

Abstract
Several real-time PCR assays are currently used for detection of pathogenic Leptospira spp.; however, few methods have been described for the successful evaluation of clinical urine samples. This study reports a rapid assay for the detection of pathogenic Leptospira spp. in California sea lions Zalophus californianus using real-time PCR with primers and a probe targeting the lipL32 gene. The PCR assay had high analytic sensitivity—the limit of detection was 3 genome copies per PCR volume using L. interrogans serovar Pomona DNA and 100% analytic specificity; it detected all pathogenic leptospiral serovars tested and none of the non-pathogenic Leptospira species (L. biflexa and L. meyeri serovar Semaranga), the intermediate species L. inadai, or the non-Leptospira pathogens tested. Our assay had an amplification efficiency of 1.00. Comparisons between the real-time PCR assay and culture isolation for detection of pathogenic Leptospira spp. in urine and kidney tissue samples from California sea lions showed that samples were more often positive by real-time PCR than by culture methods. Inclusion of an internal amplification control in the real-time PCR assay showed no inhibitory effects in PCR negative samples. These studies indicated that our real-time PCR assay has high analytic sensitivity and specificity for the rapid detection of pathogenic Leptospira species in urine and kidney tissue samples.
Keywords: Sea lions, Pathogenic Leptospira spp., lipL32 gene, Real-time PCR, Urine, Kidney
INTRODUCTION
Evidence of past infection with Leptospira spp. has been detected in mammals worldwide (Levett 2001) including marine mammals. Prior work on marine mammal exposure and infection suggest that Leptospira infection in California sea lions Zalophus californianus, Steller sea lions Eumetopias jubatus and northern elephant seals Mirounga angustirostris is caused by L. interrogans (Vedros et al. 1971, Colegrove et al. 2005, Cameron et al. 2008, Prager et al. 2013). L. kirschneri has been detected in an elephant seal (Cameron et al. 2008).
Current methods for detection of pathogenic Leptospira spp. are limited by one or more of the following issues: methods are often labor intensive, results are not available quickly, sample collection is invasive, and/or tests lack sensitivity and specificity. Microbiological culture and isolation of Leptospira spp. can take 3 to 6 mo because of a long doubling time, ranging from 10 h to up to a few days for pathogenic strains (Schreier et al. 2013). Immunohistochemical (IHC) staining of kidney tissue is time consuming, expensive and requires invasive sampling. Fluorescent antibody testing (FAT) and dark field microscopy lack sensitivity (Miller et al. 1989) and specificity; and the microscopic agglutination test (MAT) detects anti-Leptospira antibodies in the serum, indicative of prior exposure, but is not a confirmatory diagnosis for current infection. In addition, MAT does not provide species- or strain-specific information, while PCR can provide information on the infecting Leptospira species. Real-time PCR assays for the detection of pathogenic Leptospira spp. have been reported; however, few of the methods described have evaluated performance of the assay using clinical urine samples (Levett et al. 2005, Ahmed et al. 2009, Stoddard et al. 2009). We developed a real-time PCR assay targeting the lipL32 gene for the detection of pathogenic Leptospira spp. and compared results from this real-time PCR assay with those from culture to assess the relative sensitivity of our assay. To identify false-negative results due to the presence of inhibitors in DNA extracts, we included an artificially created DNA molecule as an internal amplification control.
MATERIALS AND METHODS
Reference strains
Leptospira strains (pathogenic, non-pathogenic and intermediate Leptospira spp.) and other microorganisms were included this study (see Table 1). Leptospira and Brucella strains were obtained from the National Veterinary Services Laboratories (NVSL, Ames, Iowa, USA) and the Centers for Disease Control (CDC, Atlanta, Georgia, USA), respectively.
Table 1.
