

Abstract
IGF‐motif loops project from the hexameric ring of ClpX and are required for docking with the self‐compartmentalized ClpP peptidase, which consists of heptameric rings stacked back‐to‐back. Here, we show that ATP or ATPγS support assembly by changing the conformation of the ClpX ring, bringing the IGF loops closer to each other and allowing efficient multivalent contacts with docking clefts on ClpP. In single‐chain ClpX pseudohexamers, deletion of one or two IGF loops modestly slows association with ClpP but strongly accelerates dissociation of ClpXP complexes. We probe how changes in the sequence and length of the IGF loops affect ClpX–ClpP interactions and show that deletion of one or two IGF loops slows ATP‐dependent proteolysis by ClpXP. We also find that ClpXP degradation is less processive when two IGF loops are deleted.
Keywords: protein degradation, AAA+ protease, ATP‐fueled molecular machine, kinetics
Introduction
Within cells, ring hexamers belonging to the AAA+ (ATPases Associated with various cellular Activities) superfamily of enzymes carry out a wide variety of protein remodeling, unfolding, and degradation reactions.1 These ATP‐fueled molecular machines typically function by engaging a peptide tag and pulling the attached native protein against a narrow axial pore, eventually resulting in unfolding and subsequent translocation through the pore. This activity is essential for the biological function of AAA+ proteases, which destroy specific intracellular proteins by unfolding them and then translocating the denatured polypeptide into the chamber of a self‐compartmentalized peptidase for degradation.
For example, the ClpX unfoldase/translocase and ClpP peptidase comprise the AAA+ ClpXP protease.2 ClpXP degrades a variety of cellular proteins in addition to proteins modified by co‐translational addition of the ssrA tag in Escherichia coli and many other bacteria.3, 4, 5 ClpP consists of two heptameric rings, stacked back‐to‐back, that enclose a proteolytic chamber.6 Each heptameric ClpP ring can bind one ClpX hexamer, giving rise to symmetry mismatched singly‐capped (XP) or doubly‐capped (XPX) complexes.7 These ClpXP complexes are largely stabilized by peripheral interactions in which IGF loops from ClpX dock into hydrophobic clefts on the surface of a ClpP ring [Fig. 1(A, B)].9, 10 These loops are named for an IGF tripeptide sequence that is invariant in ClpX orthologs from γ‐proteobacteria [Fig. 1(C), top panel] but can be MGF, LGF, etc. in other bacterial phyla [Fig. 1(C), bottom panel].
Figure 1.
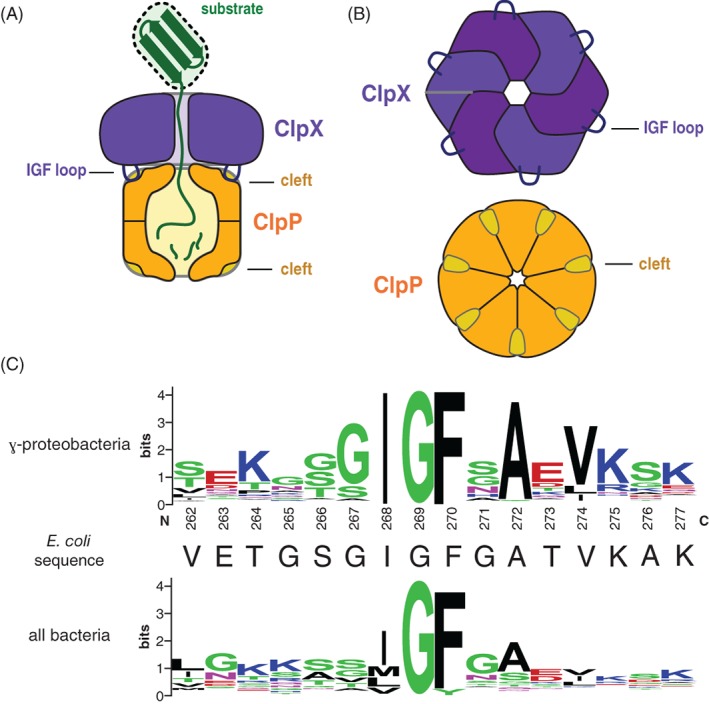
(A) Cartoon of the ClpXP protease degrading a protein substrate. The ClpX hexamer (colored light and dark purple) recognizes a protein substrate (colored green) and uses cycles of ATP hydrolysis to unfold and translocate it into the degradation chamber of the ClpP peptidase (colored dark yellow). The IGF loops of ClpX dock into hydrophobic clefts on ClpP. (B) Top views of the ClpX and ClpP rings, highlighting the six IGF loops of ClpX and seven clefts of ClpP. (C) Sequence‐logo depictions8 of sequence conservation in the IGF loops of ClpX orthologs from γ‐proteobacteria (top) and all bacteria (bottom). The sequence of the E. coli ClpX IGF loop is shown in the middle.
Previous studies show that mutating the IGF tripeptide of E. coli ClpX to EGF or IGW severely compromises ClpP binding and degradation.9 Deleting IGF loops from single‐chain pseudohexamers, which consist of genetically linked ClpX subunits, also results in ClpP‐binding defects.10 ClpX binding to ClpP requires ATP or ATPγS,11 an analog that ClpX hydrolyzes slowly,12 but the basis for this nucleoside‐triphosphate requirement is unknown. Here, we investigate the molecular mechanism that underlies the nucleotide dependence of ClpX binding to ClpP and determine how changes in the number, geometric distribution, sequence, and length of IGF loops in hexamers of E. coli ClpX impact the kinetics of ClpP binding and dissociation and the ability of ClpXP to carry out ATP‐dependent protein degradation.
