

Abstract
The objective of this study was to examine the distribution and severity of tau-PET binding in cognitively normal adults with preclinical Alzheimer’s disease as determined by positive beta-amyloid PET. 18F-AV-1451 tau-PET data from 109 cognitively normal older adults were processed with 34 cortical and 9 subcortical FreeSurfer regions and averaged across both hemispheres. Individuals were classified as being beta-amyloid positive (N = 25, A+) or negative (N = 84, A−) based on a 18F-AV-45 beta-amyloid-PET standardized uptake value ratio of 1.22. We compared the tau-PET binding in the 2 groups using covariate-adjusted linear regressions. The A+ cohort had higher tau-PET binding within 8 regions: precuneus, amygdala, banks of the superior temporal sulcus, entorhinal cortex, fusiform gyrus, inferior parietal cortex, inferior temporal cortex, and middle temporal cortex. These findings, consistent with preclinical involvement of the medial temporal lobe and parietal lobe and association regions by tauopathy, emphasize that therapies targeting tauopathy in Alzheimer’s disease could be considered before the onset of symptoms to prevent or ameliorate cognitive decline.
Keywords: Alzheimer’s disease, Tau, Positron emission tomography, Temporal lobe, Parietal lobe
1. Introduction
Alzheimer’s disease (AD), the most common cause of dementia, is characterized by extracellular beta-amyloid plaques and intraneuronal neurofibrillary tangles (NFTs); the latter contain abnormal filaments of pathologic tau protein (Braak and Braak, 1991; Braak et al., 2006). In vivo neuroimaging and cerebrospinal fluid (CSF) quantification of both beta-amyloid and tau protein can now detect pathological aggregates and abnormal fluid concentrations of these proteins as early as 2 decades before the onset of clinical symptoms (Bateman et al., 2012; Sperling et al., 2011).
Multiple clinicopathologic studies demonstrate that AD pathology is present in individuals before clinical symptoms develop; such individuals are at elevated risk for progression to AD dementia (Fagan et al., 2007; Morris et al., 2009; Vos et al., 2013). The accumulation of beta-amyloid plaques and NFTs in cognitively normal (CN) individuals has been suggested to represent preclinical AD. The 2011 National Institute of Aging (NIA) criteria, and recently updated criteria (Jack et al., 2018a), further propose that preclinical AD be divided into separate stages: stage 1 with beta-amyloidosis only (A+); stage 2 with beta-amyloidosis and indicators of neurodegenerative pathology (N+); and stage 3 with beta-amyloidosis, neurodegeneration, and subtle cognitive decline (Jack et al., 2012; Sperling et al., 2011). These 3 proposed stages of preclinical AD can be described by surrogate markers in CSF; low CSF beta-amyloid-42 is generally accepted as a marker of beta-amyloidosis, and high CSF tau is generally accepted as a marker of neurodegeneration. However, there are several limitations of CSF markers, including lack of direct information about distributions or densities of beta-amyloid or of the NFTs. In addition, increased CSF total tau levels, commonly observed in AD, are similarly observed with other dementing diseases such as frontotemporal lobar degeneration and traumatic brain injury, as well as normal aging (Franz et al., 2003; Riemenschneider et al., 2002), making it nonspecific for AD. By contrast, PET tracers thought to bind to NFTs (Lowe et al., 2016; Marquie et al., 2017) reflect both total tauopathy burden and topography, although off target binding has been noted (Lemoine et al., 2018). In addition, spatial information from tau-PET will be critical for monitoring potential tau aggregate accumulation over time and for linking the regional spread of pathologic tau to other key mechanisms involved in AD progression. Furthermore, by providing this spatial information, tau-PET may help to distinguish preclinical AD and symptomatic AD from other tauopathy-related diseases that exhibit different anatomical patterns of tau aggregates. Therefore, PET measures of tau may be more suitable than CSF measures for applying NIA-AA criteria for preclinical AD or ATN classification.
While the regional deposition of beta-amyloid plaques in vivo, as detected by beta-amyloid-PET tracers, has been well established (Klunk et al., 2004; Villemagne et al., 2011), experience with tau-PET to describe the distribution of tauopathy in vivo is more limited. Nevertheless, findings with tau-PET to date appear to be fairly consistent with the spatial distributions of tauopathy that have been as described in neuropathologic studies (Braak and Braak, 1991; Braak et al., 2006). Initial ex vivo autoradiographic studies suggest tau-PET correlates with postmortem tauopathy (Lowe et al., 2016; Marquie et al., 2015). Furthermore, in vivo studies examining individuals with symptomatic AD compared with controls have found tau-PET ligand binding in temporal as well as neocortical areas in a spatial pattern that is generally consistent with, but not identical to, advanced Braak stages V and VI (Chien et al., 2013; Cho et al., 2016; Gordon et al., 2016; Johnson et al., 2016; Schwarz et al., 2016). The spatial pattern of binding also colocalizes with changes in hypometabolism (Bischof et al., 2016; Ossenkoppele et al., 2016), and atrophy (Wang et al., 2016), observed in atypical forms of AD (Day et al., 2017; Ossenkoppele et al., 2016), and mirrors the degree of selective cognitive impairment in those cases.
While there is an established literature describing tau-PET findings in the setting of AD dementia, the investigation of regional distribution of tauopathy in CN individuals with or without preclinical AD (A+ or A−) remains relatively unexplored. Initial studies investigating tau-PET ligand binding in small cohorts of CN adults have identified several brain regions that show evidence of tauopathy in older individuals (Hanseeuw et al., 2017; Jack et al., 2018b; Jacobs et al., 2018; LaPoint et al., 2017; Lowe et al., 2018a,b; Scholl et al., 2016; Vemuri et al., 2017) and report that increased tau-PET binding inversely correlates with beta-amyloid-42 levels in the cerebrospinal fluid (Chhatwal et al., 2016; Gordon et al., 2016). This early work suggests tau-PET binding is elevated in preclinical AD, but further work is needed to define the topography of tau-PET binding early in the disease course. The characterization of regional tau accumulation in CN adults in vivo using tau-PET could be important for identifying CN individuals most at risk of cognitive decline and might encourage the evaluation of tau-related pharmacological interventions early in the disease course.
2. Materials and methods
2.1. Participants
Data from 109 participants from studies at the Knight Alzheimer’s Disease Research Center, Washington University in St Louis (including the Adult Children Study and the Healthy Aging and Senile Dementia Study) were used. Inclusion criteria included cognitive normality [Clinical Dementia Rating score equals 0 (Morris, 1997)] and completion of both beta-amyloid and tau-PET scans. The Washington University in St. Louis Institutional Review Board approved all procedures and each participant was provided signed informed consent for the study.
2.2. MRI
Data were acquired on a Siemens Biograph mMR (n = 83) or Trio 3T scanner (n = 26). T1-weighted images were acquired using a magnetization-prepared rapid acquisition gradient echo (MPRAGE) sequence with the following: repetition time = 2300 ms, echo time = 2.95 ms, flip angle = 9°, 176 slices, in plane resolution 240 × 256, slice thickness = 1.2 mm acquired in sagittal orientation. Images underwent volumetric segmentation using FreeSurfer 5.3 [mailto:http://freesurfer.net (Fischl et al., 2004)] to identify regions of interest (ROIs) used in the PET analyses.