Microorganisms used for the real-time PCR assay in this study
| Species | Serovar | Strain | Status | PCR |
|---|---|---|---|---|
| Leptospira borgpetersenii | Hardjo | JB197 | Pathogenic | + |
| L. borgpetersenii | Javanica | Veldrat Bataviae 46 | Pathogenic | + |
| L. borgpetersenii | Tarassovi | Perepelicin | Pathogenic | + |
| L. interrogans | Bratislava | Jez Bratislava | Pathogenic | + |
| L. interrogans | Canicola | Hond Utrecht IV | Pathogenic | + |
| L. interrogans | Copenhageni | M-20 | Pathogenic | + |
| L. interrogans | Grippotyphosa | Andaman | Pathogenic | + |
| L. interrogans | Hardjo | Hardjoprajtino | Pathogenic | + |
| L. interrogans | Pomona | Pomona | Pathogenic | + |
| L. kirschneri | Bim | 1051 | Pathogenic | + |
| L. kirschneri | Grippotyphosa | Moskova V | Pathogenic | + |
| L. kirschneri | Grippotyphosa | 82 | Pathogenic | + |
| L. noguchii | Panama | CZ 214 K | Pathogenic | + |
| L. santarosai | Alexi | HS 616 | Pathogenic | + |
| L. weilii | Celledoni | Celledoni | Pathogenic | + |
| L. inadai | Kaup | LT 64–68 | Intermediate | − |
| L. biflexa | Andaman | CH 11 | Non-pathogenic | − |
| L. meyeri | Semaranga | Veldrot Semarang 173 | Non-pathogenic | − |
| Brucella abortus | 544 | Other microorganism | − | |
| B. ceti | B1/94 | Other microorganism | ||
| B. pinnipedialis | B2/94 | Other microorganism | − | |
| Enterococcus faecalis | Other microorganism | − | ||
| Escherichia coli | Other microorganism | − | ||
| Pseudomonas aeruginosa | Other microorganism | − | ||
| Staphylococcus aureus | Other microorganism | − | ||
| Cryptosporidium parvum | Other microorganism | − | ||
| Giardia lamblia | Other microorganism | − | ||
| Toxoplasma gondii | Other microorganism | − |
Sample collection
Urine was collected by sterile urethral catheterization or cystocentesis from 56 wild-caught, free-ranging California sea lions. In addition, 79 urine samples were collected by sterile urethral catheterization, cystocentesis or at necropsy from 64 individual California sea lions that stranded and were brought to The Marine Mammal Center (TMMC), Sausalito, California, USA for rehabilitation. Kidney samples were collected at necropsy within 36 h of death from 40 stranded California sea lions at TMMC that did not survive rehabilitation. All samples were collected under Marine Mammal Protection Act Permit No. 932–1905-00/MA-009526 issued by the National Marine Fisheries Service (NMFS). Urine was processed as described previously (Prager et al. 2013). A section of kidney including renal cortex, medulla and pelvis was collected aseptically during necropsy and stored in a sterile whirl-pack (Nasco). All samples were stored at −80°C for 7 to 11 mo until shipment to the Hollings Marine Laboratory, Charleston, South Carolina, USA, for analysis by PCR.
Culture and isolation of Leptospira
Culture and isolation of Leptospira from urine specimens were performed as described previously (Prager et al. 2013). Culture and isolation of Leptospira from kidney specimens was as follows: 1 g of kidney was collected aseptically from the renal cortex and was homogenized in Ellinghausen, McCullough, Johnson and Harris (EMJH) medium (Ellinghausen & McCullough 1965, Johnson & Harris 1967) at a 1:10 dilution and 100 µl samples of this dilution were inoculated into either T80/40/LH, a modified semi-solid EMJH medium (Zuerner 2006) supplemented with 0.4% heat-inactivated rabbit sera, or EMJH supplemented with 5-fluorouracil. These cultures were held in a 30°C incubator at TMMC for 1 to 14 d prior to overnight shipment to CDC or the National Animal Disease Center (NADC) for isolation. Once at CDC or NADC, cultures were incubated at 29°C for up to 6 mo and received periodic examination using dark field microscopy before being considered negative for Leptospira.
