

Abstract
Though making up nearly half of the known CRISPR–Cas9 family of enzymes, the Type II-C CRISPR–Cas9 has been underexplored for their molecular mechanisms and potential in safe gene editing applications. In comparison with the more popular Type II-A CRISPR–Cas9, the Type II-C enzymes are generally smaller in size and utilize longer base pairing in identification of their DNA substrates. These characteristics suggest easier portability and potentially less off-targets for Type II-C in gene editing applications. We describe identification and biochemical characterization of a thermophilic Type II-C CRISPR–Cas from Acidothermus cellulolyticus (AceCas9). We describe several library-based methods that enabled us to identify the PAM sequence and elements critical to protospacer mismatch surveillance of AceCas9
1. Introduction
The genetic locus termed clustered regularly interspaced short palindromic repeat (CRISPR) and the CRISPR-associated (cas) genes encode the RNA-guided nucleic acid recognition systems that confer immunity against invading viruses for many bacteria and archaea (Koonin, Makarova, & Zhang, 2017; Marraffini, 2015; Mohanraju et al., 2016; Sorek, Lawrence, & Wiedenheft, 2013). There is an intense interest in all aspects of the CRISPR–Cas immunity, but special focus is given to CRISPR–Cas machineries due to their utility in the genome sciences and clinic applications. Understanding the molecular mechanism of the CRISPR–Cas machineries serves the purpose of safe and efficient gene editing in a variety of cells and organisms.
There are two main classes of CRISPR–Cas systems that are further divided into several subtypes (Koonin et al., 2017). Class 1 is defined by multi-subunit CRISPR–Cas effector complexes, whereas Class 2 is characterized by single-subunit effector complexes (Koonin et al., 2017). Due to its simplicity, Class 2 is the easiest to be implemented in gene editing as well as manipulation of cell-free nucleic acids (Barrangou & Doudna, 2016; Gootenberg et al., 2017; Komor, Badran, & Liu, 2017; Koonin & Makarova, 2009; Lee et al., 2017; Wang, La Russa, & Qi, 2016). The Class 2 systems primarily include the Types II (Cas9), V (Cas12), and VI (Cas13) systems where Cas9 and Cas12 are RNA-guided DNA cleavage enzymes and Cas13 is RNA-guided RNA cleavage enzymes, although some DNA cleavage activities have been reported (Gao, Yang, Rajashankar, Huang, & Patel, 2016; Makarova, Zhang, & Koonin, 2017; Smargon et al., 2017). Regardless of type or nucleic acid substrate, the Class 2 enzymes are comprised of the effector protein and the CRISPR RNA (crRNA), and in the case of Type II, a trans-activating RNA (tracrRNA) (Barrangou & Marraffini, 2014; Deltcheva et al., 2011; Garcia-Doval & Jinek, 2017; Nunez, Bai, Harrington, Hinder, & Doudna, 2016; Sorek et al., 2013; Tsui & Li, 2015). The crRNA or tracrRNA (Type II) must contain a scaffold that interacts with the effector protein and an antisense region of 20–24 (Type II), 23–25 (Type V), or 28–30 (Type VI) nucleotides (nts) that recognize the substrate DNA or RNA via Watson–Crick base-pairing (Garcia-Doval & Jinek, 2017; Lewis & Ke, 2017). Importantly, the nucleic acid substrates, whether DNA or RNA, must also bear an enzyme-specific activation motif adjacent to but outside the paired region. The juxtaposition of the protein, the guide RNA and the nucleic acid substrate duplex, and the activation motif lead to specific cleavage of the substrate (Anders, Niewoehner, Duerst, & Jinek, 2014; Garcia-Doval & Jinek, 2017; Smargon et al., 2017; Sternberg, Redding, Jinek, Greene, & Doudna, 2014). The abundance as well as the diverse activities of the Class 2 enzymes presents an exceptional opportunity for creation of gene editing and other nucleic acid manipulation tools. This chapter details our efforts in the discovery and mechanistic studies of a thermophilic Cas9 enzyme.
1.1. CRISPR–Cas9
CRISPR–Cas9 is a leading family of Class 2 enzymes for mechanistic studies due to its widespread use in gene editing and cell-free DNA detection (DiCarlo et al., 2013; Ding et al., 2014; Han, Slivano, Christie, Cheng, & Miano, 2015; Heler et al., 2017; Jinek et al., 2014; Long et al., 2014; Niu et al., 2014; Sander & Joung, 2014; Shen et al., 2013; Strong & Musunuru, 2017; Wang et al., 2015; Zetsche et al., 2017). Cas9 may be further divided into three subtypes depending on the sequences of Cas9 and the association with proteins involved in spacer acquisition (Chylinski, Le Rhun, & Charpentier, 2013; Mir, Edraki, Lee, & Sontheimer, 2018; Shmakov et al., 2017). The Type II-A and the Type II-B CRISPR–Cas systems contain Csn2 and Cas4, respectively, which are hypothesized spacer acquisition factors, whereas the Type II-C lacks both (Chylinski et al., 2013; Shmakov et al., 2017). The Type II-A Cas9, exemplified by Streptococcus pyrogens Cas9 (SpyCas9), was the first characterized Cas9 and currently the most popular choice for gene engineering.
Since its first report in 2012 as a potential gene-editing tool, Cas9 has been the subject of many biochemical and biophysical studies, with the ultimate goal to create powerful yet accurate gene editing platforms by harnessing their physicochemical properties. Crystal structures (Anders et al., 2014; Hirano, Gootenberg, et al., 2016; Jiang et al., 2016; Nishimasu et al., 2015; Yamada et al., 2017), single molecular fluorescence (Singh, Sternberg, Fei, Doudna, & Ha, 2016; Sung, Park, Kim, Lee, & Kim, 2018; Yang et al., 2018), mutagenesis (Anders et al., 2014; Chen et al., 2017; Hirano, Nishimasu, Ishitani, & Nureki, 2016; Kleinstiver et al., 2016), directed evolution (Gaudelli et al., 2017; Hu et al., 2018; Lee et al., 2018), cryoEM studies (Huai et al., 2017; Shin et al., 2017), enzyme kinetics (Bisaria, Jarmoskaite, & Herschlag, 2017), and theoretical studies (Zheng, 2017) revealed the architecture and dynamics of Cas9. These studies also suggest that Cas9s from different organisms may differ in the mechanism of activation, substrate recognition, optimal reaction temperature, and tolerance to guide-substrate mismatches. Detailed studies of Cas9 enzymes of different origin are, therefore, important to maximize the potential for each unique application.
