

Abstract
The macula, located near the center of the retina in the human eye, is responsible for providing critical functions, such as central, sharp vision. Structural changes in the macula are associated with many ocular diseases, including age-related macular degeneration (AMD) and glaucoma. Although macular thickness is a highly heritable trait, there are no prior reported genome-wide association studies (GWASs) of it. Here we describe the first GWAS of macular thickness, which was measured by spectral-domain optical coherence tomography using 68 423 participants from the UK Biobank cohort. We identified 139 genetic loci associated with macular thickness at genome-wide significance (P < 5 × 10−8). The most significant loci were LINC00461 (P = 5.1 × 10−120), TSPAN10 (P = 1.2 × 10−118), RDH5 (P = 9.2 × 10−105) and SLC6A20 (P = 1.4 × 10−71). Results from gene expression demonstrated that these genes are highly expressed in the retina. Other hits included many previously reported AMD genes, such as NPLOC4 (P = 1.7 × 10−103), RAD51B (P = 9.1 × 10−14) and SLC16A8 (P = 1.7 × 10−8), further providing functional significance of the identified loci. Through cross-phenotype analysis, these genetic loci also exhibited pleiotropic effects with myopia, neurodegenerative diseases (e.g. Parkinson’s disease, schizophrenia and Alzheimer’s disease), cancer (e.g. breast, ovarian and lung cancers) and metabolic traits (e.g. body mass index, waist circumference and type 2 diabetes). Our findings provide the first insight into the genetic architecture of macular thickness and may further elucidate the pathogenesis of related ocular diseases, such as AMD.
Introduction
The macula, located near the center of the retina in the human eye, is responsible for providing critical functions, such as central, sharp vision. Structural changes in the macula, such as macular thickness, are associated with many ocular diseases, including age-related macular degeneration (AMD) (1) and glaucoma (2). Macular thickness is an important quantitative trait for studying AMD and glaucoma and can be measured non-invasively using spectral domain optical coherence tomography (SD-OCT). Despite the significant role of macular thickness in these diseases, no genome-wide association study (GWAS) has been performed. Identifying genetic factors that influence macular thickness not only aids in uncovering the biological mechanisms regulating this trait but may also help improve our understanding of related ocular diseases.
Findings from epidemiological studies have identified many factors associated with macular thickness, including age, gender, body mass index, smoking and myopia (3,4). Moreover, racial differences in macular thickness have been observed in several studies. Overall, whites tend to have the thickest macula, followed by individuals of Asian and African descent, respectively (4,5). Furthermore, similar to other ocular parameters, macular thickness has a strong genetic component. Heritability estimates for this trait were over 0.80 (6), indicating that macular thickness is mainly influenced by genetic rather than environmental factors. However, there is still a significant lack of genetic studies of macular thickness, possibly because of the limited available sample size and special imaging equipment, i.e. OCT, that is required.
Recent establishment of large-scale biobank repositories and advancements in imaging technology have greatly expanded our knowledge and understanding of complex diseases and traits. As one of the largest biobanks with both genetic and ophthalmologic data, the UK Biobank (UKB) provides the opportunity to further uncover the missing heritability of many ocular traits (7) and is useful for genetic studies on previously unexamined traits, such as macular thickness. The large sample size of the UKB (n = 500 000) and uniform phenotype measurement provide significant advantages in genetic association studies. Therefore, the purpose of this study is to perform a GWAS of macular thickness using data from the UKB. Findings from our analysis will provide the first insight into the genetic architecture of macular thickness and may elucidate the pathogenesis of related ocular diseases.
Results
Study sample
Table 1 presents the study sample characteristics for the discovery and replication data sets, as well as the overall study population. A total of 68 423 UKB participants were used to assess the associations between genetic variants and macular thickness, of which 59 814 unrelated subjects were used as the discovery set and 8609 related subjects (independent of the discovery set) were used as the replication set. The overall mean age [standard deviation (SD)] is 58.2 (8.0), with the discovery set and replication set having mean ages of 58.2 (7.9) and 58.0 (8.4), respectively. The proportion of females in the study sample is 53.3%, with the discovery and replication sets consisting of 53.0% and 56.0%, respectively. The macular thickness values are normally distributed with an overall mean (SD) of 261.9 (13.6). The average macular thickness in the discovery and replication sets is 262.0 (13.6) μm (range: 190.9–328.0) and 261.8 (13.9) μm (range: 191.4–327.7), respectively.
Table 1.
Study sample characteristics
| Study | Sample size | Age (years), mean (SD) | Females, % | Macular thickness (μm), mean (SD) | Macular thickness range (μm) |
|---|---|---|---|---|---|
| Discovery set | 59 814 | 58.2 (7.9) | 53.0 | 262.0 (13.6) | 190.9–328.0 |
| Replication set | 8609 | 58.0 (8.4) | 56.0 | 261.8 (13.9) | 191.4–327.7 |
| Total | 68 423 | 58.2 (8.0) | 53.3 | 261.9 (13.6) | 190.9–328.0 |
Genome-wide association results
Supplementary Material, Figure S1 shows a quantile–quantile (Q–Q) plot displaying the observed P-values against the expected P-values for the discovery data set. The genomic control inflation factor λ was 1.14. Inflation of λ may be because of the large sample size and polygenicity. We conducted LD (linkage disequilibrium) score regression and observed that the regression intercept (an alternative way to measure genomic inflation) was 1.03, suggesting that the genomic control deviation was mainly because of polygenicity rather than confounding. Hence, our results are properly controlled for population stratification and cryptic relatedness.
We identified 14 889 genome-wide significant (P < 5 × 10−8) variants associated with macular thickness using the imputed data set from the discovery set. Figure 1 presents a Manhattan plot of the genome-wide −log10(P)-values from the discovery set association analysis. These significant genetic variants map to 113 independent loci. Table 2 shows the results for the top 20 hits. The most significant genetic variant is rs7405453 (P = 1.6 × 10−106, GRCh37/hg19 position 79 615 572, chromosome 17), located in the 3′-UTR of the Tetraspanin 10 gene (TSPAN10). The effect allele A [allele frequency (AF) = 0.36] is associated with a decrease in macular thickness (β = −1.58). NPLOC4-TSPAN10 is a known AMD locus (8). The second most significant variant, rs17421627 (P = 1.8 × 10−101, position 87 847 586, chromosome 5), is located in the downstream (variant overlaps 1 kb region downstream of transcription) of long intergenic non-protein coding RNA 461 (LINC00461). The effect allele T (AF = 0.93) is associated with a decrease in macular thickness (β = −2.96). The third most significant variant is rs3138142 (P = 1.3 × 10−96, position 56 115 585, chromosome 12), located in an exon of the Retinol Dehydrogenase 5 gene (RDH5). The effect allele C (AF = 0.76) is associated with a decrease in macular thickness (β = −1.73). RDH5 is a known AMD locus (8). The fourth most significant variant, rs17279437 (P = 2.8 × 10−63, position 45 814 094, chromosome 3), is located in an exon of the Solute Carrier Family 6 Member 20 gene (SLC6A20). The effect allele G (AF = 0.89) is associated with an increase in macular thickness (β = 2.00).