DNA extraction
Genomic DNA was extracted from reference Leptospira cultures from NVSL using the DNeasy tissue kit (Qiagen). DNA from non-Leptospira spp. and DNA from kidney samples were extracted with the QIAamp DNA mini kit (Qiagen). Isolation of genomic DNA from urine was accomplished using the QIAamp viral RNA mini kit (Qiagen). Each urine sample was concentrated by centrifugation (20 000 × g, 10 min). The resulting pellet was washed with 10 mM phosphate-buffered saline and then centrifuged again. Each sample was processed further according to the manufacturer’s instructions and 50 µl of DNA in nuclease-free water was eluted per sample. DNA was stored at −20°C until use.
Design of PCR primers and TaqMan® probe
Sequences of the lipL32 gene from L. interrogans strain Jez Bratislava (GenBank accession number AY461901), L. interrogans serovar Canicola (AY609321), L. interrogans serovar Copenhageni (AE016823), L. interrogans serovar Lai (AE010300), L. interrogans serovar Grippotyphosa (AY609327), L. interrogans serovar Hardjo (AY442332), L. interrogans serovar Icterohaemorrhagiae (AB094433) and L. interrogans serovar Pomona (AY776293) were aligned and a consensus sequence was selected using ClustalW2 (Larkin et al. 2007, Goujon et al. 2010). The primers and probe were designed using PrimerQuest (Integrated DNA Technologies [IDT], www.idtdna.com/primerquest/home/index). The designed primers LipL32F (5’-GGA TCC GTG TAG AAA GAA TGT CGG-3’) and LipL32R (5’-GTC ACC ATC ATC ATC ATC GTC C-3’) were used to amplify a 101 bp fragment of the lipL32 gene, which was detected by the probe LipL32P (6-carboxyfluorescein [FAM]-5’-ATG CCT GAC CAA ATC GCC AAA GCT GCG AAA-3’-Black Hole Quencher 1 [BHQ1]). The primers and probe were synthesized by Integrated DNA Technologies. Primer and probe specificities of the lipL32 gene in L. interrogans were verified by BLASTN analysis against all GenBank entries. Primer specificity was evaluated using extracted template DNA from all of the reference strains listed above.
Construction of internal control DNA
To assess whether inhibitors from urine and kidney samples were present in the extracted DNA and causing false-negative PCR results, we constructed an internal control DNA (147 bp) consisting of a fragment of the phyB gene-coding region of potato root (Nolan et al. 2006) flanked by the primers LipL32F and LipL32R. An amplicon was constructed from an oligonucleotide of the phyB gene (synthesized by Integrated DNA Technologies) in a conventional PCR using overhanging primers (LipL32IACF 5’-GGA TCC GTG TAG AAA GAA TGT CGG AAC TTG GCT TTA ATG GAC CTC CA-3’ and LipL32IACR 5’-GTC ACC ATC ATC ATC ATC GTC CAC ATT CAT CCT TAC ATG GCA CCA-3’). The overhanging primers were synthesized by IDT with the binding site for the lipL32 primers added to the 5’ end of SPUD primer sites (Nolan et al. 2006). PCR was performed in total reaction volumes of 20 µl with 10 µl of 2× AmpliTaq Gold® 360 Master Mix (Applied Biosystems), 100 nM of each primer and 2 µl of 1 µM SPUD-A (Nolan et al. 2006). The conditions used were 5 min at 95°C, followed by 35 cycles of denaturation for 15 s at 95°C, annealing for 15 s at 69°C, and elongation for 30 s at 72°C, and a final extension step of 72°C for 7 min. The amplicon from the first PCR was purified using a QIAquick PCR purification kit (Qiagen) and diluted at 1:1000 with nuclease-free water. The diluted amplicon was reamplified with primers LipL32F and LipL32R using the process described above with 1 modification: the annealing temperature was 59°C instead of 69°C. The final amplicon was purified using the QIAquick PCR purification kit and analyzed on an Agilent 2100 Bioanalyzer (Agilent Technologies) to confirm the presence of a single amplicon. The artificially created DNA was used as an internal amplification control (IAC) in every reaction mixture. The IAC was detected by the real-time PCR assay using the probe SPUD-T (hexacholrofluorescein [Hex]-5’-TGC ACA AGC TAT GGA ACA CCA CGT-3’-BHQ1) (Nolan et al. 2006).
