, as experience with such dialogs grows, the types of incentives and compromises that will make this approach succeed should become more apparent. Table 1 lists some of the concerns about benefits and risks that various stakeholders might have regarding EE2 trials.
Table 1.
Stakeholders’ potential concerns about benefits and risks related to efficacy and effectiveness too trials
| Benefits | Risks | |
|---|---|---|
| Patients and clinicians |
Data to inform their individual treatment choices, which will lead to better outcomes and avoid therapeutic misadventures. Evidence to inform shared patient‐physician decision making. |
Misleading/incomplete information. Information that is hard to interpret. Information overload. |
| Clinicians as medical group leaders |
Data to inform individual and population health management. Financial stewardship. |
Misleading/incomplete information. Information that is hard to interpret. Information overload. |
| Guideline developers | Additional granular data to inform guidelines (including cost‐effectiveness). |
Misleading/incomplete information. Information that is hard to interpret. Information overload. |
| Payers/health technology assessment | Evidence and data to inform adoption (add to formulary) and diffusion (coverage) decisions, including what patient characteristics make a difference in outcomes, and the consequences of a new agent replacing a currently used agent, including actual use and impact. | Additional costs due to increased coverage of medications shown to be efficacious. |
| Sponsors |
Earlier and wider coverage by payers. Earlier, more detailed detection of safety signals. Better understanding of market penetration. Increased sales. |
Additional upfront costs. Dilution of efficacy message. Reduced sales. Delays in approval. |
| Regulators |
Earlier/additional safety information. Preserves access to robust efficacy data. Better understanding of heterogeneity. |
Additional complex analyses. Negative effects on timelines. |
| Public/societal costs and benefits |
Data to inform their treatment choices for groups and populations, which can lead to better health and cost outcomes. Informs dialogue about healthcare delivery and resource allocation. |
Misleading/incomplete information. Information that is hard to interpret. Information overload. |
Although experience, attention to win‐win solutions, and good policy making will be key to this success, there may be specific strategies that will help. For example, as implied above, analysis of an EE2 trial could be done in two steps, first considering the efficacy effect, and then the effectiveness effect. This will be based on the fact that the inclusion criteria and settings for the efficacy cohort will be a subset of the broader effectiveness trial inclusion criteria and settings, with the primary end point being in the efficacy cohort. A hierarchical analysis might be used, starting with the efficacy sample, and if the results are significant, then the same hypothesis would be tested in the effectiveness sample, along with other preplanned analyses. Finding statistical significance for the primary end points for both analyses would be the best case—indicating that a drug was efficacious in the target population and also effective in a broader real‐world population.
However, a drug might have a significant effect in the efficacy target sample but not achieve statistical significance in the broader effectiveness sample. In this case, the more heterogeneous effectiveness sample presumably still would allow informative exploratory analyses of the factors that mitigated the expected effect, to inform broader use of the medication, and to suggest additional studies. In such a case, an argument could be made that regulatory approval be given for the efficacy outcome, so as to not prevent the manufacturer from providing the treatment to those who would benefit but that follow‐up investigation would be warranted, and approaches such as adaptive approval19 or an analogous track, might be considered. It is also possible that the reverse could happen: that the efficacy analysis does not reach statistical significance, but the effectiveness trial does. This would be helpful in directing follow‐on investigation, and depending on the specifics, might also lead to consideration of adaptive licensing.
Regulatory issues
In a regulatory context, if the efficacy trial component is positive and the toxicity profile looks favorable, the product could be licensed. However, it could develop that as the license is being issued, the effectiveness results become available, and they do not reach statistical significance. Because the agreement with the sponsor is that the effectiveness results would not affect the regulatory decision‐making process, the regulatory agency would move ahead with the approval. However, this creates a challenge for the regulators, as it is already known that the treatment does not seem to work in likely real‐world use. The way this situation is avoided today is that only efficacy results are available at the time of approval, and regulatory agencies do not need to address the lack of effectiveness, as such data are not available when approval is granted. From a societal perspective, having such data available and having a viable plan around the best use of the drug seems attractive. Moreover, potentially later, when an important effect, interaction, or toxicity is revealed in the effectiveness component, then some regulatory action to protect the public would be warranted.