Figure 1.
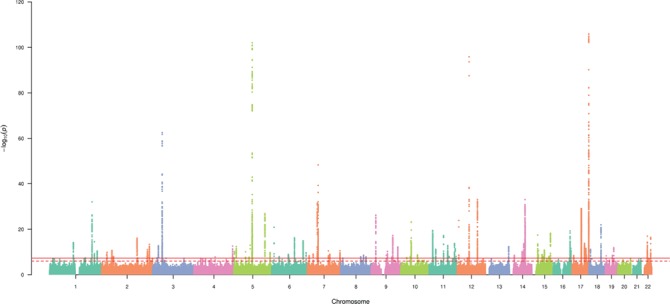
Manhattan plot displaying the –log10 (P-values) for the association between macular thickness in the discovery data set. The solid and dotted horizontal lines represent genome-wide significant associations (P = 5 × 10−8) and suggestive associations (P = 1.0 × 10−6), respectively. Genetic variants are plotted by genomic position.
Table 2.
Summary results for the top-ranking loci associated with macular thickness
| SNP | Chr | Position | Locus | Alleles | AF1 | Discovery | Replicate | Entire sample | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta | SE | P | Beta | SE | P | Beta | SE | P | ||||||
| rs7405453 | 17 | 79615572 | TSPAN10 | A/G | 0.36 | −1.58 | 0.08 | 1.6E-106 | −1.60 | 0.21 | 3.2E-14 | −1.57 | 0.07 | 1.2E-118 |
| rs17421627 | 5 | 87847586 | LINC00461 | T/G | 0.93 | −2.96 | 0.14 | 1.8E-101 | −3.18 | 0.40 | 5.3E-16 | −2.99 | 0.13 | 5.1E-120 |
| rs3138142 | 12 | 56115585 | RDH5 | C/T | 0.76 | −1.73 | 0.09 | 1.3E-96 | −1.51 | 0.24 | 7.5E-11 | −1.69 | 0.08 | 9.2E-105 |
| rs17279437 | 3 | 45814094 | SLC6A20 | G/A | 0.89 | 2.00 | 0.12 | 2.8E-63 | 1.87 | 0.33 | 8.6E-09 | 1.96 | 0.11 | 1.4E-71 |
| rs12719025 | 7 | 51100190 | COBL | A/G | 0.54 | −1.06 | 0.07 | 5.3E-49 | −0.81 | 0.21 | 5.9E-05 | −1.04 | 0.07 | 1.1E-54 |
| rs887595 | 14 | 74666641 | LIN52 | A/G | 0.18 | −1.12 | 0.10 | 9.1E-34 | −1.05 | 0.27 | 9.2E-05 | −1.12 | 0.09 | 7.9E-36 |
| rs55971426 | 12 | 96204536 | LINC02410 - LOC105369921 | A/G | 0.81 | 1.08 | 0.09 | 5.1E-33 | 1.02 | 0.26 | 8.7E-05 | 1.08 | 0.09 | 1.5E-37 |
| rs11576909 | 1 | 202812654 | MGAT4EP - LOC148709 | T/C | 0.51 | −0.89 | 0.07 | 9.6E-33 | −0.86 | 0.21 | 1.2E-05 | −0.89 | 0.07 | 1.7E-38 |
| rs11769224 | 7 | 46630354 | IGFBP3 - LOC730338 | A/C | 0.41 | −0.88 | 0.08 | 3.6E-32 | −0.50 | 0.21 | 7.5E-03 | −0.82 | 0.07 | 2.3E-32 |
| rs2732631 | 17 | 44289232 | KANSL1 | G/T | 0.77 | −0.95 | 0.09 | 1.1E-29 | −1.05 | 0.25 | 1.1E-05 | −0.96 | 0.08 | 3.0E-35 |
| rs62064364 | 17 | 43654468 | LRRC37A4P - MAPK8IP1P2 | C/T | 0.78 | −0.97 | 0.09 | 1.7E-29 | −1.06 | 0.25 | 1.6E-05 | −0.98 | 0.08 | 4.4E-35 |
| rs73089379 | 3 | 45183414 | CDCP1 | C/G | 0.93 | 1.59 | 0.15 | 2.1E-28 | 1.33 | 0.41 | 6.4E-04 | 1.54 | 0.14 | 2.5E-30 |
| rs13171669 | 5 | 148601243 | ABLIM3 | A/G | 0.57 | −0.76 | 0.07 | 1.4E-27 | −0.99 | 0.21 | 3.4E-06 | −0.78 | 0.07 | 6.8E-32 |
| rs7033598 | 9 | 21565674 | MIR31HG - MTAP | C/T | 0.35 | 0.79 | 0.08 | 6.3E-27 | 0.24 | 0.22 | 2.6E-01 | 0.72 | 0.07 | 8.2E-26 |
| rs5442 | 12 | 6954864 | GNB3 | G/A | 0.93 | 1.43 | 0.14 | 1.3E-24 | 0.97 | 0.41 | 1.5E-02 | 1.37 | 0.13 | 6.4E-26 |
| rs1947075 | 10 | 49741135 | ARHGAP22 | C/T | 0.35 | 0.75 | 0.08 | 6.2E-24 | 0.37 | 0.22 | 6.6E-02 | 0.70 | 0.07 | 8.8E-24 |
| rs973879 | 14 | 75297727 | YLPM1 | T/C | 0.46 | −0.71 | 0.07 | 1.2E-23 | −0.66 | 0.21 | 2.1E-03 | −0.69 | 0.07 | 3.5E-25 |
| rs10043238 | 5 | 87154361 | CCNH - TMEM161B | G/C | 0.69 | 0.82 | 0.08 | 1.5E-23 | 0.40 | 0.23 | 7.5E-02 | 0.76 | 0.07 | 4.5E-24 |
| rs199456 | 17 | 44797919 | NSF | C/T | 0.79 | −0.85 | 0.09 | 2.9E-23 | −0.98 | 0.25 | 5.3E-05 | −0.86 | 0.08 | 3.0E-28 |
| rs17507554 | 6 | 11394287 | NEDD9 - TMEM170B | G/A | 0.95 | −1.57 | 0.17 | 1.4E-21 | −1.93 | 0.47 | 1.7E-05 | −1.59 | 0.16 | 2.7E-25 |
Abbreviations: SNP, single-nucleotide polymorphism; Chr, chromosome; AF1, allele 1 frequency; SE, standard error.