The Cas9 ribonucleoprotein particles (RNPs) share the same components that include a Cas9 protein, a crRNA, and a tracrRNA (Charpentier, Richter, van der Oost, & White, 2015; Chylinski et al., 2013; Deltcheva et al., 2011). The tracrRNA forms an extensive interface with the Cas9, whereas the crRNA is associated with the enzyme complex through a long base-paired region with the tracrRNA (Anders et al., 2014; Garcia-Doval & Jinek, 2017; Hirano, Gootenberg, et al., 2016; Hirano, Nishimasu, et al., 2016; Huai et al., 2017; Jiang et al., 2016; Jiang, Zhou, Ma, Gressel, & Doudna, 2015; Jinek et al., 2014; Nishimasu et al., 2015; Yamada et al., 2017). The tracrRNA and crRNA can be covalently linked at the base-paired region to form a single guide RNA (sgRNA) (Esvelt et al., 2013; Hou et al., 2013; Jinek et al., 2012; Ran et al., 2015; Tsui, Hand, Duboy, & Li, 2017) (Fig. 1). Each Cas9 RNP identifies its DNA substrate through the complementarity in the guide region of the crRNA (or sgRNA) to the target DNA (protospacer) and a short DNA sequence immediately outside the complementary region called the protospacer adjacent motif (PAM) (Fig. 1).
Fig. 1.
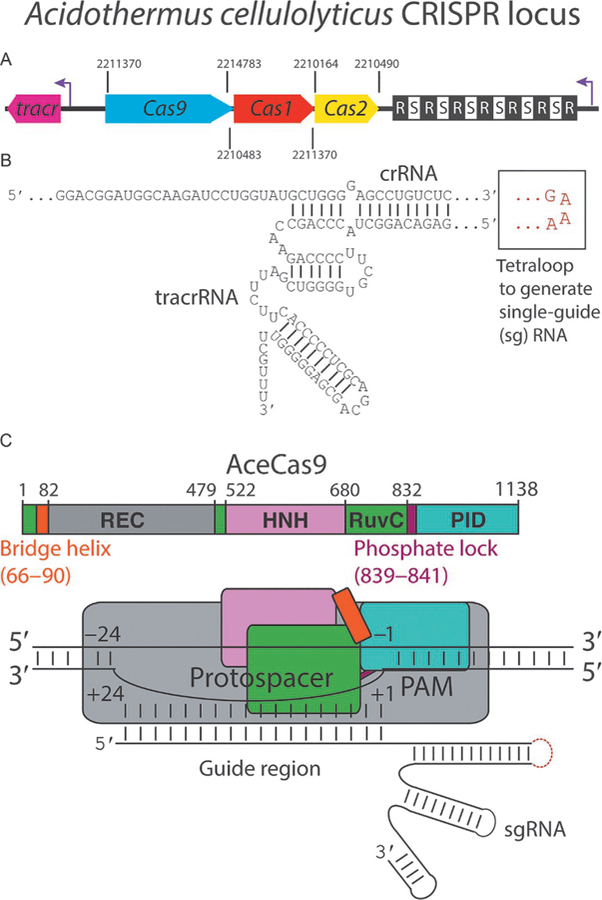
Characteristics of Acidothermus cellulolyticus CRISPR Cas9 locus. (A) A cartoon representation of the CRISPR locus of AceCas9 with all identifiable components colored coded and labeled. CRISPR repeats and spacers are depicted as black and white squares, respectively. Purple arrows represent transcription directionality. Numbers separating the components represent the known genome locations. (B) Sequence and predicted secondary structure of crRNA and tracrRNA, how they base pair, and where the tetraloop (red) is inserted to form the single guide RNA (sgRNA) used in subsequent experiments. (C) Domain architecture of AceCas9 represented either on a linear bar or with blocks. Numbers represent amino acid positions within AceCas9 or nucleotide positions within the protospacer.
Studies of SpyCas9 show that conformational change is a theme along Cas9 functional steps. Cas9 scans the DNA through 3D diffusion (Sternberg et al., 2014), quickly dissociates from nontarget regions but spends more time when the PAM sequence is found (Sternberg et al., 2014). Upon locating the target adjacent to the PAM, DNA starts unwinding sequentially from PAM toward the 5′ end followed by guide RNA base pairing with the seed region (8–10 nt at the PAM proximal region) and the positioning of the HNH domain (Sternberg et al., 2014). The two DNA strands are cleaved by the two nuclease domains (HNH and RuvC), respectively (Anders et al., 2014). Apo SpyCas9 stays in an autoinhibited stage where the active site of the HNH domain faces outward in a position that does not interact with the incoming nucleic acid. Binding of nucleic acid repositions the HNH domain to a productive conformation (Hirano, Nishimasu, et al., 2016; Slaymaker et al., 2016). An arginine-rich α helical linker between the two nuclease domain acts as an allosteric switch for this transition (Zheng, 2017). These studies suggest that not only stably folded domains but also linkers connecting them are involved in orchestrating the actions of substrate binding and cleavage.
Mechanistic studies of Cas9 address several important questions: (1) how Cas9 recognizes its PAM and how this specificity may be altered; (2) how Cas9 controls its catalytic efficiency; and (3) how Cas9 depends on the guide-protospacer base pairing in stabilizing substrate binding, and how Cas9 may be engineered to discriminate mismatched substrates. Most Cas9s studied so far have a strict dependence on the PAM but can tolerate mismatches in the guide-protospacer pairing. The tolerance of mismatches causes unwanted off-target cleavage in gene editing applications (Anderson et al., 2015; Boyle et al., 2017; Chen et al., 2017; Cho et al., 2014; Frock et al., 2015; Fu et al., 2013; Tsai et al., 2015). Biochemically, it has been established that the rates of DNA binding (kon) and release (koff), as well as that of each domain movement, all impact the catalytic efficiency and fidelity of DNA cleavage by Cas9 (Bisaria et al., 2017; Boyle et al., 2017; Raper, Stephenson, & Suo, 2018). It is believed that the conformational changes positioning the nuclease domains are a checkpoint for cleavage (Gong, Yu, Johnson, & Taylor, 2018; Sternberg et al., 2014). However, only limited information, mostly from studies of SpyCas9, is known on how each structural element within Cas9 RNPs contributes to these kinetic parameters, and thereby the catalytic efficiency and fidelity. Given the differences in the primary and tertiary structures among the diverse members of CRISPR–Cas9, the principles learned so far may not universally apply.
