

Abstract
Novel Ca2+‐independent C‐type lectins, SPL‐1 and SPL‐2, were purified from the bivalve Saxidomus purpuratus. They are composed of dimers with either identical (SPL‐2 composed of two B‐chains) or distinct (SPL‐1 composed of A‐ and B‐chains) polypeptide chains, and show affinity for N‐acetylglucosamine (GlcNAc)‐ and N‐acetylgalactosamine (GalNAc)‐containing carbohydrates, but not for glucose or galactose. A database search for sequence similarity suggested that they belong to the C‐type lectin family. X‐ray crystallographic analysis revealed definite structural similarities between their subunits and the carbohydrate‐recognition domain (CRD) of the C‐type lectin family. Nevertheless, these lectins (especially SPL‐2) showed Ca2+‐independent binding affinity for GlcNAc and GalNAc. The crystal structure of SPL‐2/GalNAc complex revealed that bound GalNAc was mainly recognized via its acetamido group through stacking interactions with Tyr and His residues and hydrogen bonds with Asp and Asn residues, while widely known carbohydrate‐recognition motifs among the C‐type CRD (the QPD [Gln‐Pro‐Asp] and EPN [Glu‐Pro‐Asn] sequences) are not involved in the binding of the carbohydrate. Carbohydrate‐binding specificities of individual A‐ and B‐chains were examined by glycan array analysis using recombinant lectins produced from Escherichia coli cells, where both subunits preferably bound oligosaccharides having terminal GlcNAc or GalNAc with α‐glycosidic linkages with slightly different specificities.
Keywords: bivalve, carbohydrate, C‐type lectin, X‐ray crystallographic analysis
Short abstract
Abbreviations
- CHES
N‐Cyclohexyl‐2‐aminoethanesulfonic acid
- CRD
carbohydrate‐recognition domain
- DLS
dynamic light scattering
- EDTA
ethylenediamine tetraacetate
- TBS
Tris‐buffered saline
- PEG
polyethylene glycol
- PVDF
polyvinylidene difluoride
- SAD
single‐wavelength anomalous diffraction
Introduction
Animal lectins play important roles in various molecular recognition processes. They are grouped into several families based on amino acid sequence homologies.1, 2 Among them, C‐type lectins constitute a major lectin family, commonly containing C‐type carbohydrate‐recognition domains (CRDs) composed of 110–130 amino acid residues.3, 4 While C‐type CRDs are well known to be involved in various molecular recognition processes in vertebrates, especially as receptors for surface molecules of foreign pathogens,5 those in invertebrates have also been found to play important roles in recognition of foreign microorganisms in either soluble or membrane‐bound form. In contrast to vertebrate C‐type lectins, most of which are composed of multiple domains, invertebrate C‐type lectins are generally smaller consisting only of C‐type CRD in oligomeric forms. Evidence for their involvement in innate immune systems has recently accumulated,6, 7 whereas detailed information on their structures and functions is very limited.
We have been investigating several marine invertebrate lectins, including C‐type lectins.8, 9 During these studies, we found four Ca2+‐dependent, galactose/N‐acetylgalactosamine (GalNAc)‐specific lectins (CEL‐I–CEL‐IV) in the sea cucumber Cucumaria echinata. Among them, CEL‐I and CEL‐IV are C‐type lectins composed of dimer and tetramer C‐type CRDs, respectively. They recognize specific carbohydrates through coordinate bonds with bound Ca2+ ions and hydrogen bond networks with nearby amino acid residues.10, 11 Carbohydrate specificities of galactose‐ or mannose‐binding C‐type CRDs are generally associated with three‐amino‐acid motifs, the QPD (Gln‐Pro‐Asp) and EPN (Glu‐Pro‐Asn) sequences, respectively. In the case of C. echinata lectins, this rule is applicable to CEL‐I, which contains the QPD motif in its binding site, though CEL‐IV uses the EPN motif to recognize galactose by changing the orientation of the bound galactose through a stacking interaction with a tryptophan residue.10 On the other hand, CEL‐III is a Ca2+‐dependent hemolytic lectin, which lyses erythrocytes and other susceptible cells by forming transmembrane pores after binding to cell surface carbohydrate chains.9, 12, 13 Although CEL‐III has two ricin‐type (R‐type) CRDs, instead of a C‐type CRD,14 its binding mode is very similar to that of C‐type lectins, which use coordinate bonds with Ca2+ along with a hydrogen bond network with nearby residues. Recently, we determined the crystal structure of another lectin, CGL1, in the pacific oyster Crassostrea gigas,15 which recognizes mannose monomer with very high specificity. This lectin is composed of two identical subunits, each of which contains two carbohydrate binding sites. The amino acid sequence of the subunit shows sequence similarity between its N‐ and C‐terminal halves, suggesting that it has been evolved by gene duplication. However, a structurally similar lectin has not been found in the database search.
As seen for the abovementioned lectins, marine invertebrates could be important resources for exploring novel lectins given their phylogenetic diversity. They are expected to provide information useful not only to understanding evolution of innate immunity, but also to engineering artificial molecular recognition proteins for practical uses. In the present study, we have isolated two lectins, SPL‐1 and SPL‐2, from the bivalve Saxidomus purpuratus, which recognize N‐acetylglucosamine (GlcNAc) and GalNAc, carbohydrates that are usually discriminated by ordinary lectins because of the different configurations of the hydroxyl groups. Here, we describe the structural and functional characterization of SPL‐1 and SPL‐2, including X‐ray crystallographic analysis. The crystal structures of the lectins reveal that they belong to the C‐type lectin family. However, their carbohydrate‐recognition mode is considerably different from those of typical C‐type CRDs, indicating architectural versatility of binding modes of marine invertebrate C‐type lectins.
Results
Purification of native S. purpuratus lectins and their carbohydrate‐binding properties
While isolation of GlcNAc‐specific lectins in S. purpuratus has been previously reported,16 there is only limited structural information beyond their amino acid compositions. In the present study, we tried to isolate and determine the structures of the lectins from S. purpuratus. As shown in Figure 1(A), when the crude extract from S. purpuratus was applied to the GlcNAc‐immobilized column (GlcNAc‐Cellufine) in the presence of 10 mM CaCl2, two protein fractions were serially obtained by elution with ethylenediamine tetraacetate (EDTA) and GlcNAc, suggesting that there are at least two types of GlcNAc‐binding lectins, which may be different in Ca2+‐dependency. These lectins were named SPL‐1 (S. purpuratus lectin‐1) and SPL‐2 (S. purpuratus lectin‐2), after separation by ion‐exchange chromatography on a HiTrap Q column [Fig. 1(B)]. As seen with the SDS‐PAGE gels [Fig. 1(C)], SPL‐2 showed a band at 27.6 kDa that was reduced to 15.3 kDa in the presence of 2‐mercaptoethanol. On the other hand, SPL‐1 showed a broadband around 23.4–33.7 kDa that was reduced to two distinct bands at 18.5 and 15.3 kDa in the presence of 2‐mercaptoethanol. These results suggest that they are composed of disulfide‐linked dimers of 15–18 kDa subunits. To determine the N‐terminal amino acid sequences of the lectins, the protein bands [a, b, and c in Fig. 1(C)] isolated on SDS‐PAGE under the reducing conditions were recovered after electroblotting onto a polyvinylidene difluoride (PVDF) membrane and subjected to N‐terminal amino acid sequence analysis. The resulting N‐terminal sequences of the bands a, b, and c were XXKXDXQSGW (band a), XXSEDDXPSG (band b), and XXSEDDXPSGWKF (band c), where “X” denotes no detection of amino acid. These results suggested that SPL‐1 is composed of two different chains (A‐ and B‐chains: band a and band b, respectively), while SPL‐2 is composed of two identical subunits (two B‐chains: band c).