We then analyzed these significant genetic variants in the replication data set. Among the 14 889 significant variants from the discovery set, 14 725 exhibited the same direction of effect in the replication set as the discovery set, among which 7975 (54.2%) were replicated at the P < 0.01 significance level. The aforementioned top four hits were all replicated with P < 10−8 (Table 2). When the discovery and replication data sets were analyzed together, most of the associations increased in significance. Supplementary Material, Figures S2 and S3 present the Q–Q and Manhattan plots for the association analysis of the entire data set. The LD score regression intercept was 1.04. In total, we identified 17 677 variants significantly associated with macular thickness, annotating to 478 unique genes in 139 independent genomic loci (Supplementary Material, Table S1). The last column of Table 2 shows the results of the entire sample for the top 20 hits. The top four novel loci remained the same, i.e. LINC00461 (rs17421627, β (SE) = −2.99 (0.13), P = 5.1 × 10−120), TSPAN10 (rs7405453, β (SE) = −1.57 (0.07), P = 1.2 × 10−118), RDH5 (rs3138142, β (SE) = −1.69 (0.08), P = 9.2 × 10−105) and SLC6A20 (rs17279437, β (SE) = 1.96 (0.11), P = 1.4 × 10−71). Figure 2 presents regional association plots for the top four genomic regions. In each plot, several imputed genetic variants were more significant than the directly genotyped variants.
Figure 2.
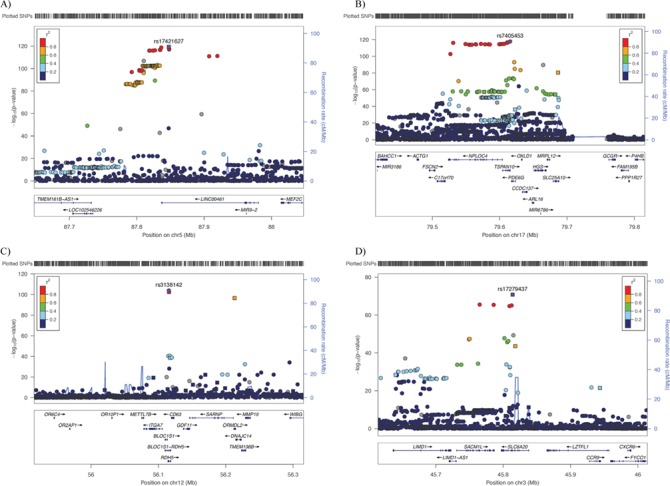
Regional association plots for the top loci associated with macular thickness. For (A)LINC00461, (B)TSPAN10, (C)RDH5 and (D)SLC6A20, the most significant SNP is plotted in purple. Squares and circles represent genotyped and imputed genetic variants, respectively. Genes are shown below the SNPs. The color coding in each plot represents the level of linkage disequilibrium with the lead SNPs.
Conditional analyses
We performed conditional analyses for the top four identified genomic regions to determine whether additional genetic variants contribute to the macular thickness associations. Supplementary Material, Figure S2 presents the regional association plots of the conditional analyses. After conditioning on the most significant genetic variant in each region, i.e. TSPAN10 and RDH5, all of the neighboring associations reduced toward the null, suggesting that the identified genetic variants are the lead markers of the macular thickness associations. We identified additional independent variants, rs199502002 and rs34106801, for the LINC00461 and SLC6A20 conditional analyses, respectively, also associated with macular thickness.
Macular thickness heritability estimates
We estimated the heritability of macular thickness among the study participants using the program BOLT-REML (9). The estimated heritability of macular thickness was 49.2% based on all genotyped genetic variants (n = 784 256). When restricting the estimation to only genome-wide significant variants (directly genotyped, n = 637), these variants explain 10.2% of macular thickness variability.
Functional relevance
To seek biological support for the top four genes associated with macular thickness, we evaluated their gene expression using Genevestigator, an online database to verify gene expression in different tissues. Figure 3 presents the gene expression data for the top 10 tissues for each of the four genes. All four genes are highly expressed in either the retina or the retinal pigment epithelium, indicating the important biological roles these macular thickness genes play in that area of the eye. To examine the association of macular thickness variants with AMD, we queried the Fritsche et al. AMD summary statistics (8). Eight out of the 34 reported AMD loci (23.5%) overlap with our macular thickness loci. Furthermore, among the 13 042 significant macular thickness variants (P < 5 × 10−8) overlapping with the AMD summary statistics, 414 (3.2%) showed associations with AMD at the 5 × 10−5 significance level (Supplementary Material, Table S2), which was significantly higher than the amount expected by chance (0.005%).
Figure 3.
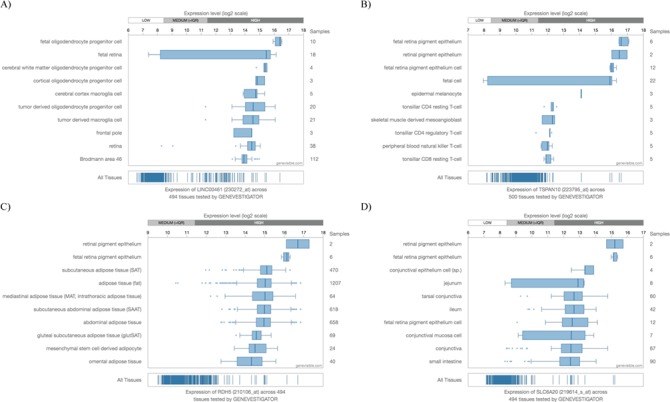
Gene expression for the top four genes associated with macular thickness from Genevestigator. All four genes, (A)LINC00461, (B)TSPAN10, (C)RDH5 and (D)SLC6A20, are highly expressed in either the retina or the retinal pigment epithelium.
Pleiotropy among significant loci
To assess the pleiotropic effect of the genome-wide significant macular thickness variants, we examined these markers in the GWAS Catalog (May 29, 2018 version). Among the 17 677 significant genetic variants identified, 274 had exact matches to the previously reported variants in the GWAS Catalog. Figure 4 presents the network of the associated traits and diseases for these 274 genetic variants (Supplementary Material, Table S3 shows the detailed mapping information). Numerous ocular-related diseases/traits were directly matched to the included single nucleotide polymorphisms (SNPs), including AMD (e.g. rs6565597 in NPLOC4, P = 1.7 × 10−103; rs8017304 in RAD51B, P = 9.1 × 10−14; rs621313 in TYR, P = 8.3 × 10−12), primary open-angle glaucoma (e.g. rs33912345 in SIX6, P = 4.4 × 10−11; rs7865618 in CDKN2B-AS1, P = 4.6 × 10−10; rs11129176 in RARB, P = 1.3 × 10−8), myopia, axial length and optic nerve head measurements. Additionally, many macular thickness SNPs mapped to neurological diseases, (e.g. Parkinson’s disease, schizophrenia and Alzheimer’s disease), cancer (e.g. breast, ovarian and lung cancers) and metabolic traits (e.g. body mass index, waist circumference and type 2 diabetes).