Real-time PCR analyses
Different primer and probe concentrations were tested to determine the optimal conditions for our real-time PCR assay. Real-time PCR was performed in a total volume of 15 µl with primer and probe concentrations of 300 nM and 150 nM, respectively, 7.5 µl of TaqMan® Universal PCR master mix (Applied Biosystems), 1.5 µl IAC (200 copies per reaction) and 1.5 µl DNA template in an Eco™ real-time PCR system (Illumina). The PCR conditions were as follows: 10 min at 95°C, followed by 40 cycles of 10 s at 95°C and 30 s at 60°C. DNA from L. interrogans serovar Pomona was used as the positive template control. Nuclease-free water instead of DNA extract was used as the no template or negative control. All samples and both negative and positive controls were run in triplicate for reproducibility. Amplification efficiency (E) was estimated by the formula E = 10−1/slope − 1 (Bustin et al. 2009).
Analytical sensitivity and specificity
Genomic DNA isolated from L. interrogans serovar Pomona strain Pomona was used to determine the analytic sensitivity of the assay. The quantity of Leptospira genomic DNA was estimated by measuring the absorbance of DNA using the Spectrophotometer ND-1000 (Nanodrop Technologies). Assuming a genome size of 4.627 Mb, we considered 5.07 fg of Leptospira DNA to be equivalent to 1 genomic DNA copy. A standard curve was developed using a 10-fold serial dilution of DNA from L. interrogans serovar Pomona, ranging from 15.2 to 1.52 × 106 fg DNA (3 to 3 × 105 genome copies) per PCR volume. To determine the analytic specificity of our real-time PCR assay, we tested pathogenic Leptospira spp. strains, the non-pathogenic strains L. biflexa serovar Andaman strain CH 11 and L. meyeri serovar Semaranga strain Veldrot Semarang 173, the intermediate strain L. inadai serovar Kaup strain LT 64–68 from NVSL, and non-leptospiral pathogens (see Table 1).
Diagnostic sensitivity and specificity
We used culture as the gold standard for the estimation of the diagnostic sensitivity and specificity of our real-time PCR assay.
Sequencing amplicons from PCR-positive urine samples
The amplicons from PCR-positive urine samples were purified using the QIAquick PCR purification kit (Qiagen) and then run by the Agilent 2100 Bioanalyzer to confirm the presence of a single amplicon prior to sequencing by SeqWright with primers LipL32F and LipL32R. The amplicon sequences were analyzed using BLASTN to assess homologies with sequences in the NCBI database.
Statistical analysis
Cohen’s kappa value was used to estimate agreement between real-time PCR and culture results with a 95% confidence interval (CI). The ranges considered for interpretation of the kappa value (Landis & Koch 1977)were poor agreement: < 0.00; slight agreement: 0.00 to 0.20; fair agreement: 0.21 to 0.40; moderate agreement: 0.41 to 0.60; substantial agreement: 0.61 to 0.80; almost perfect: 0.81 to 1.00. A 2 sample t-test was used to compare the effect of urine and kidney samples on the threshold cycle (Ct) value of the IAC. All p-values < 0.05 were considered to be significant.
RESULTS
Analytic sensitivity and specificity of the real-time PCR assay
We developed a real-time PCR assay for rapid detection of Leptospira interrogans and identified the primers and probe concentrations that optimize the amplification of the lipL32 gene fragment. The amplification efficiency of the assay was 1.00 with a correlation coefficient of 0.99 (Fig. 1). The limit of detection of the PCR assay was determined by testing serial dilutions of genomic DNA from L. interrogans serovar Pomona, and was determined to be 3 genome copies per PCR volume (Fig. 1), indicating high analytic sensitivity. All pathogenic leptospiral serovar controls included in this study were positive using our assay (Table 1), indicating 100% analytic specificity. Real-time PCR of genomic DNA from the non-pathogenic species L. biflexa serovar Andaman strain CH 11 and L. meyeri serovar Semaranga strain Veldrot Semarang 173, the intermediate species L. inadai serovar Kaup strain LT 64–68 and the non-leptospiral pathogens, using the described primers and probe, did not result in any amplification products (Table 1).
