

Abstract
Urine dipstick tests are widely used in routine medical care to diagnose kidney and urinary tract and metabolic diseases. Several environmental factors are known to affect the test results, whereas the effects of genetic diversity are largely unknown. We tested 32.5 million sequence variants for association with urinary biomarkers in a set of 150 274 Icelanders with urine dipstick measurements. We detected 20 association signals, of which 14 are novel, associating with at least one of five clinical entities defined by the urine dipstick: glucosuria, ketonuria, proteinuria, hematuria and urine pH. These include three independent glucosuria variants at SLC5A2, the gene encoding the sodium-dependent glucose transporter (SGLT2), a protein targeted pharmacologically to increase urinary glucose excretion in the treatment of diabetes. Two variants associating with proteinuria are in LRP2 and CUBN, encoding the co-transporters megalin and cubilin, respectively, that mediate proximal tubule protein uptake. One of the hematuria-associated variants is a rare, previously unreported 2.5 kb exonic deletion in COL4A3. Of the four signals associated with urine pH, we note that the pH-increasing alleles of two variants (POU2AF1, WDR72) associate significantly with increased risk of kidney stones. Our results reveal that genetic factors affect variability in urinary biomarkers, in both a disease dependent and independent context.
Introduction
The urine, a final product of nephron function which consists of glomerular filtration, tubular reabsorption and secretion of water and solutes, provides valuable information about numerous physiological and pathophysiological processes (1). Urine dipstick tests are routinely used as a diagnostic tool in medical practice, facilitating early detection of diseases, such as urinary tract infections (UTI), and renal and metabolic disorders (1–4). Urine dipstick measurements are semiquantitative traits; positive urine dipstick results are expressed as intervals (e.g. pH and specific gravity) or graded according to severity on a scale from 1 to 4 plus (written as + to ++++; e.g. proteinuria and hematuria), and require medical interpretation (3,4). Apart from various diseases, environmental factors, such as exercise and food supplements, are known to affect urine dipstick results (3). However, it is an open question as to how genetic variation influences urine dipstick test results, both in a disease dependent and independent context.
Here we describe a genome-wide association study (GWAS) of urine dipstick measurements, testing for associations of variants in the sequence of the genome with glucosuria, ketonuria, proteinuria, hematuria, urine pH, signs of UTI (i.e. nitrites and leukocyte esterase) and urine specific gravity in a set of 150 274 Icelanders. In addition, we assess the variants detected for association with a broad set of diseases and other biological traits. By doing so, we attempt to shed light on whether the variants that affect urine dipstick test results are associated with specific diseases, or if they simply affect urinary traits separate from everything else.
Results
Study design
To search for sequence variants associating with glucosuria, ketonuria, proteinuria, hematuria, urine pH, UTI and urine specific gravity, we analyzed 32.5 million sequence variants detected through whole-genome sequencing of 15 220 Icelanders that were subsequently imputed into 151 677 chip-typed, as well as 1st- and 2nd- degree relatives of chip-typed individuals (5; Materials and Methods). In total, our association analysis is based on 150 274 individuals with urine dipstick measurements (44.2% male; mean age at measurement = 52 years old, SD = 27 years), of whom up to 149 899 individuals were included in the analysis of each individual trait (Table 1). Many of the individuals had several measurements, the geometric mean of the number of measurements per individual was 1.9 for glucose and ketones, 2.0 for protein, 2.8 for blood, 2.0 for urine pH, 1.8 for nitrites and leukocyte esterase and 2.0 for specific gravity. In categorical analysis, the maximum value per individual was used with the exception of low urine pH for which the minimum was used. Quantitative traits were rank-based inverse normal transformed separately for each sex and adjusted for age. For individuals with multiple measurements, values were averaged after transformation.
Table 1.
Genotyped individuals with urine dipstick measurements
| Cases | Controls | ||||||
|---|---|---|---|---|---|---|---|
| Phenotype | Total | Chip | Family imputed | Total | Mild | Moderate/severe | Negative |
| Total | 150 274 | 79 646 | 70 628 | - | - | - | - |
| Categorical traits | |||||||
| Glucosuria | 146 369 | 78 335 | 68 034 | 10 857 | 4 748 | 6 109 | 135 512 |
| Ketonuria | 136 698 | 74 228 | 62 470 | 41 130 | 19 327 | 21 803 | 95 568 |
| Proteinuria | 145 547 | 77 913 | 67 634 | 54 009 | 28 692 | 25 317 | 91 538 |
| Hematuria | 136 954 | 74 355 | 62 599 | 68 051 | 18 839 | 49 212 | 68 903 |
| Low urine pH | 148 199 | 79 389 | 68 810 | 35 897 | - | - | 112 302 |
| UTI | 79 850 | 43 421 | 36 429 | 13 322 | - | - | 66 528 |
| Quantitative traits | |||||||
| Urine pH | 149 899 | 79 416 | 70 483 | - | - | - | - |
| Urine specific gravity | 139 555 | 75 072 | 64 483 | - | - | - | - |
For each phenotype, we show the division of genotyped individuals into chip-typed (Chip) and family imputed, who are untyped 1st- and 2nd-degree relatives of chip-typed individuals for which genotype probabilities have been computed based on genealogy. For categorical traits we show how many of the individuals (both chip-typed and family imputed) are cases and controls, as well as the division of cases into mild and moderate/severe when applicable.
For glucosuria (Ncases = 10 857; Ncontrols = 135 512), ketonuria (Ncases = 41 130; Ncontrols = 95 568), proteinuria (Ncases = 54 009; Ncontrols = 91 538) and hematuria (Ncases = 68 051; Ncontrols = 68 903), cases were identified according to at least one positive urine dipstick reading and controls as individuals with only negative urine dipstick readings. The cases were also classified as mild when at least one urine dipstick reading was + with no greater reading and moderate/severe when at least one urine dipstick reading was ++ or greater (Materials and Methods). Cases with signs of UTI, were defined as individuals with positive readings for both nitrites (indicative of nitrate-reducing bacteria) and leukocyte esterase (indicative of neutrophils) on the same day, whereas controls were individuals with only negative readings for both (Ncases = 13 322; Ncontrols = 66 528). Low urine pH cases were defined as individuals with at least one pH reading of 5.0 or below, and controls were individuals with only pH readings above 5.0 (Ncases = 35 897; Ncontrols = 112 302). Additionally, urine pH (N = 149,899; mean = 6.13; SD = 0.78) and specific gravity (N = 139 555; mean = 1.02; SD = 0.01) were assessed as quantitative traits (Supplementary Material, Fig. S1). In summary, one to three phenotypes (groups/subgroups) were tested for each of the seven urinary traits: glucosuria, ketonuria, proteinuria, hematuria, UTI, urine pH and urine specific gravity (Table 1; Supplementary Material, Table S1). There were 16 phenotypes tested in total. Detailed demographic information about study subjects can be found in Supplementary Material, Table S2.
Sequence variant associations with urinary traits
We detected a total of 20 distinct association signals with urinary traits using weighted genome-wide significance thresholds for different sequence variant annotation classes (6; Table 2, Fig. 1 and Supplementary Material, Tables S3–5; Materials and Methods). Of these, six have been reported, one with the same trait (CUBN) and five with highly correlated traits. To search for independent association signals at each locus we performed a step-wise conditional analysis (Materials and Methods). In instances where several independent signals were observed at a locus, we present the adjusted odds ratios (ORs) and P-values for each signal. The novel association signals include three variants that associated with glucosuria, one variant with ketonuria, one with proteinuria, five with hematuria and four with urine pH. No significant associations were detected with UTI or urine specific gravity (Supplementary Material, Fig. S1). Nine of the 14 novel variants are common (minor allele frequency, MAF > 5%) and five are rare (MAF < 0.5%), while none are low-frequency variants (0.5% < MAF < 5%). The leading variants in six of the novel signals are coding variants or are highly correlated with one or more coding variants (r2 > 0.8; Supplementary Material, Table S4). Nine out of the 14 novel associations are further supported by external sources; seven are at loci that harbor rare variants causing Mendelian diseases with symptoms including the urinary trait in question and two associate with a related disease (i.e. kidney stones) both in Iceland and the UK (Table 2).
Table 2.
