

Abstract
Background
The barred knifejaw (Oplegnathus fasciatus), a member of the Oplegnathidae family of the Centrarchiformes, is a commercially important rocky reef fish native to East Asia. Oplegnathus fasciatus has become an important fishery resource for offshore cage aquaculture and fish stocking of marine ranching in China, Japan, and Korea. Recently, sexual dimorphism in growth with neo-sex chromosome and widespread biotic diseases in O. fasciatus have been increasing concern in the industry. However, adequate genome resources for gaining insight into sex-determining mechanisms and establishing genetically resistant breeding systems for O. fasciatus are lacking. Here, we analyzed the entire genome of a female O. fasciatus fish using long-read sequencing and Hi-C data to generate chromosome-length scaffolds and a highly contiguous genome assembly.
Findings
We assembled the O. fasciatus genome with a total of 245.0 Gb of raw reads that were generated using both Pacific Bioscience (PacBio) Sequel and Illumina HiSeq 2000 platforms. The final draft genome assembly was approximately 778.7 Mb, which reached a high level of continuity with a contig N50 of 2.1 Mb. The genome size was consistent with the estimated genome size (777.5 Mb) based on k-mer analysis. We combined Hi-C data with a draft genome assembly to generate chromosome-length scaffolds. Twenty-four scaffolds corresponding to the 24 chromosomes were assembled to a final size of 768.8 Mb with a contig N50 of 2.1 Mb and a scaffold N50 of 33.5 Mb using 1,372 contigs. The identified repeat sequences accounted for 33.9% of the entire genome, and 24 003 protein-coding genes with an average of 10.1 exons per gene were annotated using de novo methods, with RNA sequencing data and homologies to other teleosts. According to phylogenetic analysis using protein-coding genes, O. fasciatus is closely related to Larimichthys crocea, with O. fasciatus diverging from their common ancestor approximately 70.5–88.5 million years ago.
Conclusions
We generated a high-quality draft genome for O. fasciatus using long-read PacBio sequencing technology, which represents the first chromosome-level reference genome for Oplegnathidae species. Assembly of this genome assists research into fish sex-determining mechanisms and can serve as a resource for accelerating genome-assisted improvements in resistant breeding systems.
Keywords: Oplegnathus fasciatus, chromosome-level genome assembly, Hi-C assembly, sex-determining mechanism
Data Description
Introduction of O. fasciatus
The Oplegnathidae family belongs to the order Centrarchiformes, including only one genus Oplegnathus, which is comprised of seven species (Oplegnathus conwayi, Oplegnathus fasciatus, Oplegnathus insignis, Oplegnathus peaolopesi, Oplegnathus punctatus, Oplegnathus robinsoni, and Oplegnathus woodwardi), two of which (O. fasciatus and O. punctatus) are commercially valuable in East Asia. The barred knifejaw O. fasciatus (NCBI:txid163134, Fishbase ID: 1709) (Temminck and Schlegel, 1844) is one of the two species in the Oplegnathus that is commonly found at the depth of 1 to 10 meters in association with rocky reefs [1, 2] and is distributed across a wide range of shallow waters around Korea, Japan, China, and Hawaii [1, 3, 4] (Fig. 1). Oplegnathus fasciatus has become an important fishery resource for offshore cage aquaculture and fish stocking of marine ranching in China, Japan, and Korea [5]. It has been reported that the male of Oplegnathus possesses a neo-sex chromosome, possibly a sex chromosome Y. The sex chromosome system for Oplegnathus is considered to be X1 X1 X2 X2/X1 X2 Y based on karyotype analyses [6, 7]. Furthermore, sexual dimorphism in growth has been detected in the O. fasciatus, with male fish exhibiting faster growth than females, possibly due to the sex chromosome system in Oplegnathus [8]. Oplegnathusfasciatus is vulnerable to viruses (e.g., iridovirus), and genetic degradation caused by inbreeding has led to higher susceptibility to diseases [9, 10]. It is vital to develop genomic resources to gain insight into sex-determining mechanisms and to accelerate the genome-assisted improvements in resistant breeding systems.
Figure 1:

A representative individual of O. fasciatus.
To date, a genome sequence with chromosomal assembly of O. fasciatus has not been reported. Here, we constructed a high-quality chromosome-level reference genome assembly for O. fasciatus using long reads from the Pacific Biosciences (PacBio) DNA sequencing platform and a genome assembly strategy taking advantage of the genome assembly program Canu [11]. This genome assembly of O. fasciatus is the first chromosome-level reference genome constructed for the Oplegnathidae family. The completeness and continuity of the genome will provide high-quality genomic resources for studies on sex-determining mechanisms and for accelerating the genome-assisted improvements in resistant breeding systems.