Figure 4.
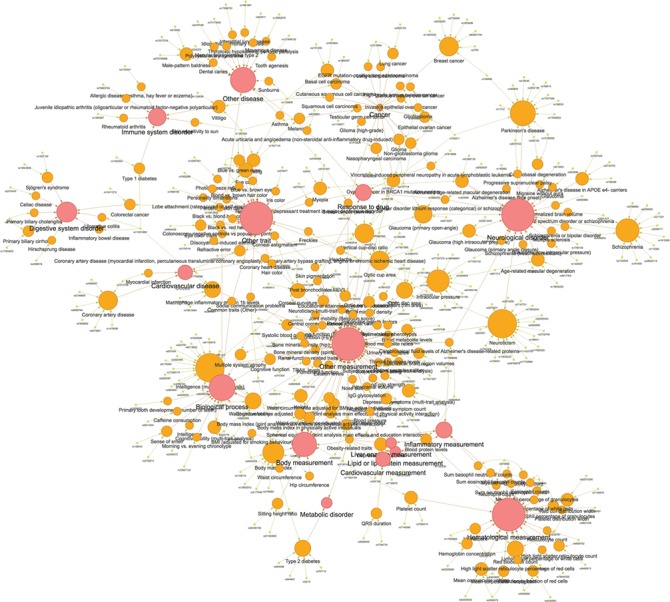
A network of the pleiotropic effects for significant macular thickness SNPs in the GWAS Catalog. A network of traits and diseases for 274 SNPs that matched directly to other phenotypes through the GWAS Catalog. Yellow, orange and red ovals denote SNPs, individual traits and categories, respectively.
Enrichment analysis
Table 3 presents the significant enrichment results for macular thickness using WebGestalt [false discovery rate (FDR) < 0.05]. The most significant GLAD4U disease enriched for macular thickness genes is eye diseases (FDR = 2.3 × 10−8). Many retinal-related GLAD4U diseases are also significant, including retinal diseases (FDR = 6.4 × 10−4), retinal degeneration (FDR = 1.1 × 10−3), retinitis NOS (not otherwise specified) (FDR = 2.0 × 10−3) and retinitis pigmentosa (FDR = 2.2 × 10−3). The most significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway associated with macular thickness is colorectal cancer (FDR = 2.4 × 10−4). Interestingly, the colorectal cancer drug, Avastin (bevacizumab), has been used to treat AMD (10). Many other cancer pathways are also enriched, including endometrial cancer (FDR = 8.5 × 10−4), prostate cancer (FDR = 2.0 × 10−3) and small cell lung cancer (FDR = 3.0 × 10−3).
Table 3.
Top enrichment results associated with macular thickness
| Term | Description | Observed/total genes | P | FDR | |
|---|---|---|---|---|---|
| Disease | |||||
| PA444119 | Eye diseases | 70/437 | 1.1E-11 | 2.3E-08 | |
| PA443521 | Blindness | 36/96 | 3.4E-08 | 3.8E-05 | |
| PA443320 | Amblyopia | 15/45 | 1.3E-07 | 9.5E-05 | |
| PA444117 | Eye abnormalities | 32/189 | 1.3E-06 | 6.4E-04 | |
| PA445532 | Retinal diseases | 42/285 | 1.4E-06 | 6.4E-04 | |
| PA445530 | Retinal degeneration | 36/234 | 2.9E-06 | 1.1E-03 | |
| PA165108137 | Retinitis NOS | 31/194 | 6.4E-06 | 2.0E-03 | |
| PA443989 | Ectopia lentis | 7/13 | 7.5E-06 | 2.1E-03 | |
| PA445537 | Retinitis pigmentosa | 31/197 | 8.8E-06 | 2.2E-03 | |
| PA447310 | Vision disorders | 21/112 | 1.7E-05 | 3.8E-03 | |
| KEGG | |||||
| hsa05210 | Colorectal cancer––Homo sapiens (human) | 18/62 | 8.0E-07 | 2.4E-04 | |
| hsa05213 | Endometrial cancer––Homo sapiens (human) | 15/52 | 7.3E-06 | 8.5E-04 | |
| hsa05161 | Hepatitis B––Homo sapiens (human) | 28/146 | 8.5E-06 | 8.5E-04 | |
| hsa05200 | Pathways in cancer––Homo sapiens (human) | 56/397 | 1.2E-05 | 8.8E-04 | |
| hsa05203 | Viral carcinogenesis––Homo sapiens (human) | 34/205 | 2.6E-05 | 1.5E-03 | |
| hsa01521 | EGFR tyrosine kinase inhibitor resistance––Homo sapiens (human) | 18/81 | 4.7E-05 | 2.0E-03 | |
| hsa05230 | Central carbon metabolism in cancer––Homo sapiens (human) | 16/67 | 4.9E-05 | 2.0E-03 | |
| hsa05215 | Prostate cancer––Homo sapiens (human) | 19/89 | 5.3E-05 | 2.0E-03 | |
| hsa04915 | Estrogen signaling pathway––Homo sapiens (human) | 20/100 | 9.0E-05 | 3.0E-03 | |
| hsa05222 | Small cell lung cancer––Homo sapiens (human) | 18/86 | 1.1E-04 | 3.0E-03 |
Discussion
To our knowledge, this is the first GWAS report on macular thickness. Using available SD-OCT and genetic data from the UKB, we identified 139 loci significantly associated with macular thickness. These loci represent the first genomic regions associated with macular thickness. The most significant loci are LINC00461, TSPAN10, RDH5 and SLC6A20. Findings from gene expression data provide additional biological support for the presence of these genes in the retina. Moreover, results from our cross-phenotype and enrichment analyses identified many biologically relevant diseases. Our study provides the first insight into the genetic architecture of macular thickness, highlighting both the polygenic and pleiotropic nature of this trait.