A different perspective for the effectiveness component, perhaps as part of a drug development program, is that a positive overall result is not necessarily expected. The goal is not doing the usual statistical tests for overall effects in control vs. experimental groups, as in the efficacy part. The goal is to include more patients to explore the variations and limits of treatment effects. In any case, the safety database will be broadened, subgroups of interest are enriched, and other confounders may be discovered that diminish or augment the response.
From a regulatory perspective, the effectiveness portion of the study cannot be required for approval of a new treatment under the current law. However, if the effectiveness component could be designed to address likely regulatory postmarketing requirements, it could deliver knowledge about residual uncertainties considered by the regulators to be important to address and to potentially provide that information in an expedited manner and potentially to provide the sponsor commercially desirable labeling supplements. In order for this scheme to be actionable, there would need to be advance discussions with some specificity between the sponsor and the regulator about potential postmarketing commitments in the event of a product approval. Although we believe that much of the pressure for EE2 trials will come from the market and patient representatives, it will be important that regulators ensure that regulatory goals are not undermined by conducting such trials.
Industry perspective
For a pharmaceutical sponsor, the first goal (based on the efficacy component of the EE2 study) is to demonstrate efficacy as is written in the statute. The second goal is to collect information relevant to patients, physicians, and payers earlier and reduce time to reimbursement. A sponsor who does not have assurance that their product would still get approved, even if the effectiveness portion is negative, would never agree to perform an EE2 study. However, the effectiveness evidence could inform the conversation. In some cases, sponsors might have incentives to use such data depending on the potential magnitude of the public health impact of the treatment and representation of patient populations that may have been under‐represented in the clinical trials, aside from regulatory considerations.
More specifically, EE2 data may play a critical role in the receptiveness of the market, and especially payers (including insurers and governments), to a new drug. One aspect of this might be the benefit of earlier access to reimbursement, particularly in countries with health technology assessment (HTA) agencies, which could counter the concern about the additional cost of doing the EE2 trial. Historically, the mature HTA markets have been Europe, Australia, and Canada, but more recently the Institute for Clinical and Economic Review (ICER) has become established as an independent HTA agency in the United States, and there has been discussion about establishing an HTA system in Japan. Since 2014, ICER reviews have impacted coverage and reimbursement decisions in the United States.20 The implication of this trend is that most major markets for new pharmaceutical products either have, or may soon have, HTA systems in place to assess the clinical and economic value of these products to inform coverage and reimbursement decisions by appraisal committees. In many cases, the appraisal committees using these assessments conclude that the product has low or intermediate value because of the uncertainty in the comparative effectiveness clinical data and target populations.21 Anticipating such demands, manufacturers are now looking for ways to provide such data in a timely manner. Pressure is increasing for pharmaceutical manufacturers to provide evidence that their agent is effective in the wide swath of patients to whom they would like to market it—and for whom they desire other than third or fourth tier placement in payers’ formularies. Further below, among examples of possible EE2 trials, we note that in the example of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, wider access to these drugs might by now have been accepted had they been tested on a wider population, as would have been the case in an EE2 trial. In addition, this is suggested by current economic analyses and market responses in the case of PCSK9 inhibitors.22, 23 This may be an example of where market forces and payers (including government payers), even in the absence of pressure from regulators, could provide incentives for the conduct of an EE2 trial. Indeed, we believe that such pressure from the marketplace, rather than from regulators, will be key motivation for doing more effectiveness trials, and EE2 trials may be the most efficient way to do this.
Another critical factor might be having earlier evidence on subgroup‐specific issues with respect to safety that would allow for partial, rather than wholesale, withdrawals of drugs from market. A negative outcome for sponsors could be more limited reimbursement for specific subsets of populations.