Genomic DNA extraction and genome size estimation
High-quality genomic DNA for sequencing using the Illumina platform (Illumina Inc., San Diego, CA) and PacBio Sequel sequencing (Pacific Biosciences of California, Menlo Park, CA) was extracted from fresh muscle tissue and blood samples from a single female O. fasciatus. The fish was collected from the near-shore area of Qingdao City (Yellow Sea), Shandong Province, China. The whole-genome size of O. fasciatus was estimated based on Illumina DNA sequencing technology. A short-insert library (300∼350 bp) was constructed and generated a total of ∼90.7 Gb of raw reads using the standard protocol provided by the Illumina HiSeq X Ten platform (Illumina Inc., San Diego, CA). After the removal of low-quality and redundant reads, we obtained ∼80.8 Gb of clean data for de novo assembly to estimate the whole-genome size (Supplementary Table S1, Fig. 2). All cleaned reads were subjected to 17-mer frequency distribution analysis [12]. As the total number of k-mers was approximately 8.09 × 1010 and the peak of k-mers was at a depth of 100, the genome size of O. fasciatus was calculated to be 777.5 Mb using the following formula with amendment: G = (Nk-mer—Nerror_k-mer)/D, where G is genome size, Nk-mer is the number of k-mers, Nerror_k-mer is the number of k-mers with the depth of 1, and D is the k-mer depth (Fig. 2). Meanwhile, an estimated heterozygosity of 0.29% and a repeat content of 38.46% were detected for O. fasciatus in this work. A pilot genome assembly was approximately 744.5 Mb with a contig N50 of 7.2 kb and a scaffold N50 of 84.1 kb using the Illumina data and the assembly program Platanus [13] (Supplementary Table S2). The GC content was 41% (Supplementary Fig. S1). This first attempt at a genome assembly was of low quality, partly due to its high genomic repeat content.
Figure 2:
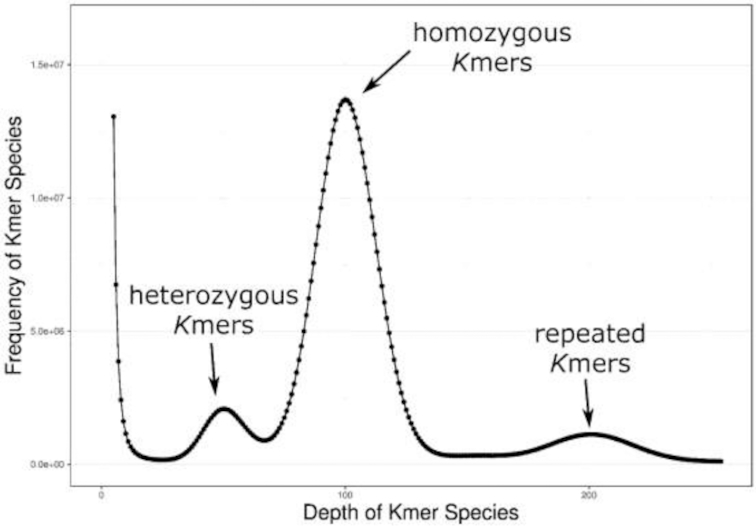
k-mer distribution of the O. fasciatus genome.
Genome assembly using PacBio long reads
Two 20 kb genomic DNA libraries were constructed and sequenced using the PacBio Sequel platform, generating 62.9 Gb raw DNA reads. We obtained 4.8 million subreads (62.8 Gb in total) with an N50 read length of ∼22 kb after removing adaptor (Supplementary Table S1).
Canu v1.4 (Canu, RRID:SCR_015880) was first used to assemble the genome with the Corrected-Error-Rate parameter set at 0.040 [11]. As a result, a genome assembly with a total length of 875.9 Mb was constructed for O. fasciatus, slightly higher than the genome size estimated by 17-mer analysis based on the Illumina data (Supplementary Table S2). The genome complexity, such as structural variants and heterozygosity, might be possible reasons to explain the relatively large genome size in the assembly. We therefore applied Redundans v0.13c [14] to remove the sequence redundancy and obtain a genome assembly size of 778.0 Mb. We then used the Arrow tool in SMRT Link 5.0 software with the minCoverage parameter set at 15 to implement error correction based on the PacBio long reads data (Table 1). The resulting genome assembly was further polished using Illumina Next-generation sequencing (NGS) data, which were used in the genome survey analysis above. The final draft genome assembly was 778.7 Mb, which reached a high level of continuity with a contig N50 length of 2.1 Mb (Table 1). The contig N50 of O. fasciatus was much higher than those of previous fish genome assemblies constructed using NGS DNA sequencing technologies and is comparable to those of recently reported model fish species (Supplementary Table S3). Previous studies illuminated the relationship between read length and genome assembly; therefore, we attributed the continuity of the genome primarily to the application of long reads in the assembly.