1.2. Type II-C CRISPR–Cas9
Among the thousands of Cas9s detectable in the published prokaryote genomes, nearly half belong to Type II-C (Chylinski et al., 2013; Mir et al., 2018; Shmakov et al., 2017). Initial research suggests that Type II-C Cas9s have weaker activities than those of Type II-A Cas9s (Ma, Harrington, O’Connell, Zhou, & Doudna, 2015). However, recent works reveal that Type II-C Cas9s are much more diverse than Type II-A Cas9 and possess unique qualities that are worthy further exploration. Type II-C Cas9s are small in size, which makes in vivo delivery using a viral vector easier for genome engineering (Kim et al., 2017; Mir et al., 2018; Tsui et al., 2017). Both the PAMs and the required guide-target base pairing for Type II-C Cas9s are longer than those of other subtypes, which make them inherently more specific (Mir et al., 2018). Genome editing in mammalian cells with three Type II-C Cas9s indeed showed a low off-target activity (Esvelt et al., 2013; Harrington et al., 2017; Hou et al., 2013; Kim et al., 2017; Lee, Cradick, & Bao, 2016). Additionally, many organisms that host Type II-C exist in extreme conditions such as Yellowstone national park acidic hot springs (Acidothermus cellulolyticus), pig respiratory tracts (Pasteurella multocida), and waste water land from Thailand (Tisterilla mobilis), and their Cas9s are stable and suited for genome editing under unusual conditions. For example, the Type II-C Cas9 from Geobacillus stearothermophilus (GeoCas9) has been shown to have a higher lifetime in human plasma making it a better tool as an RNP to inject in human bloodstream (Harrington et al., 2017).
1.3. Mechanistic questions addressed for AceCas9
The available three-dimensional structures of four Cas9s representing all three subtypes provide a good guideline to model structures for Cas9s including AceCas9 (Hirano, Gootenberg, et al., 2016; Hirano, Nishimasu, et al., 2016; Nishimasu et al., 2015; Yamada et al., 2017). AceCas9 consists of the two well-conserved nuclease domains, HNH and RuvC, responsible for cleaving the target and the nontarget strand of the target nucleic acid, respectively, the PAM interacting domain (PID) that recognizes its PAM sequence, and the REC domain that recognizes the DNA–RNA heteroduplex (Tsui et al., 2017) (Fig. 1). AceCas9 also contains the highly arginine-rich bridge helix as seen in other Cas9s that interacts extensively with the tracrRNA ( Jinek et al., 2014), the phosphate lock residues responsible for stabilizing the S-turn in the target strand, and the polar residues that potentially interact with its PAM (Anders et al., 2014; Nishimasu et al., 2015). The tracrRNA of AceCas9 is predicted to form two stem-loops. The last stem-loop as well as the linker between stem-loop 2 and stem-loop 3 are longer than those of other known Cas9s (Tsui et al., 2017) (Fig. 1).
AceCas9 provides an excellent model for the Type II-C as well as thermophilic Cas9s. It may undergo similar conformational changes and domain rearrangements as seen in other Cas9s. However, its molecule basis for recognitions of the 5′-NNNCC-3′ PAM and a long target-guide base pairing (24 nts), the dependence of superhelicity in substrate DNA, and the tolerance of target-guide mismatches requires detailed mechanistic studies.
2. Characterization of AceCas9
2.1. Acidothermus cellulolyticus CRISPR locus
A. cellulolyticus (11B) contains a single CRISPR locus that includes sequences encoding Cas1 and Cas2 proteins that are hypothesized for acquisition of protospacers and Cas9 that is involved in the interference step. In the sequenced strain of A. cellulolyticus (11B), there are 23 repeats of 36 nucleotides in length interspaced with unique spacers ranging from 27 to 29 nts that are transcribed in the opposite direction as that of Cas9. The tracrRNA was found in the noncoding region upstream of the cas9 gene and contains 16 nts with a near perfect complementarity to the direct repeat (with one mismatch) and is also transcribed opposite to Cas9.
2.2. Characterization of AceCas9 PAM
Owing to the lack of matched sequences to the potential protospacers associated with AceCas9 in database by nucleotide blast or by using CRISPRtarget, a consensus sequence of PAM could not be identified based on sequence search alone. The PAM sequence had to be identified experimentally. We accomplished this by a combination of plasmid library treatment by AceCas9 and verification by in vitro cleavage assays. Other methods include the use of consensus flanking sequence around identifiable protospacers or deep sequencing the input of the uncleaved libraries (Fonfara et al., 2014; Karvelis et al., 2015; Leenay et al., 2016; Ran et al., 2015). Though our method is less thorough, it is suited to laboratories who have no access to deep sequencing facilities. We constructed a PAM library containing seven randomized nucleotides at the 3′ end of the protospacer incorporated onto pUC19 (5′-NNNNNNN-3′) and ~500 bps away from a downstream BamHI site (Fig. 2). Treatment of the plasmid library with both AceCas9 RNP and BamHI would release ~500 bps fragments for the cognate PAM sequences. We cloned and sequenced the released fragments to look for a consensus PAM sequence. All positive clones had cytosine at positions 4 and 5 on the nontarget strand. We subsequently confirmed the 5′-NNNCC-3′ PAM by both dsDNA oligos (fluorescently labeled) and plasmid DNA cleavage that (Fig. 2).
Fig. 2.
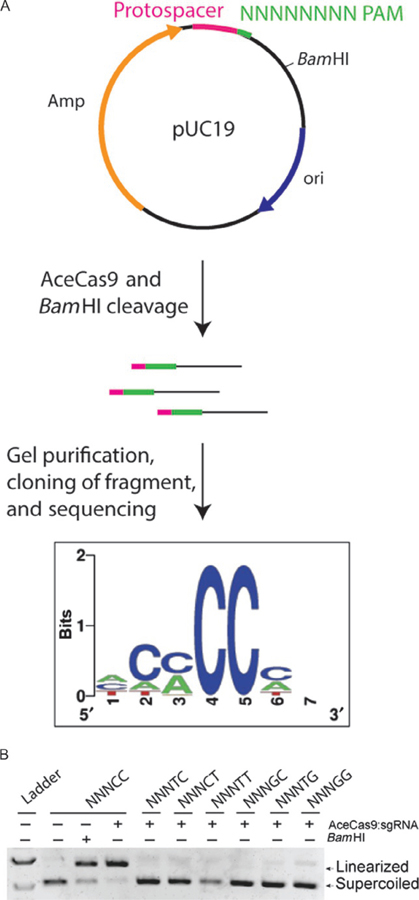
Schematics of the library-based method for identification of AceCas9 PAM. (A) Schematic of pUC19 library containing randomized PAM region and a BamHI cleavage site, the products generated by AceCas9 and BamHI cleavage, and the sequenced consensus at the PAM. (B) Agarose gel analysis of AceCas9 cleavage of the pUC19 vector with the identified PAM sequence (5′-NNNCC-3′) and its variants.