There are examples in which a drug has been withdrawn because there was not sufficient information to identify subpopulations that would react adversely to the drug. In addition, in some cases, a drug has avoided this by the regulator's understanding of who should not be treated. The more granular information the regulators have, especially on toxicity, the more likely they are to authorize marketing (or at least not be worried about authorizing and then having to withdraw).
Examples of possible EE2 trials
The basic features of EE2 trials and their envisioned advantages for efficiency in generating efficacy and effectiveness evidence are outlined above. Several examples of the possible use of EE2 trials may illustrate some more specific advantages of the addition of the broader enrollment in expanding understanding and applicability of treatments.
Understanding the behavior of a drug in the face of comorbidities is an explicit aim of EE2 trials. However, the inclusion of patients with comorbidities may uncover more. An example can be found in the case of the use of methotrexate for RA.
Long known to be effective in autoimmune and chronic inflammatory disorders, including for RA, methotrexate has been demonstrated to improve symptoms, quality of life, and disease progression.24 Of note, meta‐analyses have shown that methotrexate treatment is associated with a lower risk of cardiovascular events when compared with other disease‐modifying antirheumatic drugs, suggesting a protective effect against atherosclerosis.25, 26 Cardioprotective effects of methotrexate may be due to reducing systemic inflammation and affecting some cellular mechanisms that lead to atherosclerosis. Thus, because the comorbidity was present in trial cohorts with RA, it has been discerned that it reduces the overall cardiovascular disease (CVD) burden in patients with RA. Based on this, now, years after this observation, trials are now completing of low‐dose methotrexate in patients from the general population with CVD but who do not have RA.
In initial trials of methotrexate for RA, had there been an explicit aim to include patients with common comorbidities, it is conceivable that this cardioprotective effect might have been seen earlier. In addition, as is now being explored in patients without RA, exploratory analyses arising from such an EE2 trial might have led to further pathways of exploration of the use of the drug for cardioprotection beyond RA. As methotrexate is also used in other autoimmune rheumatic conditions and in some malignancies, it is conceivable that a similar beneficial effect might be found with patients who also have, or have a propensity for, CVD—patients who otherwise might have been avoided in efficacy trials for the primary target condition.
The fact that an anti‐inflammatory effect might be a common feature of the benefits of methotrexate in both RA and cardioprotection raises the conceptual model of “basket trials” in cancer, in which a shared molecular target found in cancers of different organs or types is exploited by using an agent in seemingly diverse cancers for its specific effect on that target.27 The case of methotrexate may not be analogous, as the specific mechanisms in the divergent conditions may be different, even if linked by some features. However, some other examples might be closer to the basket trial conceptual model (e.g., recently, tofacitinib, has been approved by the US Food and Drug Administration (FDA) and other regulators for RA, psoriatic arthritis, and ulcerative colitis).28, 29, 30, 31, 32 Given the commonality of the biology of these diseases, with appropriate forethought, the sponsor could have conducted a trial that confirmed the efficacy of tofacitinib in RA while expanding the cohort to psoriatic arthritis and/or ulcerative colitis populations to assess potential benefit in these populations (i.e., signal‐finding cohorts) and set the table for confirmatory efficacy studies to support supplemental indications. The benefits of time and cost in leveraging existing clinical trial infrastructure would have to be balanced against overall costs and other risks. Another example of this might be in the case of TNF inhibitors, which historically were tested in one indication (e.g., Crohn's disease), before then doing additional trials in RA, psoriasis, and psoriatic arthritis. For example, apremilast, which is a selective inhibitor of the enzyme phosphodiesterase 4 and inhibits spontaneous production of TNF‐alpha from human rheumatoid synovial cells, started with an indication for psoriasis, and now trials are ongoing for an indication for Behcet's disease.33 Therefore, although the explicit purpose of basket trials is to allow for cross‐condition testing, understanding the overlap of and co‐existence of rheumatic conditions, it is conceivable that the broad enrollment allowed by an EE2 trial might help generate signals that could lead to broader applications even prior to the conduct of basket‐like trials.