Fig. 1.
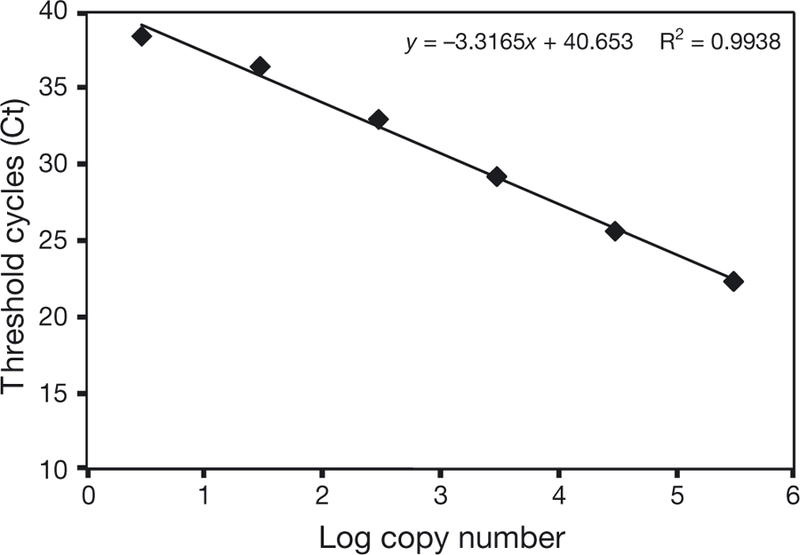
Standard curve developed using a 10-fold serial dilution of DNA from Leptospira interrogans serovar Pomona, ranging from 3 to 3 × 105 genome copies per PCR volume. The regression line is for data that are in the linear range. The graph shows the mean values from 3 replicates for each dilution
Diagnostic sensitivity and specificity
A total of 135 urine and 40 kidney samples from California sea lions were analyzed using real-time PCR and direct culture, and results from the 2 methods were compared. We detected leptospiral DNA in 41.5% (56/135) of the urine samples using our real-time PCR assay versus 8.9% (12/135) by culture (Table 2). Real-time PCR and culture of urine showed fair agreement, with a kappa value of 0.21 (95% CI, 0.02 to 0.39). When using culture as the gold standard for comparison, our real-time PCR had a diagnostic sensitivity of 92% and diagnostic specificity of 63% for urine samples. One urine sample was culture-positive but real-time PCR-negative. This urine sample was not centrifuged before being stored at −80°C for 8 mo and the sample volume was small (< 0.5 ml) relative to other urine samples tested (1 to 10 ml). We detected leptospiral DNA in 57.5% (23/40) of the kidney samples by real-time PCR, versus 27.5% (11/40) by culture (Table 3). Moderate agreement was observed between real-time PCR and culture of kidney tissues, with a kappa value of 0.44 (95% CI, 0.17 to 0.70). When using culture as the gold standard for comparison, our real-time PCR had a diagnostic sensitivity of 100% and diagnostic specificity of 59% for kidney samples.
Table 2.
Comparison of culture and real-time PCR results for Leptospira spp. in 135 urine samples from California sea lions Zalophus californianus
| Culture | ———PCR——— | Total | |
|---|---|---|---|
| Positive | Negative | ||
| Positive | 11 | 1 | 12 |
| Negative | 45 | 78 | 123 |
| Total | 56 | 79 | 135 |
Table 3.