Table 1:
Summary of Oplegnathus fasciatus genome assembly and annotation
| Genome assembly | ||
|---|---|---|
| Draft scaffolds | Chromosome-length scaffolds based on Hi-C | |
| Length of genome (bp) | 778,731,089 | 768,808,243 |
| Number of contigs | 1,692 | 1,372 |
| Contigs N50 (bp) | 2,149,025 | 2,130,780 |
| Number of scaffolds | / | 24 |
| Scaffold N50 (bp) | / | 33,548,962 |
| Genome coverage (X) | 314.6 | |
| Number of contigs (≥100 kb) | 693 | 708 |
| Total length of contigs (≥100 kb) | 735,235,962 | 732,827,446 |
| Mapping rate of contigs (≥100kb)(%) | / | 99.67 |
| Genome annotation | ||
| Protein-coding gene number | 24,003 | |
| Mean transcript length (kb) | 16.1 | |
| Mean exons per gene | 10.1 | |
| Mean exon length (bp) | 217.7 | |
| Mean intron length (bp) | 1,527.4 | |
Hi-C library construction and chromosome assembly
Hi-C is a sequencing-based approach for determining chromosome interactions by calculating the contact frequency between pairs of loci, which are strongly dependent upon the one-dimensional distance, in base pairs, between a pair of loci [15, 16]. In this work, we used Hi-C to construct the genome assembly of O. fasciatus.
Genomic DNA was extracted for the Hi-C library from a whole-blood sample of O. fasciatus as previously described [17]. Cells were fixed with formaldehyde and lysed, and the cross-linked DNA was digested with MboI. Sticky ends were biotin-labeled and proximity ligated to form chimeric junctions and then physically sheared to a size of 300–500 bp [17]. Chimeric fragments representing the original cross-linked, long-distance physical interactions were then processed into paired-end sequencing libraries, and 629 million 150-bp paired-end Illumina reads (91.5 Gb) were produced with Q20 and Q30 of ∼94.0% (Supplementary Tables S1, S4). By mapping the Hi-C data to the PacBio-based assembly using BWA software (BWA, RRID:SCR_010910), we found that sequencing data with mates mapped to a different contig (or scaffold) and data mapped to a different contig (or scaffold) (map Q5≥ 5) were 593.7 Mb (94.4%), 240.5 Mb (40.5%), and 205.1 Mb (34.6%), respectively (Supplementary Table S4). We then employed BWA and Lachesis software to align paired-end reads to filter all base sequences more than 500 bp from each restriction site [18]. According to the conduct of clustering, ordering, and orienting to the assembly contigs (1,692), these sequences were grouped into 24 chromosome clusters and scaffolded using Lachesis software with tuned parameters [19] (Supplementary Table S4, Fig. 3). Finally, we constructed the chromosome interactions map using Juicer software and employed the JucieBox to complete the visual correction of the interaction map. We obtained 1,756 polished contigs by interrupting misassembly from 1,692 contigs. Twenty-four scaffolds were assembled corresponding to the 24 chromosomes of O. fasciatus based on the karyotype analyses [6, 7] (Supplementary Table S4, Fig. 3).
Figure 3:
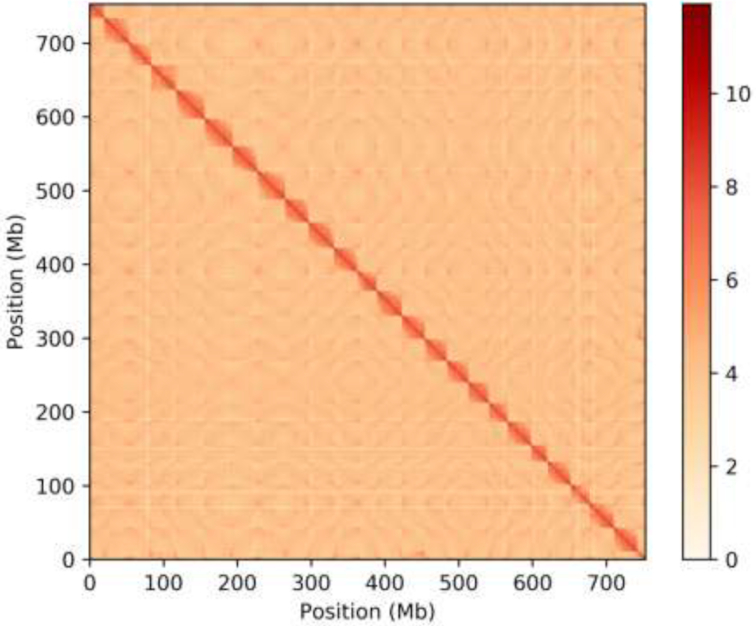
Hi-C interaction heat map for O. fasciatus reference genome showing interactions between the 24 chromosomes.