A different type of example of the potential of EE2 design could be considered in the case of PCSK9 inhibitors.34, 35, 36, 37 If the initial trials included broader inclusion, even if the primary efficacy analyses had only been done for familial hypercholesterolemia, benefit might have been more rapidly seen that those with more common causes of hypercholesterolemia, especially those having adverse effects from statins, and the broadening of accepted treatment cohorts might have been earlier. One‐third of American adults, 71 million, have high levels of low‐density lipoprotein (LDL), and about 11 million do not reach their LDL reduction goals. For some this is related to genetic conditions, but for many others this may be because of intolerance to the adverse effects of statins. It is understood that part of the strategy for targeting familial hypercholesterolemia and those with demonstrated pre‐existing CVD was because of the need to succeed in demonstrating clear effect in these high‐LDL, high‐risk groups in light of the cost of these agents, it is also conceivable that an EE2 trial could have shown opportunities for expansion of benefiting patients, assuming a practical clinical and regulatory‐sanctioned definition of statin intolerance were established.
Conclusions
An EE2 trial is intended to demonstrate efficacy rigorously, such as for regulatory approval, but also assess treatment effects that apply to real‐world clinical care. The specifics of given EE2 trial designs will depend on the interests of all stakeholders, including the sponsor, regulators, patients, clinicians, and payers. In the overall design, this could include having differing degrees of temporal overlap of efficacy and effectiveness components and the use of adaptive trial features in transitions. In the targeting of populations and settings, and in specifying analyses, the details will need to support the stakeholders’ needs. The ultimate objective will remain bringing important drugs to market in the context of valuable evidence about its best use in practice.
In proposing this approach, we are not suggesting a compromise of the two types of trials that has elements of both but does not meet usual rigorous standards. Rather, the combination is intended to accrue the intended benefits of both. The efficacy component of an EE2 study must be conducted according to strict regulatory and scientific standards, and the effectiveness component should authentically demonstrate use in usual care circumstances. In other words, considering the spectrum from efficacy to effectiveness trial procedures and execution, the EE2 design can be thought of as a model that preserves both extremes of the spectrum, the enriched stringent selection criteria that gives us evidence of efficacy and data on populations as in widespread clinical practice in the real world.
As EE2 trials are planned and executed, each stakeholder's current and future needs, incentives, and perspectives will need to be considered and addressed. Ultimately, the common purpose, as science and policies are evolving to deliver outcomes that matter to individual patients and populations, to maximize the positive impact on patients and the public's health. This will be challenging, but the benefits to all stakeholders, to the health system, and to the public, should justify this effort.
Funding
This work was supported by funding from the National Institutes of Health National Center for Advancing Translational Sciences (NIH/NCATS) Grant Number U24TR001609 in support of the Johns Hopkins‐Tufts Trial Innovation Center.
Conflict of Interest
N.E.M. is an employee of Boehringer Ingelheim and owns stock in Merck and Pfizer. W.H.D. is a shareholder and former employee of Amgen and Eli Lilly and is a current board member of BioMarin, Radius Health, Seres Therapeutics, and Mersana Therapeutics. V.S.‐M. consults for or is an external employee of Otsuka America, Sanofi S.A., Janssen Pharmaceuticals, Inc., and Univlever. P.K.H. is an employee of Pfizer, however this work was completed independently of his employment. H.P.S., H.‐G.E., N.L.S., T.C., J.K.E., K.I.K., K.A.O., and R.B.D, Sr. have no conflicts to declare. As an Associate Editor for Clinical Pharmacology & Therapeutics, P.K.H. was not involved in the review or decision process for this paper.
Disclosure
This article reflects the views of the author(s) and should not be construed to represent their institution's views or policies.
Acknowledgments
The authors are very grateful for important ideas and coordination for this work and manuscript preparation by Giuliana Green, Sheeona Gorman, PhD; William Harvey, MD, MSc; Timothy McAlindon, MD, MPH; Hanneke Parkinson, Elizabeth Patchen, and Margaret Towne, MSc, without which we would not have succeeded in this project.