2.3. PET imaging
2.3.1. Beta-amyloid-PET
Participants underwent beta-amyloid-PET imaging with 18F-AV-45 (florbetapir). Participants received a single intravenous bolus of 370 MBq (10 mCi) of florbetapir infused over 60 seconds. Scans were acquired on a Siemens Biograph mMR PET/MR scanner and attenuation corrected with a corresponding CT. Data were processed using an ROI approach using FreeSurfer software. As described previously (Gordon et al., 2016), data between the 50- to 70-minute postinjection window were examined. In each ROI, data were converted to standardized uptake value ratios (SUVRs) using the cerebellar gray as the reference region and partial volume corrected using a regional spread function approach (Rousset et al., 1998; Su et al., 2015, 2016).
2.3.2. Tau-PET
Tau-PET imaging was performed within 13 months (mean 44.0 days, range: 1–373 days) of the beta-amyloid-PET imaging session using 18F-AV-1451 (flortaucipir). Participants received a single 6.5–10 mCi intravenous bolus of flortaucipir infused over 20 seconds. Data were processed using an ROI approach using FreeSurfer and, as done in prior work using 18F-AV-1451 (Brier et al., 2016; Chien et al., 2013; Day et al., 2017; Gordon et al., 2016; Johnson et al., 2016; Wang et al., 2016), and data from the 80- to 100-minute postinjection window were examined. Scans were acquired on a Siemens Biograph 40 PET/CT scanner. As described previously, in each ROI, data were converted to SUVRs using the cerebellar gray as the reference region and partial volume corrected using a regional spread function approach (Rousset et al., 1998; Su et al., 2015, 2016). This partial volume correction method, including nonbrain region sampling, may additionally help minimize off-target binding. Tau-PET SUVRs for each cortical and subcortical ROI were extracted and averaged together from the left and right hemispheres to form 1 bilateral measure. The average amount of time between clinical assessment and tau-PET imaging session was 103 ± 57 days, and the average time between beta-amyloid-PET and tau-PET imaging sessions was 82 ± 84 days.
2.4. Amyloid positivity classification
As previously described, a composite beta-amyloid deposition measure was created using the average across the left and right lateral orbitofrontal, medial orbitofrontal, rostral middle frontal, superior frontal, superior temporal, middle temporal, and precuneus regions (Su et al., 2013, 2016). To identify individuals who were beta-amyloid positive, we split our sample based on a partial volume corrected florbetapir SUVR cutoff of 1.22 (Mishra et al., 2017). To generate a cutoff value for 18F-AV-45, a previously established cutoff using 11C-Pittsburgh compound B [(Gordon et al., 2015; Vlassenko et al., 2011)] was translated using a linear regression from a separate cohort of 100 individuals who had both 18F-AV-45 and 11C-Pittsburgh compound B imaging as part of a crossover study. Based on this cutoff, our current sample included 25 beta-amyloid-positive (A+) individuals and 84 beta-amyloid-negative (A−) participants.
2.5. Neuropsychological assessment
Each cohort at the Knight ADRC receives slightly different cognitive batteries, and in the interest of maximizing the available sample size, only tests that were common across all cohorts were considered for the present analyses. This resulted in a sample size of 108 individuals. Tests included a measure of episodic memory: the free recall score from the Free and Cued Selective Reminding Test (Grober et al., 1988); a measure of working memory: Letter Number Sequencing (Wechsler, 1997); a measure of semantic memory retrieval: category fluency for Animal Naming (Goodglass and Kaplan, 1983); a test of processing speed: Trail Making part A and a test of executive function: Trail Making part B (Armitage, 1946). Tests were standardized using the sample mean and standard deviation of the cognitive assessment that was nearest to the tau-PET scan and then averaged to form a cognitive composite score (Aschenbrenner et al., 2018).
2.6. Statistics
To test for group differences between A+ and A− cohorts on demographics, t-tests were performed for age, years of education, and Mini–Mental State Examination score; chi-squared tests were performed for sex and apolipoprotein E ε4 (APOE4) genotype (Table 1).
Table 1.
Participant characteristics
| Characteristic | A−(N= 84) | A+ (N= 25) | p-value |
|---|---|---|---|
| Age, y | 66.8 (8.6) [46–91] | 71.9 (7.4) [58–90] | 0.006 |
| Female, % (n) | 50.0 (42) | 60.0 (15) | 0.495 |
| APOE4 positive, % (n) | 23.2 (19) | 56.0 (14) | 0.003 |
| Education, y | 16.1 (2.2) [12–20] | 16.6 (1.6) [12–18] | 0.425 |
| GDS | 1.16 (1.8) [0–10] | 0.68 (1.1) [0–3] | 0.104 |
| MMSE | 29.3 (1.1) [25–30] | 29.5 (1.0) [27–30] | 0.228 |
| Clinical assessment and tau-PET time interval, d | 105.0 (54.8) [0–240] | 94.9 (65.7) [0–328] | 0.548 |
| Beta-amyloid-PET and tau-PET time interval, d | 76.4 (80.9) [1–353] | 102.8 (94.4) [1–373] | 0.212 |
Values are mean (SD) [range] unless otherwise indicated.
Key: GDS, Geriatric Depression Scale, APOE4, apolipoprotein E ε4; MMSE, Mini–Mental State Examination, A+, beta-amyloid-PET positive; A−, beta-amyloid-PET negative.
To visualize the collective anatomic distribution of tau-PET SUVR in each of the groups, we created representative group mean images. For each participant, their nonpartial volume corrected SUVR images were aligned to their individual MPRAGE using a rigid body transformation, subsequently transformed to MNI atlas space using a nonlinear warp, and resampled into a 2-mm isotropic resolution. Voxels across all participants in either the A+ or A− groups were then averaged together (Fig. 1A and B). Higher SUVRs indicate higher binding of the tracer relative to the cerebellar reference region, which reflects more tau pathology or greater nonspecific binding, in one group compared to the other.
Fig. 1.
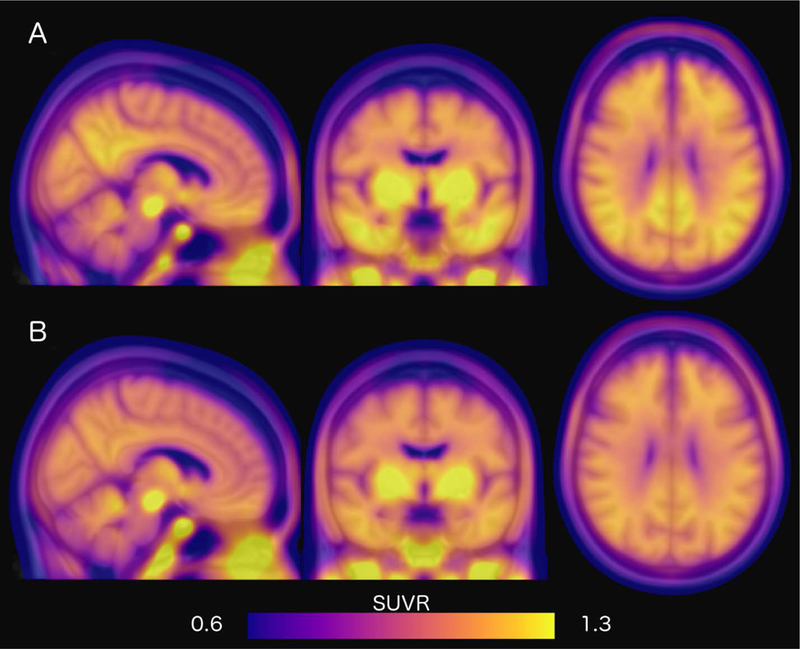
Mean tau distribution in A+ and A− cohorts. Mean tau deposition represented as SUVRs for (A) A+ and (B) A− participants. Higher SUVRs indicate higher mean binding and more tau pathology. Abbreviations: SUVR, standardized uptake value ratio; A+, beta-amyloid-PET positive; A−, beta-amyloid-PET negative.