The two most significant genomic regions associated with macular thickness are LINC00461and TSPAN10. The most significant SNP in the top region, rs17421627, locates downstream of LINC00461, a long intergenic non-coding RNA that is expressed in the retina and is conserved in the macular region across multiple species (11). A previous GWAS found LINC00461 to be associated with retinal vascular caliber (12), an ocular characteristic associated with AMD (13,14). Moreover, LINC00461 has been associated with macular telangiectasia type 2, a neurovascular degenerative retinal disease that is characterized by photoreceptor dysfunction and human retinal pigment epithelial (RPE) abnormalities (15). The RPE is a thin layer of epithelial cells that provides nutrients to the photoreceptors, and dysfunction of the RPE can lead to photoreceptor death and AMD. The lead SNP, rs7405453, in the second region is located in the 3′ UTR of TSPAN10, a family of proteins that consists of four transmembrane domains that aid in trafficking proteins required for normal organ development, including the eyes (16). Previous GWASs found TSPAN10 to be associated with several ocular diseases, including myopia (17) and AMD (8). Additionally, it is expressed in numerous ocular tissues, including the human retinal pigment epithelium, the retina and the iris (18). The results from our study, in conjunction with previous findings, suggest that these top two regions are biologically relevant and may play an important role in macular thickness determination and, subsequently, retinal disease.
The third and fourth most significant genomic regions associated with macular thickness are RDH5 and SLC6A20. The lead SNP in the third most significant region, rs3138142, resides in an exon of RDH5 and has previously been associated with AMD (8). RDH5 is a member of the retinol dehydrogenases family, which is a group of enzymes that is involved in the visual cycle. RDH5 is expressed in the RPE (19), and mutations in this gene have been associated with cone dystrophy and night blindness (20). Additionally, RDH5 has also been associated with myopia (17) and refractive error (21). The lead SNP in the fourth region, rs17279437, is located in an exon of SLC6A20 and has previously been associated with urinary and blood metabolites (22,23). SLC6A20 is a proline transporter that has been associated with iminoglycinuria, a disorder that affects the ability to reabsorb amino acids, such as proline, in the kidney and is highly expressed in the human RPE (24,25). Additionally, proline is favored by RPE cells as an energy substrate and is consumed more than any other nutrient. Proline may also play a role in AMD (26). SLC6A20 has also been associated with Hirschsprung’s disease (27), a disease that presents with ophthalmic anomalies, including microphthalmia/anophthalmia (28). Altogether, these top genes provide the first insights into the genetic factors regulating macular thickness and shed new light on the pathogenesis of related ocular diseases.
Recently, there has been growing interest in pleiotropy. Through GWASs, thousands of genetic associations have been identified. Investigating whether traits share similar genetic variants may shed new light on biological processes and may have important therapeutic implications, such as drug repositioning (29,30). For our study, the top SNPs associated with macular thickness are also related to other ocular traits, including glaucoma, AMD, myopia, axial length and optic nerve head measurements. Moreover, numerous systemic traits were also associated with the identified macular thickness variants, such as neurological diseases, cancer and metabolic factors. Among the overlapping neurological diseases, thinner retinal thickness has been observed in individuals with Parkinson’s disease (31), schizophrenia (32) and Alzheimer’s disease (33). Genetic variants associated with breast, ovarian and lung cancers also intersect with those associated with macular thickness, and previous findings identified an association between AMD and cancer mortality (34). Furthermore, several metabolic traits, including body mass index, waist circumference and type 2 diabetes, share some significant genetic variants with macular thickness. These metabolic features have previously been associated with both risk and progression of AMD (35,36). The observed pleiotropic nature of macular thickness not only aids in elucidating the effect of a single genetic variant on multiple traits but may also lead to important public health and medical implications. Knowing that certain traits are pleiotropic means that drugs used to treat one condition may be effective for treating another condition. For example, cancer drugs, such as Avastin, can also be used to treat macular disease, e.g. AMD. In addition to this advantage, we also need to be aware of possible side effects. Specifically, aromatase inhibitor use among women with hormone receptor-positive breast cancer was associated with a decrease in retinal nerve fiber layer thickness (37). Through pleiotropy, we can better understand how shared genetic factors between traits influence biological processes, such as drug metabolism, and subsequently, how they may aid in more effective treatments while also preventing negative side effects.
Our study has both strengths and limitations. We used a large sample size, advanced imaging and genomics resources in our examination of macular thickness. We further conducted a sensitivity analysis by removing subjects who self-reported macular degeneration (n = 914 overlapped with the OCT and genetics data) and obtained consistent results. Supplementary Material, Figure S5 shows the Manhattan plot of the genome-wide −log10(P)-values after removing subjects with self-reported macular degeneration. Supplementary Material, Figure S6 presents the pairwise plots of the effect sizes (a) and −log10(P)-values (b) for the 139 index SNPs from the full sample and the sample after removing self-reported macular degeneration. The results agree with each other very well with pairwise points falling at or close to the diagonal line demonstrating the robustness of our analyses. As to the limitations, we restricted the current analysis to white participants, and thus, the generalizability of these findings to other ethnic groups requires further investigation. Studies in diverse ethnic populations are needed in order to assess the transferability of our findings, as well as to potentially identify additional genetic variants. Furthermore, we excluded genetic variants with MAF < 0.5%, omitting possible rare variants associated with macular thickness in our analysis. The rare variant investigation may be more suitable for sequencing data.
It is challenging to interpret small effect sizes in genetic analyses of complex traits/diseases, which are very different from that in Mendelian diseases. Furthermore, we often see minor alleles show opposite effects for different genome-wide significant SNPs, such as in these GWASs of macular thickness and previous intraocular pressure (38–40). GWAS SNPs often show pleiotropic effects (38,41,42) and can also have antagonistic pleiotropy (43,44). A GWAS is the first step in our genetics investigation. For possible clinical utility, it would be better to consider the genes, pathways, pleiotropy and polygenic risk scores of these quantitative trait loci rather than construing each associated allele individually. The possible importance of this GWAS study or any other may fall under two major categories: association and prediction. First, all GWAS hits provide important biological insights into the genetic architecture of complex traits/diseases. In our GWAS study of macular thickness, top hits also pointed to key genes associated with AMD. Second, although the beta estimates for individual genetic variants appear to be small, the aggregative effect of these beta estimates can be quite large and hence, clinically meaningful. Many studies have built polygenic risk scores for complex diseases based on GWAS results. For example, Khera et al. (45) built polygenic risk scores for several common diseases, such as coronary artery disease, atrial fibrillation, type 2 diabetes and inflammatory bowel disease, using more than 6 million genetic variants and obtained risk predictions equivalent to monogenic mutations. Our results provide the first step for building prediction models, which require further investigation. Therefore, our GWAS results, like the other GWAS reports, are clinically relevant and meaningful. However, we acknowledge that the translation of GWAS results toward new therapeutics is a long process (46).