Results
Nucleotide effects on accessibility of the IGF loops
Prior experiments show that chymotrypsin cleaves ClpX at a single site after Phe270 in the IGF loop.11 We used chymotryptic cleavage to test if different nucleotides changed the accessibility of the IGF loops in ClpXΔN, a variant lacking the N‐domain that is still fully active in binding ClpP and supporting degradation of ssrA‐tagged substrates.11, 13 As shown in Figure 2(A), chymotrypsin cleaved ClpXΔN at one major site to generate ∼24 and ∼17 kDa fragments. These fragments were not produced when ClpXΔN harbored a deletion of the IGF loop, establishing that cleavage occurs within this loop. Moreover, cleavage was not observed for a ClpXΔN variant containing the F270A mutation, strongly supporting the Phe270–Gly271 peptide bond as the cleavage site. Cleavage of ClpXΔN after Phe270 would generate an N‐terminal fragment of 22.8 kDa and C‐terminal fragment of 16.8 kDa, which are close to the values observed. Importantly, the ClpXΔN chymotryptic fragmentation pattern and cleavage kinetics were very similar in experiments performed in the presence of ATP, ATPγS, or ADP [Fig. 2(B)]. This finding suggests that the IGF motifs in the hexamer are roughly equally accessible irrespective of the identity of bound nucleotide. ADP does not support binding of ClpX to ClpP.14, 15, 16 Our results indicate that this inability of ADP to support ClpXP assembly is not a consequence of IGF‐loop sequestration or formation of a protease‐resistant structure in the presence of ADP.
Figure 2.
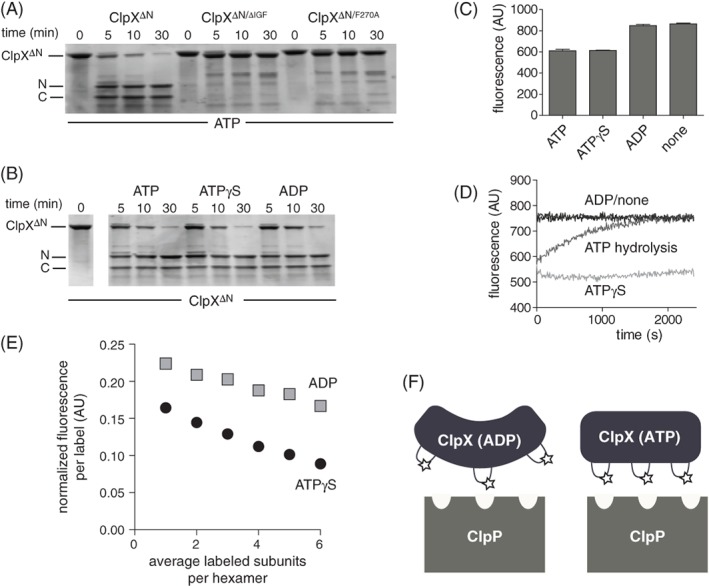
Nucleotide dependence of protease accessibility and relative distance between IGF loops. (A) As assayed by SDS‐PAGE, chymotrypsin cleaved ClpXΔN into two major fragments, labeled N and C, which were not observed following chymotrypsin incubation with ClpXΔN/ΔIGF or ClpXΔN/F270A. Experiments contained ATP (10 mM), chymotrypsin (0.01 mg/mL), and ClpXΔN variants (1 μM hexamer). (B) Chymotryptic cleavage of ClpXΔN in the presence of different nucleotides. Except for nucleotide identity, experimental conditions were the same as in Panel A. (C) Initial fluorescence of Alexa‐647 labeled T273C ClpXΔN in the presence of ATP, ATPγS, ADP, or no nucleotide. The protein concentration was 0.5 μM, and nucleotide was 1.5 mM when present. Values are averages (N = 3) ± SD. (D) Time‐dependent changes in fluorescence of Alexa‐647 labeled T273C ClpXΔN under different nucleotide conditions. Other conditions were identical to Panel C. (E) Different concentrations of unlabeled ClpXΔN and Alexa‐647 labeled T273C ClpXΔN were mixed for 1 h in the absence of nucleotide, 5 mM ADP or ATPγS was added, and fluorescence was measured. (F) Decreased fluorescence caused by increased homo quenching is consistent with the IGF loops being closer together in fluorescent T233C ClpXΔN that is bound to ATP compared to ADP. We propose that the IGF loops in ATP‐bound ClpX are properly oriented to make efficient multivalent contacts with the clefts in ClpP, whereas the IGF‐loops in ADP‐bound ClpX can only make a subset of efficient contacts.
Nucleotides affect IGF‐loop proximity
To probe the proximity of adjacent IGF loops in the ClpX ring, we sought to attach a fluorescent dye that would be sensitive to loop–loop distance. As our ClpXΔN variant contains only one cysteine, which is buried and unreactive with maleimides, we replaced the solvent exposed but non‐conserved Thr273 in the IGF loop with cysteine. The T273C variant was as active as the parent enzyme in supporting ClpP degradation of GFP‐ssrA. Next, we labeled T273CClpXΔN with Alexa‐647‐C2‐maleimide. The excitation and emission spectra of the Alexa‐647 fluorophore overlap substantially, allowing homo‐quenching if two or more labeled IGF loops are sufficiently close to each other in the hexamer. The fluorescence of labeled T273CClpXΔN was similar in the presence of 1 mM ATP or ATPγS but increased in the presence of 1 mM ADP or absence of nucleotide [Fig. 2(C)]. Fluorescence remained constant as a function of time for the no‐nucleotide or ADP experiments and increased very slowly for the ATPγS sample [Fig. 2(D)]. For the ATP sample, by contrast, fluorescence increased over the course of ∼30 min to the level of the ADP sample [Fig. 2(D)], as expected if most of the ATP initially present was hydrolyzed over this time.
