

Abstract
Machine learning (ML) is a useful tool for advancing our understanding of the patterns and significance of biomedical data. Given the growing trend on the application of ML techniques in precision medicine, here we present an ML technique which predicts the likelihood of complete remission (CR) in patients diagnosed with acute myeloid leukemia (AML). In this study, we explored the question of whether ML algorithms designed to analyze gene-expression patterns obtained through RNA sequencing (RNA-seq) can be used to accurately predict the likelihood of CR in pediatric AML patients who have received induction therapy. We employed tests of statistical significance to determine which genes were differentially expressed in the samples derived from patients who achieved CR after 2 courses of treatment and the samples taken from patients who did not benefit. We tuned classifier hyperparameters to optimize performance and used multiple methods to guide our feature selection as well as our assessment of algorithm performance. To identify the model which performed best within the context of this study, we plotted receiver operating characteristic (ROC) curves. Using the top 75 genes from the k-nearest neighbors algorithm (K-NN) model (K = 27) yielded the best area-under-the-curve (AUC) score that we obtained: 0.84. When we finally tested the previously unseen test data set, the top 50 genes yielded the best AUC = 0.81. Pathway enrichment analysis for these 50 genes showed that the guanosine diphosphate fucose (GDP-fucose) biosynthesis pathway is the most significant with an adjusted P value = .0092, which may suggest the vital role of N-glycosylation in AML.
Keywords: acute Myeloid Leukemia (AML), machine Learning (ML), gene expression profiling, remission induction
Introduction
RNA sequencing (RNA-seq) and other high-throughput next-generation sequencing platforms have emerged as powerful approaches for discovering pathogenic pathways and potential targets for clinical intervention in patients with acute myeloid leukemia (AML).1 Using whole-transcriptome sequencing, our previous work compared the profiles of core-binding factor acute myeloid leukemia (CBF-AML) cases with those characterized by normal karyotype (NK), illuminating similarities and differences with respect to gene-expression signatures and splicing events as well as RNA fusions that typify the inv(16) vs the t(8;21) AML subtypes.2
In concert with the rise of large-scale omics-oriented sequencing, machine-learning (ML) algorithms have increasingly been applied to gene-expression analysis aimed at classifying tumors, predicting survival, identifying therapeutic targets, and classifying genes according to function.3–7 Significant results have been shown for predicting outcomes of large B-cell lymphoma,8 hepatitis B virus–positive metastatic hepatocellular carcinomas9 as well as documenting diverse pathologic responses to chemotherapy in patients with breast cancer.10 Using gene-expression profiling of data generated by microarrays in conjunction with both supervised and unsupervised learning, Bullinger et al11 identified prognostic subclasses in adult AML; the research group also constructed an optimal 133-gene predictor of overall survival. Yeoh et al12 performed classification, subtype discovery, and outcome prediction in patients with pediatric acute lymphoblastic leukemia (ALL). However, no previous study has specifically addressed expression differences among large cohorts of pediatric and young-adult AML patients with regard to complete remission (CR). In this study, we compare pre-treatment gene-expression profiles using 3 supervised learning algorithms to discover predictors of CR.
Materials and Methods
We obtained 473 bone marrow specimens from 473 patients, both children and young adults with ages ranging between 8 days and 28 years who had been diagnosed with de novo AML. For comparison, we acquired an additional 20 bone marrow specimens from normal, healthy individuals. All samples were obtained by written consent from the parents/guardians of minors from the Children’s Oncology Group clinical trial AAML1031. The Institutional Review Board at Fred Hutchinson Cancer Research Center has reviewed and approved this study. It is filed under Institutional Review File #9950 (Biology of the Alterations of the Signal Transduction Pathway in Pediatric Cancer). The number of samples with clinical information regarding CR used in this study was 414. RNA sequencing was performed on all 493 samples using the Illumina platform HiSeq2000 (https://www.illumina.com). Reads were mapped to Ensembl Gene IDs (http://useast.ensembl.org/), which belong to 31 biotypes, including protein-coding sequences, non-coding sequences, and pseudogenes, among others. RPKM (reads per kilobase per million mapped reads) values were calculated for each gene. Genes that had a count of at least 1 per million (CPM) in at least 3 samples were retained. Quantile normalization was applied among all samples. Python library scikit-learn (http://scikit-learn.org/stable/) modules of commonly used statistical models and algorithms were directly implemented in the scripts. Gene set enrichment analysis (GSEA) was performed using the online tool Enrichr (http://amp.pharm.mssm.edu/Enrichr/), as well as our in-house OmicPath (v 0.1) R package.