Figure 1.
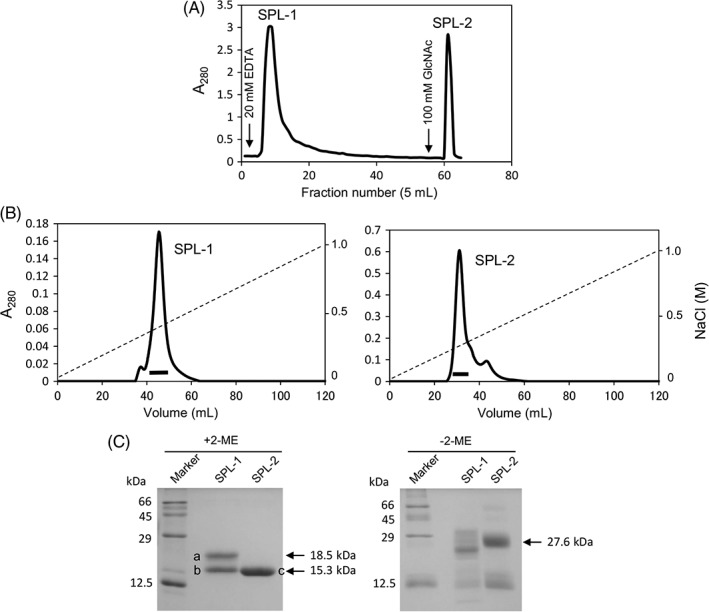
Purification of Saxidomus purpuratus lectins. (A) The crude extract of S. purpuratus was applied to the GlcNAc‐Cellufine column (1.7 × 3.0 cm) equilibrated with TBS containing 10 mM CaCl2. The adsorbed proteins were eluted with TBS containing 20 mM EDTA, followed by TBS containing 100 mM GlcNAc. (B) The eluted lectins (SPL‐1 and SPL‐2) were further separated on the HiTrap Q column (1.6 × 2.5 cm) with a linear gradient of NaCl. (C) Purified lectins were analyzed on SDS‐PAGE (12.5% gel) in the presence and absence of 2‐mercaptoethanol (2‐ME). The reduced subunits of the lectins marked a–c were subjected to N‐terminal amino acid sequence analysis after electroblotting onto PVDF membrane.
Isothermal titration calorimetry measurement for the carbohydrate binding of S. purpuratus lectins
Since hemagglutination of rabbit erythrocytes was not observed using SPL‐1 and SPL‐2 at the concentration of 1 mg/mL (approximately 6.7 μM) in Tris‐buffered saline (TBS) with 10 mM CaCl2 (data not shown), their carbohydrate‐binding activities were measured by isothermal titration calorimetry (ITC) (Fig. 2; Supporting Information Figure S1). As shown in Figure 2, both lectins, especially SPL‐2, showed higher association constants for GlcNAc than for GalNAc. Interestingly, it was significantly increased in the presence of Ca2+, although moderate affinity was observed even in the absence of Ca2+. These results appear to be consistent with the elution profiles of S. purpuratus Lectins (SPLs) from the GlcNAc‐affinity column [Fig. 1(A)] in which SPL‐2 strongly bound to the affinity column in the presence of EDTA and eluted only with GlcNAc‐containing buffer, while SPL‐1 eluted by EDTA and showed a retarded elution profile because of relatively weak interaction with the column in the absence of Ca2+.
Figure 2.
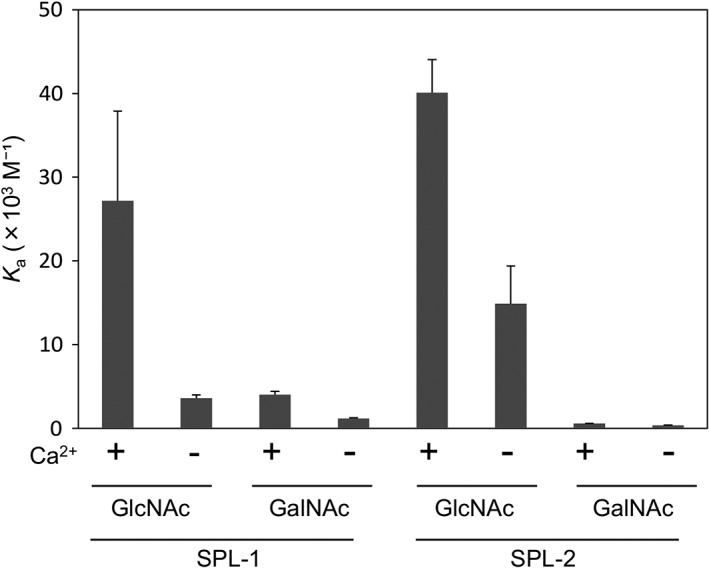
Association constants for the binding of GlcNAc and GalNAc to SPLs determined by ITC. The carbohydrate solutions were titrated into a temperature‐controlled sample cell containing SPL‐1 or SPL‐2 solution.
cDNA sequence determination
An 80 bp fragment of the cDNA for B‐chain was amplified by polymerase chain reaction (PCR) using the degenerate primers DF1 and DR1, which were designed based on the N‐terminal amino acid sequence of SPL‐2, which is composed of two B‐chains [Fig. 3(B)]. Based on the sequence of this fragment, F1 and R1 primers were designed, and 3′‐RACE and 5′‐RACE analyses were conducted with another primer R2. During the sequencing of the entire B‐chain cDNA, similar but distinct sequences corresponding to the A‐chain of SPL‐1 were also determined, and 3′‐RACE and 5′‐RACE analyses for the A‐chain cDNA was performed using the primers (F2, F3, F4, F5, R3, and F4) designed based on these sequences. Consequently, the complete sequences of the cDNAs for A‐chain (554 bp) and B‐chain (566 bp) were determined (Fig. 3) using the primers listed in Supporting Information Table S1 and define open reading frames corresponding to 158 and 159 amino acid residues, respectively, including signal sequences in the N‐terminal 21 residues. Thus, the mature A‐chain and B‐chain contain 137 and 138 amino acid residues with molecular masses of 15738.0 and 15755.2 Da, respectively. As shown in Figure 4, the alignment of the A‐ and B‐chains of SPL indicated high similarity with an identity of 68.6%. BLAST search on the UniProt database18 revealed that both chains show similarities with C‐type lectins (Fig. 5), although the similarities are relatively low; identities with these homologous proteins are below 40%. There are also conspicuous differences in that the widely shared carbohydrate‐binding motifs in C‐type lectins, QPD and EPN, are replaced by RPD (A‐chain) and KPD (B‐chain) in SPL (enclosed in a red box in Fig. 5), suggesting the SPLs recognize carbohydrates in a manner different from other ordinary C‐type lectins.