This study presents the first GWAS of macular thickness. Using data from the UKB, we identified 139 loci significantly associated with macular thickness. Gene expression data for the top macular thickness genes reveal the presence of these genes in the retina, further providing biological support to our results. Additionally, many genetic variants were also associated with AMD. We identified numerous variants and genes that exhibit pleiotropic effects with neurodegenerative diseases, cancer and metabolic traits, as well as relevant KEGG pathways that are associated with macular thickness. Our study provides the first insights into the genetic architecture of macular thickness and may further elucidate the pathogenesis of related ocular diseases, such as AMD.
Materials and Methods
Ethics statement
The UKB received approval from the North West Multi-Center Research Ethics Committee. All study participants provided informed consent. Our access to the resource was approved by UKB as complying with their Access Procedures and Ethics. We obtained fully de-identified data. Our research adheres to the tenets of the Declaration of Helsinki.
Study sample
The UKB study population has been described elsewhere (47,48). Briefly, UKB is a population-based prospective study of individuals living in the UK. The study recruited about 500 000 adult study participants registered with the National Health Service who were 40–69 years of age at enrollment. From 2006 to 2010, the biobank collected participants’ medical and family histories, lifestyle factors, physical measurements and biological specimens, including blood, saliva and urine samples. A subset of the cohort, ~118,000 study subjects, participated in the eye and vision component of the study. Within this subset, retinal photography and SD-OCT data were released on 81 807 subjects. For the current analysis, we only included participants who self-reported as white.
OCT measurements
SD-OCT was performed in each eye using the TOPCON 3D OCT 1000 Mk2 (Topcon Corporation, Tokyo, Japan) between 2009–2010 and 2012–2013. Study participants who had eye surgery 4 weeks prior to the assessment or those with possible eye infections did not undergo SD-OCT. Macular thickness was measured as the average macular thickness within the Early Treatment of Diabetic Retinopathy Study (ETDRS) grid outermost circle (diameter = 6 mm) for each eye. After removing outliers and images with image quality lower than 45, the average macular thickness of both eyes was used for downstream analysis. If only the macular thickness of one eye was available, this measurement was used as a surrogate for the final value.
Genotyping, imputation and quality control
For this analysis, we used the genetic data from the March 2018 release. Genotype calling, imputation and quality control (e.g. filtering variants based on Hardy–Weinberg equilibrium and call rate) were performed centrally by the UKB team (49). Study participants were genotyped on either the UK BiLEVE Axiom Array (807 411 markers; n = 49 950) or the UKB Axiom Array (825 927 markers; n = 438 427). After the application of both genetic variants and sample quality control processes, 488 377 participants with 805 426 single nucleotide variants were available in the release. Phasing the genotyped genetic variants was performed using SHAPEIT3 (50). Imputation was conducted using IMPUTE4, along with the 1000 Genomes Project (phase 3), UK10K and Haplotype Reference Consortium reference panels. After imputation, 93 095 623 autosomal SNPs, short indels and structural variants remained in the release. We applied additional quality control parameters to the imputed data to exclude variants with MAF < 0.5%, low imputation quality (info score < 0.3) and missing rate > 0.1. This resulted in 11.1 million autosomal variants for downstream analysis.
After genotype and phenotype quality control, a total of 68 423 self-reported white study participants with available SD-OCT measurements were included in this analysis. We further divided study participants into discovery and replication data sets based on relatedness using the KING (51) software. Unrelated participants (n = 59 814) were used as a discovery data set and related participants (n = 8609, independent of the discovery set) were used as a replication data set.
Statistical analysis
Linear mixed models were used to analyze genotyped and imputed genetic variants as implemented in BOLT-LMM (v2.3.1) (52), taking into account population structure and cryptic relatedness. We assumed an additive genetic model and adjusted for age, sex, genotyping array, study center, the first 10 principal components of genetic ancestry and refraction. We carried out LD score regression using the LDSC (LD score regression) method (53). Genetic variants with P < 5 × 10−8 in the discovery data set were analyzed in the replication data set. Genetic variants with P < 0.01 in replication results and exhibiting consistent direction of effect were declared replicated. During the analysis of the entire study sample, genetic variants with P < 5 × 10−8 were declared genome-wide significant and genetic variants with P < 1 × 10−6 were declared suggestive significance of an association. Independent loci were identified using the PLINK (54) clumping procedure (±500 kb with respect to the index SNP). To identify additional genetic variants contributing to the observed associations, conditional analyses were performed for the identified genomic loci by including the lead genetic variant in the regression models. Regional association plots were generated with LocusZoom (55). We estimated the heritability of macular thickness using BOLT-REML (9). R (v3.4.2) was used for plotting.
Enrichment analysis
To identify diseases and biological pathways associated with macular thickness, we performed enrichment analysis. We mapped all significant SNPs to genes according to their GRCh37/hg19 assembly genomic position. Using WebGestalt (56), we carried out enrichment analyses of GLAD4U, a tool that retrieves and prioritizes gene lists from PubMed literature and KEGG pathways. Biological terms with FDR < 0.05 were declared significant.
Web resources
The URLs for downloaded data and programs are the following:
ANNOVAR, http://annovar.openbioinformatics.org/
BOLT-LMM, https://data.broadinstitute.org/alkesgroup/BOLT-LMM/
Genevestigator, https://genevestigator.com/
GWAS Catalog, https://www.ebi.ac.uk/gwas/
PLINK, https://www.cog-genomics.org/plink2
WebGestalt, http://www.webgestalt.org/option.php
LocusZoom, http://locuszoom.org/.
Supplementary Material
Acknowledgements
We would like to thank the study participants from the UK Biobank and the staff who aided in data collection and processing.
Conflict of Interest statement. None declared.