Violin plots showing gene-expression distribution patterns were generated using the in-house OmicPlot (v 0.1) R package.
Feature selection
Principal components analysis (PCA) was performed to examine the general pattern of the data, remove outliers, and select algorithms appropriate for our data.
RNA sequencing expression data of m samples by n genes were used as inputs and learn the mapping using
Samples were divided into a training set (N = 331) and a test set (N = 83). Three classifiers—k-nearest neighbors algorithm (K-NN), support vector machine (SVM), and random forest (RF)—were applied to select features for the training set via 5-fold cross-validation. With the features selected, the classifier was tested on the same training set (N = 331). The classifier with the best performance was then tested on the remaining test set (N = 83).
K-NN classifier
We performed 100 iterations of a 5-fold cross-validation. In each fold, we first carried out a t test for initial feature selection to identify the 100 most statistically significant genes, ie, those that were the most differentially expressed between the CR (positive class) and non-complete remission (NCR) (negative class). We found that using more than 100 genes did not improve performance. For further feature selection out of the genes identified by t testing, we compared the performance of 2 algorithms: Hill Climbing13 (sequential feature addition) and Randomized Lasso14 (using the model’s feature weights as ranks and selecting the highest ranking feature). At each fold, an area under the curve (AUC) was computed using a selected subset of genes and the fold’s validation set. Following the 100 iterations, the features (genes) were ranked by the average of AUCs computed using those genes across different folds. Essentially, the genes that on average helped yield the best AUCs were ranked highest.
SVM classifier
To overcome the issue of class imbalance, downsampling was applied,15–17 ie, a smaller subset of 114 samples—N(CR) = 57, N(NCR) = 57; 91 for the training set and 23 for the test set—was used as input for the SVM classifier. Processes similar to those described above for K-NN were applied to SVM classifiers with 1 exception: we used a third method Recursive Feature Elimination for the second feature selection in addition to Hill Climbing and Randomized Lasso.
RF classifier
We trained RF classifiers using scikit-learn’s ensemble.RandomForestClassifier module. To select parameters for the RF classifier, we performed a grid search for the following parameters: number of trees (estimators), maximum number of features, and maximum tree depth. The remaining parameters were set to their default values. Then, the optimal parameters in terms of AUC were selected together with the best performing feature selection approach. For feature selection, we performed a comparison between 2 approaches: (1) nested 5-fold with built-in RF feature selection—we trained the classifier on 4/5 of the training set using the built-in “feature importance” attribute to rank the features (genes). Those genes were then used a second time on the same 4/5 of the training set. We then tested the classifier on the remaining 1/5 of the training set (ie, validation set) to assess performance. (2) We carried out 100 iterations of a 5-fold cross-validation while aggregating the feature importance values computed at each fold. We then computed a Spearman correlation between each gene’s importance values and the AUC computed in each fold. We used the genes with the highest correlation scores to train on the same 4/5 training set and then tested the method on the remaining validation set.
Following feature selection
At the end of each feature selection, the same cross-validation procedure was employed to generate the AUC results when testing the validation set. The final AUC result of the (chosen) K-NN classifier was a simple, 1-episode period of training on the training set with the selected genes followed by testing on the unseen test set with the same selected genes.
Results
According to PCA based on all genes, these 414 AML samples with clinical information regarding CR did not cluster by CR or NCR status, nor by age/year of diagnosis. There is no obvious outliers, so all of them were included in this study.
Area-under-the curve results from different K values were used to estimate optimal K for the K-NN classifier. Figure 1 shows that statistically significant genes identified from the t test can help improve the AUC results and that K = 27 yielded the best average AUC. With the optimal K = 27, receiver operating characteristic (ROC) curves were produced using 2 feature selection methods: Hill Climbing and Randomized Lasso (Figure 2). Overall, the Hill Climbing resulted in better results with the best AUC = 0.84.
Figure 1.