Figure 3.

The nucleotide and deduced amino acid sequences of the A‐chain (A) and B‐chain (B) of SPLs. The N‐terminal amino acid sequence of B‐chain determined from the purified proteins is indicated by dotted lines. Based on the sequence of the DNA fragment amplified with two degenerate primers, DF1 and DR1, forward primer F1 was designed, and used for 3′‐RACE of SPL‐2 cDNA. The N‐terminal amino acid of the mature protein is numbered as “+1.” The primers used for PCR are indicated by horizontal arrows. An asterisk indicates the stop codon.
Figure 4.

Comparison of the amino acid sequences of the A‐chain and B‐chain of SPLs. The alignment of the sequences was conducted by the Clustal Omega program.17 Asterisks, colons, and periods indicate the positions of identical, strongly similar, and weakly similar residues, respectively.
Figure 5.

Comparison of the amino acid sequences of SPL A‐ and B‐chains with other C‐type lectins. Alignment was conducted by the Clustal Omega program.17 The sequences are from the following species: A. limnaeus, galactose‐specific lectin nattectin (C‐type lectin)‐like protein (Austrofundulus limnaeus) (UniProt A0A2I4CJP3); M. galloprovincialis, C‐type lectin 5 (Mytilus galloprovincialis) (UniProt A0A0C5PT73); F. heteroclitus, lectin C‐type domain containing protein (Fundulus heteroclitus) (UniProt A0A146UDD9); C. teleta, Uncharacterized protein (fragment) (Capitella teleta) (UniProt R7VD19); A. mexicanus, (Astyanax mexicanus) (UniProt W5LBI3); Oreochromis niloticus, uncharacterized protein (Nematostella vectensis) (UniProt I3K683); O. aries, C‐type lectin domain containing 17A (Ovis aries) (UniProt W5P5A6); T. nigroviridis, uncharacterized protein (Tetraodon nigroviridis) (UniProt H3D482); CEL‐I, C‐type lectin from the sea cumber (Cucumaria echinata) (UniProt Q7M462).19 Asterisks, colons, and periods indicate the positions of identical, strongly similar, and weakly similar residues, respectively. Highly conserved cysteine residues, which form disulfide bonds, are enclosed in boxes.
Crystal structures of SPLs and their carbohydrate‐recognition mechanism
Crystallization of SPLs was performed using the proteins purified from S. purpuratus. As a result of screening the crystallization conditions, the crystals of SPL‐1 and SPL‐2/GalNAc complex were obtained, while crystallization of SPL‐1 complexed with specific carbohydrate has not been successful. Initial phasing of the diffraction data was determined for Pb‐derivative of SPL‐1 through the Pb‐SAD method. After that, refinement was performed using the diffraction data of native crystals of SPL‐1 collected at Photon Factory. On the other hand, the structure of SPL‐2/GalNAc complex was determined by the molecular replacement method using the structure of B‐chain of SPL‐1 as a search model. Finally, structures of SPL‐1 and SPL‐2/GalNAc complex were determined at resolutions of 1.6 and 2.0 Å, respectively (Supporting Information Table S2). Figure 6(A,B) shows dimer structures of SPL‐1 and SPL‐2/GalNAc complex. SPL‐1 is composed of two different polypeptides chains (A‐ and B‐chains), while SPL‐2 is composed of two B‐chains, as expected from the N‐terminal amino acid sequence analysis of the purified lectins. There is one Ca2+ ion bound in each subunit [magenta spheres in Fig. 6(AB)]. These Ca2+ ions are located apart from the carbohydrate‐binding sites, but may contribute to the stabilization of the proteins. These subunits are linked with two interchain disulfide bonds. In the case of SPL‐1, interchain disulfide bonds are formed between Cys2 from the A‐chain and Cys47 from the B‐chain, as well as Cys4 from the A‐chain and Cys1 from the B‐chain as illustrated in Supporting Information Figure S2. On the other hand, in SPL‐2, Cys1, and Cys47 from each chain are linked by interchain disulfide bonds. Four intrachain disulfide bonds are also present within both subunits. Only one free Cys residue (Cys130) is present in the B‐chain, where Ser129 is located as a corresponding residue in the A‐chain (Fig. 4). The intrachain disulfide bonds of Cys1‐Cys135 (A‐chain) and Cys2‐Cys136 (B‐chain) are unique for SPLs, compared with other ordinary C‐type CRDs.4
Figure 6.
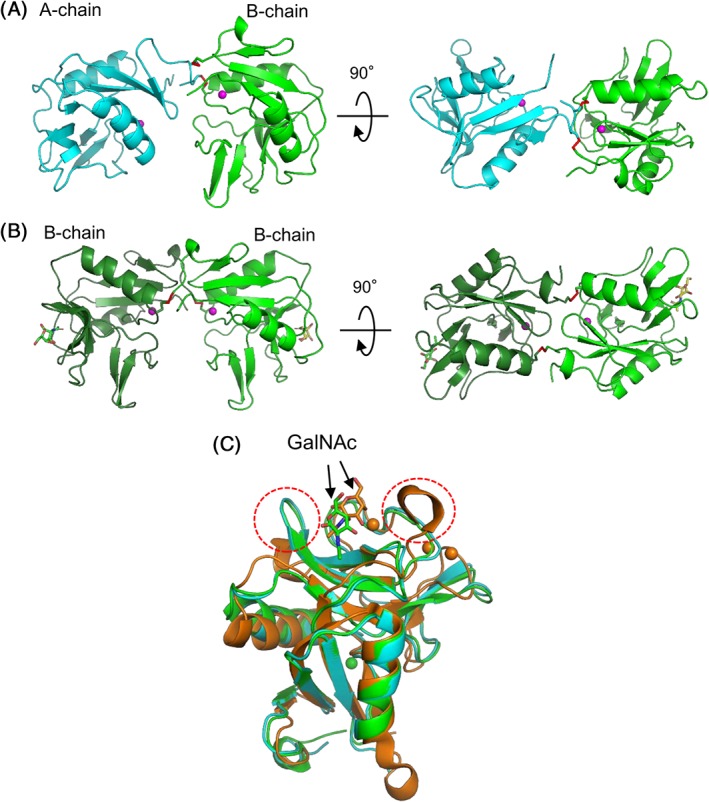
Overall structures of SPLs. (A) SPL‐1 composed of A‐chain (cyan) and B‐chain (green) (PDB ID: 6A7T). (B) SPL‐2/GalNAc complex composed of two B‐chains (PDB ID: 6A7S). (C) A‐ and B‐chains of SPLs and a subunit of C. echinata C‐type lectin, CEL‐I (PDB ID: 1WMZ) complexed with GalNAc,11 are superposed based on their main‐chain structure using PyMOL.40 Bound GalNAc molecules are depicted as stick models. Magenta spheres indicate bound Ca2+ ions.