Sequence variants associating with urinary traits
| Variant [MA] | Position (hg38) | MAF% | Locus | Coding change | Association | Supportive information | |||
|---|---|---|---|---|---|---|---|---|---|
| Glucosuria | P | OR (95%CI) | Testing | ||||||
| rs763092306[T] | chr2:181678533 | 0.01 | NEUROD1 | p.Glu110Lys | 1.34E-10 | 23.74 | (9.04,62.36) | ≥ ++ versus - | Reported diabetes variant (8) . Rare mutations in NEUROD1 cause diabetes (OMIM: 601724). |
| chr6:32664911[G] | chr6:32664911 | 25.69 | HLA-DQB1 | p.Asp89Ala | 2.43E-23 | 1.28 | (1.22,1.34) | ≥ ++ versus - | In LD with a reported diabetes signal (10) |
| rs7903146[T] | chr10:112998590 | 29.78 | TCF7L2 | intron | 3.70E-20 | 1.18 | (1.14,1.22) | ≥ + versus - | Reported diabetes variant (9) |
| rs780518365[G] | chr12:120992362 | 0.04 | HNF1A | intron | 4.30E-11 | 7.05 | (3.95,12.60) | ≥ + versus - | Rare mutations in HNF1A cause diabetes (OMIM: 142410). |
| rs766982432[G] | chr12:120994314 | 0.02 | HNF1A | p.Pro291GlnfsTer51 | 2.25E-15 | 32.09 | (13.62,75.63) | ≥ ++ versus - | Pathogenic diabetes variant (8) (Clinvar). |
| rs201938902[G] | chr16:31424046 | 31.28 | SLC5A2 | intergenic | 1.60E-13 | 1.14 | (1.10,1.18) | ≥ + versus - | Rare mutations in SLC5A2 cause renal glucosuria (OMIM: 182381). SLC5A2 encodes a protein that has been targeted to increase urinary glucose excretion in treatment of diabetes (14). |
| rs141627694[C] | chr16:31488637 | 0.1 | SLC5A2 | p.Met382Thr | 4.80E-15 | 4.17 | (2.92,5.96) | ≥ + versus - | |
| rs752992795[A] | chr16:48548859 | 0.2 | SLC5A2 | intron | 3.70E-13 | 2.95 | (2.20,3.95) | ≥ + versus - | |
| Ketonuria | P | OR (95%CI) | Testing | ||||||
| rs7712274[C] | chr5:41773628 | 22.77 | OXCT1 | intron | 1.70E-16 | 0.9 | (0.88,0.93) | ≥ + versus - | Rare mutations in OXCT1 cause SCOT deficiency, symptoms include ketonuria (OMIM: 601424). |
| Proteinuria | P | OR (95%CI) | Testing | ||||||
| rs2075252[T] | chr2:169154475 | 26.19 | LRP2 | p.Lys4094Glu | 1.40E-11 | 1.08 | (1.06,1.10) | ≥ + versus - | Rare mutations in LRP2 cause Donnai-Barrow syndrome, symptoms include proteinuria (OMIM: 600073). LRP2 and CUBN encode interacting proteins that mediate proximal tubule protein uptake together. The CUBN signal is a reported albumin to creatin ratio signal (21). |
| rs74375025[A] | chr10:16905665 | 11.85 | CUBN | intron | 3.30E-15 | 1.12 | (1.09,1.15) | ≥ + versus - | |
| Hematuria | P | OR (95%CI) | Testing | ||||||
| rs760545501[T] | chr2:224359374 | 0.17 | 2q36 | intergenic | 4.03E-10 | 2.36 | (1.80,3.09) | ≥ ++ versus - | - |
| chr2:227254752[del] | chr2:227254752 | 0.06 | COL4A3 | 2.5kb deletion | 1.62E-23 | 11.78 | (7.26,19.11) | ≥ ++ versus - | Rare mutations in COL4A3 cause familial benign hematuria and autosomal Alport syndrome (OMIM: 120070). |
| rs200287952[A] | chr2:227277511 | 0.03 | COL4A3 | p.Gly695Arg | 8.27E-08 | 5.46 | (2.94,10.16) | ≥ ++ versus - | |
| chr6:31271845[C] | chr6:31271845 | 42.75 | HLA-C | p.Asp33Tyr | 2.60E-18 | 0.92 | (0.90,0.94) | ≥ + versus - | - |
| rs56254331[C] | chr19:41320115 | 18.33 | TGFB1 | intron | 1.30E-11 | 0.92 | (0.89,0.94) | ≥ + versus - | The hematuria risk decreasing allele also associates with higher TGFB1 expression (GTEx Portal, 17.08.2017). |
| Urine pH | P | OR (95%CI) | Testing | ||||||
| rs2253944[G] | chr3:187925558 | 31.1 | 3q27 | intergenic | 3.60E-11 | 0.93 | (0.90,0.95) | ≤ 5 versus > 5 | - |
| rs12417556[A] | chr11:111352720 | 46.75 | POU2AF1 | 3 prime UTR | 2.10E-14 | 1.09 | (1.06,1.11) | ≤ 5 versus > 5 | Associates suggestively with kidney stones both in Iceland and UK (Table S12). |
| P | Effect SD (95% CI) | Testing | |||||||
| rs6790058[G] | chr3:124579018 | 36.12 | KALRN | intron | 3.88E-10 | 0.02 | (0.02,0.03) | Quant. | - |
| rs551225[A] | chr15:53710894 | 45.24 | WDR72 | p.Pro306Leu | 2.58E-15 | -0.03 | (-0.03,-0.02) | Quant. | Associates with kidney stones both in Iceland and UK (Table S12). WDR72 is highly expressed in kidney (27). Correlated to a reported creatinine signal (28). |
Effect is shown for the minor allele. Glucosuria, ketonuria, proteinuria and hematuria were tested as categorical traits for three groups of cases (all, significant and mild) and association is shown for the case group with the most significant result. Urine pH was tested both as a categorical trait (low pH) and as a quantitative trait and association is shown for the most significant one. MA: minor allele. MAF: minor allele frequency. OR: odds ratio. CI: confidence interval. ≥ + versus −: + and greater versus negatives. ≥ ++ versus −: ++ and greater versus negatives. ≤ 5 versus > 5: pH ≤ 5.0 versus pH > 5.0. Quant: Quantitative.
Figure 1.
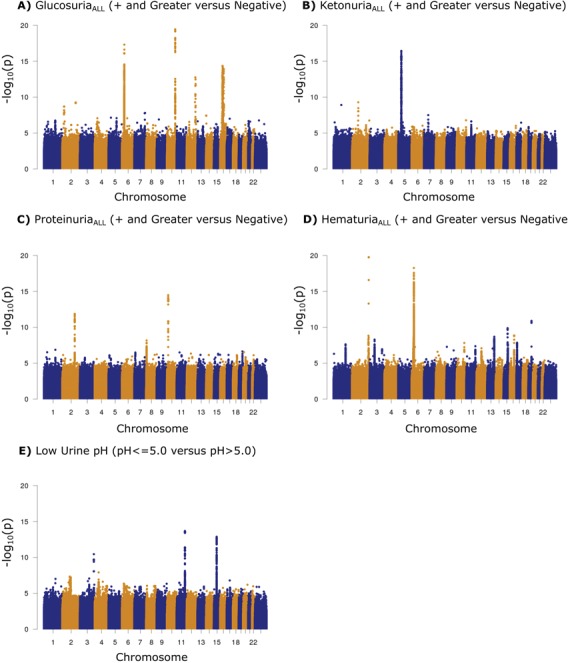
Manhattan plots for the five urinary traits with significant variant associations. Variants are plotted by chromosomal position (x-axis) and −log10P-values (y-axis). The five traits were tested as categorical traits. We divided Icelanders with urine dipstick measurements (N = 150 274) into cases and controls based on their measured strength of the trait in question: (A) glucosuriaALL (Ncases = 10 857, Ncontrols = 135 512), (B) ketonuriaALL (Ncases = 41 130, Ncontrols = 95 568), (C) proteinuriaALL (Ncases = 54 009, Ncontrols = 91 538), (D) hematuriaALL (Ncases = 68 051, Ncontrols = 68 903) and (E) low urine pH (Ncases = 35 897, Ncontrols = 112 302). A likelihood ratio test was used when testing for association.
To examine if effects of detected urinary trait variants differed by sex, we assessed the effects for men and women separately and tested them for statistical heterogeneity with a Q-test (6). None of the effects differed significantly between men and women at P < 0.05/20 (Supplementary material, Table S6).