Comparison of culture and real-time PCR results for Leptospira spp. in 40 kidney samples from California sea lions Zalophus californianus
| Culture | ———PCR——— | Total | |
|---|---|---|---|
| Positive | Negative | ||
| Positive | 11 | 0 | 11 |
| Negative | 12 | 17 | 29 |
| Total | 23 | 17 | 40 |
Performance of the IAC
The optimal IAC concentration (200 copies per reaction) was established based on the criterion that an IAC amplicon was always detected in samples containing 0 to 200 copies of the lipL32 gene per reaction. The Ct value (34.82 ± 0.4, mean ± SD) for the IAC was recorded for all 288 real-time PCR runs on the 96 urine and kidney samples where no amplification of leptospiral DNA was detected. These results did not differ from the Ct value (34.74 ± 0.3) recorded for the 42 real-time PCR runs of the 14 no template controls (p = 0.214). The IAC was amplified successfully in all negative samples, demonstrating that the real-time PCR was not inhibited in these samples. As mentioned in the above section, 1 urine sample was culture-positive but real-time PCR-negative. The Ct value of the IAC for this real-time PCR-negative sample was 34.76 ± 0.3, which did not differ from the no-template control (34.73 ± 0.3, p = 0.908), indicating that there was no PCR inhibition in this sample.
Confirmation of amplicon identity by DNA sequence analysis
PCR products from 2 positive urine samples by real-time PCR were analyzed with an Agilent 2100 Bioanalyzer and showed a single band. The purified PCR products were sequenced and the outputs were compared with the GenBank database using BLASTN. These comparisons indicated that they aligned with 100% identity with L. interrogans serovar Pomona (access number AY776293), L. borgpetersenii serovars Hardjo-bovis (DQ343231) and Mini (AY609333), L. noguchii serovar Pomona (AY609326) and uncultured Leptospira sp. clone (HQ231747).
DISCUSSION
We developed a real-time PCR assay using primers and a TaqMan probe for a lipL32 gene fragment to detect pathogenic Leptospira spp. To test the analytic specificity of our PCR method, we used known laboratory strains representing several pathogenic, intermediate and non-pathogenic Leptospira spp., and non-leptospiral pathogens. All pathogenic Leptospira serovar controls were PCR-positive and the intermediate and non-pathogenic Leptospira spp. as well as all non-leptospiral pathogens were PCR-negative, indicating that our assay had high analytic specificity. The amplicon sequences from 2 positive urine samples identified by real-time PCR aligned with 100% identity to L. interrogans serovar Pomona, L. borgpetersenii serovars Hardjo-bovis and Mini and L. noguchii serovar Pomona, further indicating the assay specificity for pathogenic Leptospira spp. The lipL32 gene has been used with real-time PCR assays for detection of pathogenic Leptospira spp., and these assays detected all pathogenic Leptospira spp. but not the intermediate pathogenic and non-pathogenic strains tested (Levett et al. 2005, Stoddard et al. 2009). Levett et al. (2005) developed a real-time PCR assay using SYBR technology which is commonly used in real-time PCR due to its low cost. A drawback of this assay is that specificity may be lower than assays that use fluorescent probes such as the TaqMan because all double-stranded DNAs formed during the PCR reaction (i.e. nonspecific PCR products and primer-dimers) are detected with the SYBR green dye, whereas only sequence-specific amplification is measured with a TaqMan probe (Valasek & Repa 2005). Stoddard et al. (2009) developed a TaqMan PCR assay that detected leptospiral DNA which had high analytic specificity, but the estimated PCR amplification efficiencies were low (0.73 to 0.76) in comparison to that of our assay (1.00). The highest attainable efficiency of 1.00 indicates that the amount of PCR product doubles with each cycle (Bustin et al. 2009), but the actual amplification efficiency of a PCR assay may be reduced below this theoretical maximum by the presence of inhibitors in DNA samples or by PCR primers and probes that do not have 100% identity with the target DNA.