As shown in Figure 6(C), when both subunits are superposed with a subunit of CEL‐I as an example of the C‐type CRD, high similarity was seen in their basic tertiary structures. On the other hand, marked differences were also observed in the loop region (enclosed in the red dotted circles) near the carbohydrate‐binding site of CEL‐I. Although CEL‐I, like other ordinary C‐type lectins, recognizes specific carbohydrates through coordinate bonds with Ca2+ ions and a hydrogen bond network with nearby residues [Fig. 7(A)], B‐chain of SPL‐2 mainly recognizes the acetamido group of GalNAc through stacking interactions with Tyr66 and His120, along with two hydrogen bonds with Asp106 and Asn118 [Fig. 7(B)] without Ca2+ ion. The 3‐OH of GalNAc also forms two hydrogen bonds with His120 and Asn118. In contrast to ordinary C‐type lectins such as CEL‐I, the 4‐OH of GalNAc, which has different configuration from that of GlcNAc, does not appear to participate in the binding. Therefore, it seems reasonable to infer that GlcNAc can bind to SPL‐2 in the same manner as GalNAc, albeit with different affinity. It is conceivable that the A‐chain of SPL‐1 recognizes the carbohydrates in the same manner as B‐chain, considering its similarity in the carbohydrate‐binding site [Fig. 7(C)] with that of B‐chain, though crystals of the SPL‐1/carbohydrate complex have not been obtained to confirm this. As seen in Figure 5, the binding motifs, QPD and EPN for canonical C‐type CRD, are replaced by RPD (A‐chain) and KPD (B‐chain) in SPLs. This fact also suggests similarities in the carbohydrate recognition modes between the A‐ and B‐chains. As seen in Figure 7(B) (Lys 97 to Asp99) and Figure 7(C) (Arg96 to Asp98), these tripeptides are distant from the binding site for GalNAc and do not have direct interaction with the carbohydrate.
Figure 7.
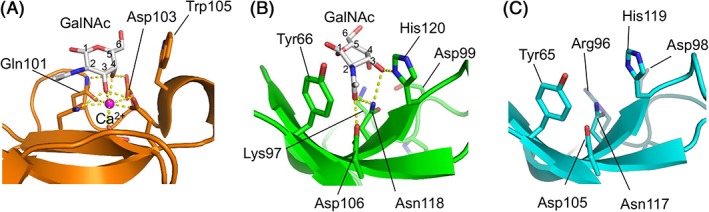
Comparison of the carbohydrate binding sites of CEL‐I/GalNAc complex (A), B‐chain of SPL‐1/GalNAc complex (B), and A‐chain of SPL‐1 (C). Hydrogen bonds and coordinate bonds are shown as yellow dotted lines.
Recombinant SPL subunits and their dimer formation
Recombinant A‐ and B‐chains of the SPLs were expressed in Escherichia coli cells to examine their carbohydrate‐binding properties and dimer formation abilities, as a homodimeric lectin composed of two A‐chains has not been isolated from S. purpuratus. Since the proteins were expressed as inclusion bodies, they were refolded after solubilization using guanidine hydrochloride. The refolding of the subunits was carried out with either single or mixed chains to produce homodimer or heterodimer lectins. As shown in the Supporting Information Figure S3(A), when the resulting proteins were applied to the GlcNAc‐immobilized column, they were bound to the column and eluted with GlcNAc, but not with EDTA. The constituent subunits of these proteins were confirmed on SDS‐polyacrylamide gel electrophoresis (PAGE) [Supporting Information Fig. S3(B)]. These results suggested that the recombinant proteins were correctly refolded and had active carbohydrate‐binding ability, while the A‐chain was not susceptible to elution by EDTA, in contrast to native SPL‐1, which was gradually eluted with EDTA [Fig. 1(A)]. The recombinant proteins were further purified by ion‐exchange chromatography on a HiTrap Q column [Supporting Information Fig. S3(C)]. The purified proteins containing B‐chain and A/B‐chains were mostly eluted as single symmetric peaks. However, refolded A‐chain was relatively unstable so as to form precipitate during multiple purification steps. Therefore, refolded A‐chain was used without purification by ion exchange chromatography hereafter. The molecular sizes of these proteins were examined by DLS. As shown in Figure 8, the hydrodynamic diameters of B‐chain and A/B‐chains were estimated to be 5.1 and 4.9 nm, respectively, corresponding to the size of dimer. By contrast, refolding of the A‐chain alone led to the formation of large particles (aggregates) with a diameter around 215 nm. These results indicate that B/B‐ and A/B‐chain dimers are preferably formed during folding process, but the A‐chain is unable to form a stable homodimer structure.
Figure 8.
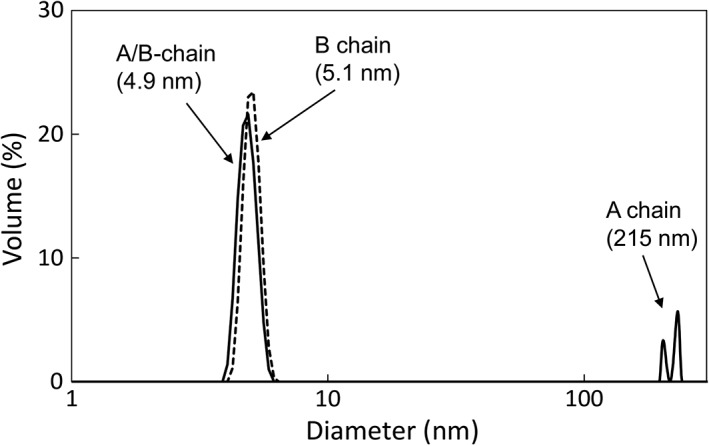
Size distribution of the recombinant chains of SPLs measured by DLS. Particle size of recombinant A‐chain, B‐chain, and mixed A‐ and B‐chains were measured in TBS at 25°C. The size distributions are expressed by volume.
Glycan array analysis of the recombinant A‐ and B‐chains
To elucidate detailed binding specificities of the SPLs for various oligosaccharides, the recombinant A‐ and B‐chains were subjected to glycan array analysis where the binding of the Cy3‐labeled lectins to various oligosaccharides immobilized on glass slides was measured by fluorescence intensity. As shown in Figure 9, both chains bound to the oligosaccharides having GlcNAc or GalNAc at their nonreducing ends. B‐chain showed binding to more oligosaccharides with GlcNAc than A‐chain, while A‐chain showed relatively higher affinity for GalNAc‐containing oligosaccharides (Nos. 44 and 57) as revealed by ITC measurement, in which SPL‐1 composed of A‐ and B‐chains indicated higher association constants for GalNAc than SPL‐2 (Fig. 2). It is also conspicuous that the branched tetrasaccharide No. 44 indicated considerable affinity, especially for the A‐chain. This suggests that these lectins may recognize multiple monosaccharide portions in the oligosaccharide chains, rather than terminal GlcNAc or GalNAc residues. Oligosaccharides containing GlcNAc or GalNAc with α‐glycosidic linkages seemed to have relatively higher affinity for both chains (Nos. 44, 54, and 81).
Figure 9.