A final size of 768.8 Mb accounting for the 98.7% draft genome was assembled, which showed a high level of continuity with a contig N50 of 2.1 Mb and a scaffold N50 of 33.5 Mb using 1,372 contigs. The anchor rate of contigs (>100 kb) to chromosomes was attained up to the 99.7% based on the Hi-C assembly (Table 1). The contig N50 and scaffold N50 of O. fasciatus were much higher than those of previous fish genome assemblies constructed using NGS DNA sequencing technologies based on the genome assembly using PacBio long reads and Hi-C assembly (Supplementary Table S3).
Genome quality evaluation
To assess the completeness of the assembled O. fasciatus genome, we subjected the assembled sequences to Benchmarking Universal Single-Copy Orthologs (BUSCO) version 3 evaluation (BUSCO. RRID:SCR_015008) (BUSCO, actinopterygii_odb9) [20]. Overall, 96.6% and 1.5% of the 4584 expected actinopterygii genes were identified in the assembled genome as complete and partial BUSCO profiles, respectively. Approximately 85 genes could be considered missing in our assembly (Supplementary Table S5). Among the expected complete actinopterygii genes, 4,259 and 171 were identified as single copy and duplicated BUSCOs, respectively (Supplementary Table S5). We then used Minimap2 to estimate the completeness and homogeneity of genome assembly based on continuous long read subreads. A high quality of completeness and homogeneity was assessed in the genome assembly, and the mapping rate, coverage rate, and average sequencing depth reached 90.2%, 99.9%, and 80.6, respectively (Supplementary Table S6). Note that the mapping ratio might be related to the repetitive content of the O. fasciatus genome, especially for the high repeat content in the sex chromosomes [6]. However, how the repetitive elements in the genome influence the karyotypes of this species needs further investigation.
To further evaluate the accuracy of the O. fasciatus genome assembly, we aligned the NGS-based short reads from the whole-genome sequencing data against the reference genome using BWA [21]. We then used GATK (GATK, RRID:SCR_001876) to implement single-nucleotide polymorphism (SNP) calling and filter work, and the results showed that 99.8% and 0.2% of the 1.6 × 106 expected SNP reads were identified in the assembled genome as heterozygous and homologous SNPs, respectively. SNP calling on the final assembly also yielded a heterozygosity rate of 0.20%, supporting the k-mer estimate analysis (0.29%) (Supplementary Table S7).
Repeat sequences within the O. fasciatus genome assembly
To identify tandem repeats, we utilized Tandem Repeat Finder to annotate repetitive elements in the O. fasciatus genome. RepeatModeler (RepeatModeler, RRID:SCR_015027) (version 1.04) and LTR_FINDER (LTR_Finder, RRID:SCR_015247) [22] were used to construct a de novo repeat library with default parameters. Subsequently, we used RepeatMasker (RepeatMasker, RRID:SCR_012954) [23] (version 3.2.9) to map our assembled sequences on the Repbase TE (version 14.04) [24] and the de novo repeat library to identify known and novel transposable elements (TEs). In addition, TE-related proteins were annotated by using RepeatProteinMask software (version 3.2.2) [23].
The identified repeat sequences accounted for 33.9% of the O. fasciatus genome, including repeat sequences with 23.6% of the genome based on the de novo repeat library (Table 2). Approximately 23.4% of the O. fasciatus genome was identified as interspersed repeats (most often TEs). Among them, DNA TEs were the most abundant type of repeat sequences, which occupied 11.5% of the whole genome. Long interspersed nuclear elements and long terminal repeats comprised 7.3% and 4.0% of the whole genome, respectively (Table 2, Supplementary Fig. S2).