2.2.1. Equipment
Agarose gel apparatus
Urea denaturing polyacrylamide gel apparatus
30°C water bath
50°C water bath
90°C heating block
Typhoon Trio (GE healthcare)
ChemiDoc XRS System (Bio-Rad)
2.2.2. Buffers and reagents
Standard cleavage buffer for Cas9 (20 mM Tris pH 7.5, 150 mM KCl, 2 mM DTT, 10 mM MgCl2, 5% glycerol)
2 × Formamide gel loading buffer (90% formamide, 50 mM EDTA pH 8.0)
15% Urea denaturing gel
5 × gel loading buffer (25 mM Tris pH 7.5, 250 mM EDTA pH 8, 1% SDS, 0.05% w/v bromophenol blue, 30% glycerol)
0.5% 1 × agarose gel
E.Z.N.A Plasmid DNA mini kit (Omega Bio-Tek)
E.Z.N.A Gel Extraction Kit (Omega Bio-Tek)
Q5 Site-Directed mutagenesis kit (New England Biolabs)
Tris–Boric acid–EDTA (TBE) buffer
EZ-Vision In-Gel solution (Thomas Scientific)
2.2.3. Procedure for PAM identification
2.2.3.1. PAM library construction
A chosen protospacer sequence followed by a 7-bp degenerate sequence (5′-NNNNNNN-3′) was cloned into pUC19 via Q5 Site-Directed mutagenesis ~ 500 bp upstream of the BamHI site;
The library was transformed in DH5α cells that were cultured in LB-medium containing appropriate antibiotics overnight;
The plasmid DNA was extracted from the overnight culture by use of the E.Z.N.A Plasmid DNA mini kit and was sequence verified to contain the 7-degenerate base pairs.
2.2.3.2. PAM identification
Incubate AceCas9 RNP as well as BamHI with the library DNA for 1 h at 37°C in the Cas9 cleavage buffer;
Separate the cleaved DNA fragment from the rest of the pUC19 plasmid on a 0.8% agarose gel;
Excise the band at the position of ~500 bps and elute the DNA using the E.Z.N.A Gel Extraction kit (Omega Bio-tek);
The extracted fragments were end-repaired and ligated with index primers from NEB Ultra II Library prep kit and NEBNext Multiplex oligos for illumine (New England Biolabs);
The fragments were PCR amplified and cloned into pCR4Blunt-TOPO vector following the manufacture’s instruction (Thermo Fisher Scientific);
Transform the ligation mixture into a high efficiency electrocompetent DH5α cells (New England Biolabs) and plate onto agar plates containing the appropriate antibiotics;
Individual colonies were picked for growth overnight, then DNA extraction the following day
Plasmid DNA was sequenced by the Sanger method using standard T3 and T7 primers.
2.2.4. Procedure for in vitro cleavage assays
2.2.4.1. Cleavage assay with oligonucleotide
DNA oligonucleotides labeled with HEX (target strand) or 6-FAM (nontarget strand) fluorescent tags were purchased from Eurofins genomics;
To generate duplex DNA target for AceCas9, the labeled DNA strand was annealed with the nonlabeled strand with a ratio of 1:20 for HEX and 1:1.2 for FAM-labeled oligos, respectively, by heating at 95°C for 5 min followed by cooling at room temperature;
AceCas9 was preincubated with the in vitro transcribed sgRNA at an equimolar ratio in a 37°C water bath for 15 min then cooled to room temperature;
The duplex DNA was added to the tube containing 3 nM AceCas9 RNP and incubated in a 50°C water bath for 1 h;
The cleavage reaction was stopped with the formamide gel loading buffer;
The samples were then loaded onto a 12% polyacrylamide urea gel and the cleavage products were visualized with the Typhoon Trio scanner with a proper wavelength filter.
2.2.4.2. Plasmid DNA cleavage assay
AceCas9 and the sgRNA were prepared as above and the plasmid containing the correct protospacer and PAM sequence was added to a final concentration of 100 ng followed by incubation for 1 h at 50°C or other temperatures tested;
The reaction was stopped by adding the 5 × gel loading buffer and separated on a 0.5% agarose 1 × TBE gel with EZ-Vision In-Gel solution;
The gel was scanned on a Chemidoc XRS System (Bio-Rad) and the products were analyzed by Image-quant software (GE Healthcare).
3. Directed evolution of AceCas9
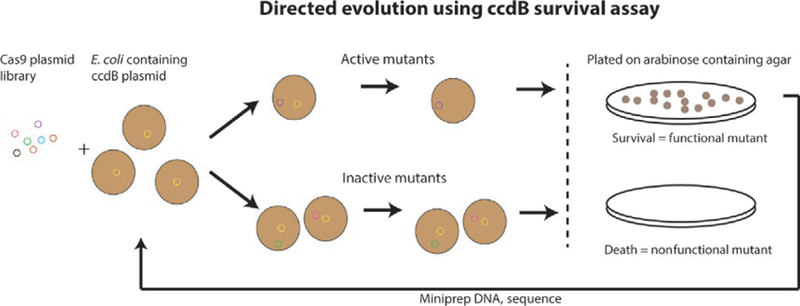
We employed a directed evolution method based on coexpressing the toxic ccdB protein inducible by arabinose and an AceCas9 library similar to that used in DNA homing endonuclease evolution (Chen & Zhao, 2005) (Fig. 3). In this method, survival of the bacterial cells transformed with both a ccdB- and an AceCas9-encoding plasmids is linked to the activity of AceCas9 (Tsui et al., 2017). Successful cleavage of the ccdB plasmid by AceCas9 removes the toxicity to cells and thus their survival in the presence of arabinose. In contrast, the inability of AceCas9 to cleave the ccdB plasmid, due either to an incorrectly matched protospacer or to a defective AceCas9, would lead to cell death (Fig. 4). Thus, by transforming a plasmid library-encoding AceCas9 variants into cells containing a noncognate ccdB plasmid, we are able to identify variants that rescue the cleavage activity. After initial screening, the survival mutants were amplified and enriched for the next rounds of survival selection until no further improvement in survival rate were observed (Fig. 4).