References
- 1. Flay, B.R. Efficacy and effectiveness trials (and other phases of research) in the development of health promotion programs. Prev. Med. 15, 451–474 (1986). [DOI] [PubMed] [Google Scholar]
- 2. Clarke, G.N. Improving the transition from basic efficacy research to effectiveness studies: methodological issues and procedures. J. Consult. Clin. Psychol. 63, 718–725 (1995). [DOI] [PubMed] [Google Scholar]
- 3. Glasgow, R.E. , Lichtenstein, E. & Marcus, A.C. Why don't we see more translation of health promotion research to practice? Rethinking the efficacy‐to‐effectiveness transition. Am. J. Public Health 93, 1261–1267 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Selker, H.P. et al A proposal for integrated efficacy‐to‐effectiveness (E2E) clinical trials. Clin. Pharmacol. Ther. 95, 147–153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selker, H.P. et al Patient‐specific predictions of outcomes in myocardial infarction for real‐time emergency use: a thrombolytic predictive instrument. Ann. Intern. Med. 127, 538–556 (1997). [DOI] [PubMed] [Google Scholar]
- 6. Selker, H.P. , Beshansky, J.R. , Griffith, J.L. & TPI Trial Investigators . Use of the electrocardiograph‐based thrombolytic predictive instrument to assist thrombolytic and reperfusion therapy for acute myocardial infarction. A multicenter, randomized, controlled, clinical effectiveness trial. Ann. Intern. Med. 137, 87–95 (2002). [DOI] [PubMed] [Google Scholar]
- 7. Kent, D.M. , Selker, H.P. , Ruthazer, R. , Bluhmki, E. & Hacke, W. The stroke‐thrombolytic predictive instrument: a predictive instrument for intravenous thrombolysis in acute ischemic stroke. Stroke 37, 2957–2962 (2006). [DOI] [PubMed] [Google Scholar]
- 8. Savvides, P. , Terrin, N. , Erban, J. & Selker, H.P. Development and validation of a patient‐specific predictive instrument for the need for dose reduction in chemotherapy for breast cancer: a potential decision aid for the use of myeloid growth factors. Support. Care Cancer 11, 313–320 (2003). [DOI] [PubMed] [Google Scholar]
- 9. Selker, H.P. , Ruthazer, R. , Terrin, N. , Griffith, J.L. , Concannon, T. & Kent, D.M. Random treatment assignment using mathematical equipoise for comparative effectiveness trials. Clin. Transl. Sci. 4, 10–16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kent, D.M. et al Comparison of mortality benefit of immediate thrombolytic therapy versus delayed primary angioplasty for acute myocardial infarction. Am. J. Cardiol. 99, 1384–1388 (2007). [DOI] [PubMed] [Google Scholar]
- 11. Selker, H.P. , Gorman, S. & Kaitin, K.I. Efficacy‐to‐effectiveness clinical trials. Trans. Am. Clin. Climatol. Assoc. 129, 279–300 (2018). [PMC free article] [PubMed] [Google Scholar]
- 12. Selker, H.P. et al Out‐of‐hospital administration of intravenous glucose‐insulin‐potassium in patients with suspected acute coronary syndromes: the IMMEDIATE randomized controlled trial. JAMA 307, 1925–1933 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Selker, H.P. et al Study design for the Immediate Myocardial Metabolic Enhancement During Initial Assessment and Treatment in Emergency Care (IMMEDIATE) trial: a double‐blind randomized controlled trial of intravenous glucose, insulin, and potassium for acute coronary syndromes in emergency medical services. Am. Heart J. 163, 315–322 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. US Food and Drug Administration (FDA) . Adaptive designs for clinical trials of drugs and biologics guidance for industry. U.S. Department of Health and Human Services, Federal Registrar. <https://www.fda.gov/downloads/drugs/guidances/ucm201790.pdf> (2018). Accessed November 19, 2018.