Our assay showed an analytic sensitivity of 3 homologous genome copies of L. interrogans serovar Pomona per reaction, a level of sensitivity that is comparable to that reported for other assays (1 to 5 copies of Leptospira spp. per reaction) developed for detection of Leptospira spp. in clinical samples (Levett et al. 2005, Ahmed et al. 2009, Bourhy et al. 2011) and higher than that (20 genomic equivalents per reaction) developed by Stoddard et al. (2009). Our PCR assay had a diagnostic sensitivity of 92% (urine) and 100% (kidney), and a specificity of 63% (urine) and 59% (kidney) when using culture as the gold standard. Culture-negative but PCR-positive samples (which cause the low specificity scores) may be due to either a higher sensitivity of the PCR assay or contamination; however, results from other studies also indicate that PCR has a higher diagnostic sensitivity than culture (Stoddard et al. 2009). PCR is a more rapid and sensitive method for detecting Leptospira DNA in samples, but bacterial culture and isolation, which can take several months, is still an important tool in evaluating leptospiral infections because it provides information that PCR cannot. In particular, bacterial isolation is necessary to obtain sufficient genetic material for identification of strain type, and while PCR detects both viable and nonviable organisms, only viable organisms can be cultured (Levett 2001).
Although our PCR was apparently more sensitive than culture, 1 urine sample was culture-positive but PCR-negative. This may have been due to inhibitory compounds that are present in biological samples such as urine, a phenomenon that is considered a major limitation to real-time PCR (Valasek & Repa 2005). However, we included an IAC in our PCR to assess the potential for PCR inhibition in false-negative samples and found no evidence of inhibition in any samples run. Low sensitivity of PCR for detecting Leptospira spp. in urine samples due to the use of uncentrifuged urine samples or storage of urine samples at −80°C for extended periods before use has been reported (Cameron et al. 2008, Stoddard et al. 2009). Therefore, the negative real-time PCR result may be due to the fact that the urine sample was not centrifuged and was stored at −80°C for 8 mo.
The main route of Leptospira transmission is through contact with infectious leptospires shed in the urine of infected individuals (Levett 2001), and for some host/strain combinations, an infected animal can remain symptom-free and shed infectious organisms in the urine for its entire lifetime (Bharti et al. 2003). Therefore, urine is an important specimen for detection of Leptospira spp. Using the real-time PCR assay we developed, we detected Leptospira DNA in 41.5% (56/135) of urine samples collected from both stranded and wild-caught, free-ranging California sea lions and demonstrated that our assay was apparently more sensitive than culture, which was only positive for 8.9% (12/135) of the urine samples. These results confirm and extend the findings of Prager et al. (2013) of leptospire carriage in both stranded and free-ranging California sea lions.
In conclusion, we developed a real-time PCR assay using primers and a probe specific for pathogenic Leptospira spp. and evaluated the assay using urine and kidney samples from California sea lions. The results in this study demonstrate that the assay is specific and more sensitive than culture for rapid detection of pathogenic Leptospira spp. in California sea lions. The inclusion of an internal amplification control showed that negative results due to PCR inhibition were not occurring. We conclude that our real-time PCR assay is a useful and reliable tool for the detection of pathogenic Leptospira spp. in urine and kidney tissue samples.
Acknowledgements.
We thank Brian Thompson, for providing non-Leptospira bacteria. We also thank the staff of The Marine Mammal Center in Sausalito, California for providing the samples from the stranded sea lions; Dr. Robert DeLong and his group at the National Marine Mammal Laboratory for their collaboration and assistance in collecting samples from wild-caught, free-ranging sea lions. This work was performed in part at the University of California Natural Reserve System Año Nuevo Island Reserve. We thank Año Nuevo State Park rangers for their logistical support and Rick Hornsby at the National Animal Disease Center for his excellent technical support. Funding for this research was provided by NOAA’s Oceans and Human Health Initiative, the John H. Prescott Marine Mammal Rescue Assistance Grant Program, and the National Science Foundation (OCE-1335657). K.C.P. and J.L.S. acknowledge financial support from the RAPIDD program of the Science and Technology Directorate, Department of Homeland Security and the Fogarty International Center, National Institutes of Health. This publication does not constitute an endorsement of any commercial product or intend to be an opinion beyond scientific or other results obtained by the NOAA or the ARS.