Fig. 3.
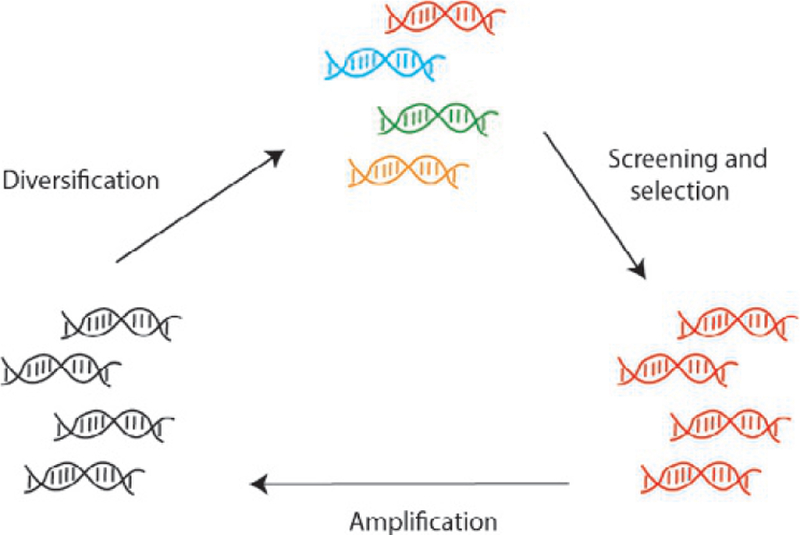
Schematic of directed evolution cell survival assay using ccdB toxic gene in E. coli strain BW25141. The plasmid library-encoding AceCas9 is transformed to the BW25141 cells containing the toxic ccdB-encoding gene inducible by arabinose. The variants capable of cleaving the ccdB-encoding plasmid remove the toxicity from the cells, leading to growth, whereas those cannot cleave the ccdB-encoding plasmid leads to cell death.
Fig. 4.
Directed evolution workflow. Wild-type plasmid encoding the enzyme targeted for directed evolution is diversified and subjected to screening. Initial hits are further enriched or diversified for additional rounds of screening. The screening process produces variants of different cleavage activities from that of the wild-type enzyme.
We showed previously that PAM-proximal mismatches between the protospacer and the guide RNA severely impaired AceCas9 cleavage activity, leading to death of the bacteria in the presence of arabinose (Hand et al., in review; Tsui et al., 2017). The lack of survival with PAM-proximal mismatches prompted us to search for mutant AceCas9s that could rescue this phenotype. To explore this possibility, we created AceCas9 libraries using two different methods. To investigate the PID and REC domains, each consisting of about 300 amino acids, we used error-prone PCR in combination with Gibson Assembly (NEB). The resulting libraries contained about 10 mutations per kilobase, which produced about 4 amino acid changes in each variant (Fig. 5). Alternatively, we used site-directed saturation mutagenesis at the linker region connecting the RuvC and PID (phosphate lock region). To accomplish this, we performed Q5 mutagenesis (NEB) in which the primers contained randomized codons at target amino acid positions (Fig. 5). Library DNA, from both Gibson Assembly and Q5 mutagenesis, was then transformed into DH5α cells and grown on agar plates containing appropriate antibiotics. The presence of the randomized sequence was confirmed by sequencing DNA extracted from several colonies, and the remaining colonies were pooled for extraction of library DNA. The pooled DNA was concentrated and subsequently used in survival assays against the cells harboring the ccdB plasmid with a − 1 position mismatch in the protospacer to screen for mutants that could rescue cell survival. We describe below the protocols for selecting AceCas9 variants that are able to rescue activity on the protospacer mismatch (T(−1)G) that is not cleaved by the wild-type AceCas9.
Fig. 5.
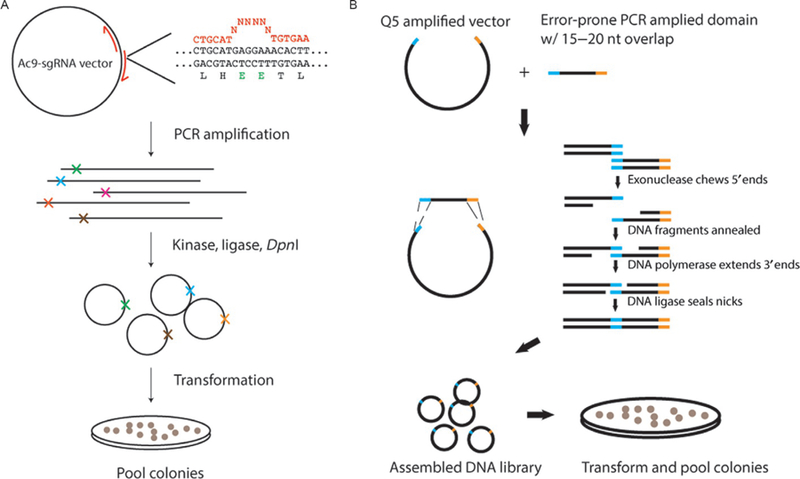
DNA diversification methods used for AceCas9 directed evolution. (A) Library creation via site-directed saturation mutagenesis by incorporating randomized codons to specific regions. (B) Library creation via error-prone PCR in combination with Gibson Assembly.
3.1. AceCas9 library construction
3.1.1. Equipment
Thermocycler
Standard agarose gel apparatus
37°C water bath and dry incubator
UV light source
Rotovap
3.1.2. Buffers and reagents
TBE buffer
Super Optimal with Catabolite repression (SOC) media
2 × Q5 Site-Directed mutagenesis kit (NEB)
E. cloni 10G chemically competent cells (Lucigen)
E.Z.N.A. Plasmid DNA mini Kit (Omega Bio-tek)
E.Z.N.A. Cycle Pure PCR clean up kit (Omega Bio-tek)
3.1.2.1. Procedure
- Perform PCR with appropriately designed DNA primers for the AceCas9-sgRNA coexpression plasmid:
- For domain library construction, error-prone PCR was used with DNA primers flanking the desired region. The number of cycles was adjusted to produce a mutation every 100 bases.
