

Abstract
Hidradenitis suppurativa (HS), or acne inversa, is a chronic inflammatory skin disorder characterized clinically with acne-like lesions in apocrine gland–bearing skin, follicular occlusion and recurrent inflammation. Thirty-four unique mutations in patients with HS have been found in three genes encoding the γ-secretase complex: nicastrin (NCSTN), presenilin 1 (PSEN1), presenilin enhancer 2 (PSENEN) and in POGLUT1, an endoplasmic reticulum O-glucosyltransferase involved in Notch signaling. We have carried out a system review and have performed a functional analysis of the 34 unique reported HS-linked mutations in NCSTN, PSEN1, PSENEN and POGLUT1. We have also examined the effects of the HS-linked PSEN1-P242LfsX11 mutation on cytokine and chemokine expression in macrophages. Mutations in NCSTN are predicted to cause loss of function, to result in loss of transmembrane (TM) domain, to affect NCSTN substrate recruitment sites, to cause loss or creation of new ligand binging sites and to alter post-translational modifications and disulfide bonds. PSEN1-P242LfsX11 occurs at the opposite side of TM5 from Alzheimer’s disease–linked PSEN1 mutations. All of the PSENEN mutations occur on TM regions that are predicted to disrupt membrane function. POGLUT1 mutations lead to an early termination of protein synthesis and are predicted to affect ligand binding function. In addition, PSEN1-P242LfsX11 mediates cytokine and chemokine expression and prolongs tumor necrosis factor α production on the inflammatory processes in THP-1 cells and phorbol-12-myristate-13-acetate–differentiated macrophages in response to lipopolysaccharide stimulation. These in silico analyses are instructive for functional studies of the HS-linked mutations. The PSEN1-P242LfsX11 mutation mediates cytokine and chemokine expression in macrophages.
Introduction
Hidradenitis suppurativa (HS), or acne inversa, is a chronic inflammatory skin disorder. It was estimated that the prevalence of HS varies from 0.05–4% in different populations or patient cohorts. A recent population study of the 47 690 patients with HS showed that the overall of sex- and age-adjusted HS population prevalence was 0.10%, or 98 per 100 000 persons in the United States. There were approximately three times the number of female HS patients (73.8% women) compared to male patients (26.2% men). The population prevalence of African American and biracial patients was more than 3- and 2-fold greater (1) than the overall population. The clinical characteristics of HS include acne-like lesions in apocrine gland–bearing skin, follicular occlusion and progressive scarring from recurrent inflammation. Treatments include antibiotics, anti-inflammation regiments, acne washes and medicines and surgical procedure such as carbon dioxide laser excision and marsupialization (2). However, the disease progression in severe HS patients who have poor response to treatments often causes keloids, contractures and immobility, severely affecting the quality of HS patients’ lives (3).
The etiology of HS is unclear. One-third of the HS patients are reported to have a family history, and the pattern of inheritance suggests a single-gene disorder inherited as an autosomal dominant trait (4). Thirty-four unique mutations in patients with familial or sporadic HS have been found in genes encoding three of the four genes comprising the γ-secretase complex: nicastrin (NCSTN), presenilin 1 (PSEN1), presenilin enhancer 2 (PSENEN) (5) and recently, in POGLUT1, an endoplasmic reticulum (ER) O-glucosyltransferase involved in Notch signaling (6). However, how these HS-linked genetic mutations lead to HS pathogenesis remains unknown.
In silico programs have emerged as a convincing approach to study the structure and function of variants and mutations. We have carried out a system review and performed a functional in silico analysis of the 34 reported HS-linked mutations in NCSTN, PSEN1, PSENEN and POGLUT1. Notably, studies have demonstrated alterations in the immune responses in HS patients (7), and HS patients have chronic or frequent bacterial infections when circulating blood monocytes mature into tissue macrophages, which actively participate in the inflammatory processes (3). The bacterial endotoxin lipopolysaccharide (LPS) has been shown to stimulate host macrophages to produce inflammatory cytokines. Human monocytic THP-1 cells differentiate into macrophages by exposure to phorbol-12-myristate-13-acetate (PMA). Here we report the effect of overexpression of wild-type (WT) PSEN1 (PSEN1-WT) and the HS-linked PSEN1-P242LfsX11 mutation on the inflammatory processes in THP-1 cells and PMA-differentiated macrophages in response to LPS.
Results
Mutation spectrum of NCSTN, PSEN1, PSENEN and POGLUT1 in HS patients and functional in silico analyses
A total of 34 unique mutations have been identified in familial or sporadic HS patients with a diversity of mutation types in Caucasian, Chinese, Japanese, Indian or African ethnic origin (Table 1). Of these, a vast majority (74%, 25/34) of the mutations were in NCSTN (six missenses, eight nonsenses, five frameshifts and six in splice sites resulted in frameshift or in-frame deletions). A single frameshift PSEN1-P242LfsX11 mutation was detected in PSEN1 (5). Six mutations were found in PSENEN (18%, 6/34) (three frameshifts, one nonsense, one splicing, one missense). Two mutations were in POGLU1 (one nonsense, one splicing). NCSTN-R117X and Q568X were identified in more than one ethnic population and multiple families; the rest of HS-linked mutations are private to each HS family or subject. NCSTN-c.1799delTG is a two-base deletion that leads to a nonsense change L600X, while two splicing site mutations in NCSTN, c.582+1delG p. F145fs_X54 and c.1551+1G>A p.A486_T517del, result in frameshifts, while the other four splicing mutations cause in-frame deletions (Table 1).
Table 1.