We tested the 20 detected urinary trait signals for association with eight kidney and urinary tract-related diseases or traits: urine albumin (N = 10 041), chronic kidney disease (CKD; Ncases = 25 608), serum creatinine (N = 241,590), end stage renal disease and/or chronic kidney disease stages 4–5 (ESRD/CKD 4-5; Ncases = 3587), glomerular diseases (Ncases = 1041), hypertension (Ncases = 54 974), kidney stones (Ncases = 5876) and calcium kidney stones (Ncases = 3948) (Supplementary Material, Table S7). Additionally, the eight variants found to associate with glucosuria were also tested for association with type 1 diabetes (T1D; Ncases = 726), type 2 diabetes (T2D; Ncases = 11 448), HbA1C (N = 67 948) and fasting serum glucose (Ncases = 110 190) (Supplementary Material, Table S8).
In summary, 12 variants were tested for association with 8 related phenotypes while the 8 glucosuria variants were tested for association with 12 related phenotypes. At a threshold of P < 0.05/(12 × 8 + 8 × 12), two of the urinary trait signals associate with at least one of the aforementioned kidney- and urinary tract-related diseases or traits: the former is a proteinuria signal that also associates with serum creatinine and the second one is a urine pH signal that associates with ESRD/CKD 4-5, kidney stones, calcium kidney stones and serum creatinine (Supplementary Material, Table S7).
Using the same significance threshold, five of the eight glucosuria signals associate with HbA1C and fasting serum glucose, four of which also associate with T2D while the fifth one associates with T1D (Supplementary Material, Table S8).
The four variants found to associate with urine pH were also tested for association with kidney stones in a combined dataset of Iceland and UK Biobank (NcasesICE = 5876; NcasesUK = 4726). Two of these four variants associate with kidney stones in the combined dataset with P < 0.05/4, one representing the previously mentioned urine pH signal associating with ESRD/CKD 4-5, kidney stones and serum creatinine in the Icelandic dataset.
Glucosuria
We identified eight signals associating with glucosuria (Table 2, Fig 1, Supplementary Material, Table S4). Glucosuria is characteristic of diabetes (7) and for five of these eight signals, the glucosuria-increasing allele also associates with increased risk of T2D (all PT2D < 6.4 × 0-4; Ncases = 11 448). Three of these variants (NEUROD1, TCF7L2 and HNF1A) represent reported diabetes signals (8,9) , one (HLA-DQB1) is in moderate linkage disequilibrium (LD) (r2 = 0.37) with a reported T1D signal (10) and the fifth signal is represented by a rare intronic variant (MAF = 0.04%) at a well-established diabetes locus (HNF1A) (8) (Supplementary Materials, Table S8 and Fig. S3). Examination of 54 previously reported T2D SNPs at established T2D loci (11) revealed a positive correlation between their T2D effects in the DIAGRAM (2012) consortium data (12) (Europeans excluding Icelanders; Ncases = 10,706; NControls = 33 668) and their glucosuria effects in the Icelandic dataset (R2 = 0.36; P = 1.6 × 10-6; t-test) (Fig. 2). This indicates that the T2D variants have an effect on glucosuria that is proportional to the effect on T2D, consistent with the fact that glucosuria is one of the signs of T2D (7).
Figure 2.
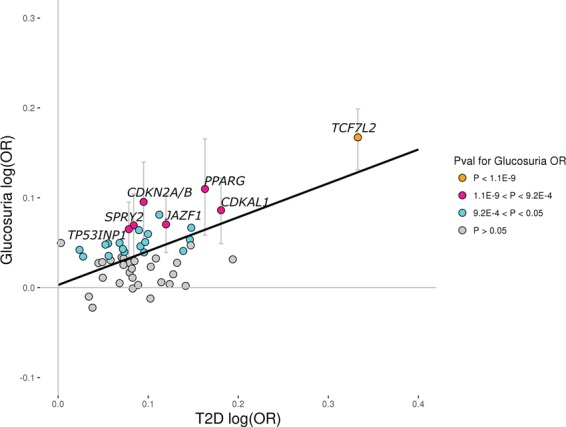
Reported T2D variants and the risk of glucosuria. Scatter plot showing 54 previously reported lead T2D SNPs at established T2D loci that, in a study by the DIAGRAM Consortium (2014) (11), associate with T2D in a subset of Europeans with P < 0.05. The x-axis shows their effect on T2D in the DIAGRAM (2012) data (12), excluding Icelanders (Ncases = 10 706; Ncontrols = 33 668), and the y-axis shows their effect on glucosuria risk (mild, moderate and severe cases (+ and greater) versus negative controls) in the Icelandic dataset (Ncases = 10 857; Ncontrols = 135 512). The black solid line (y = 0.003 + 0.38×) represents results from a simple linear regression using MAF(1-MAF) as weights with R2 = 0.36 (P = 1.6E-6; two-sided t-test). The colors of the circles represent their P-values for the glucosuria OR in the Icelandic dataset. Orange dots represent variants that associate with glucosuria with P < 1.1E-9, pink dots represent variants that associate with glucosuria with 1.1E-9 < P < 9.2E-4, cyan dots represent variants that associate with glucosuria with 9.2E-4 < P < 0.05 and gray dots represent variants that associate with glucosuria with P > 0.05. 95% confidence intervals for the glucosuria OR are shown for variants that associate with glucosuria risk in the Icelandic dataset with P < 9.2E-4 (0.05/54) and are depicted as gray vertical lines. Our data indicate that the effect on glucosuria is proportional to effect on T2D, as expected.
At the SLC5A2 locus at 16p11.2 we found three distinct signals associating with risk of glucosuria, none of which associate with T2D (PT2D > 0.14) (Supplementary Materials, Table S8 and Fig. S4). SLC5A2 encodes the main sodium-glucose transporter in the kidney (SGLT2) (13). SGLT2 inhibitors are medications that are used for improving glycemic control in diabetics, assessed by reduction in glycated hemoglobin (HbA1C), by increasing urinary glucose excretion (14). Furthermore, loss-of-function and missense mutations in SLC5A2 have been shown in case reports to cause familial renal glucosuria through both autosomal dominant and recessive modes of inheritance (Fig. 3) (15).
Figure 3.
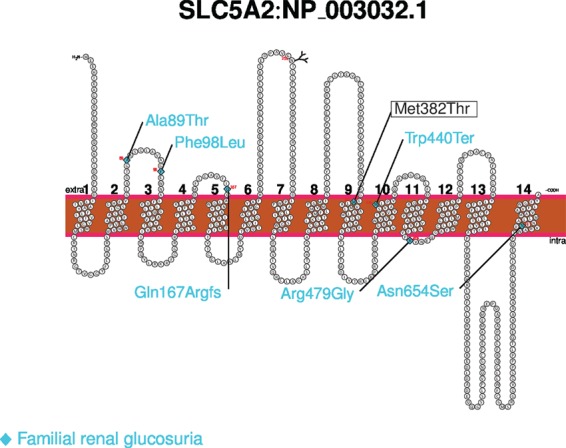
The Met382Thr variant in SLC5A2 schematic diagram showing the Met382Thr missense variant in the SLC5A2 gene product (NP_003032.1), in relation to mutations causing familial renal glucosuria classified as pathogenic and likely pathogenic according to the ClinVar database (shown in blue here).