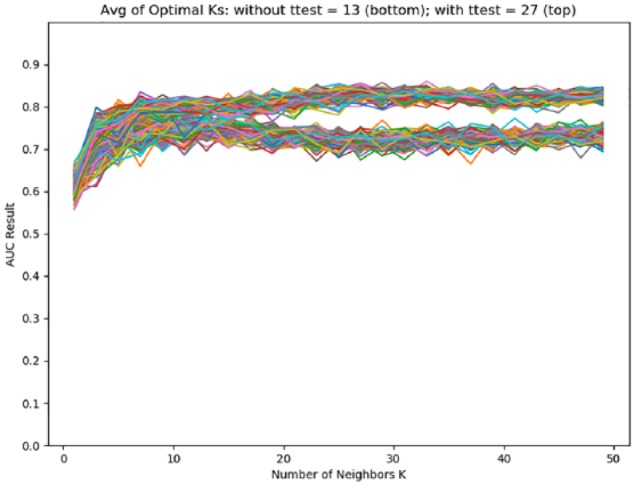
Area under the curves from different Ks used to estimate an optimal K value for K-NN classifier. AUC indicates area under the curve; K-NN, k-nearest neighbors algorithm.
Figure 2.
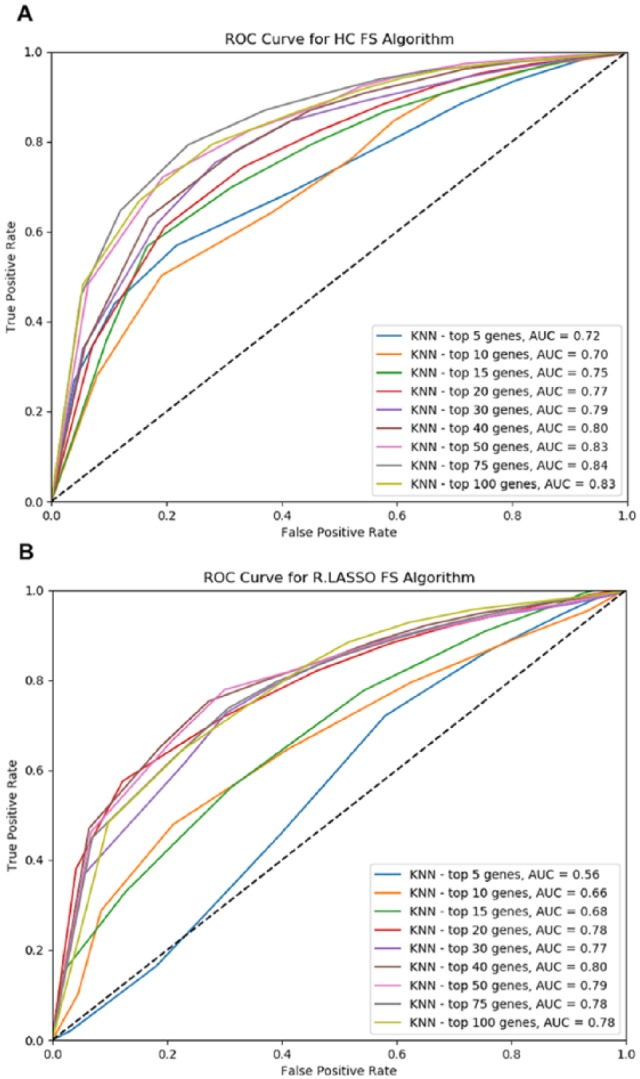
Receiver operating characteristic curves of K-NN (with the optimal K = 27) using 2 feature selection methods: (A) Hill Climbing and (B) Randomized Lasso. K-NN indicates k-nearest neighbors algorithm; ROC, receiver operating characteristic; FS, feature selection; HC, Hill Climbing; R.LASSO, Randomized Lasso.
To compare the performance of K-NN and SVM classifier, the balanced data set with N(CR) = 57 and N(NCR) = 57 was split into training set (N = 91) and the test set (N = 23). Using a 5-fold cross-validation performed on the training set, ROC curves of K-NN and SVM algorithms were calculated using 3 feature-election methods: Hill Climbing, Recursive Feature Elimination, and Randomized Lasso. The K-NN outperformed SVM, and Hill Climbing still resulted in better AUC results for K-NN (Supplemental Figure S1).
Hyperparameter tuning for RF suggested using 100 trees to have the best performance (AUC = 0.74). The simple method resulted in better results with the best (training set) AUC = 0.73 compared with the more complex approach (Supplemental Figure S2).
Based on the above observations, K-NN with Hill Climbing performed the best on the training data (N = 331), yielding an AUC score of 0.84. When we tested this model on the remaining 1/5 of the data (N = 83), using the top 50 genes with the best AUC scores from the training set yielded an AUC score of 0.81 (Figure 3).
Figure 3.