Funding
This work was supported in part by the National Institutes of Health (NIH; Bethesda, MD, USA) [R01EY022651, R01EY027315, RF1AG060472 and P30EY001792 (departmental core grant)]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- 1. Wood A., Binns A., Margrain T., Drexler W., Považay B., Esmaeelpour M. and Sheen N. (2011) Retinal and choroidal thickness in early age-related macular degeneration. Am. J. Ophthalmol., 152, 1030–1038. [DOI] [PubMed] [Google Scholar]
- 2. Greenfield D.S., Bagga H. and Knighton R.W. (2003) Macular thickness changes in glaucomatous optic neuropathy detected using optical coherence tomography. Arch. Ophthalmol., 121, 41–46. [DOI] [PubMed] [Google Scholar]
- 3. Gupta P., Sidhartha E., Tham Y.C., Chua D.K., Liao J., Cheng C.Y., Aung T., Wong T.Y. and Cheung C.Y. (2013) Determinants of macular thickness using spectral domain optical coherence tomography in healthy eyes: the Singapore Chinese Eye study. Invest. Ophthalmol. Vis. Sci., 54, 7968–7976. [DOI] [PubMed] [Google Scholar]
- 4. Patel P.J., Foster P.J., Grossi C.M., Keane P.A., Ko F., Lotery A., Peto T., Reisman C.A., Strouthidis N.G., Yang Q. et al. (2016) Spectral-domain optical coherence tomography imaging in 67 321 adults: associations with macular thickness in the UK biobank study. Ophthalmology, 123, 829–840. [DOI] [PubMed] [Google Scholar]
- 5. Kelty P.J., Payne J.F., Trivedi R.H., Kelty J., Bowie E.M. and Burger B.M. (2008) Macular thickness assessment in healthy eyes based on ethnicity using Stratus OCT optical coherence tomography. Invest. Ophthalmol. Vis. Sci., 49, 2668–2672. [DOI] [PubMed] [Google Scholar]
- 6. Chamberlain M.D., Guymer R.H., Dirani M., Hopper J.L. and Baird P.N. (2006) Heritability of macular thickness determined by optical coherence tomography. Invest. Ophthalmol. Vis. Sci., 47, 336–340. [DOI] [PubMed] [Google Scholar]
- 7. Gao X.R., Huang H., Nannini D.R., Fan F. and Kim H. (2018) Genome-wide association analyses identify new loci influencing intraocular pressure. Hum. Mol. Genet., 27, 2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fritsche L.G., Igl W., Bailey J.N., Grassmann F., Sengupta S., Bragg-Gresham J.L., Burdon K.P., Hebbring S.J., Wen C., Gorski M. et al. (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet., 48, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loh P.R., Bhatia G., Gusev A., Finucane H.K., Bulik-Sullivan B.K., Pollack S.J., Schizophrenia Working Group of Psychiatric Genomics Consortium, Candia T.R., Lee S.H., Wray N.R. et al. (2015) Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet., 47, 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mehta H., Tufail A., Daien V., Lee A.Y., Nguyen V., Ozturk M., Barthelmes D. and Gillies M.C. (2018) Real-world outcomes in patients with neovascular age-related macular degeneration treated with intravitreal vascular endothelial growth factor inhibitors. Prog. Retin. Eye Res., 65, 127–146. [DOI] [PubMed] [Google Scholar]
- 11. Mustafi D., Kevany B.M., Bai X., Maeda T., Sears J.E., Khalil A.M. and Palczewski K. (2013) Evolutionarily conserved long intergenic non-coding RNAs in the eye. Hum. Mol. Genet., 22, 2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ikram M.K., Sim X., Jensen R.A., Cotch M.F., Hewitt A.W., Ikram M.A., Wang J.J., Klein R., Klein B.E., Breteler M.M. et al. (2010) Four novel loci (19q13, 6q24, 12q24, and 5q14) influence the microcirculation in vivo. PLoS Genet., 6, 1–0.e1001184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jeganathan V.S., Kawasaki R., Wang J.J., Aung T., Mitchell P., Saw S.M. and Wong T.Y. (2008) Retinal vascular caliber and age-related macular degeneration: the Singapore Malay Eye Study. Am. J. Ophthalmol., 146, 954–959. [DOI] [PubMed] [Google Scholar]
- 14. Yang K., Zhan S.Y., Liang Y.B., Duan X., Wang F., Wong T.Y., Sun L.P. and Wang N.L. (2012) Association of dilated retinal arteriolar caliber with early age-related macular degeneration: the Handan Eye Study. Graefes Arch. Clin. Exp. Ophthalmol., 250, 741–749. [DOI] [PubMed] [Google Scholar]
- 15. Scerri T.S., Quaglieri A., Cai C., Zernant J., Matsunami N., Baird L., Scheppke L., Bonelli R., Yannuzzi L.A., Friedlander M. et al. (2017) Genome-wide analyses identify common variants associated with macular telangiectasia type 2. Nat. Genet., 49, 559–567. [DOI] [PubMed] [Google Scholar]
- 16. Charrin S., Jouannet S., Boucheix C. and Rubinstein E. (2014) Tetraspanins at a glance. J. Cell Sci., 127, 3641–3648. [DOI] [PubMed] [Google Scholar]
- 17. Pickrell J.K., Berisa T., Liu J.Z., Segurel L., Tung J.Y. and Hinds D.A. (2016) Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet., 48, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wistow G., Bernstein S.L., Wyatt M.K., Fariss R.N., Behal A., Touchman J.W., Bouffard G., Smith D. and Peterson K. (2002) Expressed sequence tag analysis of human RPE/choroid for the NEIBank project: over 6000 non-redundant transcripts, novel genes and splice variants. Mol. Vis., 8, 205–220. [PubMed] [Google Scholar]
- 19. Kiser P.D., Golczak M., Maeda A. and Palczewski K. (2012) Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta, 1821, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakamura M., Hotta Y., Tanikawa A., Terasaki H. and Miyake Y. (2000) A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Invest. Ophthalmol. Vis. Sci., 41, 3925–3932. [PubMed] [Google Scholar]
- 21. Fan Q., Verhoeven V.J., Wojciechowski R., Barathi V.A., Hysi P.G., Guggenheim J.A., Hohn R., Vitart V., Khawaja A.P., Yamashiro K. et al. (2016) Meta-analysis of gene-environment-wide association scans accounting for education level identifies additional loci for refractive error. Nat. Commun., 7, 11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raffler J., Friedrich N., Arnold M., Kacprowski T., Rueedi R., Altmaier E., Bergmann S., Budde K., Gieger C., Homuth G. et al. (2015) Genome-wide association study with targeted and non-targeted NMR metabolomics identifies 15 novel loci of urinary human metabolic individuality. PLoS Genet., 11, 1–0.e1005487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shin S.Y., Fauman E.B., Petersen A.K., Krumsiek J., Santos R., Huang J., Arnold M., Erte I., Forgetta V., Yang T.P. et al. (2014) An atlas of genetic influences on human blood metabolites. Nat. Genet., 46, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Broer S., Bailey C.G., Kowalczuk S., Ng C., Vanslambrouck J.M., Rodgers H., Auray-Blais C., Cavanaugh J.A., Broer A. and Rasko J.E. (2008) Iminoglycinuria and hyperglycinuria are discrete human phenotypes resulting from complex mutations in proline and glycine transporters. J. Clin. Invest., 118, 3881–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Strunnikova N.V., Maminishkis A., Barb J.J., Wang F., Zhi C., Sergeev Y., Chen W., Edwards A.O., Stambolian D., Abecasis G. et al. (2010) Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum. Mol. Genet., 19, 2468–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chao J.R., Knight K., Engel A.L., Jankowski C., Wang Y., Manson M.A., Gu H., Djukovic D., Raftery D., Hurley J.B. et al. (2017) Human retinal pigment epithelial cells prefer proline as a nutrient and transport metabolic intermediates to the retinal side. J. Biol. Chem., 292, 12895–12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim J.H., Cheong H.S., Sul J.H., Seo J.M., Kim D.Y., Oh J.T., Park K.W., Kim H.Y., Jung S.M., Jung K. et al. (2014) A genome-wide association study identifies potential susceptibility loci for Hirschsprung disease. PLoS One, 9, 1–0.e110292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moore S.W. (2006) The contribution of associated congenital anomalies in understanding Hirschsprung's disease. Pediatr. Surg. Int., 22, 305–315. [DOI] [PubMed] [Google Scholar]