In our data, the most common SLC5A2 signal is represented by an intergenic variant rs201938902[G] (MAF = 31.28%) located 60 Kb upstream of SLC5A2 (ORadj (95% confidence interval, CI) = 1.14 (1.10, 1.18), Padj = 1.5×10-12). The second signal consists of 91 highly correlated markers (r2 > 0.8 to leading variant) distributed over a 17.3 Mb region, spanning the centromere on chromosome 16, including the SLC5A2 gene. This signal is represented by the rare intron variant rs752992795[A] (MAF = 0.20%, ORadj (95% CI) = 3.14 (2.33, 4.23), Padj = 5.0 × 10-14) (Supplementary Material, Fig. S4). The third signal is represented by the rare missense variant rs141627694[C] (MAF = 0.10%, Met382Thr) in SLC5A2 (ORadj (95% CI) = 3.74 (2.64, 5.29), Padj = 1.0×10-13). In Iceland, 1 in 500 individuals is heterozygous for the missense variant Met382Thr, while it is essentially absent from other populations (gnomAD database; http://gnomad.broadinstitute.org; accessed 20.09.2017) (16). The Met382Thr missense variant has been reported as a compound heterozygous variant in a single case of severe renal glucosuria (17). No homozygotes were observed among chip-typed and imputed individuals in Iceland (N = 151 677) due to the low frequency of the variant. The amino acid substitution is located in the ninth transmembrane domain, and the change in coding sequence results in a substitution of the hydrophobic methionine with a polar threonine at position 382 (Supplementary Material, Fig. S5) (PolyPhen-2: probably damaging, SIFT: deleterious). For this variant, we observe a lowering effect on both fasting serum glucose (Effect (95% CI) = −0.19 SD (−0.33, −0.05), P = 0.0074, N = 110 190) and HbA1C levels (Effect (95% CI) = −0.29 SD (−0.45, -0.13), P = 3.7 × 10-4, N = 67 948). The lowering effect on HbA1C, which is a measure of long-term glucose homeostasis, is consistent with higher glucose excretion mediated by SGLT2 in rs141627694[C] carriers, and the variant is therefore likely to result in reduced SGLT2 function, consistent with what has been observed for loss-of-function mutations in mice, and decreased-function mutations seen in familial glucosuria in humans. However, the other two distinct SLC5A2 glucosuria signals mentioned above, rs201938902 and rs752992795, do not associate with fasting serum glucose or HbA1C (all P > 0.20) (Supplementary Material, Table S8).
Stratification of our glucosuria study group into T2D cases (glucosuria; Ncases = 4015; Ncontrols = 5002) and those who have not been diagnosed with T2D (glucosuria; Ncases = 6842; Ncontrols = 130 510), demonstrated that carriers of the two rare SLC5A2 variants are at comparable risk of glucosuria whether or not they have been diagnosed with T2D (heterogeneity P = 0.42 and 0.45, respectively; Supplementary Material, Table S8). This shows that the effects of these variants on glucose excretion is also present among T2D cases, who commonly experience glucosuria.
We calculated a genetic risk score using the three glucosuria-associating SLC5A2 variants. When tested for association with glucosuria and other related phenotypes (CAD, MI, T2D, UTI and HbA1C), we only observed a significant association with increased risk of glucosuria (OR = 2.62, P = 1.80 × 10-35), as well as a suggestive association with decreased HbA1C levels (Effect = 0.08 SD, P = 8.96 × 10-3) (Supplementary Material, Table S9).
Ketonuria
We identified one novel signal associating with ketonuria, represented by a group of 83 correlated, common non-coding variants (r2 > 0.8 to leading variant), spanning the entire OXCT1 gene at 5p13.1 (Table 2, Fig. 1; Supplementary Material, Table S4). The variant demonstrating the strongest association is rs7712274[C], located in intron 13 of OXCT1 (MAF = 22.77%, OR (95% CI) = 0.90 (0.88, 0.93), P = 1.7 × 10-16). OXCT1 encodes succinyl-CoA-3-oxaloacid CoA transferase (SCOT), a mitochondrial enzyme catalyzing the rate-limiting step of extrahepatic ketone body catabolism. Biallelic mutations in OXCT1 are known to cause SCOT deficiency (OMIM: 245050) which is characterized by severe ketoacidosis (18) (Supplementary Material, Table S10). Starvation and diabetes are common causes of increased ketone body production (19). In our dataset, the variant rs7712274 is not associated with T1D (P = 0.31, OR (95% CI) = 0.93 (0.80, 1.07), Ncases = 726), T2D (P = 0.34, OR (95% CI) = 0.98 (0.94, 1.02), Ncases = 11 448) or BMI (P = 0.30, Effect (95% CI) = −0.08SD (−0.07, 0.23), N = 72 747).
Proteinuria
We identified two signals associating with proteinuria (Table 2, Fig. 1). The first signal is represented by the missense variant rs2075252[T] (MAF = 26.19%, Lys4094Glu) in LRP2 at 2q31.1 (OR (95% CI) = 1.08 (1.06, 1.10), P = 1.4 × 10-11). This missense variant is moderately correlated (r2 = 0.39) with rs4667594[T], a common intronic variant (MAF = 47.7%) reported to associate with estimated glomerular filtration rate (eGFR) (20). rs4667594[T] has a suggestive association with risk of proteinuria in our dataset (OR (95% CI) = 1.04 (1.02, 1.06), P = 3.5 × 10-4). However, the missense variant we report herein, Lys4094Glu, fully accounts for its effect on proteinuria (Padj = 0.37), but not vice versa.
The second proteinuria signal is represented by the common intronic variant rs74375025[A] (MAF = 11.9%) in CUBN at 10p13 (OR (95% CI) = 1.12 (1.09,1.15), P = 3.3 × 10-15). The SNP rs74375025 is correlated (r2 = 0.85) with rs1801239, a missense variant (Ile2984Val) in CUBN reported to associate with alteration in urine albumin-to-creatinine ratio (ACR) (21). LRP2 and CUBN encode the interacting endocytic receptors megalin and cubilin, respectively, that are highly expressed in the renal proximal tubule (22). Loss-of-function and missense variants in LRP2 have been reported to cause the autosomal recessive Donnai–Barrow syndrome, a severe rare disorder with features that include proteinuria (OMIM: 222448).
Additionally, we detect a 2.5 kb deletion in COL4A3 that associates suggestively with increased risk of proteinuria (OR (95% CI) = 2.17 (1.52, 3.09), P = 1.9 × 10-5) (Supplementary Material, Table S5). This deletion also associates with hematuria (see below).
Hematuria
We found five novel signals associating with hematuria (two in COL4A3 and one at each of 2q36, HLA-C and TGFB1). Two of these signals associate with moderate/severe hematuria only (++ and greater versus negative test) (Table 2; Supplementary Materials, Table S4 and Fig. S6). Common causes of hematuria include UTI, kidney stones and urinary tract malignancy (23). None of the five hematuria signals associate with UTI or kidney stones in our dataset (Supplementary Material, Tables S5 and S7), nor are they strongly correlated with variants reported to associate with cancer (all r2 < 0.2) (GWAS catalog, accessed 29.05.2017). In women, hematuria might be due to menstruation (3) although when assessing the effect of these five variants for men and women separately no difference between the sexes was detected when tested for heterogeneity (heterogeneity P > 0.05/20) (Supplementary Material, Table S6).
A rare 2475 bp deletion–insertion spanning exons 16 and 17 of COL4A3 (chr2:227254752 – 227257226; hg38) results in a large in-frame deletion of the gene product (Gly289_Lys330del). COL4A3 is one of six alpha chains that form heterotrimeric type IV collagen molecules, the major structural component of glomerular basement membranes (OMIM: 120070). All 220 whole-genome sequenced carriers of the deletion share a break-point sequence (Fig. 4). This deletion associates with increased risk of hematuria (MAF = 0.06%, ORadj (95% CI) = 10.71 (6.47, 17.70), Padj = 2.5 × 10-20) and proteinuria (see above). Mutations in COL4A3 have been reported to cause both autosomal dominant benign familial hematuria (OMIM: 141200) as well as autosomal dominant and recessive Alport syndrome (OMIM: 203780 and OMIM: 104200), a rare disorder which commonly leads to kidney failure and deafness (Supplementary Materials, Tables S10 and S11 and Figure S7). Hematuria is the hallmark of Alport syndrome at an early age while proteinuria occurs at later stages of the disease (24). In Iceland, approximately 1 in 800 individuals carry this large deletion, and no homozygous carrier was observed as would be expected due to its low frequency. This deletion has not been detected outside of Iceland (ExAC, http://exac.broadinstitute.org/, accessed 18.08.2017) (16).
Figure 4.
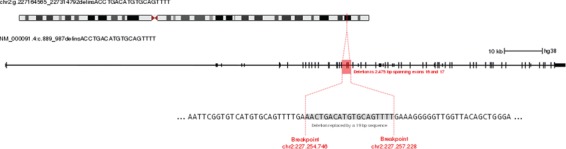
The 2.5 kb deletion in COL4A3 Schematic diagram showing the location of the COL4A3 deletion in the context of the exon structure of the transcript NM_00091.
We identified two additional distinct signals in the same region associating with increased risk of moderate/severe hematuria (Supplementary Material, Table S4 and Fig. S6) (all pairwise r2 < 0.01). These are the rare intergenic variant rs760545501[T] at 2q36 (MAF = 0.17 %, OR adj (95% CI) = 2.47 (1.69, 3.25), Padj = 5.7 × 10-10), and the rare COL4A3 missense variant rs200287952[A] (Gly695Arg, MAF = 0.03%, ORadj (95% CI) = 5.74 (3.66, 7.83), Padj = 6.8 × 10-8). We did not observe homozygous carriers of either variant.