- For site-directed saturation library construction, DNA primers containing randomized codons NNS of a desired length, where N represents any nucleotide and S represents either G or C, flanked by 15–20 nts complementary to the region around the mutagenized site were purchased;
Remove the extra salts and proteins from the amplified PCR product the E.Z.N.A. Cycle Pure PCR clean up kit;
Concentrate the PCR product, now in water, in a rotovap down to a small volume (2–5 μL in our case);
Ligate all the PCR product using KLD ligation mixture in multiple reactions following its recommended procedure;
Transform all ligated plasmids to the E. cloni 10G chemically competent cells in multiple reactions following a standard chemical transformation protocol;
Plate all the transformed cells onto agar plates containing the correct antibiotics. We plated 200 μL each on five separate agar plates;
Incubate the plates at 37°C overnight;
Collect the colonies the following day by adding 5 mL of prewarmed SOC media followed by transferring them into a 250-mL Erlenmeyer flask;
Grow the culture and extract the plasmid DNA by E.Z.N.A Plasmid DNA mini kit;
Estimate the library size by transforming a defined amount of library DNA and calculating colony formation units (CFU) per μg DNA. The size of the library is typically 104–107.
3.2. Iterative rounds of survival assays
3.2.1. Equipment
Strategene Electroporator 1000 or other Electroporation device
Bulldog Bio 1-mm electroporation cuvettes
P-10, P-200, and P-1000 pipettes and tips
15-mL culture tubes
37°C plate incubator
37°C shaking incubator
Tabletop centrifuge
Disposable L-bar spreading device
Bunsen burner
3.2.2. Buffers and reagents
SOC media
Agar plates with Luria-Bertani (LB) and appropriate antibiotics;
50 mM Isopropyl β-d-1 thiogalactopyranoside (IPTG) solution;
E.Z.N.A. Plasmid DNA mini Kit (Omega Bio-tek).
3.2.3. Procedure for preparing CcdB-containing electrocompetent cells
Inoculate a 50 mL LB culture containing 0.2% glucose and antibiotics with a single colony from a fresh transformation of BW25141 cells with a desired ccdB plasmid. Incubate at 37°C with 220 rpm shaking;
Inoculate 1 L of media with the entirety of the starter culture when the OD600 reaches 0.5. Return to incubator with shaking;
When the OD600 of the culture reaches 0.5, remove the culture from the incubator and place on ice for 15 min;
Transfer the culture to an ice-cold centrifuge bottle and spin at 4000 × g for 15 min at 4°C;
Decant the supernatant and resuspend the cell pellet in 1 L of ice-cold distilled water. Return to centrifuge and pellet the cells as before;
Decant the supernatant and resuspend the cell pellet in 500 mL of ice-cold distilled water. Return to centrifuge and pellet the cells as before.
Decant the supernatant and resuspend the cell pellet in 40 mL of ice-cold sterile 10% glycerol. Return to centrifuge and pellet the cells as before.
Decant the supernatant and resuspend the cell pellet in 2 mL of ice-cold sterile 10% glycerol.
Aliquot 50 μL of cells to Eppendorf tubes on ice and flash-freeze with liquid nitrogen. Store cells at −80°C.
Procedure notes: It is critical that the OD600 of the cell cultures never be allowed to go above 0.5.
3.2.4. Procedure for survival assay
Remove previously made ccdB-containing (either WT or protospacer mutants) electrocompetent cells from −80°C storage and thaw on ice for 30 min. Place appropriate number of electroporation cuvettes on ice;
Add 100–200 ng of library or single plasmid DNA to the cells on ice, and gently mix by tapping;
Place the cell/DNA mixture at the bottom of the chilled electroporation cuvettes with a P-200 pipette, being careful to avoid bubbles;
Transfer 1 mL of prewarmed SOC into a 15-mL culture tube;
Place the cuvette into the electroporator and transform DNA into the ccdB-containing cells following the manufactures instructions;
After electroporation, add part of the 1 mL prewarmed SOC media to the cuvette, making sure cells are well suspended and then transfer the entire content to the rest of the 1 mL SOC in the 15 mL culture tube;
Place the culture tube in a 37°C incubator with shaking for 30 min;
-
Add 0.5 mM IPTG to the culture tube and return to shaking for additional 60 min;
For library screening: take 10 μL from the culture and add to 190 μL of prewarmed SOC for plating on a plate without arabinose for transformation control. Centrifuge the remaining culture at 3000 × g for 5 min at 23°C. Decant supernatant, and gently resuspend the cells in 200 μL of prewarmed SOC and plate on an agar plate with arabinose;
Incubate the plates at 37°C;
Harvest the colonies on the arabinose+ plates, if present, by swirling with 5 mL fresh LB, and extract DNA by use of the E.Z.N.A Plasmid DNA mini kit. Repeat transformation in step 1 for iterative rounds of selection, if necessary. The survival plasmid DNA may be used for Illumina or individual sequencing to identify the variations.
4. Functional regions of AceCas9 identified by directed evolution
We identified a thermophilic Type II-C Cas9 for mechanistic study and development of gene editing and nucleic acid detection tools. We employed the ccdB-based directed evolution, which offers an effective method for identifying functional regions in AceCas9 on a large scale. By pairing AceCas9 variant libraries with the ccdB plasmid containing various protospacers, we are able to select the variants with defined activities. In this work, we paired an AceCas9 library-encoding AceCas9 variants in its phosphate lock region with the ccdB plasmid containing mismatched protospacer at a PAM-proximal site. We identified phosphate lock residues that are able to rescue the otherwise defective protospacer mismatches. Our results connect phosphate lock residues to the step of protospacer recognition by AceCas9.