- 29. Rastegar-Mojarad M., Ye Z., Kolesar J.M., Hebbring S.J. and Lin S.M. (2015) Opportunities for drug repositioning from phenome-wide association studies. Nat. Biotechnol., 33, 342–345. [DOI] [PubMed] [Google Scholar]
- 30. Robinson J.R., Denny J.C., Roden D.M. and Van Driest S.L. (2018) Genome-wide and phenome-wide approaches to understand variable drug actions in electronic health records. Clin. Transl. Sci., 11, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hajee M.E., March W.F., Lazzaro D.R., Wolintz A.H., Shrier E.M., Glazman S. and Bodis-Wollner I.G. (2009) Inner retinal layer thinning in Parkinson disease. Arch. Ophthalmol., 127, 737–741. [DOI] [PubMed] [Google Scholar]
- 32. Lee W.W., Tajunisah I., Sharmilla K., Peyman M. and Subrayan V. (2013) Retinal nerve fiber layer structure abnormalities in schizophrenia and its relationship to disease state: evidence from optical coherence tomography. Invest. Ophthalmol. Vis. Sci., 54, 7785–7792. [DOI] [PubMed] [Google Scholar]
- 33. Haan J., Verbraak F.D., Visser P.J. and Bouwman F.H. (2017) Retinal thickness in Alzheimer’s disease: a systematic review and meta-analysis. Alzheimers Dement. (Amst), 6, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheung N., Shankar A., Klein R., Folsom A.R., Couper D.J., Wong T.Y. and Atherosclerosis Risk in Communities (ARIC) Study Investigators (2007) Age-related macular degeneration and cancer mortality in the atherosclerosis risk in communities study. Arch. Ophthalmol., 125, 1241–1247. [DOI] [PubMed] [Google Scholar]
- 35. Seddon J.M., Cote J., Davis N. and Rosner B. (2003) Progression of age-related macular degeneration: association with body mass index, waist circumference, and waist–hip ratio. Arch. Ophthalmol., 121, 785–792. [DOI] [PubMed] [Google Scholar]
- 36. Chen X., Rong S.S., Xu Q., Tang F.Y., Liu Y., Gu H., Tam P.O., Chen L.J., Brelen M.E., Pang C.P. et al. (2014) Diabetes mellitus and risk of age-related macular degeneration: a systematic review and meta-analysis. PLoS One, 9, 1–0.e108196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moschos M.M., Chatziralli I.P., Sergentanis T., Zagouri F., Chrysikos D., Ladas I. and Zografos G. (2016) Electroretinographic and optical coherence tomography findings in breast cancer patients using aromatase inhibitors. Cutan. Ocul. Toxicol., 35, 13–20. [DOI] [PubMed] [Google Scholar]
- 38. Gao X.R., Huang H., Nannini D.R., Fan F. and Kim H. (2018) Genome-wide association analyses identify new loci influencing intraocular pressure. Hum. Mol. Genet., 27, 2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Khawaja A.P., Bailey J.N.C., Wareham N.J., Scott R.A., Simcoe M., Igo R.P., Song Y.E., Wojciechowski R., Cheng C.Y., Khaw P.T. et al. (2018) Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat. Genet., 50, 778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. MacGregor S., Ong J.S., An J.Y., Han X.K., Zhou T., Siggs O.M., Law M.H., Souzeau E., Sharma S., Lynn D.J. et al. (2018) Genome-wide association study of intraocular pressure uncovers new pathways to glaucoma. Nat. Genet., 50, 1067–1071. [DOI] [PubMed] [Google Scholar]
- 41. Chesmore K., Bartlett J. and Williams S.M. (2018) The ubiquity of pleiotropy in human disease. Hum. Genet., 137, 39–44. [DOI] [PubMed] [Google Scholar]
- 42. Visscher P.M. and Yang J. (2016) A plethora of pleiotropy across complex traits. Nat. Genet., 48, 707–708. [DOI] [PubMed] [Google Scholar]
- 43. Grassmann F., Kiel C., Zimmermann M.E., Gorski M., Grassmann V., Stark K., International AMD Genomics Consortium (IAMDGC), Heid I.M. and Weber B.H. (2017) Genetic pleiotropy between age-related macular degeneration and 16 complex diseases and traits. Genome Med., 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stearns F.W. (2010) One hundred years of pleiotropy: a retrospective. Genetics, 186, 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khera A.V., Chaffin M., Aragam K.G., Haas M.E., Roselli C., Choi S.H., Natarajan P., Lander E.S., Lubitz S.A., Ellinor P.T. et al. (2018) Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet., 50, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A. and Yang J. (2017) 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet., 101, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Allen N.E., Sudlow C., Peakman T., Collins R. and Biobank U.K. (2014) UK biobank data: come and get it. Sci. Transl. Med., 6, 224ed224. [DOI] [PubMed] [Google Scholar]
- 48. Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M. et al. (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, 1–0.e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J. et al. (2017) Genome-wide genetic data on ~500,000 UK biobank participants. bioRxiv, Preprint at bioRxiv 10.1101/166298. [DOI]
- 50. O’Connell J., Sharp K., Shrine N., Wain L., Hall I., Tobin M., Zagury J.F., Delaneau O. and Marchini J. (2016) Haplotype estimation for biobank-scale data sets. Nat. Genet., 48, 817–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M. and Chen W.M. (2010) Robust relationship inference in genome-wide association studies. Bioinformatics, 26, 2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Loh P.R., Tucker G., Bulik-Sullivan B.K., Vilhjalmsson B.J., Finucane H.K., Salem R.M., Chasman D.I., Ridker P.M., Neale B.M., Berger B. et al. (2015) Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet., 47, 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics C onsortium, Patterson N., Daly M.J., Price A.L. and Neale B.M. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M. and Lee J.J. (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R. and Willer C.J. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics, 26, 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang J., Vasaikar S., Shi Z., Greer M. and Zhang B. (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res., 45, W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.