Approximately 1 out of 600 Icelanders carries either the 2.5 kb deletion or the missense variant Gly695Arg in COL4A3. For carriers of either variant, our data are not compatible with manifestations of Alport syndrome other than hematuria. To our knowledge, none of the observed carriers are deaf (excluding age-related hearing loss) and none have been diagnosed with ESRD/CKD 4-5 at an early age. In Iceland, there are four documented Alport syndrome families, but none of them carry either of these two COL4A3 mutations.
The common intronic variant rs56254331[C] at 19q13.2 associates with a reduced risk of hematuria (MAF = 18.33%, OR (95% CI) = 0.92 (0.89, 0.94), P = 1.3 × 10-11). This sequence variant is the strongest cis-eQTL in GTEx, affecting the expression of the neighboring TGFB1 which associates significantly with expression in several tissues (The GTEx Portal; https://www.gtexportal.org/; accessed 17.08.2017). The allele associating with higher expression of TGFB1 associates with a lower risk of hematuria.
In the human leukocyte antigen (HLA) region, we observe a signal associating with risk of hematuria. The signal is represented by a missense variant at chr6:31271845[C] (Asp33Tyr; MAF = 42.75%), in HLA-C (OR (95% CI) = 0.92 (0.90, 0.94), P = 2.6 × 10-18). We note that the genes TGFB1 and HLA-C both play a role in the immune response. TGFB1 encodes transforming growth factor beta 1, a pro-inflammatory cytokine (25), and HLA-C encodes a major histocompatibility complex class 1 molecule (26). There are no reported autoimmune disease-associated signals at TGFB1 (GWAS catalog, accessed 29.05.2017). Rare missense mutations in TGFB1 cause autosomal dominant Camurati–Engelmann disease (OMIM: 131300). Several variants at the HLA region have been reported to associate with autoimmune diseases but Asp33Tyr is not strongly correlated with the top HLA signals in any of these disorders (r2< 0.15).
Urine pH
We found four signals (11q23, 15q21, 3q27 and 3q21) associating with urine pH as a categorical and a quantitative trait (Table 2, Fig. 1; Supplementary Material, Table S4).
At 11q23, we detected a signal represented by a common 3' UTR variant rs12417556[A] (MAF = 46.7%) in POU2AF1 that associates with increased risk of low urine pH (OR (95% CI) = 1.09 (1.06, 1.11), P = 2.1 × 10-14).
At 15q21, we observed a signal consisting of 76 highly correlated variants (r2 > 0.8 to leading variant) associating with urine pH. The signal is represented by the common missense variant rs551225[A] (MAF = 45.2%, Pro306Leu) in WDR72 (OR (95% CI) = 1.08 (1.06, 1.10), P = 6.5 × 10-13) that associates with low urine pH. WDR72 encodes WD repeat-containing protein 72, a poorly characterized protein that is highly expressed in the kidney (27). We note that rs551225 is moderately correlated (r2 = 0.25) with rs491567, a variant reported to associate with serum creatinine (28).
At 3q27, the common intergenic variant rs2253944[G] (MAF = 31.1%) associates with decreased risk of low urine pH (OR (95% CI) = 0.93 (0.90, 0.95), P = 3.6 × 10-11).
At 3q21, a signal represented by the intergenic variant rs6790058[G] (MAF = 36.1%) associates at genome-wide significance with urine pH as a quantitative trait (Effect (95% CI) = 0.02 SD (0.02, 0.03), P = 3.9 × 10-10). The variant also associates with decreased risk of low urine pH (OR (95% CI) = 0.95 (0.93, 0.97), P = 6.9 × 10-6) (Supplementary Material, Table S6[S5]).
Urine pH is a major risk factor for the formation of several types of kidney stones. High urine pH promotes the formation of calcium phosphate containing stones (29), whereas low pH favors the formation of uric acid stones (30). In a combined kidney stone cohort from Iceland (Ncases = 5876, Ncontrols = 322 970) and the UK Biobank database (individuals of white British ancestry: Ncases = 4726, Ncontrols = 403 841) (31) (Materials and Methods), the pH-increasing alleles of the SNPs at POU2AF1 and WDR72 associate with increased risk of kidney stones (ORMETA (95% CI) = 1.06 (1.03, 1.09) and 1.09 (1.06, 1.12), respectively, PMETA = 2.3 × 10-4 and 4.8 × 10-8; Supplementary Material, Table S12). The other urine pH signals at 3q21 and 3q27 did not associate with kidney stones (PMETA = 0.087 and 0.40, respectively; Supplementary Material, Table S12).
Discussion
In this study, we report associations of sequence variants with biomarkers routinely assessed in clinical practice using urine dipstick tests. We identified 14 novel associations with 5 urinary traits in the Icelandic population.
Most notably, we describe novel association signals at SLC5A2, one common and two rare, influencing the risk of glucosuria in the general population. SLC5A2 encodes SGLT2, the principal sodium-glucose transporter in the kidney, responsible for the bulk of the tubular reabsorption of glucose (13). Increased glucosuria among these subjects is due to a lowering of the renal threshold for glucose reabsorption (32), rather than being a result of increased glycemia such as in diabetics. SGLT2 inhibitors are a new class of diabetes drugs that are used to regulate blood glucose levels in patients by enhancing the urinary glucose excretion. The effect of the rare missense variant Met382Thr in the Icelandic population corresponds to a decrease of 0.40% in HbA1C (P = 3.7 × 10-4; population mean = 6.44%, SD = 1.48%), mimicking the effect of the SGLT2 inhibitor dapagliflozin (15).
Approximately 1 out of 80 glucosuria cases are carriers of one of the two rare SLC5A2 glucosuria variants, the aforementioned Met382Thr variant and the rare intron variant rs752992795, while the carriers account for 1 out of 340 individuals without glucosuria (Supplementary Material, Fig. S8). Of carriers of one of the two variants with urine dipstick measurements (N = 523), 24% have glucosuria while the fraction is 7% for the total dataset (N = 146 639) (Supplementary Material, Fig. S9). Although UTI is a reported adverse effect of SGLT2 inhibitor drugs (33), the SLC5A2 variants do not associate with UTI in our dataset (Supplementary Material, Table S5).
The Met382Thr substitution is located in a transmembrane domain and results in a substitution of a hydrophobic methionine with a polar threonine, a substitution likely to affect the integrity of the transmembrane domain (34). The genomic position is highly conserved across mammalian species (GERP = 4.7), indicating an important biological role conserved through mammalian evolution. Furthermore, the observed effect of the minor allele of the variant (increased glucosuria) is consistent with what has been described for loss-of-function alleles in Slc5a2 deficient mice, and for mutations decreasing glucose transporter activity in familial renal glucosuria in humans (OMIM: 182381). The mutation is therefore likely to result in reduced SGLT2 transport capacity.
We detected two common proteinuria associations of variants in the multiligand endocytic co-receptors LRP2 and CUBN that encode megalin and cubilin, respectively, which play an essential role in proximal tubule protein uptake (35). Urine dipstick tests are intended for measurement of total protein, the reagent pad is primarily sensitive to albumin and reacts much less with other proteins, including globulins and immunoglobulin light chains (36). The fact that only the CUBN signal was identified in a previous study of albuminuria determined by immunoassay using albumin specific antibodies (21), while the LRP2 signal was not, could indicate that the LRP2 signal is associating with low molecular weight proteinuria. This finding would be consistent with previous studies demonstrating that megalin-deficient mice excrete low-molecular-mass plasma proteins in the urine (37) and that selective low molecular weight proteinuria is one of the principal clinical manifestation of Donnai–Barrow syndrome (38), which is caused by mutations in LRP2. On the other hand, the LRP2 signal associates with eGFR in a previous study of kidney function (20), whereas the CUBN signal does not. This might suggest that LRP2 plays a larger part in glomerular filtration than CUBN and that the role of CUBN is more specific for protein uptake
Until now, most GWAS' have focused on SNPs and small indels. We also call large structural variants (Materials andMethods), allowing us to capture signals such as the rare 2.5 kb deletion in COL4A3 that associates with hematuria and proteinuria. COL4A3 also harbors a rare missense variant, Gly695Arg, that associates with risk of hematuria in our dataset. In the ClinVar database, this variant is classified as `likely pathogenic’ and has been detected in a single case of autosomal dominant Alport syndrome (Variation ID: 369964) (Supplementary Material, Fig. S7). Our data do not suggest that the two COL4A3 variants cause Alport syndrome. These variants could be associating with benign familial hematuria, also known as thin basement membrane nephropathy, a disease with a benign course where minimal proteinuria is sometimes observed (39,40).