Two models could explain why ATP/ATPγS‐bound ClpX hexamers have lower fluorescence than ADP‐bound or nucleotide‐free hexamers. First, the IGF loops could be closer to each other in ATP/ATPγS‐bound hexamers, allowing more efficient auto‐quenching, and farther apart in ADP‐bound or nucleotide‐free hexamers. Second, decreased fluorescence in the presence of ATP/ATPγS could result from changes in the local environment of individual IGF loops rather than their proximity to one another. To distinguish between these models, we mixed different concentrations of unlabeled protein and fluorescently labeled protein for 1 h in the absence of nucleotide to allow subunit exchange, and then added ATPγS or ADP before assaying fluorescence. For a given nucleotide, the local‐environment model predicts that the relative fluorescence per fluorophore will remain constant regardless of the fraction labeling per hexamer. Instead, we observed a decrease in normalized fluorescence per label with increasing label density, and also observed proportionally greater fluorescence quenching with increasing label density in the presence of ATPγS [Fig. 2(E)]. We conclude that the IGF‐loops in a ClpX hexamer are closer together, on average, in ATP‐bound or ATPγS‐bound enzymes and farther apart in ADP‐bound or nucleotide‐free enzymes, implying a substantial ATP‐dependent conformational change in the ClpX hexamer that positions multiple IGF‐loops for efficient docking with ClpP [Fig. 2(F)].
Effects of IGF‐loop removal on ClpX association with ClpP
Bio‐layer interferometry (BLI) allows the kinetics of ClpX binding to ClpP to be followed in real time.14 We constructed variants in which we replaced one or two IGF loops with GGSSGG linkers in single‐chain ClpXΔN pseudohexamers with a biotinylation site near the C‐terminus.13 In the single‐chain hexamer, Subunit A is at the N‐terminus, subunits B, C, D, and E follow in order, and Subunit F is at the C‐terminus. The single IGF deletion/substitution was in Subunit B, and the double deletion/substitutions were in Subunit AB, BC, BD, or BE.
We attached biotinylated ClpX variants to streptavidin‐coated biosensors, moved them into buffer containing ClpP, and assayed association via changes in BLI‐response signal. Under these conditions, association should be pseudo first‐order. Figure 3(A) shows typical trajectories. The variant with six IGF loops showed a major fast phase (amplitude ∼80%) and minor slow phase (amplitude ∼20%) when fit to a double‐exponential function. This behavior was observed previously,14 suggesting that there are fast‐binding and slow‐binding conformations of ClpXΔN that interconvert on a timescale slower than the fast‐binding phase. By contrast, association trajectories for variants with four or five IGF loops were slower and fit well to a single‐exponential function. It is possible that removing one or two IGF loops destabilizes the slow‐binding conformation or that fast‐binding and slow‐binding conformations now interconvert on a timescale comparable to binding. For each variant, values of the apparent association rate constant (k app) were determined at different concentrations of ClpP, and these data were then fit to obtain an association‐rate constant (k assn). Figure 3(B) shows this fit for the AB variant missing two IGF loops. Dissociation occurs on the same time scale as association for this variant, and the positive Y‐intercept reflects the dissociation‐rate constant.
Figure 3.
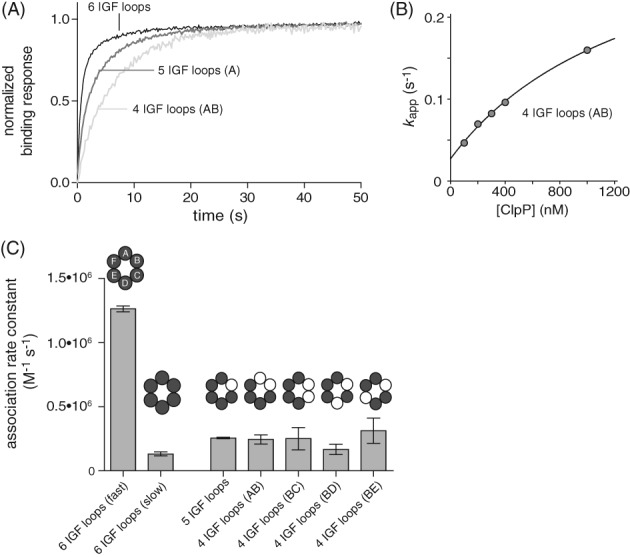
Effects of IGF‐loop deletion on ClpP association with ClpX. (A) Normalized BLI association trajectories for experiments performed using 100 nM ClpP. (B) For single‐chain ClpXΔN with four IGF loops (loops in subunits AB deleted), k app varied hyperbolically with ClpP concentration. The line is a non‐linear‐least‐squares fit to the equation k app = intercept + max•[ClpP]/(K 1/2 + [ClpP]), where k assn = max/K 1/2. (C) Second‐order rate constants for ClpP association to single‐chain ClpXΔN variants with different numbers and configurations of IGF‐loop deletions determined from experiment like those shown in Panels A and B. Values are averages (N = 3) ± SD. All association experiments in this panel contained 2 mM ATP.