Table 2:
Detailed classification of repeat sequences of Oplegnathus fasciatus
| Repbase TEs | TE proteins | De novo | Combined TEs | |||||
|---|---|---|---|---|---|---|---|---|
| Type | Length (bp) | % in genome | Length (bp) | % in genome | Length (bp) | % in genome | Length (bp) | % in genome |
| DNA | 39,147,527 | 5.03 | 5,390,266 | 0.69 | 93,089,344 | 11.95 | 124,417,402 | 15.98 |
| Long interspersed nuclear element | 23,983,322 | 3.08 | 16,460,762 | 2.11 | 57,167,551 | 7.34 | 85,761,250 | 11.01 |
| Short interspersed nuclear element | 875,585 | 0.11 | 0 | 0.00 | 914,559 | 0.12 | 1,747,250 | 0.22 |
| Long terminal repeat | 10,163,601 | 1.31 | 5,770,483 | 0.74 | 31,126,639 | 4.00 | 42,465,968 | 5.45 |
| Satellite | 2,028,992 | 0.26 | 0 | 0.00 | 2,613,480 | 0.34 | 4,361,048 | 0.56 |
| Simple_repeat | 1,556,026 | 0.20 | 0 | 0.00 | 5,179,965 | 0.67 | 6,386,303 | 0.82 |
| Other | 6,545 | 0.00 | 0 | 0.00 | 0 | 0.00 | 6,545 | 0.00 |
| Unknown | 331,430 | 0.04 | 0 | 0.00 | 20,636,768 | 2.65 | 20,967,052 | 2.69 |
| Total | 73,544,786 | 9.44 | 27,613,880 | 3.55 | 183,954,095 | 23.62 | 250,611,845 | 32.18 |
RNA preparation and sequencing
We sequenced cDNA libraries prepared from the eggs of O. fasciatus that were used for genome annotation using Illumina sequencing technology. RNA quality was determined based on the estimation of the ratio of absorbance at 260 nm/280 nm (OD = 2.0) and the RIN (value = 9.2) by using a Nanodrop ND-1000 spectrophotometer (LabTech, USA) and a 2100 Bioanalyzer (Agilent Technologies, USA), respectively. We used the Clontech SMARTer cDNA synthesis kit to complete reverse transcription. A paired-end library was prepared following the Paired-End Sample Preparation Kit manual (Illumina Inc., San Diego, CA). Finally, a library with an insert length of 300 bp was sequenced by Illumina HiSeq X Ten in 150PE mode (Illumina Inc., San Diego, CA). As a result, we obtained ∼42.2 Gb high-quality transcriptome data from RNA-seq (Supplementary Tables S1, S8).
Gene annotation
Gene annotation of the O. fasciatus genome was performed using de novo, homology-based, and transcriptome sequencing-based predictions. We employed Augustus (Augustus, RRID:SCR_008417) (version 2.5.5) [25] and GenScan (GENSCAN, RRID:SCR_012902) (version 1.0) [26] software to predict protein-coding genes in the O. fasciatus genome assembly. Protein sequences of closely related fish species including Larimichthys crocea, Lates calcarifer, Gasterosteus aculeatus, Paralichthys olivaceus, Cynoglossus semilaevis, and Gadus morhua were downloaded from Ensembl [27] and aligned against the O. fasciatus genome using TBLASTN (TBLASTN, RRID:SCR_011822) software [28]. Subsequently, Genewise2.2.0 (GeneWise, RRID:SCR_015054) software [29] was employed to predict potential gene structures on all alignments.
We also mapped these NGS transcriptome short reads onto our genome assembly using TopHat1.2 (TopHat, RRID:SCR_013035) software [30], and then we employed Cufflinks (Cufflinks, RRID:SCR_014597) [31] to predict gene structures (Supplementary Table S9). All gene models were then integrated using MAKER (MAKER, RRID:SCR_005309) to obtain a consensus gene set [32]. The final total gene set was composed of 24,003 genes with an average of 10.1 exons per gene in the O. fasciatus genome (Table 1). The gene number, gene length distribution, coding sequence length distribution, exon length distribution, and intron length distribution were all comparable with those of other teleost fish species (Supplementary Table S9, Fig. S3).
To obtain further functional annotation of the protein-coding genes in the O. fasciatus genome, we employed the local BLASTX (BLASTX, RRID:SCR_001653) and BLASTN (BLASTN, RRID:SCR_001598) programs and the Swiss-prot database with an e-value ≤ 1e-5 [33] to align the non-redundant nucleotide and non-redundant protein, respectively. We also used Blast2GO (Blast2GO, RRID:SCR_005828) software to search the Gene Ontology, and Kyoto Encyclopedia of Genes and Genomes pathway databases [34, 35, 36]. Ultimately, 97.3% (23,364 genes) of the 24,003 genes were annotated by at least one database (Supplementary Table S10). Four types of non-coding RNAs (microRNAs, transfer RNAs, ribosomal RNAs, and small nuclear RNAs) were also annotated using the tRNAscan-SE (tRNAscan-SE, RRID:SCR_010835) and the Rfam database [37, 38] (Supplementary Table S11).