Next, to statistically compare regional tau-PET SUVRs in the A+ and A− groups, we ran linear regressions for each of the 34 cortical and 9 subcortical regions examined (see Table 2 for a list of regions), including age and sex as covariates in the model. Covariates were selected based on their established association with AD and brain measures. To correct for multiple comparisons, we implemented a Benjamini-Hochberg procedure with a false discovery rate of 5%.
Table 2.
Regional tau-PET SUVR in A+ and A− cohorts
| Region | A− mean SUVR (SD) |
A+ mean SUVR (SD) |
Unstandardized B (SE) |
95% CI | Partial eta squared |
p- value |
|---|---|---|---|---|---|---|
| Accumbens | 1.35 (0.30) | 1.40 (0.26) | −0.004 (0.07) | −0.138 to 0.130 | 0.000 | 0.958 |
| Amygdala | 1.08 (0.17) | 1.24 (0.28) | 0.136 (0.05) | 0.044 to 0.227 | 0.076 | 0.004 |
| Caudate | 1.33 (0.25) | 1.44 (0.26) | 0.044 (0.06) | −0.069 to 0.157 | 0.006 | 0.441 |
| Choroid plexus | 1.82 (0.65) | 1.94 (0.93) | 0.063 (0.17) | −0.274 to 0.400 | 0.001 | 0.712 |
| Banks of superior temporal sulcus | 1.23 (0.13) | 1.36 (0.19) | 0.094 (0.03) | 0.027 to 0.160 | 0.069 | 0.006 |
| Caudal anterior cingulate | 1.11 (0.17) | 1.10 (0.20) | −0.006 (0.04) | −0.089 to 0.076 | 0.000 | 0.879 |
| Caudal middle frontal | 1.00 (0.15) | 1.08 (0.23) | 0.082 (0.04) | 0.003 to 0.162 | 0.039 | 0.042 |
| Cuneus | 1.22 (0.15) | 1.21 (0.16) | −0.009 (0.04) | −0.080 to 0.061 | 0.001 | 0.792 |
| Entorhinal cortex | 1.03 (0.22) | 1.21 (0.31) | 0.168 (0.06) | 0.055 to 0.281 | 0.077 | 0.004 |
| Frontal pole | 0.91 (0.34) | 0.85 (0.26) | 0.006 (0.06) | −0.120 to 0.133 | 0.000 | 0.921 |
| Fusiform | 1.22 (0.12) | 1.33 (0.12) | 0.095 (0.03) | 0.04 to 0.150 | 0.100 | 0.001 |
| Inferior parietal | 1.23 (0.15) | 1.35 (0.20) | 0.100 (0.04) | 0.030 to 0.170 | 0.070 | 0.006 |
| Inferior temporal | 1.26 (0.15) | 1.39 (0.18) | 0.111 (0.04) | 0.040 to 0.181 | 0.085 | 0.002 |
| Insula | 0.98 (0.13) | 0.98 (0.14) | 0.005 (0.03) | −0.056 to 0.066 | 0.000 | 0.878 |
| Isthmus cingulate cortex | 1.20 (0.16) | 1.32 (0.23) | 0.090 (0.04) | 0.010 to 0.170 | 0.045 | 0.028 |
| Lateral occipital cortex | 1.24 (0.23) | 1.30 (0.22) | 0.038 (0.05) | −0.062 to 0.139 | 0.005 | 0.450 |
| Lateral orbital frontal cortex | 1.18 (0.15) | 1.25 (0.15) | 0.053 (0.04) | −0.016 to 0.122 | 0.021 | 0.134 |
| Lingual cortex | 1.16 (0.14) | 1.18 (0.14) | 0.008 (0.03) | −0.056 to 0.072 | 0.001 | 0.808 |
| Medial orbital frontal cortex | 1.02 (0.16) | 1.06 (0.17) | 0.034 (0.04) | −0.043 to 0.111 | 0.007 | 0.382 |
| Middle temporal cortex | 1.21 (0.14) | 1.31 (0.14) | 0.098 (0.03) | 0.039 to 0.158 | 0.092 | 0.001 |
| Paracentral cortex | 1.06 (0.18) | 1.06 (0.19) | −0.005 (0.04) | −0.090 to 0.079 | 0.000 | 0.900 |
| Parahippocampal gyrus | 1.01 (0.15) | 1.09 (0.17) | 0.086 (0.04) | 0.014 to 0.159 | 0.051 | 0.019 |
| Pars opercularis | 1.03 (0.15) | 1.05 (0.19) | 0.019 (0.04) | −0.055 to 0.092 | 0.002 | 0.617 |
| Pars orbitalis | 1.21 (0.23) | 1.19 (0.19) | −0.004 (0.05) | −0.096 to 0.089 | 0.000 | 0.928 |
| Pars triangularis | 1.14 (0.18) | 1.14 (0.22) | 0.014 (0.04) | −0.071 to 0.099 | 0.001 | 0.747 |
| Peri calcarine cortex | 1.19 (0.14) | 1.21 (0.16) | 0.001 (0.03) | −0.066 to 0.067 | 0.000 | 0.985 |
| Post central | 0.98 (0.13) | 0.98 (0.15) | 0.003 (0.03) | −0.058 to 0.064 | 0.000 | 0.927 |
| Posterior cingulate cortex | 1.23 (0.14) | 1.31 (0.19) | 0.075 (0.04) | 0.004 to 0.146 | 0.040 | 0.038 |
| Precentral cortex | 0.97 (0.12) | 0.98 (0.17) | 0.019 (0.03) | −0.044 to 0.082 | 0.003 | 0.556 |
| Precuneus | 1.20 (0.15) | 1.32 (0.21) | 0.110 (0.04) | 0.034 to 0.187 | 0.072 | 0.005* |
| Rostral anterior cingulate cortex | 1.06 (0.17) | 1.05 (0.17) | −0.011 (0.04) | −0.090 to 0.068 | 0.001 | 0.784 |
| Rostral middle frontal cortex | 0.94 (0.18) | 0.95 (0.17) | −0.029 (0.04) | −0.042 to 0.100 | 0.006 | 0.419 |
| Superior frontal cortex | 0.88 (0.15) | 0.91 (0.18) | 0.024 (0.04) | −0.049 to 0.098 | 0.004 | 0.428 |
| Superior parietal cortex | 1.05 (0.17) | 1.14 (0.24) | 0.072 (0.04) | −0.015 to 0.160 | 0.025 | 0.105 |
| Superior temporal cortex | 1.01 (0.12) | 1.03 (0.13) | 0.033 (0.03) | −0.021 to 0.087 | 0.014 | 0.225 |
| Supramarginal gyrus | 1.16 (0.13) | 1.22 (0.15) | 0.053 (0.03) | 0.006 to 0.112 | 0.029 | 0.079 |
| Temporal pole | 1.00 (0.17) | 1.06 (0.22) | 0.077 (0.04) | 0.002 to 0.157 | 0.034 | 0.057 |
| Transverse temporal cortex | 0.98 (0.16) | 1.01 (0.23) | 0.036 (0.04) | −0.048 to 0.120 | 0.007 | 0.395 |
| Hippocampus | 1.21 (0.19) | 1.24 (0.15) | 0.005 (0.04) | −0.078 to 0.088 | 0.000 | 0.905 |
| Pallidum | 1.91 (0.34) | 1.98 (0.32) | −0.039 (0.07) | −0.177 to 0.100 | 0.003 | 0.643 |
| Putamen | 1.52 (0.24) | 1.62 (0.23) | 0.015 (0.05) | −0.079 to 0.109 | 0.001 | 0.751 |
| Thalamus | 1.25 (0.15) | 1.28 (0.13) | 0.020 (0.03) | −0.047 to 0.087 | 0.003 | 0.557 |
| Ventral diencephalon | 1.49 (0.21) | 1.46 (0.18) | −0.041 (0.05) | −0.137 to 0.055 | 0.007 | 0.401 |
Key: SUVR, standardized uptake value ratio; SE, standard error; CI, confidence interval, A+, beta-amyloid-PET positive; A−, beta-amyloid-PET negative.