Despite the large size of our GWAS of urinary traits there are several noteworthy limitations. Our study is based on urine dipstick measurements obtained in the clinical setting. It is therefore possible that a random sample of healthy individuals would give different association results for variants associating with diseases that result in elevated urinary biomarker levels. The lack of replication in other populations is a limitation of the study. For the signals represented by common variants, it is likely that the same variant would associate with the same trait and possibly related traits in other European populations. In support of this notion, 5 of the 12 common urinary trait signals (MAF > 5%) detected in the present study associated with a phenotypically related trait in other populations. These include the two glucosuria signals at TCF7L2 and HLA-DQB1 that associate with diabetes in British and French cohorts (9,10), the CUBN proteinuria signal that has been reported to associate with ACR (21) and the two urine pH signals at POU2AF1 and WDR72 that associate with risk of kidney stones, both in Iceland and the UK (S12 Table). In addition, three common signals implicate genes in which rare mutations have been reported to cause Mendelian conditions: OXCT1 in SCOT deficiency (ketonuria; OMIM: 601424), LRP2 in Donnai–Barrow syndrome (proteinuria; OMIM: 600073) and SLC5A2 in renal glucosuria (OMIM: 182381). In the case of rare variant associations (MAF < 0.5%), one would expect the detection of other rare variants at the same loci in other populations. This is already the case for seven of the eight rare urinary trait signals. Four implicate genes that harbor rare mutations reported to cause Mendelian conditions that have the associated urinary trait as a clinical manifestation, two in COL4A3 (Alport syndrome and benign familial hematuria; OMIM: 120070) and two in SLC5A2 (renal glucosuria; OMIM: 182381). Three are rare glucosuria signals in genes harboring variants reported to cause diabetes, two in/at HNF1A (OMIM: 142410) and one in NEUROD1 (OMIM: 11719843). Two of those three signals are represented by variants that have been reported to cause diabetes in Iceland, Glu110Lys in NEUROD1 and Pro291GlnfsTer51 in HNF1A (8).
We observed common and rare variants associating with urinary traits, some of which are apparently not disease-related. Our findings support the notion that genetic factors affect variation of urinary traits. In the clinical practice, urine dipstick tests are routinely used in the diagnostic evaluation of diseases of the kidney and urinary tract and diabetes (3). The interpretation of these tests could be improved by the inclusion of genetic information, as this would facilitate the differentiation between a pathological condition and an innocent finding. Additionally, the genes in which these sequence variants occur are potential drug targets, as is already the case for SGLT2 inhibitor drugs.
Materials and Methods
Study population
This study is based on whole-genome sequence data of 15 220 Icelanders participating in various disease projects at deCODE genetics (mean YOB = 1954, SD = 22 years). A total of 151 677 Icelanders have been genotyped using Illumina SNP chips (mean YOB = 1959, SD = 21 years) and, in addition, genotype probabilities for untyped 1st and 2nd degree relatives of chip-typed individuals have been calculated based on the Icelandic genealogy database. The process of whole-genome sequencing the Icelandic population, and the subsequent imputation from which the data for this analysis were generated has been extensively described in a recent publication (41).
The National Bioethics Committee approved the study (ref: VSN-15-023, VSNb2015010033-03.12) including the protocol, methodology and all documents presented to the participants. All participating individuals who donated samples, or their guardians, provided written informed consent. All sample identifiers were encrypted in accordance with the regulations of the Icelandic Data Protection Authority. Personal identities of the participants and biological samples were encrypted by a third-party system and approved and monitored by the Icelandic Data Protection Authority. All methods were performed in accordance with the relevant guidelines and regulations.
Phenotypic information
To perform GWAS for glucosuria, ketonuria, proteinuria, hematuria, urine pH, UTI and specific gravity, we examined urine dipstick tests obtained at two laboratories in Iceland: the Biochemistry Laboratory of Landspitali—The National University Hospital of Iceland and the Icelandic Medical Center Laboratory in Mjodd.
The data span the period from 1993–2015 and include measurements of 159 625 Icelanders. Of those, 150 274 were included in the present analysis (Mean YOB = 1963, SD = 27 years), a group that consists of 79 646 genotyped individuals and 70 628 untyped individuals, imputed on the basis of genotype information of close relatives.
The urine dipstick markers tested in this analysis are based on the results of Siemens Multistix 10 SG Reagent Strips (Siemens Healthcare diagnostics Ltd. [Sir William Siemens Sq.,] Frimely, Camberly, UK GU168QD).
For glucosuria, ketonuria, proteinuria and hematuria, we performed three different categorical analyses for each trait: (1) All positive results: cases with at least one measurement of + or greater versus negative controls; (2) Mild: cases with at least one + measurement, but never ++ or greater, versus negative controls; (3) Moderate/Severe: cases with at least one measurement of ++ or greater versus negative controls (Table 1). Results that read `trace’ were not included in the analysis. For these phenotypes, we also performed separate analysis for each sex and examined if the association results were different between the sexes.
Glucosuria cases and controls were categorized as + cases (N = 4748), ++/+++/++++ cases (N = 6109) and − controls (N = 135 512).
Ketonuria cases and controls were categorized as + cases (N = 19 327), ++/+++/++++ cases (N = 21 803) and − controls (N = 95 568).
Proteinura cases and controls were categorized as + cases (N = 28 692), ++/+++/++++ cases (N = 25 317) and − controls (N = 91 538)
Hematuria cases and controls were categorized as + cases (N = 18 839), ++/+++/++++ cases (N = 49 212) and − controls (N = 68 903).
For urine pH, we performed categorical analysis in which low pH cases (N = 35 897) were those with at least one measurement of pH ≤ 5, while controls (N = 112 302) were individuals who had only measurements of ≥5.5. We also tested urine pH as a quantitative trait (N = 149 899) with the measurements rank-standard normalized separately for each sex, using age as a covariate. Multiple measurements per individual were averaged after standardization.
UTI cases (N = 13 322) were individuals who had a positive measurement of both nitrite and leukocyte esterase on the same day, at least once. Controls (N = 66 528) were individuals who had only negative measurements of nitrite and leukocyte esterase.
Urine specific gravity was tested as a quantitative trait among 139 555 individuals with available measurements. The specific gravity values were rank-standard normalized separately for each sex, using age as a covariate. Multiple measurements per individual were averaged after standardization.
When assessing the effect of glucosuria variants on T2D, we performed case-control analysis where 11 448 T2D cases were tested against 278 376 population controls. Determination of T2D case status was based on medical records, use of oral diabetes medications or HbA1C levels >6.5%, excluding T1D cases.
T1D cases (N = 726) are individuals with clinically confirmed T1D, diagnosed at the National Pediatric Diabetes Center at the University Hospital (ICD-9: 250 or ICD-10: E10).
CKD (N = 25 608) was defined based on the discharge diagnoses N18 (ICD-10) or 585 (ICD-09) and eGFR, derived from serum creatinine using the Chronic Kidney Disease Epidemiology Collaboration equation, of <60 mL/min/1.73 m2 for 3 months or longer. Serum creatinine measurements from the time period 1993–2017 were obtained at the University Hospital, the Icelandic Medical Center Laboratory in Mjodd, the Department of Clinical Biochemistry at Akureyri Hospital and the in-house laboratory at deCODE genetics. CKD stages 4 and 5 was defined as eGFR < 30 mL/min/1.73 m2 for 3 months or longer. End-stage renal disease (ESRD) was defined as treatment with maintenance dialysis or kidney transplantation. For the analysis ESRD and CKD stages 4 and 5 were combined (N = 3587). The data on ESRD were obtained from the Icelandic End-Stage Renal Disease Registry at the University Hospital and cover the period from August 1968 to April 2017.
The glomerular diseases group (N = 1041) included all individuals with ICD-10 codes N0 to N08 or ICD-09 codes 580–583.
To identify Icelandic kidney stone cases (N = 5876), we searched for patients with ICD codes (ICD-9: 592.0, 592.1, 592.9, 270.0, 271.8; ICD-10: N20.0 N20.1, N20.2, N20.9, N21.0, N21.1, N21.8, N21.9, N22.0, N22.8, E72.0, E74.8, E79.8), radiology diagnosis codes and surgical procedure codes indicative of kidney stones. Patients with calcifications other than kidney stones and asymptomatic kidney stones were excluded.