For the parental enzyme with six IGF loops, k assn for the major phase was ∼1.2 × 106 M−1 s−1. This value was reduced ∼6‐fold for the variants with four or five IGF loops [Fig. 3(C)], a value larger than the 1.16–1.33 decrease expected from the simple fraction of IGF loops remaining in the hexamer. This finding suggests that binding is a multistep process with formation of metastable complexes preceding formation of a stable complex (see Discussion). We also tested a ClpXΔN variant in which three IGF loops in Subunits A, B, and C were deleted, but no significant ClpP binding was detected to this mutant in BLI experiments.
Dissociation kinetics
To assay dissociation kinetics, we allowed ClpXP complexes to assemble, and then shifted the BLI biosensor into buffer containing no ClpP [Fig. 4(A)]. No detectable dissociation of complexes with six IGF loops was observed, as observed previously.14 The deletion of one IGF loop resulted in dissociation with a half‐life of ∼2000 s [Fig. 4(A)], but the amplitude of the single‐exponential fit was ∼50% of the expected value, indicating that there are two populations of complexes, one longer lived and one shorter lived. We obtained the same result in experiments using a different preparation of this variant, suggesting that biphasic dissociation was not a result of enzyme heterogeneity. The deletion of two IGF loops reduced the half‐life of the complex to ∼30–40 s, irrespective of the configuration of the subunits containing the deletions [Fig. 4(A,B)]. Thus, the ClpXP complex becomes dramatically less stable as an increasing number of IGF‐loops are removed from the hexamer.
Figure 4.
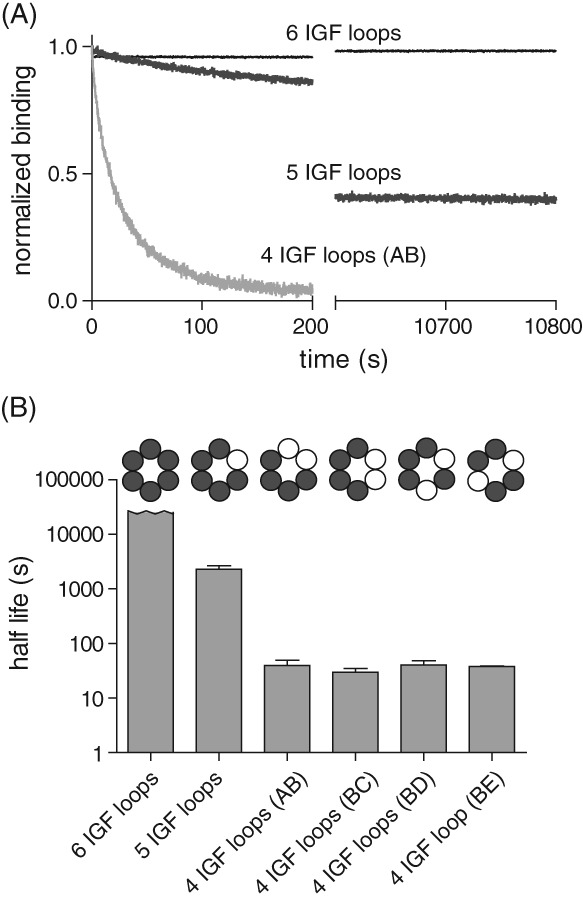
Effect of IGF‐loop deletion on dissociation kinetics. (A) Dissociation kinetics for complexes of ClpP and different variants of single‐chain ClpXΔN were measured in 1 mM ATP by monitoring changes in BLI response following transfer of the biosensor into buffer lacking ClpP. (B) Half‐lives were calculated from single exponential fits of dissociation experiments like those shown in Panel A. The half‐life for the variant with six IGF loops is a lower limit. Values are averages (N = 3) ± SD.
ATP‐dependent proteolysis
We tested the ability of variants to support ClpP degradation of GFP‐ssrA [Fig. 5(A)]. Compared with the parental ClpXΔN pseudohexamer, deletion of one loop decreased the degradation rate ∼2‐fold, as observed previously,10 whereas the deletion of two loops decreased the degradation rate ∼5‐fold. The reduced activity of the deletion mutants is unlikely to result solely from an inability to form ClpXP complexes, as affinities predicted from the association and dissociation kinetics were 120 nM or tighter, whereas the ClpP concentration in degradation experiments was 900 nM. To test if reduced degradation is a consequence of reduced rates of unfolding or translocation by the mutant ClpX enzymes in the absence of ClpP, we assayed unfolding of Arc‐GCN4‐ssrA dimers.17 Single‐chain ClpXΔN variants with four, five, or six IGF loops unfolded this protein substrate at similar rates [Fig. 5(B)]. Although this assay detects unfolding, the rates measured involve multiple turnovers, and thus unfolding and/or translocation could, in principle, be rate limiting. Nevertheless, our variants missing one or two IGF loops had no obvious defects in mechanical processing of this substrate. Under single‐turnover conditions, prior studies show that ClpX hexamers missing one or six IGF loops unfold GFP‐ssrA at almost the same rate as the parental enzymes in the absence of ClpP.10, 15 Hence, any unfolding or translocation defect associated with removal of IGF loops is likely to be a property of the ClpXP complex and not of free ClpX (see Discussion).
Figure 5.