Next, because APOE4 status was significantly different between the A− and A+ groups, we repeated these multivariable linear regressions, comparing regional tau-PET SUVRs in the A+ and A− groups, for each cortical and subcortical region found to be significant in the primary analyses, but added APOE4 status as a covariate, along with age and sex. To correct for multiple comparisons, we implemented a Benjamini-Hochberg procedure with a false discovery rate of 5%.
Since the A+ classification was derived from a composite beta-amyloid-PET SUVR, we wanted to explore further the relationship between regional tau-PET and this composite beta-amyloid-PET SUVR value. We ran a multivariate model for each of the 8 regions identified as having significantly higher tau-PET SUVRs in the A+ cohort compared to controls, controlled for age and sex. We additionally report the within-group Pearson’s correlation of regional tau-PET and beta-amyloid-PET for these 8 regions. To correct for multiple comparisons, we implemented a Benjamini-Hochberg procedure with a false discovery rate of 5%.
Finally, to determine whether the regional tau deposition is related to cognition, we ran linear regression models between 18F-AV-1451 and the global cognition score, adjusting for age and sex. To correct for multiple comparisons, we implemented a Benjamini-Hochberg procedure with a false discovery rate of 5%.
3. Results
3.1. Participant characteristics
As shown in Table 1, participants in the A+ cohort were older (mean age = 71.9 years) compared to A− individuals (mean age = 66.8 years, Fdf = 1.2591,107, p = 0.008) and had a higher percentage of APOE4carriers (56.0%) than the A− group (23.2%, X2 = 9.681, p = 0.002).
3.2. Tau distribution in A+ cohort
Linear regressions evaluating group differences between the A+ and A− groups show that the A+ cohort had significantly higher tau-PET SUVR in 9 regions (Table 2), including within the amygdala (B = 0.136, p = 0.004), banks of the superior temporal sulcus (B = 0.094, p = 0.006), entorhinal cortex (B = 0.168, p = 0.004), fusiform gyrus (B = 0.095, p = 0.001), inferior parietal cortex (B = 0.100, p = 0.006), inferior temporal cortex (B = 0.111, p = 0.002), parahippocampal gyrus (B = 0.086, p = 0.019), middle temporal cortex (B = 0.098, p = 0.001), precuneus (B = 0.110, p = 0.005). Effect size maps of regression coefficients (B values) from significant linear regressions are presented in Figure 2. Violin plots show the distribution of AV1451 in 9 significant regions of interest (Supplementary Material (SM1)). Furthermore, 18F-AV-1451 signal in these regions is highly correlated across all regions (Supplementary Material (SM2)).
Fig. 2.
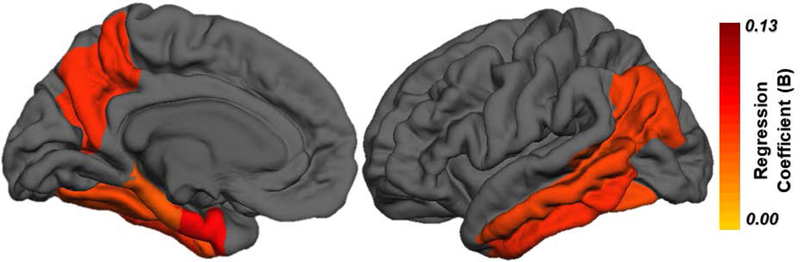
Difference in tauopathy in A+ cohort compared to A−. Effect size maps, depicting regression coefficients (B values), from significant linear regressions, adjusting for age and sex, comparing A− and A+ participants. Abbreviations: AD, Alzheimer’s disease, A+, beta-amyloid-PET positive; A−, beta-amyloid-PET negative.
When APOE4 status was included as a covariate, regional tau-PET SUVRs for the A+ cohort remained significantly higher compared to the A− group in 8 of the 9 regions including the amygdala (B = 0.141, p = 0.005), banks of the superior temporal sulcus (B = 0.091, p = 0.013), entorhinal cortex (B = 0.152, p = 0.014), fusiform gyrus (B = 0.084, p = 0.005), inferior parietal cortex (B = 0.101, p = 0.009), inferior temporal cortex (B = 0.103, p = 0.008), middle temporal cortex (B = 0.098, p = 0.003), and precuneus (B = 0.109, p = 0.010), with the exception being the parahippocampal gyrus (p = 0.110). Furthermore, in these models, APOE4 status is not significantly associated with tau-PET SUVRs in any regions examined. Full models are presented in Supplementary Material (SM3).
3.3. Relationship between beta-amyloid- and tau-PET
There were significant associations between regional tau-PET SUVR and composite beta-amyloid SUVR levels in all 8 regions examined (Fig. 3), including the amygdala (B [standard error {SE} = 0.153 [0.04], p = <0.001, ηp2 = 0.121), banks of the superior temporal sulcus (B[SE] = 0.090[0.03], p = 0.003, ηp2 = 0.080), entorhinal cortex (B[SE]= 0.168[0.05], p = 0.001, ηp2 = 0.098), fusiform gyrus (B[SE] = 0.087[0.03], p = 0.001, ηp2 = 0.106), inferior parietal (B[SE] = 0.102[0.03], p = 0.002, ηp2 = 0.092), inferior temporal (B[SE] = 0.106[0.03], p = 0.001, ηp2 = 0.098), middle temporal (B[SE] = 0.093[0.03], p = 0.001, ηp2 = 0.104), and precuneus (B[SE] = 0.135[0.03], p = <0.001, ηp2 = 0.136). In addition, there were within-group correlations between tau-PET and beta-amyloid-PET in A+ individuals in the inferior parietal (r = 0.498, p = 0.011), inferior temporal (r = 0.453, p = 0.023), and middle temporal (r = 0.531, p = 0.006). There was no correlation in A− individuals between tau-PET and beta-amyloid-PET in any region examined.
Fig. 3.