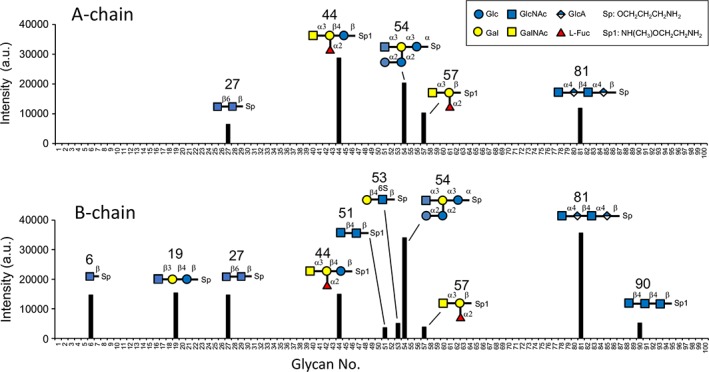
Glycan array analysis of recombinant A‐chain and B‐chain. The binding specificities of Cy3‐labeled lectins for 100 oligosaccharides were measured. All the glycoconjugate structures are listed in Supporting Information Table S3.
Discussion
The amino acid sequences of SPL‐1 and SPL‐2 deduced from cDNA indicated their homology with C‐type lectins, although sequence identities were relatively low. Structural similarities of SPLs with C‐type lectins were further confirmed by X‐ray crystallographic analysis. While most of the oligomeric C‐type lectins consist of identical protomers, SPLs have either homodimeric (SPL‐2 with two B‐chains) or heterodimeric (SPL‐1 with A‐ and B‐chains) forms. The two constituent chains show closely related amino acid sequences, but are different in length by one residue at the N‐terminus (Fig. 4). Since homodimeric lectin containing the A‐chain has not been obtained from S. purpuratus, dimerization of the individual chains was examined using recombinant proteins and confirmed that the A‐chain is not able to form stable homodimer. Figure 10 shows dimer structures of SPL‐1, SPL‐2, and a putative A‐chain dimer model, in which two A‐chain subunits are placed to form two interchain disulfide bonds between Cys residues (Cys2 of one subunit and Cys4 of the other subunit) involved in the interchain disulfide bonds in SPL‐1 (Supporting Information Fig. S2). As seen in these figures, dimerization of SPL‐1 and SPL‐2 allows significant contact area between the subunits [Fig. 10(A,B)], leading to stable dimer structures, while there would be little contact between the subunits in the A‐chain homodimer [Fig. 10(C)]. This model seems to explain why A‐chain homodimer was not obtained from the recombinant A‐chain or the extract of S. purpuratus.
Figure 10.
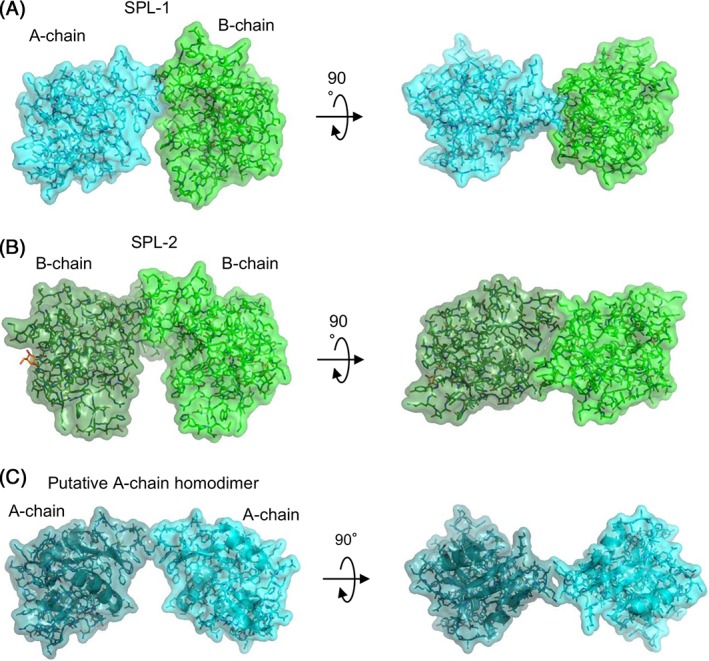
Dimer structures of SPL‐1 (A), SPL‐2/GalNAc complex (B), and a putative A‐chain homodimer model (C). In the putative A‐chain homodimer model, two A‐chains are positioned so as to form disulfide bonds between Cys2 and Cys4 of different subunits.
One of the most conspicuous features of SPLs is their Ca2+‐independent carbohydrate‐binding ability. As seen in Figure 1(A), while native SPL‐1 was gradually eluted from the GlcNAc‐immobilized column with EDTA, SPL‐2 was only eluted with GlcNAc, suggesting that SPL‐1 and SPL‐2 are Ca2+‐dependent and Ca2+‐independent lectins, respectively. However, ITC measurement revealed that both chains indeed had carbohydrate‐binding ability in the absence of Ca2+, although binding was enhanced by Ca2+. This suggests that the apparent Ca2+‐dependency of SPL‐1 in the affinity column chromatography may have been caused by its relatively weak binding ability. In fact, recombinant A‐chains and A/B‐chain dimers remained bound to the GlcNAc‐immobilized column during the elution with EDTA [Supporting Information Fig. S3(A)], which might be caused by subtle differences between the native SPL‐1 and these recombinant proteins. On the other hand, the reason for the enhancement of the association constants of the lectins in the presence of Ca2+ is not clear, though it might be related to the Ca2+‐binding sites found in the crystal structures of both chains, which are remote from the carbohydrate‐binding site [magenta spheres in Fig. 6(A,B)]. These Ca2+ ions might make weak interactions with carbohydrates, although discernible electron density of carbohydrate was not found around the Ca2+ ion in the crystal structure of SPL‐2/GalNAc. Alternatively, binding of Ca2+ ion in these sites might indirectly affect carbohydrate‐binding sites to enhance their binding affinity.
As revealed by the crystal structure of SPL‐2/GalNAc complex, binding of GalNAc is mostly mediated by stacking interactions between its acetamido group and the side chains of tyrosine and histidine residues, in contrast to ordinary C‐type lectins, which recognize carbohydrates through coordinate bonds with Ca2+ and a hydrogen bond network with nearby residues. This is closely related to the fact that the binding sites of SPLs contain RPD or KPD sequences, instead of the QPD and EPN motifs. In ordinary C‐type lectins, the latter motifs are essentially important not only to bind a Ca2+ ion in the carbohydrate‐binding site, but also to discriminate the orientations of the hydroxyl groups at the C‐3 and C‐4 positions of glucose or galactose by forming hydrogen bonds. The corresponding KPD in SPL B‐chain [Lys97 to Asp99 in Fig. 7(B)], and most probably RPD in SPL A‐chain [Arg96 to Asp98 in Fig. 7(C)], is not directly involved in the binding of GalNAc.
Like SPLs, the recognition of acetamido groups in carbohydrates using aromatic amino acids or histidine has also been observed in other lectins, such as wheat (Triticum aestivum) germ agglutinin21 and pokeweed (Phytolacca americana) lectin.22 In the case of pokeweed lectin, the side chain of a histidine residue (His72) in the binding site forms a hydrogen bond with the acetamido group of a GlcNAc residue in the bound tri‐N‐acetylchitotriose, which resembles SPL‐B chain recognizing GalNAc [Fig. 7(B)].