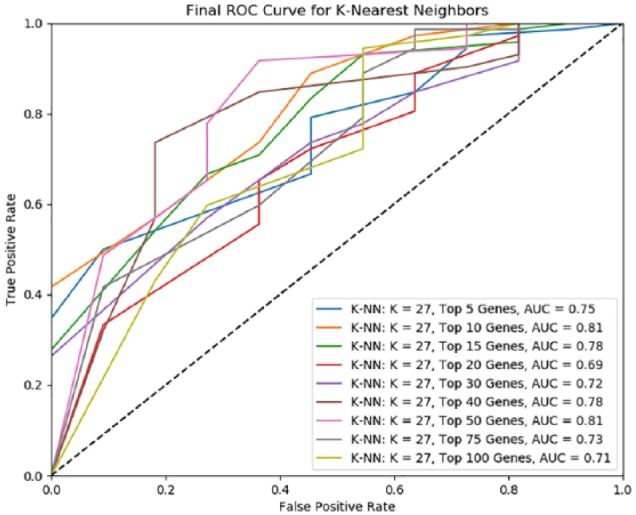
Final K-NN model performance on test data (N = 83). ROC indicates receiver operating characteristic.
Based on using these top 50 genes, our GSEA analysis using OmicPath showed that BATF (basic leucine zipper transcriptional factor ATF-like) and RAC2 (Ras-related C3 botulinum toxin substrate 2) are related to a decreased IgM (Immunoglobulin M) level with FDR (false discovery rate) =0.0073. TSTA3 (GDP-l-fucose synthase) and RAC2 are related to an increased neutrophil cell number (FDR = 0.0073). Pathway enrichment analysis using Enrichr showed that TSTA3 and FPGT (fucose-1-phosphate guanylyltransferase) were mapped to the GDP-fucose biosynthesis pathway (Reactome 2016; https://reactome.org) with an adjusted P value of .0092. These 2 genes were also mapped to the pathway’s parent terms “Synthesis of substrates in N-glycan biosynthesis” and “Biosynthesis of the N-glycan precursor (dolichol lipid-linked oligosaccharide, LLO) and transfer to a nascent protein.” This indicates the vital role of N-glycosylation in AML pathology and patient prognosis. The expression of these top 50 genes compared with normal bone marrow (NBM) samples are shown in violin plot (Figure 4).
Figure 4.

Expressions of top 50 genes with the best AUC scores from K-NN with Hill Climbing. AML indicates acute myeloid leukemia; AUC, area under the curve; CR, complete remission; K-NN, k-nearest neighbors algorithm; NBM, normal bone marrow; NCR, non-complete remission; RPKM, reads per kilobase per million.
Discussion
This study explored and evaluated different ML algorithms for predicting CR in AML patients based on their pre-treatment gene-expression signatures. It revealed a significant underlying genetic difference between patients with contrasting outcomes following treatment. Gene set enrichment analysis results highlighted specific biological features that carry prognostic value for further exploration. For example, low IgM and leukocyte count >50 × 109/liter have been demonstrated as 2 of the adverse predictors for the duration of complete continuous remission in childhood ALL.18 Fucose-containing glycans play important roles in selectin-mediated leukocyte-endothelial adhesion as well as various immunity and signaling processes. Alterations in expression or structure of fucosylated oligosaccharides have also been observed in cancer pathology. Conditional impairment in fucosylated glycan expression in mice exhibited altered myeloid development including aberrant proliferation of myeloid progenitors and an increased production of granulocytes which leads to neutrophilia. The loss of AB blood group antigen expression along with the increases in H and Lewisy expression are associated with poor prognosis. Increased expression of Lewisx/a structures, Tn/sialyl-Tn/T antigens, and β1,6 GlcNAc branching of N-linked core structures were observed in advanced cancers and related with poor prognosis.19–22 This information may help physicians select more suitable courses of treatment, whether the treatment be more aggressive chemotherapy or an altogether novel alternative therapy.
Supplemental Material
Supplemental material, Predict_CR_AML_Supplemental_xyz13921555f86f3 for Predicting Complete Remission of Acute Myeloid Leukemia: Machine Learning Applied to Gene Expression by Ophir Gal, Noam Auslander, Yu Fan and Daoud Meerzaman in Cancer Informatics
Acknowledgments
The authors wish to acknowledge Dr Meshinchi’s group from Fred Hutchinson Cancer Research Center for data provision and Laura K Fleming, PhD, for editorial critique.