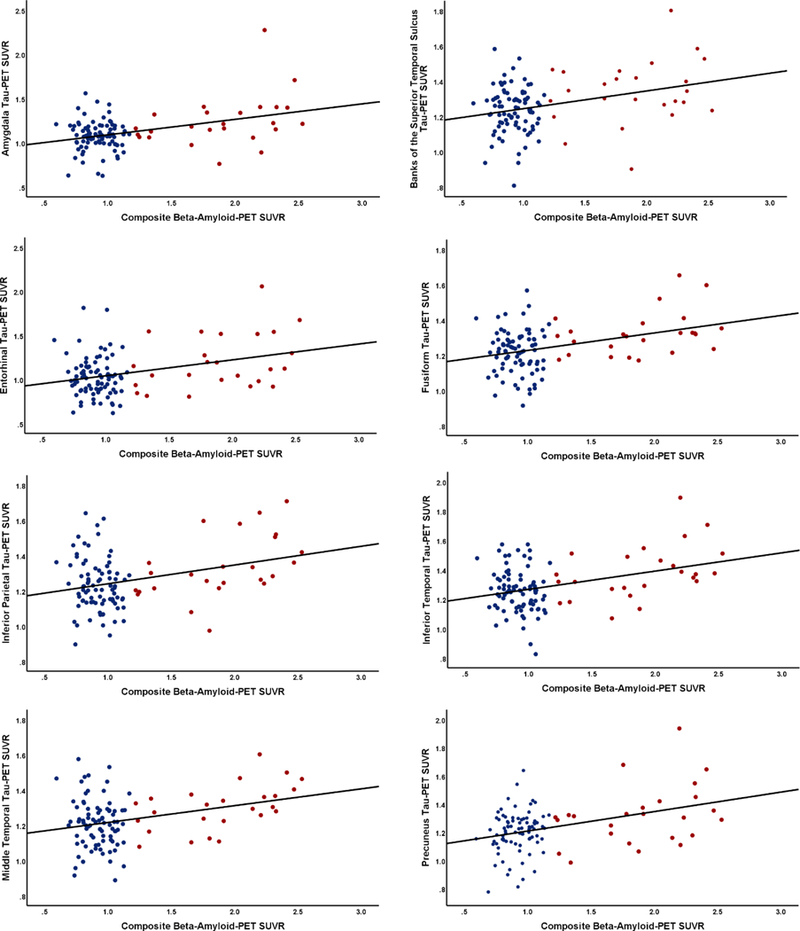
Association between regional tau-PET and beta-amyloid-PET. Relationship between composite beta-amyloid-PET measure and regional tau-PET SUVR from (A) amygdala, (B) banks of superior temporal sulcus, (C) entorhinal cortex, (D) fusiform, (E) inferior parietal, (F) inferior temporal, (G) middle temporal, and (H) precuneus. Abbreviations: Red, beta-amyloid-PET positive; A+ and blue, beta-amyloid-PET negative; A− PET, positron emission tomography; SUVR, standardized uptake value ratio.
3.4. Relationship between cognition and tau-PET
There was no relationship between tau-PET and the cognitive composite score in any of the 8 regions examined (p >0.101, Table 3).
Table 3.
Associations between regional tau-PET SUVR and cognition
| Region | Unstandardized B (SE) |
p- value |
95% CI | Partial eta squared |
|---|---|---|---|---|
| Amygdala | 0.049 (0.03) | 0.101 | −0.010 to 0.108 | 0.026 |
| Banks of superior temporal sulcus | 0.035 (0.02) | 0.102 | −0.007 to 0.078 | 0.026 |
| Entorhinal | 0.010 (0.04) | 0.789 | −0.064 to 0.083 | 0.001 |
| Fusiform | −0.012 (0.02) | 0.529 | −0.048 to 0.025 | 0.004 |
| Inferior parietal | −0.007 (0.02) | 0.755 | −0.053 to 0.039 | 0.001 |
| Inferior temporal | −0.008 (0.02) | 0.735 | −0.054 to 0.038 | 0.001 |
| Middle temporal | −0.001 (0.02) | 0.943 | −0.041 to 0.038 | 0.000 |
| Precuneus | 0.024 (0.03) | 0.327 | −0.025 to 0.073 | 0.009 |
Key: SUVR, standardized uptake value ratio; SE, standard error; CI, confidence interval.
4. Discussion
Clinicopathologic studies of AD propose a stereotypical spread of tauopathy based on postmortem pathological studies. The Braak and Braak staging scheme envisions spread of NFTs from the brainstem and transentorhinal cortex to entorhinal cortex, then into neocortical regions, including the fusiform gyrus, medial temporal gyrus, and insular cortex, and, later, into frontal, parietal, and occipital cortices (Braak and Braak, 1991; Braak et al., 2006). Recent studies (Lowe et al., 2016; Marquie et al., 2017) comparing 18F-AV-1451 autoradiography with tau immunohistochemistry have found high colocalization of 18F-AV-1451 with tauopathy in AD compared to other non-AD tauopathies, particularly in the brains with advanced Braak NFT stages, supporting the use of this tracer to map AD-associated tauopathy.
Other studies have investigated the spatial pattern of 18F-AV-1451 binding tauopathy in vivo, using 18F-AV-1451 and PET, comparing CN individuals to older adults with symptomatic AD. Johnson and colleagues (2016) found that participants with mild cognitive impairment and AD had significantly higher tau-PET SUVRs in regions including the inferior temporal lobe, fusiform gyrus, posterior cingulate cortex, occipital cortex, parahippocampal gyrus, and entorhinal cortex compared with CN peers. Similarly, our group (Brier et al., 2016; Gordon et al., 2016) and others (Cho et al., 2016; Johnson et al., 2016) have shown patterns of increased tau-PET SUVRs in cognitively impaired individuals compared to controls in temporal and occipital neocortical areas. This homogeneity across multiple centers suggests a consistent spatial pattern of tau-PET binding in symptomatic AD cohorts relative to cognitively normal controls. This spatial pattern of tau-PET binding in symptomatic AD has been stereotyped into an estimated tau-PET Braak staging scheme (Maass et al., 2017; Scholl et al., 2016; Schwarz et al., 2016) in which the regional tau burden in symptomatic AD participants appears consistent with higher Braak stages (V and VI).
This study adds to converging literature examining tau-PET in A+ and A− CN individuals (Gordon et al., 2016; Hanseeuw et al., 2017; Jack et al., 2018b; Jacobs et al., 2018; Lowe et al., 2018a,b; Mishra et al., 2017; Scholl et al., 2016; Sepulcre et al., 2016; Vemuri et al., 2017; Villemagne et al., 2017). For example, Schöll and colleagues (2016) examined 18F-AV-1451 tau-PET, mapped onto the Braak NFT staging scheme, being distributed across all Braak stages, in a cohort of 5 CN young adults, 33 CN older adults, and 15 symptomatic AD patients. Among them, CN older adults ranged across stages 0, I/II, and III/IV. Other studies also described regional associations with other core AD biomarkers including beta-amyloid-PET (Brier et al., 2016; Lockhart et al., 2017; Sepulcre et al., 2016), CSF tau levels (Chhatwal et al., 2016; Gordon et al., 2016; Wang et al., 2016), gray matter volumes and cortical thinning (LaPoint et al., 2017; Sepulcre et al., 2016), and functional connectivity MRI in CN individuals (Schultz et al., 2017). Specifically, a study investigating difference in inherent tau-PET signal across the brain in younger adults proposed implementation of region-specific z-score values to assess severity of NFT burden (Vemuri et al., 2017). Results focus on the entorhinal cortex as an AD-specific tau-PET signature. Similarly, we find an increase in tau-PET signal in the entorhinal cortex in A+ individuals compared to A−. However, we importantly show that there are a number of other regions also significantly elevated in this A+ cohort, which extend outside the medial temporal lobe. Recent studies investigating the cross-sectional and longitudinal tau-PET signal in CN and cognitively impaired individuals additionally support our results. Using a meta-region of interest approach to assess longitudinal change in tau, results were in alignment with the current findings, suggesting increase in the rates of tau accumulation in regions other than the entorhinal cortex, including midtemporal, retrosplenial, and posterior cingulate (Jack et al., 2018b). However, the regions characterized as an ”early-AD” meta-region for longitudinal analyses included the fusiform and posterior cingulate gyrus, therefore not completely consistent with our results.