To investigate more detailed carbohydrate‐binding specificities of the individual subunits of SPLs, glycan array analysis was performed using the recombinant A‐ and B‐chains. The results indicated that both chains preferably bound the oligosaccharides having GlcNAc or GalNAc at the nonreducing end. They also showed relatively higher affinity for terminal GlcNAc or GalNAc with α‐glycosidic linkages. Higher affinities for larger oligosaccharides were also observed (especially in A‐chain) strongly suggesting that they recognize larger portions of oligosaccharides, rather than the terminal GlcNAc and GalNAc.
Two S. purpuratus lectins (SPA‐I and SPA‐III) have been reported by Tatsumi et al.16 These lectins showed the highest binding specificity toward GlcNAc, followed by GalNAc and ManNAc, and their binding affinity was enhanced in the presence of Ca2+. These characteristics were similar to those of SPLs, suggesting that SPAs may be the same lectins as SPLs. However, the molecular masses of SPAs are somewhat higher (40 kDa) than those of SPLs (31.5 kDa) and SPAs were shown to be glycoproteins, in contrast to SPLs, which were confirmed not to be glycoproteins by the periodic acid–Schiff staining23 on SDS‐PAGE (data not shown).
Evidence has been accumulated that lectins in invertebrates play important roles in innate immunity,6, 24 while information about their structures and functions is very limited. Many of the C‐type lectins have been known to play important roles in immune systems by recognizing foreign substances.5, 25, 26, 27 This is presumably based on their structural versatility to recognize various molecules by changing only limited regions. In this study, SPL‐1 and SPL‐2 have been shown to recognize acetamido groups of the carbohydrates in a novel fashion, without the Ca2+ utilized by ordinary C‐type CRDs. Recently, another Ca2+‐independent C‐type lectin ScCTL‐2 from the bivalve Sinonovacula constricta has also been reported.27 This lectin lacks common carbohydrate‐recognition motifs, QPD or EPN, but instead contains GAN sequence at the corresponding position. Because of the great diversity of the species, other marine invertebrate lectins having unique characteristics are likely to be discovered, providing important information concerning the nature of protein–carbohydrate interactions between various cells and molecules in marine invertebrates.
Materials and Methods
Materials
Oligonucleotides were purchased from Sigma‐Aldrich. Oligotex‐dT30 mRNA Purification Kit was obtained from Takara (Otsu, Japan). Plasmid vector pTAC‐2 was obtained from BioDynamics Laboratory (Tokyo, Japan). Plasmid vector pET‐3a and E. coli BL21(DE3)pLysS were obtained from Novagen. E. coli JM109 cells, SMARTer cDNA Cloning Kit, and In‐Fusion HD Cloning Kit were obtained from Clontech. All other chemicals were of analytical grade for biochemical use. S. purpuratus specimens harvested in Mikawa Bay (Aichi Prefecture, Japan) were purchased from a local dealer and stored at −20°C.
Purification of SPLs
Proteins were extracted from S. purpuratus bodies in TBS (Tris‐buffered saline; 10 mM Tris–HCl, pH 7.6, 150 mM NaCl) containing 10 mM CaCl2, and applied to the GlcNAc‐Cellufine column (3 × 10 cm), in which GlcNAc was immobilized on Cellufine gel (JNC Corp., Tokyo, Japan) with the cross‐linking reagent divinyl sulfone.28 After washing with this buffer, bound lectins were eluted with TBS containing 20 mM EDTA, followed by TBS containing 100 mM GlcNAc. The lectins were further purified by ion‐exchange chromatography on a HiTrap Q column (1.6 × 2.5 cm) (GE Healthcare) and gel filtration on a Superdex 200 column (2.3 × 60 cm) (GE Healthcare UK Ltd, Buckinghamshire, England) equilibrated with TBS using an ÄKTAprime plus apparatus (GE Healthcare).
N‐terminal amino acid sequence analysis
The N‐terminal amino acid sequences of the lectins were determined using a protein sequencer, PPSQ‐21 (Shimadzu, Kyoto, Japan). For sequencing, the proteins separated by SDS‐PAGE were transferred onto the PVDF membrane in the transfer buffer (48 mM Tris, 39 mM glycine, 0.1% [w/v] SDS, 20% [v/v] methanol) for 80 min at 160 mA. Protein bands were stained with Ponceau S solution (0.1% [w/v] Ponceau S in 5% [v/v] acetic acid) and their N‐terminal amino acid sequences were analyzed using PPSQ‐21.
cDNA cloning of SPL
The whole body of S. purpuratus was flash frozen in liquid nitrogen and ground to form a powder. Total RNA was extracted using Isogen solution (Nippon Gene, Tokyo, Japan). Poly(A) RNA was collected using the Oligotex‐dT30 mRNA Purification Kit, and cDNA was synthesized using the PrimeScript II 1st strand cDNA Synthesis Kit (Takara, Otsu, Japan). A DNA fragment corresponding to the N‐terminal region of SPL‐2, which had been determined by N‐terminal amino acid sequence analysis, was amplified by PCR using two degenerate primers, DF1: 5′‐CCI(A/T)(C/G)IGGITGGAA(A/G)TT(C/T)TT(C/T)GG‐3′ and DR1: 5′‐(A/G)AAIGC(C/T)TTI(C/G)(A/T)ICC(C/T)TCCCAICC‐3′, where “I” represents deoxyinosine, and the letters in parentheses represent mixed bases. An amplified DNA fragment of approximately 80 bp was cloned into a pTAC‐2 vector using E. coli JM109 cells and was sequenced using an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems). Based on the sequence of this fragment, specific primers were designed, and 3′‐ and 5′‐rapid amplification of cDNA ends (3′‐RACE and 5′‐RACE) were performed using the SMARTer cDNA Cloning Kit. During this process, two similar but distinct sequences corresponding two different proteins were observed and cloned separately. Finally, cDNAs encoding these two lectins (SPL‐1 and SPL‐2) were cloned by comparing the N‐terminal amino acid sequences. The entire amino acid sequences of SPL‐1 and SPL‐2 deduced from the cDNA sequences were compared with sequences in the UniProt database (www.uniprot.org)18 by performing a BLAST search.29 Multiple sequence alignments were performed using Clustal Omega.17 These nucleotide sequences were deposited in DDBJ/GenBankTM/EBI (accession numbers: LC388679 (SPL‐A‐chain) and LC388680 (SPL‐B‐chain). Chemical and physical parameters of the lectins were calculated from the deduced sequence using the ProtParam tool in ExPASy Bioinformatics Resource Portal (www.expasy.org).30, 31
Isothermal titration calorimetry
ITC for analyzing the interaction between the lectins and GlcNAc or GalNAc was performed at 25°C using iTC200 (MicroCal, GE Healthcare). Aliquots of GlcNAc or GalNAc solutions (5 or 25 mM) were injected into the SPL‐1 or SPL‐2 (2.6 mg/mL, 81 μM) solution in TBS in the presence and absence of 10 mM CaCl2 using a cell with a volume of 200 μL at 2‐minute intervals. The data were analyzed using ORIGIN software, version 7.0. Control experiments were carried out to measure ligand dilution‐related heat, which were subsequently subtracted from the ligand binding thermograms.