Mutation spectrum of NCSTN, PSEN1, PSENEN and POGLUT1 in HS patients
| ID | Mutation category | Nucleotide change | Amino acid change | TM | Ethnic origin | Reference |
|---|---|---|---|---|---|---|
| NCSTN | ||||||
| 1 | Missense | c. 223G>A | p.V75I | Yes | Chinese | (13) |
| 2 | c.553G>A | p.D185N | Yes | Caucasian | (9) | |
| 3 | c.632C>G | p.P211R | Yes | Chinese | (14) | |
| 4 | c.647A>C | p.Q216P | Yes | Chinese | (13) | |
| 5 | c.944C>T | p.A315V | Yes | Chinese | (15) | |
| 6 | c.1229C>T | p.A410V | Yes | Chinese | (16) | |
| 7 | Nonsense | c.349C>T | p.R117X | No | Chinese, Caucasian, African | (5,16,17) |
| 8 | c.477C>A | p.C159X | No | Chinese | (18) | |
| 9 | c.497C>A | p.S166X | No | Chinese | (19) | |
| 10 | c.1258C>T | p.Q420X | No | Chinese | (54) | |
| 11 | c. 1300C>T | p.R434X | No | Caucasian | (20) | |
| 12 | c. 1695T>G | p.Y565X | No | Chinese | (14) | |
| 13 | c.1702C>T | p.Q568X | No | Caucasian, Japanese | (55) | |
| 14 | c.1799delTG | p.L600X | No | Indian | (21) | |
| 15 | Frameshift | c.210_211delAG | p.T70fsX18 | No | Chinese | (10) |
| 16 | c.487delC | p.Q163SfsX39 | No | Chinese | (5) | |
| 17 | c.687insCC | p.C230PfsX31 | No | Indian | (21) | |
| 18 | c.1752delG | p.E584DfsX44 | No | Chinese | (5) | |
| 19 | c.1768A>G | p.590AfsX3 | No | Caucasian | (20) | |
| 20 | Splice site | c.582+1delG | p.F145fs_X54 | No | Japanese | (55) |
| 21 | c.996+7G>A | p.L282_G332del | Yes | Caucasian | (9) | |
| 22 | c.1101+1G>A | p.E333_Q367del | Yes | Caucasian | (22) | |
| 23 | c.1101+10A>G | p.E333_Q367del | Yes | African | (9) | |
| 24 | c.1352+1G>A | p.Q393fs_X9 | No | Chinese | (10) | |
| 25 | c.1551+1G>A | p.A486_T517del | No | Chinese | (5) | |
| PSEN1 | ||||||
| 26 | Frameshift | c.725delC | P242LfsX11 | Chinese | (5) | |
| PSENEN | ||||||
| 27 | Frameshift | c.66delG | p.F23LfsX46 | Chinese | (5,23) | |
| 28 | c.66_67insG | p.F23VfsX98 | Caucasian | (9) | ||
| 29 | c.279delC | p.P94SfsX51 | Chinese | (5) | ||
| 30 | Nonsense | c.168T>G | p.Y56-101Pdel | Caucasian | (24) | |
| 31 | Splicing | c.167-2A>G | p.G55-101Pdel | Chinese | (25) | |
| 32 | Missense | c.194T>G | p.L65R | Chinese | (25) | |
| POGLUT1 | ||||||
| 33 | Nonsense | c.814C>T | p.R272* | Caucasian | (26) | |
| 34 | Splicing | c.430-1G>A | p.K246_392Ldel | Caucasian | (6) | |
We have performed a functional in silico analysis of the HS-linked mutations using a variety of programs. Of the HS-linked mutations in NCSTN, PSEN1, PSENEN and POGLUT1, there are 29% (10/34) nonsense, 26% frameshift (9/34) and 24% splicing site changes (8/34). By SWISS-MODEL, most of the HS-linked nonsense, frameshift and splice site mutations resulted in marked 3D structure changes (Fig. 1). Notably, a C-terminal end frameshift mutation NCSTN-E584DfsX44 resulted in a striking 3D structural change (Fig. 1), while another nearby downstream frameshift mutation p.590AfsX3 (six amino acids apart) caused only a minor 3D change (Fig. 1).
Figure 1.
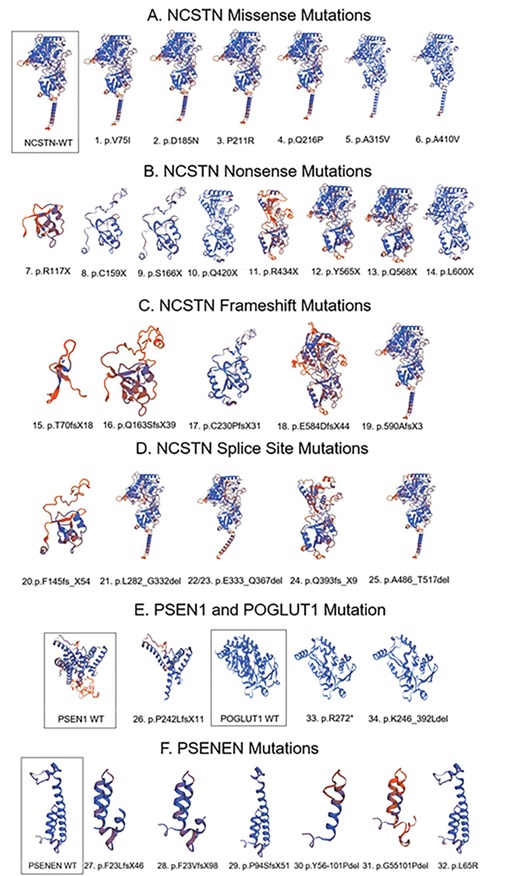
3D structures of 34 unique HS-linked mutations in NCSTN, PSEN1, PSENEN and POGLUT1 by SWISS-MODEL. Control WT protein is framed. Most of the HS-linked nonsense, frameshift and splice site mutations resulted in marked 3D structure change, consistent with loss of function. A C-terminal end frameshift mutation NCSTN-E584DfsX44 resulted in striking 3D structural change, while another nearby downstream frameshift mutation p.590AfsX3 (six amino acids apart) caused only minor 3D change.
By PolyPhen-2, SNP & Go and Proven prediction, among six NCSTN missense mutations, NCSTN-P211R and Q216P were most deleterious; V75I is probably damaging by PolyPhen-2; and D185N, A315V and A410V are predicted to have benign or neutral effects (Table 2). Sixty percent (15/25) of NCSTN mutations are nonsense or frameshift mutations that cause a truncation of the protein product. Structurally, NCSTN contains a large extra cellular domain and a single transmembrane (TM) (8), which is located at amino acid position 670–692 by Transmembrane Region Structure (THMEM). Forty percent (10/25) of NCSTN mutations (six missense mutations and four splicing site mutations) retain the TM region, while 60% (15/25) of other NCSTN nonsense, frameshift mutations and c.582+1delG (9) and c.1352+1G>A (experimental confirmed) (10) lose the TM domain to become cytosolic proteins that cannot enter the cell to initial signaling (Table 1). Among four splicing site mutations that do not affect TM regions, three potentially affect two key NCSTN substrate recruitment sites: Gry333 and Tyr337. p. L282_G332del occurs next to the residue of NCSTN substrate recruitment site G333, and E333_Q367del and E333_Q367del completely abolish two NCSTN substrate recruitment sites Gry333 and Tyr337 (8), which suggests that these NCSTN mutations affect important substrate recruitment structures. Fifty percent (3/6) of the NCSTN splicing site mutations affect substrate recruitment.
Table 2.
Predictions of functional effects of the NCTSN and POGLUT1 missense mutations
| ID | Gene | Mutations | PolyPhen-2 | SNP&Go | Proven |
|---|---|---|---|---|---|
| 1 | NCSTN | p.V75I | Probably damaging | Neutral | Neutral |
| 2 | p.D185N | Benign | Neutral | Neutral | |
| 3 | p.P211R | Probably damaging | Disease | Deleterious | |
| 4 | p.Q216P | Probably damaging | Disease | Deleterious | |
| 5 | p.A315V | Benign | Neutral | Neutral | |
| 6 | p.A410V | Benign | Neutral | Neutral | |
| 32 | PSENEN | p.L65R | Possibly damaging | Disease | Deleterious |
Moreover, NCSTN mutations also led to altered post-translational modifications. Y565X occurs on a tyrosine phosphorylation site, and R434X occurs on a glycosylation site. NCSTN-R434X disrupts the protein immediately before Asn435, one of the two NCSTN prominent glycans Asn55 and Asn435 (8). Twenty-one percent (5 of 24) of the NCSTN mutations, NCTSN-P211R, L600X, C230PfsX31, P590AfsX3 and F145fs_X54, occur at cysteine residues participating in disulfide bonds (11,12). Six potential NCSTN ubiquitination sites are predicted: K78, T127, K386, K403, K591 and K597. Six residues in NCSTN undergo sumoylation: G146, S341, K386, P423, T459 and D476. NCSTN-P590AfsX3 occurs immediately before the predicted ubiquitination site K591 and abolishes two ubiquitination sites—K591 and K597. F145fs_X54 abolishes sumoylation site G146. Both NCSTN-E333_Q367del and E333_Q367del abolish sumoylation site S341. NCSTN-T70fsX18 and R117X abolish all the ubiquitination and sumoylation sites, and C159X and S166X abolish four of the six ubiquitination sites and five of the six sumoylation sites.