The present study extends the work of those studies in several critical ways. First, it focuses on the generation of a topographical map of 18F-AV-1451 signal in A+ CN individuals. Such work aids in our interpretation of tau accumulation in preclinical AD. Second, with its restriction specifically on a large A+ cohort, characterized clinically by CDR and for AD pathology by beta-amyloid-PET, this study provides strong evidence that regions of tauopathy detected by tau-PET in A+ are comparable to those identified in recent studies of AD dementia and are comparable, but not identical to, Braak NFT staging. It is possible that the discrepancy in our results compared to Braak NFT staging is due to differences in specificity of 18F-AV-1451 binding of certain tau isoforms compared to classical histochemical staining or signal contamination in 18F-AV-1451 from nonspecific binding in nearby regions. Alternatively, this discordance could be related to subtle difference in the cohorts studied and comparisons examined. While Braak NFT staging is primarily a classification of presence or absence of regional NFT, our analyses more specifically examined not only the distribution of NFT in A+ CN individuals but also the density of NFTs in these regions, as compared to an A−cohort. We therefore provide evidence for significant regional differences in 18F-AV-1451 signal in A+ CN individuals, compared to A− individuals.
We additionally report that APOE e4 may not be associated with tau-PET signal above and beyond that of A− and A+ classification. This is in contrast to recent studies in animal models suggesting a role of APOE e4 exacerbating NFT accumulation (Shi et al., 2017). It is possible, however, that the present study may be underpowered to detect a modulating effect of APOE e4 status within A− and A+ groups. Larger studies with a sufficient sample of APOE e4 A− individuals are needed to determine whether there are interactions between APOE e4 status and beta-amyloid load on regional tau-PET signal.
Furthermore, we report no associations between regional 18F-AV-1451 and global cognition in our cohort. These results support findings by Schöll and colleagues (2016) who similarly reported a lack of association between cross-sectional global cognition and 18F-AV-1451.
Overall, results from prior studies, in addition to the present study, provide evidence that there are associations with tau-PET and established biomarkers of AD in preclinical stage, further strengthening the utility of tau-PET in AD research and clinical trials.
Overall, by emphasizing the presence of widespread tauopathy early in the disease course, these findings should inform treatment strategies for preclinical AD. Having learned from previous clinical trials that unsuccessfully targeted mild-to-moderate AD dementia (Wang et al., 2017), ongoing clinical trials are now administering anti-beta-amyloid therapies in asymptomatic individuals who are determined to be positive by beta-amyloid-PET. The results of those trials will provide insight into the potential benefit of targeting beta-amyloid pathology early in the disease course. However, the results of this present study demonstrate that many clinical trial participants who are beta-amyloid-PET positive are also likely to have a significant tauopathy burden. Given that both tauopathy and beta-amyloid deposits are present in presymptomatic stages, it may be worthwhile to consider anti-tau therapies at this early stage—either alone, or in combination with anti-beta-amyloid therapy. Indeed, combined therapy might be more effective than either single therapy alone.
A particular strength of the present study is its large, well-characterized sample compared with prior studies (Johnson et al., 2016; Ossenkoppele et al., 2016). However, its sample size remains relatively modest for evaluating effects at a whole-brain level, underscoring the future need for a increasingly larger sample.
There remain several limitations of the present study. First, the current interpretation of NFT pathophysiology through utilization of 18F-AV-1451-PET in CN individuals is a potential limitation. It has been reported that 18F-AV-1451 may bind nonspecifically to neuromelanin, MAO, and iron deposits, in regions including putamen and thalamus. Furthermore, most investigations on off-target binding of 18F-AV-1451 have been conducted primarily in individuals with impairment or dementia, and little is known about the contribution of off-target binding in studies of CN individuals. However, the largest factor so far tied to this nonspecific binding has been age; thus, we included age as a covariate. Even so, there is evidence, as seen in Figure 1, of off-target binding, including parts of the basal ganglia and brainstem. Strong correlations of 18F-AV-1451 between brain regions suggest that associations of primary interest are not due to off-target binding of the basal ganglia and brainstem, yet we acknowledge there may be minimal bleeding effect of off-target binding on some regions examined, but largely controlled by partial volume correction methods including nonbrain region sampling.
Another potential limitation of our study is that the tau-PET imaging sessions were more likely to occur after the collection of beta-amyloid-PET data and clinical examination; however, we limited this interval to 12 months, so any overestimation of tauopathy in relation to beta-amyloid and clinical data should be very modest. In addition, similar to many other studies in preclinical AD and AD dementia, our A+ group was older than A− group. We accounted for this difference by including age as a covariate in the model; however, future studies would be improved by age-matching A− and A+ groups.
Finally, because we classified our participants categorically as A+ or A− based on beta-amyloid status, it is possible that some of our beta-amyloid-negative participants are actually subtly beta-amyloid-positive, but below threshold (Palmqvist et al., 2016; Vlassenko et al., 2016). Furthermore, as depicted in Figure 3, there are a few participants who may have elevated 18F-AV-1451 levels in contrast to their low beta-amyloid-PET levels, representing a potential primary age-related tauopathy subset in our A− group or generally noisy binding properties of the tracer in the absence of AD pathology. In addition, it is possible that accumulation of tau pathology might be happening in parallel, and A− individuals who present with high AV1451 might convert to A+ in the future. Longitudinal beta-amyloid- and tau-PET studies would support more confident identification of participants with preclinical AD.
In summary, our findings contribute to the understanding of tau pathology and illustrate in vivo that tauopathy is widespread in preclinical AD, encompassing both the temporal and parietal lobes. This and future studies of pathologic tau-PET in preclinical AD will be useful in designing clinical trials for AD dementia, especially when tauopathy-related therapies are administered. These results suggest that it may be worthwhile to consider antitauopathy therapies early in the disease course to prevent cognitive decline due to Alzheimer’s disease.
Supplementary Material
Acknowledgements
The authors acknowledge the financial support of Fred Simmons and Olga Mohan, the Charles F. and Joanne Knight Alzheimer’s Research Initiative, the Hope Center for Neurological Disorders, the Mallinckrodt Institute of Radiology, the American Society for Neuroradiology, and the Barnes-Jewish Hospital Foundation (BJHF), the BJHF Paula and Rodger Riney Fund, and the BJHF Willman Scholar Fund. This research was additionally funded by the National Institutes of Health grants P50AG005681, P01AG026276, P01AG003991, R01AG043434, UL1TR000448, R01EB009352, 1P30NS098577, and K01AG053474–01A1. Avid Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly) provided doses of 18F-florbetapir, partial funding for 18F-florbetapir scanning, precursor for 18F-flortaucipir, and technology transfer for manufacturing of 18F-flortaucipir. The authors thank their participants, without whom this study would not have been possible.
Footnotes
Disclosure
John C. Morris, Tammie L.S. Benzinger, Richard J. Perrin, and Brian A. Gordon report participation in clinical trials sponsored by Eli Lilly, Roche, and Biogen. Avid Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly provided doses of 18F-florbetapir, partial funding for 18F-florbetapir scanning, precursor for 18F-flortaucipir and technology transfer for manufacturing of 18F-flortaucipir). None of the authors, nor their family members, own stock or have equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company.