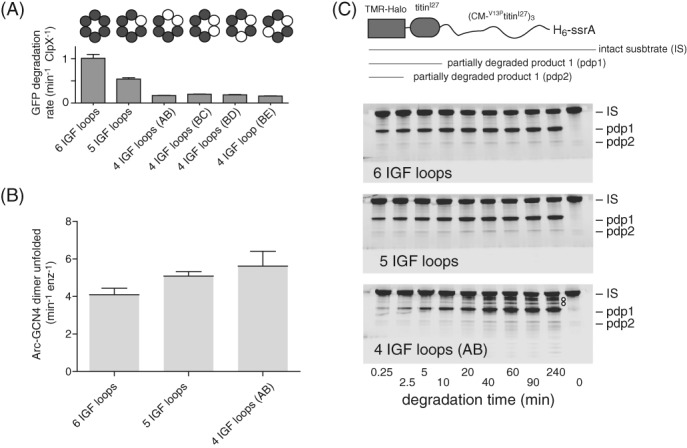
IGF‐loop deletion affects degradation rates and processivity. (A) Rates of degradation of GFP‐ssrA (20 μM) by ClpP (0.9 μM) and different variants of ClpX (0.3 μM pseudohexamer) were measured by monitoring loss of GFP fluorescence. Values are averages (N = 3) ± SD. (B) Rates of unfolding of a fluorescent Arc‐GCN4‐ssrA (5 μM) by ClpX variants (0.3 μM pseudohexamer) were measured in the presence of 10 mM ATP. (C) Top; cartoon of a substrate containing a TAMRA‐labeled Halo domain, a native titinI27 domain, three V13PtitinI27 domains unfolded by carboxymethylation (CM), and a H6‐ssrA degron. Bottom; SDS‐PAGE assays of the ClpP degradation of this substrate by ClpXΔN variants with six, five, or four IGF loops. Note that the variant with four IGF loops shows multiple additional bands between IS and pdp1, indicative of poorly processive degradation. For the enzyme with six IGF‐loops, little accumulation of the pdp1 product or loss of IS occurred after 20 min, probably because the ssrA tag is missing from the majority of substrate molecules because of exopeptidase clipping during purification. Reactions contained substrate (10 μM), ClpP (0.9 μM), single‐chain ClpXΔN variants (0.3 μM hexamer equivalents), and ATP (10 mM). Gels were imaged for fluorescence of the TAMRA dye.
We tested degradation of a multidomain ssrA‐tagged substrate consisting of an N‐terminal Halo domain labeled with a fluorescent dye, a native titinI27 domain, and three V13PtitinI27 domains unfolded by carboxymethylation of cysteines normally buried in the hydrophobic core [Fig. 5(C), top].18 Previous studies show that ClpXΔN and ClpP efficiently degrade the unfolded domains of this substrate but a partially degraded product (pdp1) consisting of the Halo and native titinI27 domains accumulates as a consequence of some enzymes dissociating after failing to unfold native titinI27.18, 19 As assayed by SDS‐PAGE and fluorescence imaging, the pdp1 product was observed during ClpP degradation supported by single‐chain ClpXΔN variants with six, five, or four IGF loops, but this product accumulated more slowly as more IGF loops were removed [Fig. 5(C)]. For example, the pdp1 product initially accumulated at 57% of the wild‐type rate for the single IGF deletion and at 34% of this rate for the double IGF deletion, supporting the idea that ClpXP translocation may slow as more IGF loops are deleted. Notably, fragments intermediate in size between the intact substrate (IS) and pdp1 product were observed for the ClpXΔN variant containing four IGF loops [marked by circles in the lower gel of Fig. 5(C)] but not for enzymes containing five or six IGF loops. These results suggest that degradation by the variant with four IGF loops is less processive and that some enzymes dissociate during translocation of unfolded portions of the substrate.
Loop‐length and substitution mutations
In an unlinked ClpXΔN background, we found that some changes in IGF‐loop length were tolerated, whereas others were not [Fig. 6(A)]. For example, the deletion of three residues from the C‐terminal part of the IGF‐loop did not substantially impact the rate of ClpP degradation of GFP‐ssrA, but degradation defects were observed after deletion of two residues from the N‐terminal part of the loop or insertion of two alanines in either the N‐terminal or C‐terminal parts of the loop. The ClpX IGF loop is 16 residues in length. When we calculated IGF‐loop lengths for a large number of ClpX orthologs, the most common lengths were 14 or 15 residues with a range from 8 to 16 residues.
Figure 6.
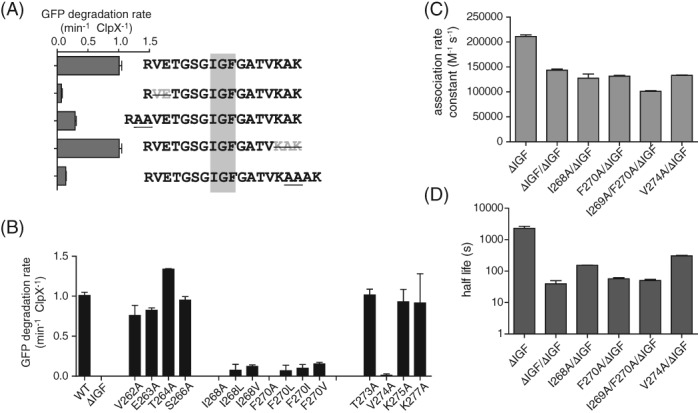
Effects of mutations in the IGF loop. (A) Rates of degradation of GFP‐ssrA (20 μM) by ClpP (0.9 μM) and ClpXΔN variants containing longer or shorter IGF loops (0.3 μM hexamer). (B) Rates of degradation of GFP‐ssrA (20 μM) by ClpP (0.9 μM) and ClpXΔN variants (0.3 μM hexamer) with single‐ or double‐residue substitutions in the IGF loop. (C) Association rate constants determined by BLI experiments using 1 μM ClpP and 2 mM ATP. (D) Dissociation half lives determined by BLI experiments in the presence of 2 mM ATP. In Panels C and D, single‐chain ClpXΔN variants had the IGF‐loop deleted from Subunit B and had a wild‐type or mutant IGF motif in Subunit A. In all panels, values are averages (N = 3) ± SD.