Footnotes
Declaration of conflicting interest:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:The author(s) received no financial support for the research, authorship, and/or publication of this article.
Author Contributions: OG and NA planned the analyses; OG performed analyses and provided the guidelines for the manuscript. YF wrote the manuscript. OG and NA edited the manuscript. DM provided advice for the manuscript. All authors reviewed the manuscript.
Data Availability: All data used or generated during this study have been deposited at the database of Genotypes and Phenotypes (dbGaP, http://www.ncbi.nlm.nih.gov/gap) under study accession phs000465.
Supplemental material: Supplemental material for this article is available online.
ORCID iD: Yu Fan  https://orcid.org/0000-0002-7473-6104
https://orcid.org/0000-0002-7473-6104
References
- 1. Tarlock K, Meshinchi S. Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am. 2015;62:75–93. [DOI] [PubMed] [Google Scholar]
- 2. Hsu CH, Nguyen C, Yan C, et al. Transcriptome profiling of pediatric core binding factor AML. PLoS ONE. 2015;10:e0138782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan AC, Gilbert D. Ensemble machine learning on gene expression data for cancer classification. Appl Bioinformatics. 2003;2:S75–S83. [PubMed] [Google Scholar]
- 4. Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16:906–914. [DOI] [PubMed] [Google Scholar]
- 5. Bair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2:E108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee JS, Thorgeirsson SS. Genome-scale profiling of gene expression in hepatocellular carcinoma: classification, survival prediction, and identification of therapeutic targets. Gastroenterology. 2004;127:S51–S55. [DOI] [PubMed] [Google Scholar]
- 7. Brown MP, Grundy WN, Lin D, et al. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A. 2000;97:262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shipp MA, Ross KN, Tamayo P, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8:68–74. [DOI] [PubMed] [Google Scholar]
- 9. Ye QH, Qin LX, Forgues M, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–423. [DOI] [PubMed] [Google Scholar]
- 10. Ayers M, Symmans WF, Stec J, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–2293. [DOI] [PubMed] [Google Scholar]
- 11. Bullinger L, Dohner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–1616. [DOI] [PubMed] [Google Scholar]
- 12. Yeoh EJ, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–143. [DOI] [PubMed] [Google Scholar]
- 13. Tsamardinos I, Brown LE, Aliferis CF. The max-min hill-climbing Bayesian network structure learning algorithm. Mach Learn. 2006;65:31. [Google Scholar]
- 14. Meinshausen N. Stability selection. J Roy Statistical Society B. 2010;72:417–473. [Google Scholar]
- 15. Longadge R, Dongre SS, Malik L. Class imbalance problem in data mining: review. Int J Comput Sci Netw. 2013;2:84. [Google Scholar]
- 16. Galar M, Fernandez A, Barrenechea E, Bustince H, Herrera F. A review on ensembles for the class imbalance problem: bagging-, boosting-, and hybrid-based approaches. IEEE T Syst Man Cyb: Part C. 2012;42:463–484. [Google Scholar]
- 17. Drummond C, Holte RC. C4.5, class imbalance, and cost sensitivity: why under-sampling beats over-sampling. In: Proceedings of the workshop on learning from imbalanced data sets II, international conference on machine learning; August 21, 2003; Washington, DC. [Google Scholar]
- 18. Miller DR, Leikin S, Albo V, Sather H, Karon M, Hammond D. Prognostic factors and therapy in acute lymphoblastic leukemia of childhood: CCG-141. A report from childrens cancer study group. Cancer. 1983;51:1041–1049. [DOI] [PubMed] [Google Scholar]
- 19. Becker DJ, Lowe JB. Fucose: biosynthesis and biological function in mammals. Glycobiology. 2003;13:41R–53R. [DOI] [PubMed] [Google Scholar]
- 20. Smith PL, Myers JT, Rogers CE, et al. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 2002;158:801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim YJ, Varki A. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj J. 1997;14:569–576. [DOI] [PubMed] [Google Scholar]
- 22. Orntoft TF, Vestergaard EM. Clinical aspects of altered glycosylation of glycoproteins in cancer. Electrophoresis. 1999;20:362–371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Predict_CR_AML_Supplemental_xyz13921555f86f3 for Predicting Complete Remission of Acute Myeloid Leukemia: Machine Learning Applied to Gene Expression by Ophir Gal, Noam Auslander, Yu Fan and Daoud Meerzaman in Cancer Informatics