We transformed the AceCas9 plasmid library randomized at the phosphate lock region into the cell containing the ccdB plasmid with a mismatched protospacer at the first position (T-1G) (Fig. 6). Unlike the wild-type AceCas9 that did not produce any survival colonies, the phosphate lock variants yielded significant survival after the first round of survival assay. Following iterative rounds of selection, when no further improvement in survival rate was observed, 10 colonies were picked and their DNA were sequenced. Invariably, each of the surviving mutants contained arginine in place of glutamate at position 839 (Glu839). In addition, at position 840, Glu840 was replaced with either tyrosine, leusine, or histidine (Fig. 6). The activity of these variants was confirmed both by testing them individually in the survival assay against the −1 position mismatch ccdB plasmid, and via in vitro cleavage assays with purified variant AceCas9 proteins in complex with a guide RNA (Fig. 6). These variants, but not the WT AceCas9, yielded high cell survival rates in the presence ofa −1 position mismatch. This led us to hypothesize the role of charge and size in influencing AceCas9 sensitivity to mismatches at PAM-proximal locations (Hand et al., in review) (Fig. 6). Site-directed mutation of Glu840 to a smaller amino acid, alanine, indeed had a substantially reduced survival rate (Hand et al., in review) (Fig. 6). Removal of the negative charge from the phosphate lock residues significantly decreases sensitivity to the guide-DNA mismatches. An increase in size of the substituted residues further reduces the sensitivity to mismatches. Our findings identify the phosphate lock residues as an important site for tuning the specificity and catalytic efficiency of Cas9.
Fig. 6.
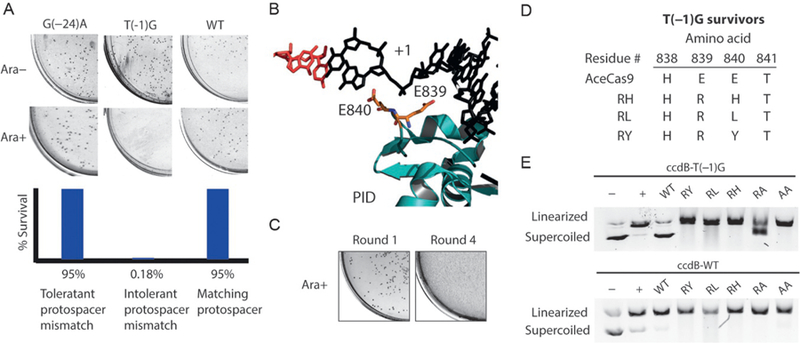
AceCas9 variants identified by directed evolution that impact protospacer recognition specificity. (A) Bacterial survival assay results and quantification for the wild-type AceCas9 targeting ccdB plasmids harboring no mismatches (WT), a PAM-proximal mismatch (T(– 1)G), and a PAM-distal mismatch (G(– 24)A). (B) Structural model of AceCas9 zoomed into the phosphate lock region (teal) around the first position of the target DNA strand (black). The two phosphate lock residues, Glu839 and Glu840, are represented by stick models. (C) Bacterial survival assay results after the first round and fourth round of survival with the phosphate lock library. (D) Resulting mutants obtained from iterative rounds of survival from the phosphate lock library against the ccdB T(– 1)G target. (E) DNA cleavage assay results of the wild-type and five phosphate lock variants on the wild-type (bottom) and the T(– 1)G protospacer (top) plasmids.
We believe that the described directed evolution method used in AceCas9 studies is well suited to explore the structural parameters that impact the specificity of other Cas9s. By identifying variants that are able to cleave altered protospacers, one can identify critical roles of Cas9 structural elements in each of the functional steps.
Acknowledgments
We thank all Li Lab members for helpful discussions.
Funding
This work was supported by NIH Grant R01 GM099604 to H.L.
References
- Anders C, Niewoehner O, Duerst A, & Jinek M (2014). Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature, 513, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EM, Haupt A, Schiel JA, Chou E, Machado HB, Strezoska Ž, et al. (2015). Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity. Journal of Biotechnology, 211, 56–65. [DOI] [PubMed] [Google Scholar]
- Barrangou R, & Doudna JA (2016). Applications of CRISPR technologies in research and beyond. Nature Biotechnology, 34, 933–941. [DOI] [PubMed] [Google Scholar]
- Barrangou R, & Marraffini LA (2014). CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Molecular Cell, 54, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaria N, Jarmoskaite I, & Herschlag D (2017). Lessons from enzyme kinetics reveal specificity principles for RNA-guided nucleases in RNA interference and CRISPR-based genome editing. Cell Systems, 4, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EA, Andreasson JOL, Chircus LM, Sternberg SH, Wu MJ, Guegler CK, et al. (2017). High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding. Proceedings of the National Academy of Sciences of the United States of America, 114, 5461–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier E, Richter H, van der Oost J, & White MF (2015). Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiology Reviews, 39, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, et al. (2017). Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature, 550, 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, & Zhao H (2005). A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Research, 33, e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. (2014). Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Research, 24, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylinski K, Le Rhun A, & Charpentier E (2013). The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biology, 10, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, et al. (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature, 471, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, & Church GM (2013). Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Research, 41, 4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, et al. (2014). Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circulation Research, 115, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, & Church GM (2013). Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nature Methods, 10, 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lecrivain AL, Bzdrenga J, et al. (2014). Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Research, 42, 2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, & Alt FW (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology, 33, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology, 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Yang H, Rajashankar KR, Huang Z, & Patel DJ (2016). Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Research, 26, 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Doval C, & Jinek M (2017). Molecular architectures and mechanisms of class 2 CRISPR-associated nucleases. Current Opinion in Structural Biology, 47, 157–166. [DOI] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. (2017). Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature, 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Yu HH, Johnson KA, & Taylor DW (2018). DNA unwinding is the primary determinant of CRISPR-Cas9 activity. Cell Reports, 22, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science, 356, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Slivano OJ, Christie CK, Cheng AW, & Miano JM (2015). CRISPR-Cas9 genome editing of a single regulatory element nearly abolishes target gene expression in mice—Brief report. Arteriosclerosis, Thrombosis, and Vascular Biology, 35, 312–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TH, Das A, Roth MO, Smith CL, Jean-Baptiste UL, & Li H Phosphate lock residues of Acidothermus cellulolyticus Cas9 are critical to its substrate specificity, ACS Synthetic Biology (in review). [DOI] [PMC free article] [PubMed]
- Harrington LB, Paez-Espino D, Staahl BT, Chen JS, Ma E, Kyrpides NC, et al. (2017). A thermostable Cas9 with increased lifetime in human plasma. Nature Communications, 8, 1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Wright AV, Vucelja M, Bikard D, Doudna JA, & Marraffini LA (2017). Mutations in Cas9 enhance the rate of acquisition of viral spacer sequences during the CRISPR-Cas immune response. Molecular Cell, 65, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano H, Gootenberg JS, Horii T, Abudayyeh OO, Kimura M, Hsu PD, et al. (2016). Structure and engineering of Francisella novicida Cas9. Cell, 164, 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Nishimasu H, Ishitani R, & Nureki O (2016). Structural basis for the altered PAM specificities of engineered CRISPR-Cas9. Molecular Cell, 61, 886–894. [DOI] [PubMed] [Google Scholar]
- Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, et al. (2013). Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proceedings of the National Academy of Sciences of the United States of America, 110, 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, et al. (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature, 556, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huai C, Li G, Yao R, Zhang Y, Cao M, Kong L, et al. (2017). Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nature Communications, 8, 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Taylor DW, Chen JS, Kornfeld JE, Zhou K, Thompson AJ, et al. (2016). Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science, 351, 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Zhou K, Ma L, Gressel S, & Doudna JA (2015). A Cas9-guide RNA complex preorganized for target DNA recognition. Science, 348, 1477–1481. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, & Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, et al. (2014). Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science, 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvelis T, Gasiunas G, Young J, Bigelyte G, Silanskas A, Cigan M, et al. (2015). Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biology, 16, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, et al. (2017). In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nature Communications, 8, 14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature, 529, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, & Liu DR (2017). CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell, 169, 559. [DOI] [PubMed] [Google Scholar]
- Koonin EV, & Makarova KS (2009). CRISPR-Cas: An adaptive immunity system in prokaryotes. F1000 Biology Reports, 1, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, & Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Current Opinion in Microbiology, 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Cradick TJ, & Bao G (2016). The Neisseria meningitidis CRISPR-Cas9 system enables specific genome editing in mammalian cells. Molecular Therapy, 24, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim YH, et al. (2018). Directed evolution of CRISPR-Cas9 to increase its specificity. Nature Communications, 9, 3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Yu J, Hwang GH, Kim S, Kim HS, Ye S, et al. (2017). CUT-PCR: CRISPR-mediated, ultrasensitive detection of target DNA using PCR. Oncogene, 36, 6823–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, et al. (2016). Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Molecular Cell, 62, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KM, & Ke A (2017). Building the class 2 CRISPR-Cas arsenal. Molecular Cell, 65, 377–379. [DOI] [PubMed] [Google Scholar]
- Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, & Olson EN (2014). Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science, 345, 1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma E, Harrington LB, O’Connell MR, Zhou K, & Doudna JA (2015). Single-stranded DNA cleavage by divergent CRISPR-Cas9 enzymes. Molecular Cell, 60, 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Zhang F, & Koonin EV (2017). SnapShot: Class 2 CRISPR-Cas systems. Cell, 168, 328–328.e1. [DOI] [PubMed] [Google Scholar]
- Marraffini LA (2015). CRISPR-Cas immunity in prokaryotes. Nature, 526, 55–61. [DOI] [PubMed] [Google Scholar]
- Mir A, Edraki A, Lee J, & Sontheimer EJ (2018). Type II-C CRISPR-Cas9 biology, mechanism, and application. ACS Chemical Biology, 13, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, & van der Oost J (2016). Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science, 353, aad5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Cong L, Yan WX, Ran FA, Zetsche B, Li Y, et al. (2015). Crystal structure of Staphylococcus aureus Cas9. Cell, 162, 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell, 156, 836–843. [DOI] [PubMed] [Google Scholar]
- Nunez JK, Bai L, Harrington LB, Hinder TL, & Doudna JA (2016). CRISPR immunological memory requires a host factor for specificity. Molecular Cell, 62, 824–833. [DOI] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature, 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper AT, Stephenson AA, & Suo Z (2018). Functional insights revealed by the kinetic mechanism of CRISPR/Cas9. Journal of the American Chemical Society, 140, 2971–2984. [DOI] [PubMed] [Google Scholar]
- Sander JD, & Joung JK (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nature Biotechnology, 32, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, et al. (2013). Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Research, 23, 720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Jiang F, Liu JJ, Bray NL, Rauch BJ, Baik SH, et al. (2017). Disabling Cas9 by an anti-CRISPR DNA mimic. Science Advances, 3, e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, et al. (2017). Diversity and evolution of class 2 CRISPR-Cas systems. Nature Reviews. Microbiology, 15, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Sternberg SH, Fei J, Doudna JA, & Ha T (2016). Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nature Communications, 7, 12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, & Zhang F (2016). Rationally engineered Cas9 nucleases with improved specificity. Science, 351, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargon AA, Cox DBT, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS, et al. (2017). Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Molecular Cell, 65, 618–630. e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Lawrence CM, & Wiedenheft B (2013). CRISPR-mediated adaptive immune systems in bacteria and archaea. Annual Review of Biochemistry, 82, 237–266. [DOI] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, & Doudna JA (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature, 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong A, & Musunuru K (2017). Genome editing in cardiovascular diseases. Nature Reviews. Cardiology, 14, 11–20. [DOI] [PubMed] [Google Scholar]
- Sung K, Park J, Kim Y, Lee NK, & Kim SK (2018). Target specificity of Cas9 nuclease via DNA rearrangement regulated by the REC2 domain. Journal of the American Chemical Society, 140, 7778–7781. [DOI] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology, 33, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui TKM, Hand TH, Duboy EC, & Li H (2017). The impact of DNA topology and guide length on target selection by a cytosine-specific Cas9. ACS Synthetic Biology, 6, 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui TK, & Li H (2015). Structure principles of CRISPR-Cas surveillance and effector complexes. Annual Review of Biophysics, 44, 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Du Y, Shen B, Zhou X, Li J, Liu Y, et al. (2015). Efficient generation of gene-modified pigs via injection of zygote with Cas9/sgRNA. Scientific Reports, 5, 8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, La Russa M, & Qi LS (2016). CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry, 85, 227–264. [DOI] [PubMed] [Google Scholar]
- Yamada M, Watanabe Y, Gootenberg JS, Hirano H, Ran FA, Nakane T, et al. (2017). Crystal structure of the minimal Cas9 from Campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Molecular Cell, 65, 1109–1121. e1103. [DOI] [PubMed] [Google Scholar]
- Yang M, Peng S, Sun R, Lin J, Wang N, & Chen C (2018). The conformational dynamics of Cas9 governing DNA cleavage are revealed by single-molecule FRET. Cell Reports, 22, 372–382. [DOI] [PubMed] [Google Scholar]
- Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, et al. (2017). Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nature Biotechnology, 35, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W (2017). Probing the structural dynamics of the CRISPR-Cas9 RNA-guided DNA-cleavage system by coarse-grained modeling. Proteins, 85, 342–353. [DOI] [PubMed] [Google Scholar]