The IGF motif of E. coli ClpX includes Ile268 and Phe270. Mutating Ile268 to Ala, Val, or Leu, or changing Phe270 to Ala, Val, Ile, or Leu, severely compromised or eliminated the ability of unlinked ClpXΔN to collaborate with ClpP in degrading GFP‐ssrA [Fig. 6(B)]. Replacing Val274 with Ala was also highly deleterious, but other amino‐acid substitutions at non‐alanine and non‐glycine positions in the IGF loop of ClpXΔN—including V262A, E263A, T264A, S266A, T273A, K275A, and K277A—had little effect on degradation [Fig. 6(B)].
To further probe the relative importance of Ile268, Phe270, and Val274, we constructed single‐chain ClpXΔN pseudo hexamers containing an IGF‐loop deletion in subunit B and I268A, F270A, I268A/F270A, or V274A mutations in subunit A. Because the deletion of a single IGF‐loop accelerates dissociation to a measurable rate, we reasoned that effects of the mutations in subunit A on association and dissociation kinetics could be detected in BLI experiments. The single‐ and double‐point mutations decreased the rate of association less than 2‐fold [Fig. 6(C)] but increased the rate of dissociation by ∼5 to ∼50‐fold [Fig. 6(D)]. Specifically, faster dissociation followed the trend ΔIGF >I268A/F270A >F270A >I268A >V274A.
Discussion
The IGF loops of ClpX are disordered in most crystal structures, suggesting that they are statically or dynamically disordered.20, 21 Indeed, flexibility of these loops was initially postulated as a mechanism to overcome the symmetry mismatch between the hexameric ring of ClpX and heptameric rings of ClpP.9 If these loops are flexible, however, then why is ATP or ATPγS needed to support ClpXP assembly? Our results indicate that the IGF loops are equally accessible to chymotryptic cleavage in the presence of ATP, ATPγS, or ADP. Thus, the inability of ADP to support assembly is not a consequence of the IGF loops being hidden or sequestered. Rather, fluorescence homo‐quenching studies indicate that the IGF loops are farther apart, on average, in the presence of ADP or absence of nucleotide and closer together in the presence of ATP or ATPγS. Thus, it seems likely ATP and ATPγS alter the conformation of the hexameric ClpX ring, which places the IGF loops in positions that allow efficient multivalent binding to the clefts on the ClpP ring.
The precise affinity of single‐chain ClpXΔN for ClpP is not known because the complex is too kinetically stable to determine an accurate dissociation rate constant in the presence of ATP. Nevertheless, this affinity appears to be ∼100 pM or less.14 Based on the studies here, removal of one IGF loop reduces this affinity to ∼5 nM, whereas removal of two IGF loops reduces it to ∼100 nM. We were unable to detect binding of a ClpXΔN variant with three IGF loops to ClpP, but transient binding could occur at higher concentrations.
In previous studies, we found that the rate of association saturates hyperbolically,14 suggesting that ClpP initially forms an unstable encounter complex with ClpX, which then is stabilized by unimolecular docking of more IGF loops. Based on those studies, the major encounter complex forms with a saturated rate of 27 ± 7 s−1 (k 2), a half maximal ClpP concentration of ∼35 ± 14 μM ([k 2 + k −1]/k 1), and an apparent second‐order rate constant (k 2•k 1/[k 2 + k −1]) of ∼600,000 ± 80,000 M−1 s−1. We solved for values of k 1 and k −1 that would minimize differences with the observed values for the half‐maximal concentration and apparent second‐order rate constant. A k 1 value of 107 M−1 s−1 and k −1 value of 370 s−1 predict half‐maximal formation of the encounter complex at ∼40 μM ClpP and an apparent second‐order rate constant of ∼700,000 M−1 s−1. By this model, the encounter complex would form ∼8‐fold faster than the stable ClpXP complex but dissociate at a rate ∼14‐fold faster than the rate of conversion of the encounter complex to the stable ClpXP complex. These values seem plausible if the encounter complex involves docking of two or three ClpX IGF loops with ClpP. Dividing k 2 (the rate of unimolecular docking of IGF loops) by the second‐order association rate constants for ClpXΔN variants with six, five, or four IGF loops (bimolecular docking of IGF loops) gives effective concentrations from approximately 20 to 200 μM. These values are reasonable for intramolecular docking of flexible loops in an encounter complex. The trend we observe of faster dissociation rates as additional IGF loops are deleted is also consistent with a model in which a transient complex with two or three docked IGF loops could dissociate on the millisecond timescale. It is possible that ADP‐bound ClpX can also form a similar encounter complex but is then unable to then make additional IGF‐loop contacts because of conformational constraints.