A single frameshift PSEN1-P242LfsX11 mutation is identified in PSEN1, which is predicted to truncate the PS1 proteins after the fifth TM domain at the cytosolic region of the N-terminal, which markedly alters the 3D structure of PS1 (Fig. 1). Western blotting quantification of PS1 expression in PMA-differentiated macrophages detected a 25 kDa PS1 C-terminal fragment (Fig. 2). Compared to vector control, overexpression of PSEN1-WT or PSEN1-P242LfsX11 led to a significantly increased expression of PS1-WT (384 ± 17%) and PS1-P242LfsX11 (328 ± 11%), respectively.
Figure 2.
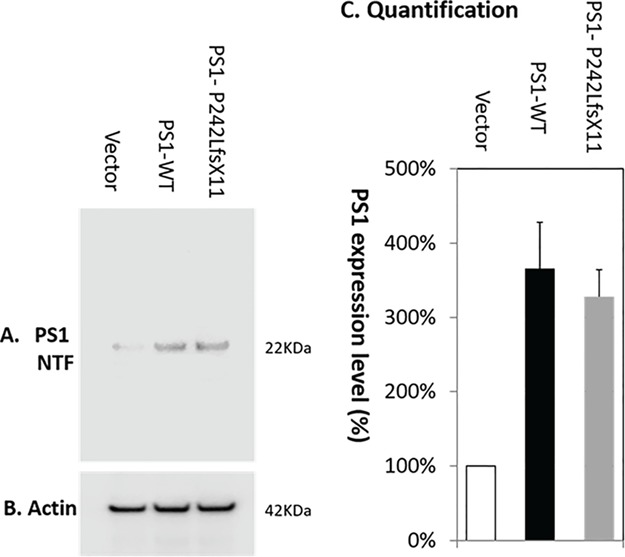
Western blotting quantification of PS1 expression in PMA-differentiated macrophages. Compared to vector control (100%), overexpression of PSEN1-WT or PSEN1- P242LfsX11 led to a significantly increased expression of PS1-WT (384 ± 11%) and PS1- P242LfsX11 (328 ± 11%), respectively. (A) Anti-PS1 N-terminal antibody detected the expression of a PS1 N-terminal product (25 kDa). (B) Anti-actin antibody detected the even expression level of β-actin (42 kDa). (C) Quantification of PS1 expression level using Image Studio Version 5.2.
PSENEN contains three TMs, at amino acid positions 18–38, 60–80 and 85–101. The PSENEN N-terminus is cytoplasmic, followed by two short helices that dip into the membrane (8). All the PSENEN mutations occur on TM regions: frameshift mutations F23LfsX46 and F23VfsX98 delete all three TM regions, while P94SfsX51 disrupts TM 3. Nonsense Y56-101Pdel and c.167-2A>G splicing site mutations lead to similar disruptions of TM 2 and 3. The missense mutation PSENEN-L65R lies in the second TM region and is predicted to be deleterious by all three programs (Table 2).
POGLUT1 is located in the lumen of the ER. Both POGLUT1-R272* and C.430-1G>A, K246* lead to an early termination of protein synthesis. POGLUT1-R272* is located in the C-terminal domain and results in a truncated form of POGLUT1 with partial loss of the C-terminal domain. The splicing site c.430-1G>A mutation was identified in exon 4 of the POFUT1 gene in patients with HS and Dowling–Degos disease (DDD) syndrome, which potentially generates aberrant splicing with loss of functionality (6). POGLUT1 is predicted to possess 17 ligand binding sites of interactions with chain A. Hydrogen bonds include A.Y117, A.S152, A.R158, A.R158, A.D196, A.V197, A.V197, A.L199, A.V214, A.A215, A.A215, A.S217, A.F218, A.R219, A.R219 and salt bridges, A.R158 and A.R219. Either POGLUT1- c.430-1G>A (K246*) or R272* completely abolished ligand binding function and shows significant alteration of global quality estimate by Qualitative Model Energy Analysis values: POGLUT1-WT: −71; POGLUT1- c.430-1G>A (K246*): 0.90; and R272* 0.45, indicating a greater deviation in mutant forms from the POGLUT1-WT.
The reported HS patients who carry a mutation in NCSTN, PSEN1, PSENEN or POGLUT1 display severe and typical symptoms of HS lesions (13,9,14–16,5,17–21,10,22–26,6). While mutations in NCSTN, PSEN1 and PSENEN occur in patients with HS only, HS patients who are carrying a POFUT1 mutation also have DDD syndrome (6).
Overexpression of PSEN1-WT or PSEN1-P242LfsX11 increased cytokine expressions in response to LPS stimulation
Compared to control, PSEN1-WT expression was increased by 297 ± 19% and PSEN1-P242LfsX11 expression was increased by 214 ± 31% as detected by RT-PCR 24 h after transfection. Overexpression of PSEN1-WT or PSEN1-P242LfsX11 was also validated by western blotting quantification of PS1 expression in PMA-differentiated macrophages. Compared to vector control, overexpression of PSEN1-WT or PSEN1- P242LfsX11 led to a significantly increased expression of PS1-WT (384 ± 17%) and PS1-P242LfsX11 (328 ± 11%), respectively, as detected with an anti-PS1 N-terminal antibody (Fig. 2). A 25 kDa PS1 N-terminal product was detected in PMA-differentiated macrophages following overexpression of either PSEN1-WT or PSEN1-P242LfsX11. Overexpression of PSEN1-WT or PSEN1-P242LfsX11 resulted in over- and under-expressed genes encoding both cytokines and chemokines (Table 3). Overexpression of PSEN1-WT or PSEN1-P242LfsX11 led to approximately four and five genes significantly overexpressed, with only the concordant-expressed gene encodes the pro-inflammatory cytokine tumor necrosis factor α (TNFα) in either the PSEN1-WT or the PSEN1-P242LfsX11 group. In contrast, LIF, IL12B, BMP2 and CSF2 were concordantly under-expressed (Table 3).
Table 3.