Gene family identification and phylogenetic tree construction
We employed the BLASTP (BLASTP, RRID:SCR_001010) program [39] with an e-value threshold of 1e-5 to identify gene families based on the transcript alignments of each gene from O. fasciatus and other fish species, which included Larimichthys crocea, Gadus morhua, Paralichthys olivaceus, Cynoglossus semilaevis, Notothenia coriiceps, Boleophthalmus pectinirostris, Lepisosteus oculatus, Gasterosteus aculeatus, Callorhinchus milii, Danio rerio, Salmo salar, and Oryzias latipes. A total of 21,528 gene families were identified by clustering the homologous gene sequences based on H-scores calculated from Bit-score using Hcluster_sg software (Supplementary Fig. S4). Subsequently, we selected 1,236 single-copy orthogroups from the above-mentioned species to construct the phylogenetic relationship between O. fasciatus and other fish species. We used the ClustalW (ClustalW, RRID:SCR_002909) program [40] to extract and align coding sequences of single-copy genes from the 1,158 orthogroups with a length filter (Supplementary Fig. S5). All the alignments were concatenated as a single dataset for each species. Nondegenerated sites extracted from the dataset were then joined into new sequences for each species to construct a phylogenetic tree based on the maximum-likelihood method implemented in the PhyML package [41] (with the -m PROTGAMMAAUTO model). We used the MCMCtree program to estimate divergence times among species based on the approximate likelihood method [42] and molecular clock data from the divergence time between medaka from the TimeTree database [43]. According to the phylogenetic analysis, O. fasciatus (Eupercaria: Centrarchiformes) clustered with Larimichthys crocea in the order Perciformes (Eupercaria), which was consistent with the new fish species taxonomy [44] (Fig. 4). The divergence time between O. fasciatus and the common ancestor with Larimichthys crocea was at approximately 70.5–88.5 million years ago.
Figure 4:

The phylogenetic relationships of O. fasciatus with other fishes. The bootstrap values (larger than 1) calculated from 1,000 bootstrap replicates and the branch lengths (smaller than 1) were labeled at and below/above each branch, respectively.
Conclusions
We successfully assembled the genome of O. fasciatus and reported the first chromosome-level genome sequencing, assembly, and annotation based on long reads from the third-generation PacBio Sequel sequencing platform. The final draft genome assembly is approximately 778.7 Mb, which was slightly higher than the estimated genome size (777.5 Mb) based on k-mer analysis. Those contigs were scaffolded to chromosomes using Hi-C data, resulting in a genome with a high level of continuity, with a contig N50 of 2.1 Mb and a scaffold N50 of 33.5 Mb. The chromosome-level genome assembly of O. fasciatus is also the first high-quality genome in the Oplegnathidae family. We also predicted 24,003 protein-coding genes from the generated assembly, and 97.3% (23,364 genes) of all protein-coding genes were annotated. We found that the divergence time between O. fasciatus and its common ancestor with Larimichthys crocea was approximately 70.5–88.5 million years ago. As far as we know, the Y chromosomes has always exhibited many specific sequence characteristics compared to X1 and X2, such as repeat content, and those differences might increase the difficulty of the sequence assembly of chromosomes X1 and X2. The chromosome-level genome assembly together with gene annotation data generated for the female fish in this work will provide a valuable resource for further research on sex-determining mechanisms, especially for obtaining an accurate assembly of the Y chromosome in male fish. These results will also accelerate genome-wide association studies in resistant breeding systems.
Supplementary Material
10/1/2018 Reviewed
1/7/2019 Reviewed
10/3/2018 Reviewed
Availability of supporting data
Supporting data and materials are available in the GigaScience GigaDB database [45], with the raw sequences deposited in the NCBI Sequence Read Archive under the accessions SRP158313 and SRP160016 .
Additional files
S Fig. 1 The GC content of O. fasciatus base on the Illumina platform for genome size survey
S Fig. 2 The content of interspersed repeats of O. fasciatus genome assembly. (a) Repbase library (b) de novo library
S Fig. 3 The gene number, gene length distribution, coding sequence length distribution, exon length distribution and intron length distribution were all comparable with those in other teleost fish species.
S Fig. 4 Comparing genome assemblies between O. fasciatus and other fish species.