X‐ray crystallographic analysis
The crystals of SPL‐1 and SPL‐2 purified from S. purpuratus were grown at 20°C by the vapor diffusion method. SPL‐1 crystals were obtained by mixing 2 μL of the protein solution with 2 μL of reservoir solution (30%(w/v) polyethylene glycol (PEG) 3000, 100 mM N‐Cyclohexyl‐2‐aminoethanesulfonic acid/NaOH pH 9.5). SPL‐2 crystals were obtained as a complex with GalNAc by mixing 2 μL of protein solution containing 100 mM GalNAc and 10 mM CaCl2 with 2 μL of reservoir solution (0.2 M lithium sulfate, 0.1 M Tris–HCl, pH 8.5, 30% PEG 4000). A heavy atom derivative was obtained by soaking a native crystal of SPL‐1 for 2 hours in the reservoir solution containing 1 mM lead(II) acetate. The data sets from SPL‐1(Pb‐SAD) were collected in‐house (Rigaku MicroMax007 & R‐AXIS IV++), whereas those from the other crystals were collected using beamline NW‐12A at the Photon Factory (KEK, Tsukuba, Japan). All crystals were frozen at 95 K before data collection. The obtained data set from SPL‐1(Pb‐SAD) was processed and scaled in the MOSFLM32 and SCALA33 software packages, respectively, and the other data sets were processed and scaled in HKL2000.34 The crystals of SPL‐1 and SPL‐2 belonged to the space groups P21 and P3121, with 2 and 1 molecules per asymmetric unit, respectively. Crystals of SPL‐1 included two polypeptides composed of A‐ and B‐chains in the asymmetric unit, in which there are two intermolecular disulfide bonds between the chains. On the other hand, in the SPL‐2/GalNAc complex crystals, the asymmetric unit included only one polypeptide of B‐chain that was disulfide bonded with the next protomer related by crystallographic twofold symmetry. The dataset of SPL‐1(Pb‐SAD) was used for phase calculation by the single‐wavelength anomalous diffraction method in the PHENIX35 software. Phase improvement by density modification was also performed in PHENIX. The structure was built using the ARP/wARP36 and COOT37 software packages and refined in Refmac20 with 5% of the dataset aside as a free set. During subsequent refinement, the SPL‐1 (Pb‐SAD) dataset was replaced by the SPL‐1 dataset. The structure of SPL‐2 complexed with GalNAc was solved by the molecular replacement method using B chain in the SPL‐1 structure. The molecular replacement was performed using the Molrep CCP4 suite.38 The model of GalNAc was fitted into the carbohydrate‐binding sites according to the difference electron density map. The refinement statistics are listed in Supporting Information Table S2. All figures were produced using the PyMOL software.39 The interface surface area and assemblies of SPLs were calculated using Protein Interfaces, Surfaces and Assemblies (PISA).40
Expression and purification of recombinant SPLs
E. coli BL21(DE3)pLysS cells were transformed with the pET‐3a plasmid containing the genes encoding SPL A‐chain and SPL B‐chain and protein expression was induced with 0.4 mM isopropylthiogalactoside. The recombinant proteins were obtained as inclusion bodies after cell disruption by sonication and were subsequently solubilized in solubilization buffer (50 mM Tris–HCl, pH 8.0; 0.2 M NaCl; 1 mM EDTA; 6 M guanidine hydrochloride, 0.4% 2‐mercaptoethanol) and refolded in refolding buffer (0.1 M Tris–HCl, pH 8.0; 0.8 M L‐arginine; 2 mM EDTA; 5 mM reduced glutathione; 0.5 mM oxidized glutathione; 0.1 mM phenylmethylsulfonyl fluoride). After dialysis of the refolded proteins in TBS, the proteins were purified by affinity chromatography using the GlcNAc‐Cellufine column (3 cm × 10 cm) in TBS containing 10 mM CaCl2. Elution of the bound protein was performed with TBS containing 20 mM EDTA, followed by 100 mM GlcNAc.
Dynamic light scattering
The hydrodynamic radius of the proteins was measured by dynamic light scattering in TBS at 25°C using a Zetasizer Nano ZS (Malvern Instruments). The value was calculated as an average of six measurements.
Glycan array analysis
Binding specificities of the recombinant SPLs for various oligosaccharides were examined by glycan array analysis using the RayBio Glycan Array 100 kit (RayBiotech, Norcross, GA). Although the original protocol uses cyanine3 (Cy3) equivalent dye‐conjugated streptavidin for fluorescence‐labeling of bound lectins, in the current experiment, the lectins were directly labeled with Cy3 NHS ester (Abcam plc, Cambridge, UK) to avoid multiple washing processes and facilitate the detection of relatively weak binding of the lectins. Briefly, the lectins (0.3–0.5 mg/mL in 10 mM sodium phosphate, pH 8.0, 140 mM NaCl) were incubated with Cy3 NHS ester (0.63 mg/mL) at room temperature for 1 hour in the dark and dialyzed against TBS for 2 days to remove remaining reagent. After blocking the surface of the glycan array slide for 30 minutes with the Sample Diluent included in the glycan array kit, Cy3‐labeled lectins were added to the array, a slide with 100 oligosaccharides immobilized by the linkers (Supporting Information Table S3). The array was then washed with Wash Buffer I and Wash Buffer II from the kit, followed by water. Detection of the bound lectins was performed using an Agilent DNA microarray scanner G2565CA (Agilent, Santa Clara, CA). Fluorescence intensity analysis was performed using ImageJ.41
Accession numbers
The nucleotide sequences reported in this paper have been submitted to the DDBJ/GenBankTM/EBI Data Bank with accession numbers LC388679 and LC388680. The atomic coordinates and structure factors for SPL‐1 (PDB ID 6A7T) and SPL‐2/GalNAc complex (PDB ID 6A7S) have been deposited in the Protein Data Bank.
Conflict of interest
The authors declare that they have no conflicts of interest.
Acknowledgments
The authors thank the staff of the Gene Research Center, Nagasaki University for assistance with DNA sequence analysis. This work was supported by Grants‐in‐Aid for Scientific Research (15K06977, 16K07695, and 17K07760) from the Japan Society for the Promotion of Science (JSPS).