Differential expression of genes in cells overexpressing PSEN1 or PSEN1-P242LfsX11 in response to LPS in PMA-differentiated macrophages by PCR Array analyses
| Genes | Description | Fold change | Gene function |
|---|---|---|---|
| PSEN1 versus control overexpressed | |||
| TNF α a | Tumor necrosis factor α | 4.90 | Multifunctional proinflammatory cytokine, apoptosis |
| CCL13 | Chemokine (C-C motif) ligand 13 | 2.45 | Inducing chemotaxis, chronic inflammation |
| CXCL13 | Chemokine (C-X-C motif) ligand 13 | 2.11 | Lymphoid chemokine, chronic inflammation |
| CCL19 | Chemokine (C-C motif) ligand 19 | 2.04 | Lymphoid chemokine, chronic inflammation |
| PSEN1 versus control under-expressed | |||
| IL12A | Interleukin 12A | −3.34 | IL12 activate and link the innate and acquired immune responses |
| IL16 | Interleukin 16 | −3.18 | Binds and signals through CD4 receptor |
| LIF | Leukemia inhibitory factor | −3.14 | Mediates cell proliferation, differentiation and survival |
| IL12B | Interleukin 12B | −2.96 | Acts on T and natural killer cells |
| IL1B | Interleukin 1B | −2.64 | A subunit for IL12 and IL23; IL23 mediates late-stage inflammation |
| BMP2 | Bone morphogenetic protein 2 | −2.64 | Regulates the process of hair follicle regeneration |
| CSF2 | Colony stimulating factor 2 | −2.52 | Proliferation and differentiation of hematopoietic progenitor cell |
| CCL22 | Chemokine (C-C motif) ligand 22 | −2.16 | T-cell differentiation |
| IL23A | Interleukin 23A | −2.09 | IL23 mediates late-stage inflammation |
| IL10 | Interleukin 10 | −2.05 | Arrests and reverses the chronic inflammatory response |
| PSEN1-P242LfsX11 versus control overexpressed | |||
| CX3CL1 | Chemokine (C-X3-C motif) ligand 1 | 4.43 | The molecular control of leukocyte traffic at the endothelium |
| TNFSF11 | Tumor necrosis factor 11 | 2.55 | Regulates dendritic cell and osteoclast function; T-cell activation |
| IL11 | Interleukin 11 | 2.52 | The pleiotropic effects on hematopoietic cells |
| CCL17 | Chemokine (C-C motif) ligand 7 | 2.19 | Recruits selected subsets of leukocytes in inflammation |
| TNF α a | Tumor necrosis factor α | 2.00 | Multifunctional proinflammatory cytokine; apoptosis |
| PSEN1-P242LfsX11 versus control under-expressed | |||
| CSF2 | Colony stimulating factor 2 | −3.50 | Proliferation and differentiation of hematopoietic progenitor cell |
| IL5 | Interleukin 5 | −2.71 | Growth and differentiation factor for B cells and eosinophils |
| BMP2 | Bone morphogenetic protein 2 | −2.52 | Regulates the process of hair follicle regeneration |
| IL12B | Interleukin 12B | −2.10 | IL12 activate and link the innate and acquired immune responses |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 | −2.09 | Directs and confines neutrophil influx to sites of injury |
| ADIPOQ | Adiponectin | −2.05 | Attenuate the inflammatory response |
| IFNG | Interferon γ | −2.05 | Critical for innate and adaptive immunity against bacterial infections |
| IFNA2 | Interferon α2 | −2.05 | Reduces inflammation |
| MSTN | Myostatin | −2.05 | Regulates embryonic development and maintains tissue homeostasis |
| THPO | Thrombopoietin | −2.05 | The megakaryocyte proliferation and lineage |
| XCL1 | Chemokine (C motif) ligand 1 | −2.05 | Induces the migration of cells expressing XCR1, including lymphocytes |
| NODAL | Nodal homolog | −2.05 | A long-range signaling molecule |
| IL4 | Interleukin 4 | −2.05 | B-cell stimulatory factor; T-cell and mast cell growth factor activities |
| IL21 | Interleukin 21 | −2.05 | Stimulates B-cell proliferation |
| IL3 | Interleukin 3 | −2.05 | The proliferation of hematopoietic cell |
| IL2 | Interleukin 2 | −2.05 | The T-cell growth factor maintaining the immune system |
| IL17A | Interleukin 17A | −2.05 | Stimulates cells to produce inflammatory mediators including IL1 |
| BMP7 | Bone morphogenetic protein 7 | −2.05 | Osteoblast differentiation in pluripotential and mesenchymal stem cells |
| CCL11 | Chemokine (C-C motif) ligand 11 | −2.05 | Recruitment of leukocytes to inflammatory lesions |
| LIF | Leukemia inhibitory factor | −2.02 | Mediates cell proliferation, differentiation and survival |
a TNF α exhibited overexpression, and LIF, IL12B, BMP2 and CSF2 exhibited under-expression in both PSEN1 and PSEN1-P242LfsX11 groups.
To validate the observed increase in TNFα expression, time-dependent TNFα levels in the culture supernatant of THP-1 cells or PMA-differentiated macrophages at 0, 4, 6, 8 and 24 h after LPS exposure were analyzed by ELISA. Consistent with the increased TNFα expression detected by PCR Array analysis, TNFα levels were statistically significantly increased from 4 h with peak level at 6–8 h after LPS exposure in either THP-1 cells or PMA-differentiated macrophages by analysis of variance (ANOVA) and student’s t-test (Table 4). Compared to control, overexpression of either PSEN1-WT or PSEN1-P242LfsX11 significantly increased TNFα levels at 6, 8 or 24 h. PSEN1-P242LfsX11 led to a 3.2-fold increase of TNFα at 24 h. Moreover, overexpression of PSEN1-P242LfsX11 led to a statistically significant 1.9–2.4-fold higher TNFα level than overexpression of PSEN1-WT from 4 h through 24 h after LPS stimulation in PMA-differentiated macrophages. In addition, at 24 h after LPS stimulation, the TNFα level declined in control but remained at peak in either PSEN1-WT or PSEN1-P242LfsX11 PMA-differentiated macrophages. This indicates that overexpression of PSEN1 or PSEN1-P242LfsX11 not only increased but also prolonged TNFα production in response to LPS stimulation.
Table 4.
Time-dependent TNFα levels in the culture media in THP-1 cells and PMA-differentiated macrophages in response to LPS stimulation
| Cells | Group/mean ± SEM (pg/ml) | 0 h | 4 h | 6 h | 8 h | 24 h | P ANOVA time |
|---|---|---|---|---|---|---|---|
| Control | 51.5 ± 17.7 | 1125.9 ± 95.9 | 1279.9 ± 31.9 | 1041.5 ± 67.0 | 309.0 ± 23.7 | <0.001 | |
| PSEN1-WT | 58.1 ± 19.2 | 957.5 ± 19.6 | 1235.5 ± 29.6 | 1215.5 ± 18.4 | 425.2 ± 17.4 | <0.001 | |
| PSEN1-P242LfsX11 | 104.0 ± 21.4 | 1314.8 ± 106.6 | 1555.5 ± 167.7 | 1343.6 ± 72.6 | 314.2 ± 21.1 | <0.001 | |
| THP-1 | P PSEN1-WT versus Ctrl (Fold change) | NS | NS | NS | 0.01 (1.2) | <0.001 (0.98) | |
| P P242LfsX11 versus Ctrl (Fold change) | 0.04 (2.0) | NS | NS | 0.003 (1.3) | NS | ||
| P P242LfsX11 versus PSEN1-WT (Fold change) | NS | 0.002 (1.4) | 0.04 (1.3) | 0.05 (1.1) | <0.001 (0.74) | ||
| Control | 481.0 ± 68.8 | 758.0 ± 67.7 | 1190.3 ± 153.6 | 1075.5 ± 157.9 | 653.8 ± 77.9 | <0.001 | |
| PSEN1-WT | 690.9 ± 29.7 | 782.9 ± 86.8 | 849.7 ± 60.1 | 1105.0 ± 99.5 | 1102.3 ± 176.7 | <0.001 | |
| PMA- | PSEN1-P242LfsX11 | 252.2 ± 29.1 | 1563.7 ± 205.1 | 2065.3 ± 274.3 | 2067.5 ± 279.5 | 2087.9 ± 352.9 | <0.001 |
| differentiated | P PSEN1-WT versus Ctrl (Fold change) | 0.006 (1.4) | NS | 0.03 | NS | 0.01 (1.7) | |
| macrophages | P P242LfsX11 versus Ctrl (Fold change) | 0.003 (0.50) | 0.0008 (2.1) | 0.006 (1.7) | 0.003 (1.9) | 0.001 (3.2) | |
| P P242LfsX11 versus PSEN1-WT (Fold change) | <0.001 (0.37) | 0.001 (2.0) | <0.001 (2.4) | 0.002 (1.87) | 0.03 (1.89) |
Discussion
We have performed an extensive analysis of 34 HS-linked mutations in NCSTN, PSEN1, PSENEN and POGLUT1 in HS patients. The results of this analysis are an important instructive tool for functional studies of these HS-linked mutations.