S Fig. 5 Orthologous gene families across four fish genomes (Oplegnathus fasciatus, Larimichthys crocea, Gadus morhua and Salmo salar)
S Table 1 Summary of sequence data from O. fasciatus
S Table 2 Genome assembly statistics for O. fasciatus
S Table 3 Comparing genome assemblies between O. fasciatus and other fish species. Some data were cited from the reference (Gaorui Gong, Cheng Dan, Shijun Xiao, Wenjie Guo, Peipei Huang, Yang Xiong, Junjie Wu, Yan He, Jicheng Zhang, Xiaohui Li, Nansheng Chen, Jian-Fang Gui, Jie Mei; Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis, GigaScience, Volume 7, Issue 11, 1 November 2018, giy120, doi.org/10.1093)
S Table 4 Alignment of clean reads for Hi-C data and Hi-C libraries for chromosome-scale assembly
S Table 5 Genome quality of Oplegnathus fasciatus based on the BUSCO assessment
S Table 6 The estimation of the completeness for O. fasciatus genome assembly based on CLR (Continuous Long Reads) subreads mapping
S Table 7 The estimation of the accuracy for O. fasciatus genome assembly based on SNP calling
S Table 8 Transcriptome data from RNA-seq for O. fasciatus
S Table 9 Gene annotation of the O. fasciatus genome
S Table 10 Functional annotation of the protein-coding genes in O. fasciatus genome
S Table 11 The annotation of non-coding RNAs of O. fasciatus genome
Abbreviation
BUSCO: Benchmarking Universal Single-Copy Orthologs; NGS: Next Generation Sequencing; PacBio: Pacific Bioscience; SNP: single-nucleotide polymorphism; TE: transposable element.
Competing interests
The authors declare that they have no competing interests.
Funding
This study was supported by a grant from the National Natural Science Foundation of China (41506170, 31672672, and 31872195), Shandong Province Key Research and Invention Program (2017GHY15102, 2017GHY15106), Qingdao Source Innovation Program (17-1-1-57-jch), Marine Fishery Institute of Zhejiang Province, Key Laboratory of Mariculture and Enhancement of Zhejiang Province (2016KF002), National Key Research and Development Program (2018YFD0901204), STS project (KFZD-SW-106, ZSSD-019), Qingdao National Laboratory for Marine Science and Technology (2015ASKJ02), and China Agriculture Research System (CARS-47).
Ethics Statement
This research was approved by the Animal Care and Use Committee of the Chinese Academy of Science.
Author contributions
Y.S.X. conceived the project. Z.Z.X. and D.Y.M. collected the samples and extracted the genomic DNA. Y.S.X., J.Li, and J.Liu performed the genome assembly and data analysis. Y.S.X., Z.Z.X., J.Li, D.Y.M., and J.Liu wrote the paper.
References
- 1. Schembri PJ, Bodilis P, Francour P, et al.. Occurrence of barred knifejaw, Oplegnathusfasciatus (Actinopterygii: Perciformes: Oplegnathidae), in Malta (Central Mediterranean) with a discussion on possible modes of entry. Acta Ichthyol Piscat. 2010;40:101–4. [Google Scholar]
- 2. Mundy BC. Checklist of the fishes of the Hawaiian Archipelago. Bishop Mus Bull Zool. 2005;6:1–704. [Google Scholar]
- 3. An HS, Hong SW. Genetic diversity of rock bream Oplegnathusfasciatus in Southern Korea. Genes Genom. 2008;30:451–9. [Google Scholar]
- 4. Xiao YS, Li J, Ren GJ, et al.. Pronounced population genetic differentiation in the rock bream Oplegnathusfasciatus inferred from mitochondrial DNA sequences. Mitochondrial DNA A. 2016;27:2045–52. [DOI] [PubMed] [Google Scholar]
- 5. Park HS, Kim CG, Par YJ et al.. Population genetic structure of rock bream (Oplegnathusfasciatus Temminck & Schlegel, 1844) revealed by mtDNA COI sequence in Korea and China. Ocean Sci J. 2018;53:261–74. [Google Scholar]
- 6. Xu DD, Lou B, Bertollo LAC, et al.. Chromosomal mapping of microsatellite repeats in the rock bream fish Oplegnathusfasciatus, with emphasis of their distribution in the neo-Y chromosome. Mol Cytogenet. 2013;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xue R, An H, Liu QH, et al.. Karyotype and Ag-Nors in male and female of Oplegnathuspunctatus. Oceanol Limnol Sin. 2016;47:626–32. [Google Scholar]
- 8. Xiao ZZ, . Study on population genetics and culture biology of Oplegnathusfasciatus. Doctoral thesis. 2015, Ocean University of China, Qingdao: P. 162–76. [Google Scholar]
- 9. Zhang BC, Zhang J, Xiao ZZ, et al.. Rock bream (Oplegnathusfasciatus) viperin is a virus-responsive protein that modulates innate immunity and promotes resistance against megalocytivirus infection. Dev Comp Immunol. 2014;45:35–42. [DOI] [PubMed] [Google Scholar]
- 10. Li H, Sun ZP, Jiang YL, et al.. Characterization of an iridovirus detected in rock bream (Oplegnathusfasciatus; Temminck and Schlegel). Chin J Virol. 2011;27:158–64. [PubMed] [Google Scholar]