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.neurobiolaging.2018.08.022.
References
- Armitage S, An analysis of certain psychological tests used for the evaluation of brain injury, Psychol. Monogr. 60, 1946, 1–48. [Google Scholar]
- Aschenbrenner A, Gordon B, Benzinger T, Morris J and Hassenstab J, Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease, Neurology. 91 2018, e859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S and Morris JC, Dominantly Inherited Alzheimer N, Clinical and biomarker changes in dominantly inherited Alzheimer’s disease, N. Engl. J. Med. 367, 2012, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof GN, Jessen F, Fliessbach K, Dronse J, Hammes J, Neumaier B, Onur O, Fink GR, Kukolja J, Drzezga A and van Eimeren T, Alzheimer’s Disease Neuroimaging I, Impact of tau and amyloid burden on glucose metabolism in Alzheimer’s disease, Ann. Clin. Transl. Neurol. 3, 2016, 934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H and Del Tredici K, Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry, Acta Neuropathol. 112, 2006, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H and Braak E, Neuropathological stageing of Alzheimer-related changes, Acta Neuropathol. 82, 1991, 239–259. [DOI] [PubMed] [Google Scholar]
- Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, Owen C, Aldea P, Su Y, Hassenstab J, Cairns NJ, Holtzman DM, Fagan AM, Morris JC, Benzinger TL and Ances BM, Tau and Abeta imaging, CSF measures, and cognition in Alzheimer’s disease, Sci. Transl. Med. 8, 2016, 338ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Schultz AP, Marshall GA, Boot B, Gomez-Isla T, Dumurgier J, LaPoint M, Scherzer C, Roe AD, Hyman BT, Sperling RA and Johnson KA, Temporal T807 binding correlates with CSF tau and phospho-tau in normal elderly, Neurology 87, 2016, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A and Kolb HC, Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807, J. Alzheimers Dis. 34, 2013, 457–468. [DOI] [PubMed] [Google Scholar]
- Cho H, Choi JY, Hwang MS, Kim YJ, Lee HM, Lee HS, Lee JH, Ryu YH, Lee MS and Lyoo CH, In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum, Ann. Neurol. 80, 2016, 247–258. [DOI] [PubMed] [Google Scholar]
- Day GS, Gordon BA, Jackson K, Christensen JJ, Rosana Ponisio M, Su Y, Ances BM, Benzinger TL and Morris JC, Tau-PET binding distinguishes patients with early-stage posterior cortical atrophy from amnestic Alzheimer disease dementia, Alzheimer Dis. Assoc. Disord. 31, 2017, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC and Holtzman DM, Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults, Arch. Neurol. 64, 2007, 343–349. [DOI] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B and Dale AM, Automatically parcellating the human cerebral cortex, Cereb. Cortex 14, 2004, 11–22. [DOI] [PubMed] [Google Scholar]
- Franz G, Beer R, Kampfl A, Engelhardt K, Schmutzhard E, Ulmer H and Deisenhammer F, Amyloid beta 1–42 and tau in cerebrospinal fluid after severe traumatic brain injury, Neurology 60, 2003, 1457–1461. [DOI] [PubMed] [Google Scholar]
- Goodglass H and Kaplan E, In: Febiger PL, (Ed), Boston diagnostic aphasia examination Booklet, III: Oral Expression: Animal Naming (Fluency in Controlled Association), 1983, Lea & Febiger; Philadelphia. [Google Scholar]
- Gordon BA, Friedrichsen K, Brier M, Blazey T, Su Y, Christensen J, Aldea P, McConathy J, Holtzman DM, Cairns NJ, Morris JC, Fagan AM, Ances BM and Benzinger TL, The relationship between cerebrospinal fluid markers of Alzheimer pathology and positron emission tomography tau imaging, Brain 139 (Pt 8), 2016, 2249–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BA, Najmi S, Hsu P, Roe CM, Morris JC and Benzinger TL, The effects of white matter hyperintensities and amyloid deposition on Alzheimer dementia, Neuroimage Clin. 8, 2015, 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grober E, Buschke H, Crystal H, Bang S and Dresner R, Screening for dementia by memory testing, Neurology 38, 1988, 900–903. [DOI] [PubMed] [Google Scholar]
- Hanseeuw BJ, Betensky RA, Schultz AP, Papp KV, Mormino EC, Sepulcre J, Bark JS, Cosio DM, LaPoint M, Chhatwal JP, Rentz DM, Sperling RA and Johnson KA, Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline, Ann. Neurol. 81, 2017, 583–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R and Contributors, NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease, Alzheimers Dement. 14, 2018a, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Weigand SD, Wiste HJ, Vemuri P, Lowe V, Kantarci K, Gunter JL, Senjem ML, Ivnik RJ, Roberts RO, Rocca WA, Boeve BF and Petersen RC, An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease, Ann. Neurol. 71, 2012, 765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, Weigand SD, Therneau TM, Knopman DS, Gunter JL, Jones DT, Graff-Radford J, Kantarci K, Roberts RO, Mielke MM, Machulda MM and Petersen RC, Longitudinal tau PET in ageing and Alzheimer’s disease, Brain 141, 2018b, 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs HIL, Hedden T, Schultz AP, Sepulcre J, Perea RD, Amariglio RE, Papp KV, Rentz DM, Sperling RA and Johnson KA, Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals, Nat. Neurosci. 21, 2018, 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, Mormino E, Chhatwal J, Amariglio R, Papp K, Marshall G, Albers M, Mauro S, Pepin L, Alverio J, Judge K, Philiossaint M, Shoup T, Yokell D, Dickerson B, Gomez-Isla T, Hyman B, Vasdev N and Sperling R, Tau positron emission tomographic imaging in aging and early Alzheimer disease, Ann. Neurol. 79, 2016, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA and Langstrom B, Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B, Ann. Neurol. 55, 2004, 306–319. [DOI] [PubMed] [Google Scholar]
- LaPoint MR, Chhatwal JP, Sepulcre J, Johnson KA, Sperling RA and Schultz AP, The association between tau PET and retrospective cortical thinning in clinically normal elderly, Neuroimage 157, 2017, 612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine L, Leuzy A, Chiotis K, Rodriguez-Vieitez E and Nordberg A, Tau positron emission tomography imaging in tauopathies: the added hurdle of off-target binding, Alzheimers Dement. (Amst) 10, 2018, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart SN, Scholl M, Baker SL, Ayakta N, Swinnerton KN, Bell RK, Mellinger TJ, Shah VD, O’Neil JP, Janabi M and Jagust WJ, Amyloid and tau PET demonstrate region-specific associations in normal older people, Neuroimage 150, 2017, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Bruinsma TJ, Min HK, Lundt ES, Fang P, Senjem ML, Boeve BF, Josephs KA, Pandey MK, Murray ME, Kantarci K, Jones DT, Schwarz CG, Knopman DS, Petersen RC and Jack CR Jr., Elevated medial temporal lobe and pervasive brain tau-PET signal in normal participants, Alzheimers Dement. (Amst) 10, 2018a, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, Kantarci K, Boeve BF, Pandey MK, Bruinsma T, Knopman DS, Jones DT, Petrucelli L, Cook CN, Graff-Radford NR, Dickson DW, Petersen RC, Jack CR Jr. and Murray ME, An autoradiographic evaluation of AV-1451 Tau PET in dementia, Acta Neuropathol. Commun. 