Hypertension diagnoses (N = 54 974) were obtained from the primary health care clinics of the Reykjavik area, or from the University Hospital.
Fasting serum glucose (N = 110 190), HbA1C (N = 67 948), serum creatinine (N = 241 590), urine albumin (concentration) (N = 10 041) and BMI (N = 72 747) effects were attained by testing those traits on a quantitative scale with the values being rank-standard normalized separately for each sex, using age as a covariate. Information about number of genotyped Icelanders used in the analysis of the aforementioned traits and diseases have been summarized in Supplementary Material, Table S13.
UK Biobank kidney stones cohort
The UK Biobank project is a large prospective cohort study of ~500 000 individuals from across the United Kingdom, aged between 40–69 at recruitment (31). The UK Biobank kidney stone cohort examined in the present study, is based on individuals determined to be of white British ancestry (42): 4726 kidney stone cases (ICD-10: N20) and 403 841 controls. We do not exclude related individuals from the analysis but use LD score regression (43) to account for inflation in test statistics due to relatedness.
In the UK Biobank, genotyping was performed using a custom-made Affimetrix chip, UK BiLEVE Axiom (44), in the first 50 000 participants, and with Affimetrix UK Biobank Axiom array in the remaining participants; (45) 95% of the signals are on both chips. Imputation was performed by Wellcome Trust Centre for Human Genetics using a combination of 1000 Genomes phase 3, (46) UK10K (47) and HRC (48) reference panels, for up to 92 693 895 SNPs (42).
Association testing
For case-control analysis, we used logistic regression to test for association under the additive model. We used a likelihood ratio test to compute P-values. We treated trait status as the response, expected genotype counts from imputation as covariates and adjusted for sex, age and county. To further account for inflation in test statistics due to relatedness and stratification, we applied the method of LD score regression (43). The estimated correction factors for glucosuria, ketonuria, proteinuria, hematuria, low urine pH and UTI were between 1.03 and 1.18.
Quantitative traits were tested using a linear mixed model implemented in BOLT-LMM (49).
The threshold for genome-wide significance was corrected for multiple testing with a weighted Bonferroni adjustment using as weights the enrichment of variant classes with predicted functional impact among association signals (50). With 32.5 million sequence variants being tested, the significance threshold was 2.6 × 10-7 for high-impact variants (N = 8474), 5.1 × 10-8 for moderate-impact variants (N = 149 983), 4.6 × 10-9 for low-impact variants (N = 2 283 889), 2.3 × 10-9 for other DNase I hypersensitivity sites (DHS) variants (N = 3 913 058) and 7.9 × 10-10 for other non-DHS variants (N = 26 108 039).
After testing for association, we performed conditional analysis ±1 Mb of loci containing genome-wide significant variants. All variants in the area with imputation information >0.8 were included in the analysis (41) with the lead variant (lowest P-value) as covariate. Variants were concluded to belong to an independent signal if their adjusted P-value was significant. Conditional analysis was repeated for each area until a result with no significant adjusted P-value was attained. In the case of glucosuria, ketonuria, proteinuria and hematuria, this was done for both the mild version of the trait (+ versus −) and the moderate/severe (++ or greater versus negative), since in some instances we had signals that only associated with the moderate/severe trait.
Structural variants
After the independent signals had been established with conditional analysis, we examined if the leading variants were correlated (r2 > 0.8) with structural variants from the whole-genome sequence set. Deletions where discovered using the program Delly (v. 0.7.2) (51) in a process analogous to the one described by Kehr et al. (52); deletions were discovered from whole-genome sequence data generated from blood-extracted DNA of 14,101 individuals, sequenced using Illumina HiSeq and HiSeqX instruments. Discovery of deletions was done in 391 batches of 36 samples and 1 batch of 25 samples. The deletions discovered were filtered to only include deletions between 200 bp and 1 Mb and those with alternate allele frequency in carriers above 20%. Deletions discovered in different batches with begin and end positions less than 25 bp apart were merged into a single vcf record. After merging and filtering, a total of 156 622 variants were available for genotyping and were genotyped in 15 219 samples; 14 101 with blood-extracted DNA and 1118 with buccal swap-extracted DNA. 39 884 of these variants were polymorphic with imputation information above 0.9 and used in the subsequent analysis (41).
Heritability
The SNP-heritability h2 was estimated using LD score regression (43) computed using an LD score map based on Icelandic data consisting of 8 116 865 variants (Phenotype combinations using LD score regression, Bjarni Gunnarsson, Master Thesis, http://hdl.handle.net/1946/29539). Only variants with imputation info >0.90 and MAF > 1% were considered.
Data availability
Sequence variants passing GATK filters have been deposited in the European Variation Archive, accession number PRJEB15197.
Supplementary Material
Acknowledgements
We thank the individuals who participated in the study and whose contributions made this work possible. We thank our valued colleagues who contributed to data collection, sample handling and genotyping. This research has been conducted using the UK Biobank Resource under Application Number ‘24711’.
Conflict of Interest statement. S.B., R.P.K., A.O., V.S., E.M., B.O.J., G.A.A., G.Sulem., G. Sveinbjornsson, S.K., E.V.I., V.T., B.G., J.G.A., A.M.D., O.B D., T.R., G.T., A.H., B.V.H., G.M., H.H., D.F.G., U.T., P.S. and K.S. who are affiliated with deCODE genetics/Amgen Inc. declare competing financial interests as employees. The remaining authors declare no competing financial interests.
References
- 1. Suhre K., Wallaschofski H., Raffler J., Friedrich N., Haring R., Michael K., Wasner C., Krebs A., Kronenberg F., Chang D. et al. (2011) A genome-wide association study of metabolic traits in human urine. Nat. Genet., 43, 565–569. [DOI] [PubMed] [Google Scholar]
- 2. Tesch G.H. (2010) Review: Serum and urine biomarkers of kidney disease: a pathophysiological perspective. Nephrology, 15, 609–616. [DOI] [PubMed] [Google Scholar]
- 3. Simerville J.A., Maxted W.C. and Pahira J.J. (2005) Urinalysis: a comprehensive review. Am. Fam. Physician, 71, 1153–1162. [PubMed] [Google Scholar]
- 4. Rao P.K. and Jones J.S. (2008) How to evaluate ‘dipstick hematuria’: what to do before you refer. Cleve. Clin. J. Med., 75, 227–233. [DOI] [PubMed] [Google Scholar]
- 5. Kong A., Masson G., Frigge M.L., Gylfason A., Zusmanovich P., Thorleifsson G., Olason P.I., Ingason A., Steinberg S., Rafnar T. et al. (2008) Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet., 40, 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Higgins J.P.T. and Thompson S.G. (2002) Quantifying heterogeneity in a meta-analysis. Stat. Med., 21, 1539–1558. [DOI] [PubMed] [Google Scholar]
- 7. Cowart S.L. and Stachura M.E. (1990) Glucosuria. In Walker, H.K., Hall, W.D. and Hurst, J.W. (eds), Clinical Methods: The History, Physical, and Laboratory Examinations. Butterworths, Boston, MA. [PubMed]
- 8. Kristinsson S.Y., Thorolfsdottir E.T., Talseth B., Steingrimsson E., Thorsson A.V., Helgason T., Hreidarsson A.B. and Arngrimsson R. (2001) MODY in Iceland is associated with mutations in HNF-1alpha and a novel mutation in NeuroD1. Diabetologia, 44, 2098–2103. [DOI] [PubMed] [Google Scholar]
- 9. Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S. et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature, 445, 881–885. [DOI] [PubMed] [Google Scholar]
- 10. Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J. et al. (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature, 447, 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mahajan A., Go M.J., Zhang W., Below J.E., Gaulton K.J., Ferreira T., Horikoshi M., Johnson A.D., Ng M.C.Y., Prokopenko I. et al. (2014) Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet., 46, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morris A., Voight B. and Teslovich T. (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet., 44, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vallon V., Platt K.A., Cunard R., Schroth J., Whaley J., Thomson S.C., Koepsell H. and Rieg T. (2011) SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol., 22, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chao E.C. (2011) A paradigm shift in diabetes therapy--dapagliflozin and other SGLT2 inhibitors. Discov. Med., 11, 255–263. [PubMed] [Google Scholar]
- 15. Wright E.M., Loo D.D.F. and Hirayama B.A. (2011) Biology of human sodium glucose transporters. Physiol. Rev., 91, 733–794. [DOI] [PubMed] [Google Scholar]
- 16. The Exome Aggregation Consortium (ExAC) (2015) Analysis of protein-coding genetic variation in 60,706 humans Cold Spring Harbor Labs Journals.