- 11. Koren S, Walenz BP, Berlin K, et al.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kajitani R, Toshimoto K, Noguchi H et al.. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24:1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pryszcz LP, Gabaldón T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 2016;44:e113–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simão FA, Waterhouse RM, Ioannidis P, et al.. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210. [DOI] [PubMed] [Google Scholar]
- 16. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tarailo-Graovac M, Chen NS. Using RepeatMasker to identify repetitive elements in genomic sequences. In: Editorial Board, Baxevanis, Andreas D et al.(eds). Current Protocols in Bioinformatics, 2009. p. 5: 4.10.1-4.10.14. [DOI] [PubMed] [Google Scholar]
- 19. Jurka J, Kapitonov VV, Pavlicek A et al.. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic Genome Res. 2005;110:462–7. [DOI] [PubMed] [Google Scholar]
- 20. Stanke M, Steinkamp R, Waack S, et al.. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32:309–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cai Y, González JV, Liu Z, et al.. Computational systems biology methods in molecular biology, chemistry biology, molecular biomedicine, and biopharmacy. BioMed Res Int. 2014;2014:746814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flicek P, Amode MR, Barrell D et al.. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gertz EM, Yu YK, Agarwala R, et al.. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. MBC Biology. 2006;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Birney E, Clamp M, Durbin R et al.. GeneWise and Genomewise. Genome Res. 2004;14:988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trapnell C, Pachter L, Salzberg SL, et al.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghosh S, Chan CKK. Analysis of RNA-Seq data using TopHat and Cufflinks. Methods Mole Biol. 2016;1374:339–61. [DOI] [PubMed] [Google Scholar]
- 27. Campbell MS, Holt C, Moore B et al.. Genome annotation and curation using MAKER and MAKER-P. Current Protocols in Bioinformatics. 2014;48:4.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lobo I. Basic local alignment search tool (BLAST). J Mol Biol. 2008;215:403–10. [DOI] [PubMed] [Google Scholar]
- 29. Harris MA, Clark J, Ireland A, et al.. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ogata H, Goto S, Sato K, et al.. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Conesa A, Götz S, García-Gómez JM, et al.. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674. [DOI] [PubMed] [Google Scholar]
- 32. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Griffiths-jones S, Bateman A, Marshall M, et al.. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lieberman-Aiden E, Van Berkum NL, Wiiliams L et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rao SSP, Huntley MH, Durand NC et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sandborn AL, Rao SSP, Huang SC, et al.. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A. 2015;112:E6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Flot JF, Marie-Nelly H, Koszul R, et al.. Contact genomics: scaffolding and phasing (meta) genomes using chromosome be 3D physical signatures. FEBs Letters. 2015;589:2966–74. [DOI] [PubMed] [Google Scholar]
- 38. Burton JN, Adey A, Patwardhan RP et al.. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31:1119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lobo I. Basic local alignment search tool (BLAST). J Mol Biol. 2008;215:403–10. [DOI] [PubMed] [Google Scholar]
- 40. Thompson JD, , Gibson TJ, Higgins DG et al.. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2003;2.3.1 -2.3.22, PMID 18792934. [DOI] [PubMed] [Google Scholar]
- 41. Guindon S, Dufayard JF, Hordijk W, et al.. PhyML: fast and accurate phylogeny reconstruction by maximum likelihood. Infect Genet Evol. 2009;9:384–5. [Google Scholar]
- 42. Yang Z, Rannala B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol Biol Evol. 2006;23:212–26. [DOI] [PubMed] [Google Scholar]
- 43. Hedges SB, Marin J, Suleski M, et al.. Tree of life reveals clock-like speciation and diversification. Mol Biol Evol. 2015;32:835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Betancur-R R, Wiley E, Bailly N, et al.. Phylogenetic classification of bony fishes. BMC Evol Biol. 2017;17:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiao YS, Xiao ZZ, Ma DY, et al.. Supporting data for “Genome sequence of the barred knifejaw Oplegnathus fasciatus (Temminck & Schlegel, 1844): the first chromosome-level draft genome in the family Oplegnathidae.”. GigaScience Database. 2019. 10.5524/100556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
10/1/2018 Reviewed
1/7/2019 Reviewed
10/3/2018 Reviewed
Data Availability Statement
Supporting data and materials are available in the GigaScience GigaDB database [45], with the raw sequences deposited in the NCBI Sequence Read Archive under the accessions SRP158313 and SRP160016 .