- 15. MIT Center for Biomedical Innovation . The Next Wave in Adaptive Biomedical Innovation: Advancing Platform Trials into End‐to‐End Rapid Learning Systems (MIT, Cambridge, MA, 2018). [Google Scholar]
- 16. Mega, J.L. et al Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 385, 2264–2271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schunkert, H. & Samani, N.J. Statin treatment: can genetics sharpen the focus? Lancet 385, 2227–2229 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Ingelfinger, J.R. & Rosen, C.J. Cardiac and renovascular complications in type 2 diabetes – is there hope? N. Engl. J. Med. 375, 380–382 (2016). [DOI] [PubMed] [Google Scholar]
- 19. Eichler, H.G. et al Adaptive licensing: taking the next step in the evolution of drug approval. Clin. Pharmacol. Ther. 91, 426–437 (2012). [DOI] [PubMed] [Google Scholar]
- 20. Institute for Clinical and Economic Review (ICER) . Abuse‐deterrent opioids. <https://icer-review.org/topics/#past-topics>. Accessed November 19, 2018.
- 21. Neumann, P. , Silver, M. & Cohen, J.T. . Should a drug's value depend on the disease or population it treats? Insights from ICER's value assessments. Health Affairs Blog. 10.1377/hblog20191105.38350. [e‐pub ahead of print]. [DOI] [Google Scholar]
- 22. O’Riordan, M. Alirocumab should be priced at $6,300 annually: ODYSSEY cost‐effectiveness study. tmtMD/the heart beat. Accessed December 16, 2018. [Google Scholar]
- 23. Robinson, J.G. et al Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372, 1489–1499 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Singh, J.A. et al 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease‐modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res. (Hoboken) 64, 625–639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Micha, R. et al Systematic review and meta‐analysis of methotrexate use and risk of cardiovascular disease. Am. J. Cardiol. 108, 1362–1370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marks, J.L. & Edwards, C.J. Protective effect of methotrexate in patients with rheumatoid arthritis and cardiovascular comorbidity. Ther. Adv. Musculoskelet. Dis. 4, 149–157 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trusheim, M.R. et al PIPELINEs: creating comparable clinical knowledge efficiently by linking trial platforms. Clin. Pharmacol. Ther. 100, 713–729 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sandborn, W.J. et al Tofacitinib as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 376, 1723–1736 (2017). [DOI] [PubMed] [Google Scholar]
- 29. US Food and Drug Administration (FDA) . FDA approves new treatment for moderately to severely active ulcerative colitis. [press release]. <https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609225.htm>. Accessed September 28, 2018.
- 30. Gladman, D. et al Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N. Engl. J. Med. 377, 1525–1536 (2017). [DOI] [PubMed] [Google Scholar]
- 31. Pfizer announces FDA approval of Xeljanz (tofacitinib) and Xeljanz XR for the treatment of active psoriatic arthritis [press release]. <www.drugs.com> (2017).
- 32. European Medicine Agency (EMA) . Xeljanz (tofacitinib). March 31, 2017. <https://www.ema.europa.eu/medicines/human/EPAR/xeljanz>. Accessed September 28, 2018.
- 33. Hatemi, G. et al Apremilast for Behcet's syndrome–a phase 2, placebo‐controlled study. N. Engl. J. Med. 372, 1510–1518 (2015). [DOI] [PubMed] [Google Scholar]
- 34. Sabatine, M.S. et al Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372, 1500–1509 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Ridker, P.M. et al Cardiovascular efficacy and safety of bococizumab in high‐risk patients. N. Engl. J. Med. 376, 1527–1539 (2017). [DOI] [PubMed] [Google Scholar]
- 36. Rosenson, R.S. , Hegele, R.A. , Fazio, S. & Cannon, C.P. The evolving future of PCSK9 inhibitors. J. Am. Coll. Cardiol. 72, 314–329 (2018). [DOI] [PubMed] [Google Scholar]
- 37. Cholesterol Treatment Trialists’ (CTT) Collaboration et al Efficacy and safety of more intensive lowering of LDL cholesterol: a meta‐analysis of data from 170,000 participants in 26 randomised trials. Lancet 376, 1670–1681 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]