Martin et al. found that deletion of one IGF loop from a ClpX hexamer slowed ClpXP degradation of native GFP‐ssrA or of an unfolded titinI27 substrate to ∼50–60% of the parental rate.10 Here, we find that the deletion of two IGF loops slows ClpXP degradation of GFP‐ssrA or of the unfolded portion of a multidomain substrate to ∼20–35% of the parental rate. In the absence of ClpP, experiments performed previously10, 15 and here show that ClpX variants with zero, four, five, or six IGF loops unfold native protein substrates at similar rates. Hence, the ClpXP degradation defects associated with IGF‐loop deletions are likely to result from a reduced rate of polypeptide translocation and not from slower substrate unfolding. The ClpP axial channel, which gates access into the degradation chamber, is widened substantially upon binding of ClpX or small‐molecule acyldepsipeptides (ADEPs), which mimic the IGF loops of ClpX.22, 23, 24 As some ClpX or ADEP binding energy must be used to stabilize the ClpP open‐channel conformation, ClpX variants with fewer IGF loops may not fully open the channel or result in equilibration between open and closed channel conformations. Either model could explain slower substrate translocation and thus slower degradation for ClpX variants with less than a full complement of IGF loops. Our ClpX variant with four IGF loops mediated less processive ClpP degradation of unfolded portions of a substrate compared with variants with five or six IGF loops. This result suggests that ClpP dissociates from the four‐loop variant in the midst of polypeptide translocation. By contrast, ClpX with a wild‐type complement of IGF loops rarely dissociates from ClpP during translocation but can dissociate during failed unfolding of a native substrate domain.19
Our mutagenic and biochemical studies reveal that substitutions in the first and third residues of the IGF motif (Ile268 and Phe270) can be as deleterious to binding as deletion of the entire loop. This result suggests that many of the stabilizing contacts between the IGF loop and ClpP pay for the energetic cost of altering the conformation of ClpP and/or the entropic cost of ordering the loop. We find that some changes in the length of the IGF loop are tolerated but others are not, suggesting that the geometry with which the IGF motif and supporting residues are displayed is also an important determinant of ClpXP binding affinity.
In summary, our results show that a full complement of six IGF loops is required for strong ClpP affinity, for kinetic stability of the ClpXP complex, and for efficient protein degradation. Although multivalent binding of IGF loops to ClpP is clearly important, our results suggest that individual loops act relatively independently. For example, the association and dissociation rate constants were similar for ClpX variants missing two IGF loops, irrespective of the configuration of the missing loops. This result suggests that neighboring IGF loops do not contact or interact with each other in ways that influence assembly or disassembly kinetics.
Materials and Methods
Proteins
ClpP and single‐chain ClpXΔN (with C169S and K408E substitutions) pseudohexamers were expressed and purified as described.14 Unlinked ClpXΔN containing an N‐terminal His6‐TEV tag (MGSSHHHHHHDYDIPTTENLYFQGSS) was expressed from pET‐22b (EMD Millipore) and purified as described for single‐chain ClpXΔN.14 IGF‐loop deletions replaced ClpX Residues 262–277 with a GGSSGG linker. Point mutations were generated by polymerase‐chain‐reaction mutagenesis. His6‐TEV‐GFP‐ssrA was expressed from pET‐22b (EMD Millipore) and purified as described.14 Arc‐GCN4‐ssrA was purified as described.17
Biochemical assays
Unless noted, biochemical assays were performed at 25°C in PD buffer (25 mM HEPES, pH 7.5, 100 mM KCl, 10 mM MgCl2, and 10% glycerol) supplemented with ATP, ATPγS, or ADP as necessary. Enzyme concentrations refer to ClpX hexamers or ClpP 14‐mers. Rates of degradation of GFP‐ssrA (20 μM) by ClpXΔN variants (300 nM) and ClpP (900 nM) were measured in the presence of ATP (5 mM), phosphocreatine (32 mM), and creatine kinase (0.064 mg/mL) in black NBS 384‐well plates (Corning) using a SpectraMax M5 plate reader. Assays were preformed in triplicate, and the degradation rate was calculated from a linear fit of the initial loss of fluorescence (excitation 467 nm; emission 511 nm). Unfolding of Arc‐GCN4‐ssrA was assayed as described.17
Chymotrypsin cleavage
ClpXΔN, ClpXΔN/ΔIGF, or ClpXΔN/F270A (1 μM each) were mixed with ATP, ATPγS, or ADP (10 mM each), and cleavage reactions were initiated by addition of chymotrypsin (0.01 mg/mL; Worthington). Reactions were quenched after different times by the addition of phenylmethanesulfonyl fluoride (1 mM; Sigma) and mixed with 2× Sample Buffer (BioRad). Samples were analyzed after electrophoresis on a 2–20% gradient SDS‐PAGE (GenScript) in 1× MOPS buffer (GenScript), staining using Sypro Ruby Gel Stain (BioRad), and imaging using a FluorChem R ProteinSimple imager.
Alexa‐647 labeling
T273C ClpXΔN (10 μM) was labeled with Alexa Fluor 647‐C2−Maleimide (90 μM; Thermo Fisher) with an efficiency calculated to be ∼6 dyes/hexamer using spectrophotometric measurements and the manufacturer's dye‐correction factor. Fluorescence measurements (excitation 655 nm; emission 671 nm) were taken using the labeled T273C ClpXΔN enzyme (500 nM) in the absence of nucleotide or presence of ATP, ATPγS, or ADP (1 mM each). For mixing experiments, different concentrations of unlabeled ClpXΔN and Alexa‐647‐labeled T273C ClpXΔN were mixed and allowed to equilibrate for 1 h prior to addition of nucleotide and fluorescence determination.