References
- 1. Kilpatrick DC (2002) Animal lectins: a historical introduction and overview. Biochim Biophys Acta 1572:187–197. [DOI] [PubMed] [Google Scholar]
- 2. Gabius HJ (1997) Animal lectins. Eur J Biochem 243:543–576. [DOI] [PubMed] [Google Scholar]
- 3. Drickamer K (1999) C‐type lectin‐like domains. Curr Opin Struct Biol 9:585–590. [DOI] [PubMed] [Google Scholar]
- 4. Zelensky AN, Gready JE (2005) The C‐type lectin‐like domain superfamily. FEBS J 272:6179–6217. [DOI] [PubMed] [Google Scholar]
- 5. Cambi A, Koopman M, Figdor CG (2005) How C‐type lectins detect pathogens. Cell Microbiol 7:481–488. [DOI] [PubMed] [Google Scholar]
- 6. Wang W, Song X, Wang L, Song L (2018) Pathogen‐derived carbohydrate recognition in molluscs immune defense. Int J Mol Sci 19:E721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang XW, Xu WT, Wang XW, Mu Y, Zhao XF, Yu XQ, Wang JX (2009) A novel C‐type lectin with two CRD domains from Chinese shrimp Fenneropenaeus chinensis functions as a pattern recognition protein. Mol Immunol 46:1626–1637. [DOI] [PubMed] [Google Scholar]
- 8. Hatakeyama T, Kohzaki H, Nagatomo H, Yamasaki N (1994) Purification and characterization of four Ca2+‐dependent lectins from the marine invertebrate, Cucumaria echinata . J Biochem 116:209–214. [DOI] [PubMed] [Google Scholar]
- 9. Hatakeyama T, Nagatomo H, Yamasaki N (1995) Interaction of the hemolytic lectin CEL‐III from the marine invertebrate Cucumaria echinata with the erythrocyte membrane. J Biol Chem 270:3560–3564. [DOI] [PubMed] [Google Scholar]
- 10. Hatakeyama T, Kamiya T, Kusunoki M, Nakamura‐Tsuruta S, Hirabayashi J, Goda S, Unno H (2011) Galactose recognition by a tetrameric C‐type lectin, CEL‐IV, containing the EPN carbohydrate recognition motif. J Biol Chem 286:10305–10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sugawara H, Kusunoki M, Kurisu G, Fujimoto T, Aoyagi H, Hatakeyama T (2004) Characteristic recognition of N‐acetylgalactosamine by an invertebrate C‐type lectin, CEL‐I, revealed by X‐ray crystallographic analysis. J Biol Chem 279:45219–45225. [DOI] [PubMed] [Google Scholar]
- 12. Hatakeyama T, Furukawa M, Nagatomo H, Yamasaki N, Mori T (1996) Oligomerization of the hemolytic lectin CEL‐III from the marine invertebrate Cucumaria echinata induced by the binding of carbohydrate ligands. J Biol Chem 271:16915–16920. [DOI] [PubMed] [Google Scholar]
- 13. Unno H, Goda S, Hatakeyama T (2014) Hemolytic lectin CEL‐III heptamerizes via a large structural transition from α‐helices to a β‐barrel during the transmembrane pore formation process. J Biol Chem 289:12805–12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uchida T, Yamasaki T, Eto S, Sugawara H, Kurisu G, Nakagawa A, Kusunoki M, Hatakeyama T (2004) Crystal structure of the hemolytic lectin CEL‐III isolated from the marine invertebrate Cucumaria echinata: implications of domain structure for its membrane pore‐formation mechanism. J Biol Chem 279:37133–37141. [DOI] [PubMed] [Google Scholar]
- 15. Unno H, Matsuyama K, Tsuji Y, Goda S, Hiemori K, Tateno H, Hirabayashi J, Hatakeyama T (2016) Identification, characterization, and X‐ray crystallographic analysis of a novel type of mannose‐specific lectin CGL1 from the pacific oyster Crassostrea gigas . Sci Rep 6:29135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tatsumi M, Arai Y, Itoh T (1982) Purification and characterization of a lectin from the shellfish, Saxidomus purpuratus . J Biochem 91:1139–1146. [DOI] [PubMed] [Google Scholar]
- 17. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Consortium U (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res 40:D71–D75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hatakeyama T, Matsuo N, Shiba K, Nishinohara S, Yamasaki N, Sugawara H, Aoyagi H (2002) Amino acid sequence and carbohydrate‐binding analysis of the N‐acetyl‐D‐galactosamine‐specific C‐type lectin, CEL‐I, from the Holothuroidea, Cucumaria echinata . Biosci Biotechnol Biochem 66:157–163. [DOI] [PubMed] [Google Scholar]
- 20. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255. [DOI] [PubMed] [Google Scholar]
- 21. Schwefel D, Maierhofer C, Beck JG, Seeberger S, Diederichs K, Möller HM, Welte W, Wittmann V (2010) Structural basis of multivalent binding to wheat germ agglutinin. J Am Chem Soc 132:8704–8719. [DOI] [PubMed] [Google Scholar]
- 22. Hayashida M, Fujii T, Hamasu M, Ishiguro M, Hata Y (2003) Similarity between protein‐protein and protein‐carbohydrate interactions, revealed by two crystal structures of lectins from the roots of pokeweed. J Mol Biol 334:551–565. [DOI] [PubMed] [Google Scholar]
- 23. Glossmann H, Neville DM (1971) Glycoproteins of cell surfaces. A comparative study of three different cell surfaces of the rat. J Biol Chem 246:6339–6346. [PubMed] [Google Scholar]
- 24. Marques MR, Barracco FMA (2000) Lectins, as non‐self‐recognition factors, in crustaceans. Aquaculture 191:23–44. [Google Scholar]
- 25. Guo XN, Jin XK, Li S, Yu AQ, Wu MH, Tan SJ, Zhu YT, Li WW, Zhang P, Wang Q (2013) A novel C‐type lectin from Eriocheir sinensis functions as a pattern recognition receptor with antibacterial activity. Fish Shellfish Immunol 35:1554–1565. [DOI] [PubMed] [Google Scholar]
- 26. Cambi A, Figdor CG (2003) Dual function of C‐type lectin‐like receptors in the immune system. Curr Opin Cell Biol 15:539–546. [DOI] [PubMed] [Google Scholar]
- 27. Shi Y, Zhao X, Wang Z, Shao Y, Zhang W, Bao Y, Li C (2018) Novel Ca2+‐independent C‐type lectin involved in immune defense of the razor clam Sinonovacula constricta . Fish Shellfish Immunol 84:502–508. [DOI] [PubMed] [Google Scholar]
- 28. Teichberg VI, Aberdam D, Erez U, Pinelli E (1988) Affinity‐repulsion chromatography. Principle and application to lectins. J Biol Chem 263:14086–14092. [PubMed] [Google Scholar]
- 29. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. [DOI] [PubMed] [Google Scholar]
- 30. Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, Grosdidier A, Hernandez C, Ioannidis V, Kuznetsov D, Liechti R, Moretti S, Mostaguir K, Redaschi N, Rossier G, Xenarios I, Stockinger H (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40:W597–W603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gasteiger E, Hoogland C, Gattiker ASD, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. New York: Humana Press. [DOI] [PubMed] [Google Scholar]
- 32. Leslie AG (2006) The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 62:48–57. [DOI] [PubMed] [Google Scholar]
- 33. Evans P (2006) Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. [DOI] [PubMed] [Google Scholar]
- 34. Otwinowski Z, Minor W. In: Carter CW., Jr et al., Eds, (1997) Methods in enzymology: macromolecular crystallography, Part A. New York, NY: Academic Press; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 35. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS (2001) ARP/wARP and molecular replacement. Acta Crystallogr D Biol Crystallogr 57:1445–1450. [DOI] [PubMed] [Google Scholar]
- 37. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of coot. Acta Crystallogr D Biol Crystallogr 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vagin A, Teplyakov A (2010) Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66:22–25. [DOI] [PubMed] [Google Scholar]
- 39. DeLano WL. The PyMOL molecular graphics systems. San Carlos, CA: DeLano Scientific, 2002. [Google Scholar]
- 40. Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. [DOI] [PubMed] [Google Scholar]
- 41. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]