NCSTN, PSEN1 and PSENEN encode the components of the γ-secretase complex (5), and POGLUT1 is involved in Notch signaling (6). γ-secretase–deficient mice mimic histological features found in HS patients, including follicular keratinization (5). NCSTN knockdown in HaCaT cells impaired γ-secretase activity and proliferation and differentiation of keratinocytes. Expression levels of several γ-secretase substrates involved in the Notch pathway were significantly attenuated in NCSTN-silencing HaCaT cells and the lesion of the HS patient. Phosphoinositide 3-kinase (PI3K), AKT and its activated form pAKT were markedly elevated in NCSTN-silencing HaCaT cells (18). This indicates that HS-linked NCSTN mutations may impair proliferation and differentiation of keratinocytes mainly through the Notch and PI3K/AKT signaling pathways (18).
Mutations in NCSTN are predicted to cause loss of function as a result of frameshift and premature translation termination, to result in loss of TM domain, to affect NCSTN substrate recruitment sites, to cause loss or creation of new ligand binging site and to alter post-translational modifications and disulfide bonds (11,12), all of which support the notion that the NCSTN mutations result in significantly reduced levels of NCT and reduced γ-secretase–mediated processing of Notch and signaling in the skin (27). However, testing four NCSTN–missense mutations, V75I, D185N, P211R and Q216P, for their effects on mediating Notch processing and signaling demonstrated the vague role of HS-linked NCSTN mutations in HS pathogenesis. The NCSTN-V75I, D185N and P211R mutants can function in Notch signaling in vivo; in contrast, mutant Q216P failed to rescue Notch processing and nuclear signaling (28). This suggests that NCSTN-V75I, D185N and P211R mutations have a significant role in the pathogenesis of the disease but through mechanism(s) other than impaired signaling by Notch 1 (28). The C-terminal end frameshift mutation NCSTN-E584DfsX44 resulted in a striking 3D structural change, suggesting that this mutation is likely located at a critical site for NCSTN conformation. Ubiquitiation and sumoylation are involved in post-translational modification. A large number of NCSTN mutations affect predicted ubiquitiation and sumoylation sites, suggesting that post-translational modification might contribute to HS pathogenesis.
PSEN1 was a major locus for early-onset familial Alzheimer’s disease (FAD) (5). A single frameshift PSEN1-P242LfsX11 mutation was detected in familial HS patients (5). More than 185 missense or inframe deletion mutations and promoter variants in PSEN1 have previously been found in patients with FAD (http://www.alz.org/) and sporadic dilated cardiomyopathy (29), indicating the pleiotropic nature of the presenilins (30). AD-associated PSEN1 mutations alter the γ-secretases cleavage of β-APP to increase Aβ 42/40 ratio, resulting in Aβ plague formation and related AD pathology (5). Overexpression or silencing of presenilin caused cardiac dysfunction in Drosophila (31). Overexpression of PSEN1-P242LfsX11 in zebrafish embryos enhanced Notch signaling but did not affect γ-secretase cleavage of APP (32), which suggests that the involvement of the PSEN1 mutation in HS pathogenesis also has a mechanism independent of γ-secretase activity. Notably, in AD patients, only one side of each TM helix in PS1 is affected; the hot spots of Leu219, Glu222, Leu226, Ser230, Met233 and Phe237 are placed on the same side of TM5 (8), while the HS-linked PSEN1-P242LfsX11 is on the other side of TM5 in PS1. This distribution or structure of AD-linked PSEN1 mutation is significantly different from HS-linked PSEN mutations that may indicate functional importance.
POGLUT1 is an ER O-glucosyltransferase that adds glucose moieties to serine residues in EGF-like repeats, such as those found in the trans-activating NOTCH intracellular domain (33). Mutations in POGLUT1, including W4X, R218X, R279PfsX3 and R279W, have been previously described in unrelated Caucasian patients with DDD4, an abnormally dark skin coloring condition (hyperpigmentation) (26,34,35). Mutations in POGLUT1 caused an ~50% weaker POGLU1 expression in a patient’s lesional skin compared to controls by immunohistologic staining for POGLUT1 (35). In addition, a missense mutation in POGLUT1 was identified in patients with muscular dystrophy. Muscles from patients demonstrated decreased Notch signaling, dramatic reduction in satellite cell pool and a muscle-specific α-dystroglycan hypoglycosylation not present in patients’ fibroblasts, suggesting a Notch-dependent pathomechanism for this novel form of muscular dystrophy (33). Mutations in PSENEN are also identified in DDD patients (24). Evidence has suggested the association between decreased Notch activity and POFUT1 mutations (36). The finding of POGLUT1 mutations in patients with HS–DDD syndrome indicates that aberrant Notch signaling is involved in both HS and DDD pathogenesis. Notably, mutations in POGLUT1 and NCSTN are linked to dysregulation of Notch signaling that might also contribute to small vessel disease, as well as vascular cognitive impairment (37), forming the basis for developing new therapies to control neurodegeneration in Alzheimer’s patients.
An increasing amount of evidence suggests the presence of systemic inflammation associated with HS, and bacteria are likely a second driver of inflammation. Our findings demonstrate that overexpression of PSEN1-WT or PSEN1-P242LfsX11 altered four or five cytokine and chemokine expression profiles in PMA-differentiated macrophages. Overexpression of presenilin WT exerts a dominant negative effect when expressed at high levels (38). The truncated PS1 protein resulting from PSEN1-P242LfsX11 led to higher TNFα expression than PS1-WT and four different cytokine or chemokine gene expressions. The mechanisms are unclear, and it might be related to noted different structures as the PSEN1-P242LfsX11 mutation resulted in a truncated PS1 protein that did not affect γ-secretase activity as much as the PS1-WT. The increased expression of proinflammatory TNFα and the decreased expression of LIF, IL12B, CSF2, BMP2 and other genes associated with the overexpression of PSEN1-P242LfsX11 may promote inflammatory processes, impair the activation/maintenance of immune cells and reduce hair follicle regeneration. Of concordantly under-expressed genes, LIF and CSF2 are essential for the proliferation and differentiation of hematopoietic progenitor cells into granulocytes and macrophages (39,40); IL12 is critical for the activation and maintenance of immune responses (41); and BMP2 regulates stem cell activation in the process of hair follicle regeneration in the dermis (42). Finally, the altered cytokine and chemokine gene expression profile may contribute to the pathogenesis in HS patients with the PSEN1-P242LfsX11 mutation. This indicates that overexpression of PSEN1-P242LfsX11 not only increased but also prolonged TNFα production in response to LPS stimulation.