BLI assays
BLI assays were carried out using an Octet RED96 instrument (ForteBio) at 30°C in PD buffer with 0.05% TWEEN‐20 (Amresco) as described.14 Single‐chain ClpXΔN variants containing a biotin‐acceptor peptide were loaded onto streptavidin‐coated biosensors to 0.5 nm loading signal. After washing with 2 mM ATP, biosensors were transferred to buffer containing different concentrations of ClpP and ATP (2 mM) to measure association kinetics. Data were initially fit to a single exponential equation to estimate half‐lives. Data were then truncated to ∼10 half‐lives and re‐fit to a single‐exponential or double‐exponential equation to obtain apparent rate constants (k app). The association rate constant was calculated as the initial slope of a plot of k app versus ClpP concentration. To measure dissociation kinetics, biosensors were transferred to buffer containing ATP (2 mM) but no ClpP, and the dissociation rate constant was calculated from a single‐exponential fit of these data.
Acknowledgments
BLI experiments were performed in the M.I.T. Biophysical Instrument Facility. T.A.B. is an employee of the Howard Hughes Medical Institute. K.R.S. was supported by a Charles A. King Trust Postdoctoral Research Fellowship.
References
- 1. Sauer RT, Baker TA (2011) AAA+ proteases: ATP‐fueled machines of destruction. Annu Rev Biochem 80:587–612. [DOI] [PubMed] [Google Scholar]
- 2. Baker TA, Sauer RT (2012) ClpXP, an ATP‐powered unfolding and protein‐degradation machine. Biochim Biophys Acta 1823:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA (2003) Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX‐recognition signals. Mol Cell 11:671–683. [DOI] [PubMed] [Google Scholar]
- 4. Moore SD, Sauer RT (2007) The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem 76:101–124. [DOI] [PubMed] [Google Scholar]
- 5. Keiler KC (2008) Biology of trans‐translation. Annu Rev Microbiol 62:133–151. [DOI] [PubMed] [Google Scholar]
- 6. Wang J, Hartling JA, Flanagan JM (1997) The structure of ClpP at 2.3 Å resolution suggests a model for ATP‐dependent proteolysis. Cell 91:447–456. [DOI] [PubMed] [Google Scholar]
- 7. Grimaud R, Kessel M, Beuron F, Steven AC, Maurizi MR (1998) Enzymatic and structural similarities between the Escherichia coli ATP‐dependent proteases, ClpXP and ClpAP. J Biol Chem 273:12476–12481. [DOI] [PubMed] [Google Scholar]
- 8. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA (2001) Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol 8:230–233. [DOI] [PubMed] [Google Scholar]
- 10. Martin A, Baker TA, Sauer RT (2007) Distinct static and dynamic interactions control ATPase‐peptidase communication in a AAA+ protease. Mol Cell 27:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh SK, Rozycki J, Ortega J, Ishikawa T, Lo J, Steven AC, Maurizi MR (2001) Functional domains of the ClpA and ClpX molecular chaperones identified by limited proteolysis and deletion analysis. J Biol Chem 276:29420–29429. [DOI] [PubMed] [Google Scholar]
- 12. Burton RE, Baker TA, Sauer RT (2003) Energy‐dependent degradation: linkage between ClpX‐catalyzed nucleotide hydrolysis and protein–substrate processing. Protein Sci 12:893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin A, Baker TA, Sauer RT (2005) Rebuilt AAA + motors reveal operating principles for ATP‐fuelled machines. Nature 437:1115–1120. [DOI] [PubMed] [Google Scholar]
- 14. Amor AJ, Schmitz KR, Sello JK, Baker TA, Sauer RT (2016) Highly dynamic interactions maintain kinetic stability of the ClpXP protease during the ATP‐fueled mechanical cycle. ACS Chem Biol 11:1552–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joshi SA, Hersch GL, Baker TA, Sauer RT (2004) Communication between ClpX and ClpP during substrate processing and degradation. Nat Struct Mol Biol 11:404–411. [DOI] [PubMed] [Google Scholar]
- 16. Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT (2005) Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell 121:1017–1027. [DOI] [PubMed] [Google Scholar]
- 17. Baytshtok V, Baker TA, Sauer RT (2015) Assaying the kinetics of protein denaturation catalyzed by AAA+ unfolding machines and proteases. Proc Natl Acad Sci U S A 112:5377–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iosefson O, Olivares AO, Baker TA, Sauer RT (2015) Dissection of axial‐pore loop function during unfolding and translocation by a AAA+ proteolytic machine. Cell Rep 12:1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kenniston JA, Baker TA, Sauer RT (2005) Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci U S A 102:1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT (2009) Structures of asymmetric ClpX hexamers reveal nucleotide‐dependent motions in a AAA+ protein‐unfolding machine. Cell 139:744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stinson BM, Nager AR, Glynn SE, Schmitz KR, Baker TA, Sauer RT (2013) Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell 153:628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li DH, Chung YS, Gloyd M, Joseph E, Ghirlando R, Wright GD, Cheng YQ, Maurizi MR, Guarné A, Ortega J (2010) Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: a model for the ClpX/ClpA‐bound state of ClpP. Chem Biol 17:959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee BG, Park EY, Lee KE, Jeon H, Sung KH, Paulsen H, Rübsamen‐Schaeff H, Brötz‐Oesterhelt H, Song HK (2010) Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat Struct Mol Biol 17:471–478. [DOI] [PubMed] [Google Scholar]
- 24. Lee ME, Baker TA, Sauer RT (2010) Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J Mol Biol 399:707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]