LITERATURE CITED
- Ahmed A, Engelberts MF, Boer KR, Ahmed N, Hartskeerl RA (2009) Development and validation of a real-time PCR for detection of pathogenic Leptospira species in clinical materials. PLoS ONE 4:e7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti AR, Nally JE, Ricaldi JN, Matthias MA and others (2003) Leptospirosis: a zoonotic disease of global importance. Lancet Infect Dis 3:757–771 [DOI] [PubMed] [Google Scholar]
- Bourhy P, Bremont S, Zinini F, Giry C, Picardeau M (2011) Comparison of real-time PCR assays for detection of pathogenic Leptospira spp. in blood and identification of variations in target sequences. J Clin Microbiol 49: 2154–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J and others (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622 [DOI] [PubMed] [Google Scholar]
- Cameron CE, Zuerner RL, Raverty S, Colegrove KM and others (2008) Detection of pathogenic Leptospira bacteria in pinniped populations via PCR and identification of a source of transmission for zoonotic leptospirosis in the marine environment. J Clin Microbiol 46:1728–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrove KM, Lowenstine LJ, Gulland FM (2005) Leptospirosis in northern elephant seals (Mirounga angustirostris) stranded along the California coast. J Wildl Dis 41:426–430 [DOI] [PubMed] [Google Scholar]
- Ellinghausen HC Jr, McCullough WG (1965) Nutrition of Leptospira Pomona and growth of 13 other serotypes: fractionation of oleic albumin complex and a medium of bovine albumin and polysorbate 80. Am J Vet Res 26: 45–51 [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li WZ, Valentin F, Squizzato S, Paern J, Lopez R (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38(Suppl 2):W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RC, Harris VG (1967) Differentiation of pathogenic and saprophytic leptospires I. growth at low temperatures. J Bacteriol 94:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis JR, Koch GG (1977) The measurement of observer agreement for categorical data. Biometrics 33:159–174 [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R and others (2007) Clustal W and Clustal X version 2. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- Levett PN (2001) Leptospirosis. Clin Microbiol Rev 14: 296–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levett PN, Morey RE, Galloway RL, Turner DE, Steigerwalt AG, Mayer LW (2005) Detection of pathogenic leptospires by real-time quantitative PCR. J Med Microbiol 54:45–49 [DOI] [PubMed] [Google Scholar]
- Miller DA, Wilson MA, Kirkbride CA (1989) Evaluation of multivalent Leptospira fluorescent antibody conjugates for general diagnostic use. J Vet Diagn Invest 1: 146–149 [DOI] [PubMed] [Google Scholar]
- Nolan T, Hands RE, Ogunkolade W, Bustin SA (2006) SPUD: a quantitative PCR assay for the detection of inhibitors in nucleic acid preparations. Anal Biochem 351:308–310 [DOI] [PubMed] [Google Scholar]
- Prager KC, Greig DJ, Alt DP, Galloway RL and others (2013) Asymptomatic and chronic carriage of Leptospira interrogans serovar Pomona in California sea lions (Zalophus californianus). Vet Microbiol 164:177–183 [DOI] [PubMed] [Google Scholar]
- Schreier S, Doungchawee G, Chadsuthi S, Triampo D, Triampo W (2013) Leptospirosis: current situation and trends of specific laboratory tests. Expert Rev Clin Immunol 9:263–280 [DOI] [PubMed] [Google Scholar]
- Stoddard RA, Gee JE, Wilkins PP, McCaustland K, Hoffmaster AR (2009) Detection of pathogenic Leptospira spp. through TaqMan polymerase chain reaction targeting the LipL32 gene. Diagn Microbiol Infect Dis 64:247–255 [DOI] [PubMed] [Google Scholar]
- Valasek MA, Repa JJ (2005) The power of real-time PCR. Adv Physiol Educ 29:151–159 [DOI] [PubMed] [Google Scholar]
- Vedros NA, Smith AW, Schonewald J, Migaki G, Hubbard RC (1971) Leptospirosis epizootic among California sea lions. Science 172:1250–1251 [DOI] [PubMed] [Google Scholar]
- Zuerner RL (2006) Laboratory maintenance of pathogenic Leptospira. Curr Protoc Microbiol 00:E:12E.1:12E.1.1–12E.1.13 [DOI] [PubMed]