Elevated expression of TNFα has been identified in skin lesions of patients with HS, suggesting a key role for HS pathogenesis (43). The role of Th17 cells and enhanced expression of IL-17 and IL-1β have been explored (44). The analysis on lesional HS biopsy samples showed a clustering of all TH1/TH17-associated cytokines (IL-17, interferon γ, IL-12, IL-23, IL-32, IL-1β, TNFα) around overall lesional inflammation, highlighting the importance of the TH1/TH17 cytokines in HS pathogenesis (44). Macrophages are enriched in HS infiltrates and release numerous pro-inflammatory cytokines such as IL-23 and IL-1β and TNFα, exacerbating the inflammation and contributing to the pathogenesis of HS. Dysregulated matrix metalloproteases 2 and 9 overexpression, toll-like receptor upregulation, impaired Notch signaling, NLRP3 inflammasome upregulation and dysregulated keratinocyte function have also been shown to be associated with HS (45). Obesity and smoking contribute to macrophage dysfunction and correlate with HS incidence (45). Moderate to severe HS patients who received a TNFα inhibitor infliximab showed a 50% decline from the baseline HS Severity Index score (46). In addition, patients who were administered a TNFα inhibitor adalimumab by injection every 2 weeks experienced a decrease in Sartorius score after 6 weeks (47). It was reported that two Phase III clinical trials of adalimumab for treatment of HS patients compared to placebo resulted in significantly higher clinical response rates in both trials at 12 weeks (48). Our findings provide mechanistic support for these clinical trial data. HS patients with a PSEN1 mutation may benefit greatly from TNFα-inhibiting agents such as infliximab, adalimumab, rituximab and ustekinumab, in particular after anti-inflammatory regimens fail to control the disease process. In contrast, administration of the TNFα modulator etanercept in AD patients demonstrated no apparent effect on cognitive functioning though TNFα has been implicated in the pathogenesis of AD (49,50). Human-induced pluripotent stem cell (iPSC) can modulate inflammatory responses in animal models of atherosclerosis (51), inflammatory bowel disease (52), periodontal disease (53) and so on, suggesting that iPSCs may be a new optional treatment for HS. In addition, it is of importance to explore the functional effects of iPSCs in HS inflammatory pathogenesis and compare the functional effects of HS-linked mutations with AD-linked mutations in iPSC-derived neurons, astrocytes and microglia.
In summary, we have provided functional in silico analyses of HS-linked genetic mutations and also found that the PSEN1-P242LfsX11 mutation mediates cytokine and chemokine expression in LPS-stimulated macrophages. Further functional studies are warranted to understand the role of HS-linked genetic mutations in the disease etiology and pathogenesis.
Materials and Methods
Identification of reported HS-linked genetic mutations and functional in silico analysis
We performed an NCBI-Pubmed search using the terms ‘hidradenitis suppurativa’ and ‘mutation’, and all reported studies were retrieved (13,9,14–16,5,17–21,10,22–26,6).
Functional in silico analysis of the HS-linked mutations was carried out by using the following programs: SWISS-MODEL (https://swissmodel.expasy.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), PROVEAN (http://provean.jcvi.org/index.php), SNP&GO (http://snps.biofold.org/snps-and-go/snps-and-go.html), TMHMM (http://www.cbs.dtu.dk/services/TMHMM/), NetGlycate (http://www.cbs.dtu.dk/services/NetGlycate/), UniProt (https://www.uniprot.org/uniprot/) and SUMOplot Analysis Program (http://www.abgent.com/sumoplot) to predict the mutational impact on the biological function of NCSTN, PSEN1, PSENEN and POGLUT1, as well as on 3D structure, protein stability, TM domain, ligand binding, glycation, phosphorylation, ubiquitination, sumoylation and glycosylation.
Cloning, THP-1 and PMA-differentiated macrophages cell culture
We obtained PSEN1-WT cDNA by using RT-PCR amplification of human brain mRNA and PSEN1-P242LfsX11 cDNA by site-directed mutagenesis (QuikChange® Site-Directed Mutagenesis Kit, Stratagene) and subcloned PSEN1-WT or PSEN1-P242LfsX11 cDNA into pcDNA3.1 vector (Life Technologies). The constructs were sequenced on an ABI 3730 DNA analyzer (Thermo Fisher Scientific, Inc) and validated by comparison with the PSEN1 NCBI-GenBank sequence (accession number NM_000021).
THP-1 cells (TIB-202, ATCC) were cultured using the standard ATCC culture condition. THP-1 differentiation into macrophages was achieved by addition of 100 nm PMA for 72 h in culture media. THP-1 cells and PMA-differentiated macrophages were transfected with PSEN1-WT-, PSEN1-P242LfsX11- pcDNA3.1 or control pcDNA3.1 vector, respectively, using the Effectene® Transfection Reagent (Qiagen, CA). Cells were harvested 24 h after transfection. Total RNA was extracted using RNeasy® Mini Kit, and cDNA was synthesized using the RT2 First Strand Kit (Qiagen). The expression of PSEN1-WT or PSEN1-P242LfsX11 in THP-1 cells was validated using real-time RT-PCR (Qiagen) on a CFX96 Real-time PCR analyzer (BIORAD). The level of overexpression of PSEN1-WT or PSEN1-P242LfsX11 in PMA-differentiated macrophages was detected by western blotting with an anti-PS1 N-terminal antibody. PS1 expression level was quantified using Image Studio Version 5.
LPS stimulation and Human Cytokines & Chemokines RT2 Profiler PCR Array
PMA-differentiated macrophages were treated with LPS (100 ng/mL) at 24 h after transfection. At 6 h after LPS treatment, the expression of 84 key secreted proteins central to the immune response in PMA-differentiated macrophages was examined and analyzed using the Human Cytokines & Chemokines RT2 Profiler PCR Array (Qiagen). Differentially expressed genes were defined as those that were over- or under-expressed, exceeding 2-fold as compared to control in repeated experiments with a statistically significant value (P < 0.05). To validate increased TNFα expression, time-dependent TNFα levels in the culture supernatant of THP-1 cells or PMA-differentiated macrophages at 0, 4, 6, 8 and 24 h after LPS exposure were analyzed by ELISA using the Human TNFα Single-Analytic ELISA Kit (Qiagen).
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (R01AG014713 and R01MH060009 to R.E.T., R03AR063271 and R15EB019704 to A.L.); National Science Foundation (NSF1455613 to A.L.); Cure Alzheimer’s Fund (to R.E.T.).