- 17. Calado J., Sznajer Y., Metzger D., Rita A., Hogan M.C., Kattamis A., Scharf M., Tasic V., Greil J., Brinkert F. et al. (2008) Twenty-one additional cases of familial renal glucosuria: absence of genetic heterogeneity, high prevalence of private mutations and further evidence of volume depletion. Nephrol. Dial. Transplant, 23, 3874–3879. [DOI] [PubMed] [Google Scholar]
- 18. Kassovska-Bratinova S., Fukao T., Song X.Q., Duncan A.M., Chen H.S., Robert M.F., Perez-Cerda C., Ugarte M., Chartrand C., Vobecky S. et al. (1996) Succinyl CoA: 3-oxoacid CoA transferase (SCOT): human cDNA cloning, human chromosomal mapping to 5p13, and mutation detection in a SCOT-deficient patient. Am. J. Hum. Genet., 59, 519–528. [PMC free article] [PubMed] [Google Scholar]
- 19. Laffel L. (1999) Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes. Metab. Res. Rev., 15, 412–426. [DOI] [PubMed] [Google Scholar]
- 20. Pattaro C., Teumer A., Gorski M., Chu A.Y., Li M., Mijatovic V., Garnaas M., Tin A., Sorice R., Li Y. et al. (2016) Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun., 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Böger C.A., Chen M.-H., Tin A., Olden M., Köttgen A., Boer I.H., Fuchsberger C., O’Seaghdha C.M., Pattaro C., Teumer A. et al. (2011) CUBN is a gene locus for albuminuria. J. Am. Soc. Nephrol., 22, 555–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Christensen E.I. and Birn H. (2002) Megalin and cubilin: multifunctional endocytic receptors. Nat. Rev. Mol. Cell Biol., 3, 256–266. [DOI] [PubMed] [Google Scholar]
- 23. Khadra M.H., Pickard R.S., Charlton M., Powell P.H. and Neal D.E. (2000) A prospective analysis of 1,930 patients with hematuria to evaluate current diagnostic practice. J. Urol., 163, 524–527. [PubMed] [Google Scholar]
- 24. Savige J., Gregory M., Gross O., Kashtan C., Ding J. and Flinter F. (2013) Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J. Am. Soc. Nephrol., 24, 364–375. [DOI] [PubMed] [Google Scholar]
- 25. Sanjabi S., Zenewicz L.A., Kamanaka M. and Flavell R.A. (2009) Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol., 9, 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mahil S.K., Capon F. and Barker J.N. (2015) Genetics of Psoriasis. Dermatol. Clin., 33, 1–11. [DOI] [PubMed] [Google Scholar]
- 27. Kamatani Y., Matsuda K., Okada Y., Kubo M., Hosono N., Daigo Y., Nakamura Y. and Kamatani N. (2010) Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet., 42, 210–215. [DOI] [PubMed] [Google Scholar]
- 28. Kottgen A., Pattaro C., Boger C.A., Fuchsberger C., Olden M., Glazer N.L., Parsa A., Gao X., Yang Q., Smith A.V. et al. (2010) New loci associated with kidney function and chronic kidney disease. Nat. Genet., 42, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parmar M.S. (2004) Kidney stones. BMJ, 328, 1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ennis J.L. and Asplin J.R. (2016) The role of the 24-h urine collection in the management of nephrolithiasis. Int. J. Surg., 36, 633–637. [DOI] [PubMed] [Google Scholar]
- 31. Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M. et al. (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lawrence R.D. (1940) Renal thresholds for glucose: normal and in diabetics. Br. Med. J., 4140, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. FDA Drug Communication Safety Announcement (2015) FDA revises labels of SGLT2 inhibitors for diabetes to include warnings about too much acid in the blood and serious urinary tract infections .
- 34. Partridge A., Therien A. and Deber C. (2004) Missense mutations in transmembrane domains of proteins: phenotypic propensity of polar residues for human disease. Proteins, 54, 648–656. [DOI] [PubMed] [Google Scholar]
- 35. Nielsen R., Christensen E.I. and Birn H. (2016) Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int., 89, 58–67. [DOI] [PubMed] [Google Scholar]
- 36. Lamb E.J., MacKenzie F. and Stevens P.E. (2009) How should proteinuria be detected and measured? Ann. Clin. Biochem., 46, 205–217. [DOI] [PubMed] [Google Scholar]
- 37. Leheste J.R., Rolinski B., Vorum H., Hilpert J., Nykjaer A., Jacobsen C., Aucouturier P., Moskaug J.O., Otto A., Christensen E.I. et al. (1999) Megalin knockout mice as an animal model of low molecular weight proteinuria. Am. J. Pathol., 155, 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kantarci S., Ragge N.K., Thomas N.S., Robinson D.O., Noonan K.M., Russell M.K., Donnai D., Raymond F.L., Walsh C.A., Donahoe P.K. et al. (2008) Donnai–Barrow Syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am. J. Med. Genet. A, 146, 1842–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Paassen P., Van Breda Vriesman P.J.C., Van Rie H. and Tervaert J.W.C. (2004) Signs and symptoms of thin basement membrane nephropathy: a prospective regional study on primary glomerular disease—The Limburg Renal Registry. Kidney Int., 66, 909–913. [DOI] [PubMed] [Google Scholar]
- 40. Tryggvason K. (2006) Thin basement membrane nephropathy. J. Am. Soc. Nephrol., 17, 813–822. [DOI] [PubMed] [Google Scholar]
- 41. Gudbjartsson D.F., Helgason H., Gudjonsson S.A., Zink F., Oddson A., Gylfason A., Besenbacher S., Magnusson G., Halldorsson B.V., Hjartarson E. et al. (2015) Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet., 47, 435–444. [DOI] [PubMed] [Google Scholar]
- 42. Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J. et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. [DOI] [PMC free article] [PubMed]
- 43. Bulik-Sullivan B.K., Loh P.-R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N., Daly M.J., Price A.L. and Neale B.M. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wain L.V., Shrine N., Miller S., Jackson V.E., Ntalla I., Artigas M.S., Billington C.K., Kheirallah A.K., Allen R., Cook J.P. et al. (2015) Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): A genetic association study in UK Biobank. Lancet Respir. Med., 3, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Welsh S., Peakman T., Sheard S. and Almond R. (2017) Comparison of DNA quantification methodology used in the DNA extraction protocol for the UK Biobank cohort. BMC Genomics, 18, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Auton A., Abecasis G.R., Altshuler D.M., Durbin R.M., Abecasis G.R., Bentley D.R., Chakravarti A., Clark A.G., Donnelly P., Eichler E.E. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walter K., Min J.L., Huang J., Crooks L., Memari Y., McCarthy, S., Perry J.R.B., Xu C., Futema M., Lawson D. et al. (2015) The UK10K project identifies rare variants in health and disease. Nature, 526, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A.R., Teumer A., Kang H.M., Fuchsberger C., Danecek P., Sharp K. et al. (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet., 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Loh P.-R., Tucker G., Bulik-Sullivan B.K., Vilhjálmsson B.J., Finucane H.K., Salem R.M., Chasman D.I., Ridker P.M., Neale B.M., Berger B. et al. (2015) Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet., 47, 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sveinbjornsson G., Albrechtsen A., Zink F., Gudjonsson S.A., Oddson A., Másson G., Holm H., Kong A., Thorsteinsdottir U., Sulem P. et al. (2016) Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nat. Genet., 48, 314–317. [DOI] [PubMed] [Google Scholar]
- 51. Rausch T., Zichner T., Schlattl A., Stutz A.M., Benes V. and Korbel J.O. (2012) DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics, 28, i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kehr B., Helgadottir A., Melsted P., Jonsson H., Helgason H., Jonasdottir A., Jonasdottir A., Sigurdsson A., Gylfason A., Halldorsson G.H. et al. (2017) Diversity in non-repetitive human sequences not found in the reference genome. Nat. Genet., 49, 588–539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence variants passing GATK filters have been deposited in the European Variation Archive, accession number PRJEB15197.