4, 2016, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Wiste HJ, Senjem ML, Weigand SD, Therneau TM, Boeve BF, Josephs KA, Fang P, Pandey MK, Murray ME, Kantarci K, Jones DT, Vemuri P, Graff-Radford J, Schwarz CG, Machulda MM, Mielke MM, Roberts RO, Knopman DS, Petersen RC and Jack CR Jr., Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia, Brain 141, 2018b, 271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, Rabinovici GD and Jagust WJ, Alzheimer’s Disease Neuroimaging I, Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease, Neuroimage 157, 2017, 448–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquie M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, Klunk WE, Mathis CA, Ikonomovic MD, Debnath ML, Vasdev N, Dickerson BC, Gomperts SN, Growdon JH, Johnson KA, Frosch MP, Hyman BT and Gomez-Isla T, Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue, Ann. Neurol. 78, 2015, 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquie M, Siao Tick Chong M, Anton-Fernandez A, Verwer EE, Saez-Calveras N, Meltzer AC, Ramanan P, Amaral AC, Gonzalez J, Normandin MD, Frosch MP and Gomez-Isla T, [F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging, Acta Neuropathol. 134, 2017, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Gordon BA, Su Y, Christensen J, Friedrichsen K, Jackson K, Hornbeck R, Balota DA, Cairns NJ, Morris JC, Ances BM and Benzinger TLS, AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure, Neuroimage 161, 2017, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC , Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type, Int. Psychogeriatr. 9 (Suppl 1), 1997, 173–176. [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, Fagan AM, Holtzman DM and Mintun MA, Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease, Arch. Neurol. 66, 2009, 1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Schonhaut DR, Scholl M, Lockhart SN, Ayakta N, Baker SL, O’Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ and Rabinovici GD, Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease, Brain 139 (Pt 5), 2016, 1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmqvist S, Mattsson N and Hansson O, Alzheimer’s Disease Neuroimaging I, Cerebrospinal fluid analysis detects cerebral amyloid-beta accumulation earlier than positron emission tomography, Brain 139 (Pt 4), 2016, 1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemenschneider M, Wagenpfeil S, Diehl J, Lautenschlager N, Theml T, Heldmann B, Drzezga A, Jahn T, Forstl H and Kurz A, Tau and Abeta42 protein in CSF of patients with frontotemporal degeneration, Neurology 58, 2002, 1622–1628. [DOI] [PubMed] [Google Scholar]
- Rousset OG, Ma Y and Evans AC, Correction for partial volume effects in PET: principle and validation, J. Nucl. Med. 39, 1998, 904–911. [PubMed] [Google Scholar]
- Scholl M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, Rabinovici GD and Jagust WJ, PET imaging of tau deposition in the aging human brain, Neuron 89, 2016, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz AP, Chhatwal JP, Hedden T, Mormino EC, Hanseeuw BJ, Sepulcre J, Huijbers W, LaPoint M, Buckley RF, Johnson KA and Sperling RA, Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals, J. Neurosci. 37, 2017, 4323–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, Joshi AD, Devous MD Sr. and Mintun MS, Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages, Brain 139 (Pt 5), 2016, 1539–1550. [DOI] [PubMed] [Google Scholar]
- Sepulcre J, Schultz AP, Sabuncu M, Gomez-Isla T, Chhatwal J, Becker A, Sperling R and Johnson KA, In vivo tau, amyloid, and gray matter profiles in the aging brain, J. Neurosci. 36, 2016, 7364–7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging I, Fagan A.M., Miller B.L., Boxer A.L., Seeley W.W., Butovsky O., Barres B.A., Paul S.M. and Holtzman D.M., ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy, Nature 549, 2017, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr., Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV and Phelps CH, Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease, Alzheimers Dement. 7, 2011, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Blazey TM, Owen CJ, Christensen JJ, Friedrichsen K, Joseph-Mathurin N, Wang Q, Hornbeck RC, Ances BM, Snyder AZ, Cash LA, Koeppe RA, Klunk WE, Galasko D, Brickman AM, McDade E, Ringman JM, Thompson PM, Saykin AJ, Ghetti B, Sperling RA, Johnson KA, Salloway SP, Schofield PR, Masters CL, Villemagne VL, Fox NC, Forster S, Chen K, Reiman EM, Xiong C, Marcus DS, Weiner MW, Morris JC, Bateman RJ and Benzinger TL, Dominantly Inherited Alzheimer N, Quantitative amyloid imaging in autosomal dominant Alzheimer’s disease: results from the DIAN study group, PLoS One 11, 2016, e0152082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Blazey TM, Snyder AZ, Raichle ME, Marcus DS, Ances BM, Bateman RJ, Cairns NJ, Aldea P, Cash L, Christensen JJ, Friedrichsen K, Hornbeck RC, Farrar AM, Owen CJ, Mayeux R, Brickman AM, Klunk W, Price JC, Thompson PM, Ghetti B, Saykin AJ, Sperling RA, Johnson KA, Schofield PR, Buckles V, Morris JC, Benzinger TL and Dominantly Inherited Alzheimer N, Partial volume correction in quantitative amyloid imaging, Neuroimage 107, 2015, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, D’Angelo GM, Vlassenko AG, Zhou G, Snyder AZ, Marcus DS, Blazey TM, Christensen JJ, Vora S, Morris JC, Mintun MA and Benzinger TL, Quantitative analysis of PiB-PET with FreeSurfer ROIs, PLoS One 8, 2013, e73377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Lowe VJ, Knopman DS, Senjem ML, Kemp BJ, Schwarz CG, Przybelski SA, Machulda MM, Petersen RC and Jack CR Jr., Tau-PET uptake: regional variation in average SUVR and impact of amyloid deposition, Alzheimers Dement. (Amst). 6, 2017, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Dore V, Bourgeat P, Burnham SC, Laws S, Salvado O, Masters CL and Rowe CC, Abeta-amyloid and tau imaging in dementia, Semin. Nucl. Med. 47, 2017, 75–88. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, Ackermann U, Jones G, Szoeke C, Salvado O, Martins R, O’Keefe G, Mathis CA, Klunk WE, Ames D, Masters CL and Rowe CC, Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease, Ann. Neurol. 69, 2011, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, McCue L, Jasielec MS, Su Y, Gordon BA, Xiong C, Holtzman DM, Benzinger TL, Morris JC and Fagan AM, Imaging and cerebrospinal fluid biomarkers in early preclinical alzheimer disease, Ann. Neurol. 80, 2016, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, Mintun MA, Xiong C, Sheline YI, Goate AM, Benzinger TL and Morris JC, Amyloid-beta plaque growth in cognitively normal adults: longitudinal [11C]Pittsburgh compound B data, Ann. Neurol. 70, 2011, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SJ, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, Cairns NJ, Morris JC, Holtzman DM and Fagan AM, Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study, Lancet Neurol. 12, 2013, 957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, McConathy J, Cairns NJ, Fagan AM, Morris JC and Ances BM, Evaluation of tau imaging in staging alzheimer disease and revealing interactions between beta-amyloid and tauopathy, JAMA Neurol. 73, 2016, 1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yan T, Lu H, Yin W, Lin B, Fan W, Zhang X and Fernandez-Funez P, Lessons from anti-amyloid-beta immunotherapies in alzheimer disease: aiming at a moving target, Neurodegener. Dis. 17, 2017, 242–250. [DOI] [PubMed] [Google Scholar]
- Wechsler D, Wechsler memory scale, In: Administration and Scoring Manual, third ed., 1997, The Psychological Corporation; San Antonio. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.