References
- 1. Garg A., Kirby J.S., Lavian J., Lin G. and Strunk A. (2017) Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol., 153, 760–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alikhan A., Lynch P.J. and Eisen D.B. (2009) Hidradenitis suppurativa: a comprehensive review. J. Am. Acad. Dermatol., 60, 539–561. [DOI] [PubMed] [Google Scholar]
- 3. Lasko L.A., Post C. and Kathju S. (2008) Hidradenitis suppurativa: a disease of apocrine gland physiology. JAAPA, 21, 23–25. [DOI] [PubMed] [Google Scholar]
- 4. Ingram J.R. (2016) The genetics of hidradenitis suppurativa. Dermatol. Clin., 34, 23–28. [DOI] [PubMed] [Google Scholar]
- 5. Wang B., Yang W., Wen W., Sun J., Su B., Liu B., Ma D., Lv D., Wen Y., Qu T. et al. (2010) Gamma-secretase gene mutations in familial acne inversa. Science, 330, 1065. [DOI] [PubMed] [Google Scholar]
- 6. Gonzalez-Villanueva I., Gutierrez M., Hispan P., Betlloch I. and Pascual J.C. (2018) Novel POFUT1 mutation associated with hidradenitis suppurativa–Dowling–Degos disease firm up a role for Notch signalling in the pathogenesis of this disorder. Br. J. Dermatol., 178, 984–986. [DOI] [PubMed] [Google Scholar]
- 7. Dreno B., Khammari A., Brocard A., Moyse D., Blouin E., Guillet G., Leonard F. and Knol A.C. (2012) Hidradenitis suppurativa: the role of deficient cutaneous innate immunity. Arch. Dermatol., 148, 182–186. [DOI] [PubMed] [Google Scholar]
- 8. Bai Y., Dai X., Harrison A.P. and Chen M. (2015) RNA regulatory networks in animals and plants: a long noncoding RNA perspective. Brief Funct. Genomics, 14, 91–101. [DOI] [PubMed] [Google Scholar]
- 9. Pink A.E., Simpson M.A., Desai N., Dafou D., Hills A., Mortimer P., Smith C.H., Trembath R.C. and Barker J.N. (2012) Mutations in the gamma-secretase genes NCSTN, PSENEN, and PSEN1 underlie rare forms of hidradenitis suppurativa (acne inversa). J. Invest. Dermatol., 132, 2459–2461. [DOI] [PubMed] [Google Scholar]
- 10. Liu Y., Gao M., Lv Y.M., Yang X., Ren Y.Q., Jiang T., Zhang X., Guo B.R., Li M., Zhang Q. et al. (2011) Confirmation by exome sequencing of the pathogenic role of NCSTN mutations in acne inversa (hidradenitis suppurativa). J. Invest. Dermatol., 131, 1570–1572. [DOI] [PubMed] [Google Scholar]
- 11. Sun L., Zhao L., Yang G., Yan C., Zhou R., Zhou X., Xie T., Zhao Y., Wu S., Li X. et al. (2015) Structural basis of human gamma-secretase assembly. Proc. Natl. Acad. Sci. U. S. A., 112, 6003–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bai X.C., Rajendra E., Yang G., Shi Y. and Scheres S.H. (2015) Sampling the conformational space of the catalytic subunit of human gamma-secretase. Elife, 4, 11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang C., Wang L., Chen L., Ren W., Mei A., Chen X. and Deng Y. (2013) Two novel mutations of the NCSTN gene in Chinese familial acne inverse. J. Eur. Acad. Dermatol. Venereol., 27, 1571–1574. [DOI] [PubMed] [Google Scholar]
- 14. Li C.R., Jiang M.J., Shen D.B., Xu H.X., Wang H.S., Yao X., Zhang Y., Zhou W.Q. and Wang B. (2011) Two novel mutations of the nicastrin gene in Chinese patients with acne inversa. Br. J. Dermatol., 165, 415–418. [DOI] [PubMed] [Google Scholar]
- 15. Zhang S., Meng J., Jiang M. and Zhao J. (2016) Characterization of a novel mutation in the NCSTN gene in a large Chinese family with acne inversa. Acta Derm. Venereol., 96, 408–409. [DOI] [PubMed] [Google Scholar]
- 16. Liu M., Davis J.W., Idler K.B., Mostafa N.M., Okun M.M. and Waring J.F. (2016) Genetic analysis of NCSTN for potential association with hidradenitis suppurativa in familial and nonfamilial patients. Br. J. Dermatol., 175, 414–416. [DOI] [PubMed] [Google Scholar]
- 17. Chen S., Mattei P., You J., Sobreira N.L. and Hinds G.A. (2015) gamma-Secretase mutation in an African American family with hidradenitis suppurativa. JAMA Dermatol., 151, 668–670. [DOI] [PubMed] [Google Scholar]
- 18. Xiao X., He Y., Li C., Zhang X., Xu H. and Wang B. (2016) Nicastrin mutations in familial acne inversa impact keratinocyte proliferation and differentiation through the Notch and phosphoinositide 3-kinase/AKT signalling pathways. Br. J. Dermatol., 174, 522–532. [DOI] [PubMed] [Google Scholar]
- 19. Ma S., Yu Y., Yu G. and Zhang F. (2014) Identification of one novel mutation of the NCSTN gene in one Chinese acne inversa family. Dermatologica Sinica, 32, 126–128. [Google Scholar]
- 20. Miskinyte S., Nassif A., Merabtene F., Ungeheuer M.N., Join-Lambert O., Jais J.P. and Hovnanian A. (2012) Nicastrin mutations in French families with hidradenitis suppurativa. J. Invest. Dermatol., 132, 1728–1730. [DOI] [PubMed] [Google Scholar]
- 21. Ratnamala U., Jhala D., Jain N.K., Saiyed N.M., Raveendrababu M., Rao M.V., Mehta T.Y., Al-Ali F.M., Raval K., Nair S. et al. (2016) Expanding the spectrum of gamma-secretase gene mutation–associated phenotypes: two novel mutations segregating with familial hidradenitis suppurativa (acne inversa) and acne conglobata. Exp. Dermatol., 25, 314–316. [DOI] [PubMed] [Google Scholar]
- 22. Pink A.E., Simpson M.A., Brice G.W., Smith C.H., Desai N., Mortimer P.S., Barker J.N. and Trembath R.C. (2011) PSENEN and NCSTN mutations in familial hidradenitis suppurativa (acne inversa). J. Invest. Dermatol., 131, 1568–1570. [DOI] [PubMed] [Google Scholar]
- 23. Liu Y., Miao T., Ma J., Shao L., Luo S., Li Y. and Liu Q. (2016) PSENEN c.66delG in sporadic acne inversa. Eur. J. Dermatol., 26, 298–299. [DOI] [PubMed] [Google Scholar]
- 24. Pavlovsky M., Sarig O., Eskin-Schwartz M., Malchin N., Bochner R., Mohamad J., Gat A., Peled A., Hafner A. and Sprecher E. (2018) A phenotype combining hidradenitis suppurativa with Dowling–Degos disease caused by a founder mutation in PSENEN. Br. J. Dermatol., 178, 502–508. [DOI] [PubMed] [Google Scholar]
- 25. Zhou C., Wen G.D., Soe L.M., Xu H.J., Du J. and Zhang J.Z. (2016) Novel mutations in PSENEN gene in two Chinese acne inversa families manifested as familial multiple comedones and Dowling–Degos disease. Chin. Med. J., 129, 2834–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duchatelet S., Clerc H., Machet L., Gaboriaud P., Miskinyte S., Kervarrec T. and Hovnanian A. (2018) A new nonsense mutation in the POGLUT1 gene in two sisters with Dowling–Degos disease. J. Eur. Acad. Dermatol. Venereol., 32, e440–e442. [DOI] [PubMed] [Google Scholar]
- 27. Pink A.E., Simpson M.A., Desai N., Trembath R.C. and Barker J.N.W. (2013) gamma-Secretase mutations in hidradenitis suppurativa: new insights into disease pathogenesis. J. Invest. Dermatol., 133, 601–607. [DOI] [PubMed] [Google Scholar]
- 28. Zhang X. and Sisodia S.S. (2015) Acne inversa caused by missense mutations in NCSTN is not fully compatible with impairments in Notch signaling. J. Invest. Dermatol., 135, 618–620. [DOI] [PubMed] [Google Scholar]
- 29. Gianni D., Li A., Tesco G., McKay K.M., Moore J., Raygor K., Rota M., Gwathmey J.K., Dec G.W., Aretz T. et al. (2010) Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation, 121, 1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li A. and Tanzi R.E. (2012) Pleiotropy of presenilins. Hereditary Genetics, 1(4), e105. [Google Scholar]
- 31. Li A., Zhou C., Moore J., Zhang P., Tsai T.H., Lee H.C., Romano D.M., McKee M.L., Schoenfeld D.A., Serra M.J. et al. (2011) Changes in the expression of the Alzheimer’s disease–associated presenilin gene in drosophila heart leads to cardiac dysfunction. Curr. Alzheimer Res., 8, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Newman M., Wilson L., Verdile G., Lim A., Khan I., Moussavi Nik S.H., Pursglove S., Chapman G., Martins R.N. and Lardelli M. (2013) Differential, dominant activation and inhibition of Notch signalling and APP cleavage by truncations of PSEN1 in human disease. Hum. Mol. Genet., 23, 602–617. [DOI] [PubMed] [Google Scholar]
- 33. Servian-Morilla E., Takeuchi H., Lee T.V., Clarimon J., Mavillard F., Area-Gomez E., Rivas E., Nieto-Gonzalez J.L., Rivero M.C., Cabrera-Serrano M. et al. (2016) A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol. Med., 8, 1289–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mauerer A., Betz R.C., Pasternack S.M., Landthaler M. and Hafner C. (2010) Generalized solar lentigines in a patient with a history of radon exposure. Dermatology, 221, 206–210. [DOI] [PubMed] [Google Scholar]
- 35. Basmanav F.B., Oprisoreanu A.M., Pasternack S.M., Thiele H., Fritz G., Wenzel J., Grosser L., Wehner M., Wolf S., Fagerberg C. et al. (2014) Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling–Degos disease. Am. J. Hum. Genet., 94, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li M., Cheng R., Liang J., Yan H., Zhang H., Yang L., Li C., Jiao Q., Lu Z., He J. et al. (2013) Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling–Degos disease. Am. J. Hum. Genet., 92, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Montagne A., Zhao Z. and Zlokovic B.V. (2017) Alzheimer’s disease: a matter of blood–brain barrier dysfunction? J. Exp. Med., 214, 3151–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ye Y. and Fortini M.E. (1999) Apoptotic activities of wild-type and Alzheimer's disease–related mutant presenilins in Drosophila melanogaster. J. Cell. Biol., 146, 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nicola N.A. and Babon J.J. (2015) Leukemia inhibitory factor (LIF). Cytokine Growth Factor Rev., 26, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cantrell M.A., Anderson D., Cerretti D.P., Price V., McKereghan K., Tushinski R.J., Mochizuki D.Y., Larsen A., Grabstein K., Gillis S. et al. (1985) Cloning, sequence, and expression of a human granulocyte/macrophage colony-stimulating factor. Proc. Natl. Acad. Sci. U. S. A., 82, 6250–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wolf S.F., Sieburth D. and Sypek J. (1994) Interleukin 12: a key modulator of immune function. Stem Cells, 12, 154–168. [DOI] [PubMed] [Google Scholar]
- 42. Plikus M.V., Mayer J.A., Cruz D., Baker R.E., Maini P.K., Maxson R. and Chuong C.M. (2008) Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature, 451, 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mozeika E., Pilmane M., Nurnberg B.M. and Jemec G.B. (2012) Tumour necrosis factor-alpha and matrix metalloproteinase-2 are expressed strongly in hidradenitis suppurativa. Acta Derm. Venereol., 93, 301–304. [DOI] [PubMed] [Google Scholar]
- 44. Thomi R., Cazzaniga S., Seyed Jafari S.M., Schlapbach C. and Hunger R.E. (2018) Association of hidradenitis suppurativa with T helper 1/T helper 17 phenotypes: a semantic map analysis. JAMA Dermatol., 154, 592–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shah A., Alhusayen R. and Amini-Nik S. (2017) The critical role of macrophages in the pathogenesis of hidradenitis suppurativa. Inflamm. Res., 66, 931–945. [DOI] [PubMed] [Google Scholar]
- 46. Grant A., Gonzalez T., Montgomery M.O., Cardenas V. and Kerdel F.A. (2010) Infliximab therapy for patients with moderate to severe hidradenitis suppurativa: a randomized, double-blind, placebo-controlled crossover trial. J. Am. Acad. Dermatol., 62, 205–217. [DOI] [PubMed] [Google Scholar]
- 47. Miller I., Lynggaard C.D., Lophaven S., Zachariae C., Dufour D.N. and Jemec G.B. (2011) A double-blind placebo-controlled randomized trial of adalimumab in the treatment of hidradenitis suppurativa. Br. J. Dermatol., 165, 391–398. [DOI] [PubMed] [Google Scholar]
- 48. Kimball A.B., Okun M.M., Williams D.A., Gottlieb A.B., Papp K.A., Zouboulis C.C., Armstrong A.W., Kerdel F., Gold M.H., Forman S.B. et al. (2016) Two Phase 3 trials of adalimumab for hidradenitis suppurativa. N. Engl. J. Med., 375, 422–434. [DOI] [PubMed] [Google Scholar]
- 49. Tobinick E., Gross H., Weinberger A. and Cohen H. (2006) TNF-alpha modulation for treatment of Alzheimer's disease: a 6-month pilot study. MedGenMed, 8, 25. [PMC free article] [PubMed] [Google Scholar]
- 50. Butchart J., Brook L., Hopkins V., Teeling J., Puntener U., Culliford D., Sharples R., Sharif S., McFarlane B., Raybould R. et al. (2015) Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology, 84, 2161–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shi H., Liang M., Chen W., Sun X., Wang X., Li C., Yang Y., Yang Z. and Zeng W. (2018) Human induced pluripotent stem cell–derived mesenchymal stem cells alleviate atherosclerosis by modulating inflammatory responses. Mol. Med. Rep., 17, 1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Soontararak S., Chow L., Johnson V., Coy J., Wheat W., Regan D. and Dow S. (2018) Mesenchymal stem cells (MSC) derived from induced pluripotent stem cells (iPSC) equivalent to adipose-derived MSC in promoting intestinal healing and microbiome normalization in mouse inflammatory bowel disease model. Stem Cells Transl. Med., 7, 456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hynes K., Bright R., Marino V., Ng J., Verma P.J., Gronthos S. and Bartold P.M. (2018) Potential of iPSC-derived mesenchymal stromal cells for treating periodontal disease. Stem Cells Int., 2018, 2601945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang J.Q., Wu X.J., Dou T.T., Jiao T., Chen X.B., Min M., Cai S.Q. and Zheng M. (2015) Haploinsufficiency caused by a nonsense mutation in NCSTN underlying hidradenitis suppurativa in a Chinese family. Clin. Exp. Dermatol., 40, 916–919. [DOI] [PubMed] [Google Scholar]
- 55. Nomura Y., Nomura T., Suzuki S., Takeda M., Mizuno O., Ohguchi Y., Abe R., Murata Y. and Shimizu H. (2014) A novel NCSTN mutation alone may be insufficient for the development of familial hidradenitis suppurativa. J. Dermatol. Sci., 74, 180–182. [DOI] [PubMed] [Google Scholar]