

Abstract
Peptide display approaches, in which peptide epitopes of known binding activities are grafted onto stable protein scaffolds, have been developed to constrain the peptide in its bioactive conformation and to enhance its stability. However, peptide grafting can be a lengthy process requiring extensive computational modeling and/or optimisation by directed evolution techniques. In this study, we show that ultra‐stable consensus‐designed tetratricopeptide repeat (CTPR) proteins are amenable to the grafting of peptides that bind the Kelch‐like ECH‐associated protein 1 (Keap1) onto the loop between adjacent repeats. We explore simple strategies to optimize the grafting process and show that modest improvements in Keap1‐binding affinity can be obtained by changing the composition of the linker sequence flanking either side of the binding peptide.
Keywords: protein engineering, tetratricopeptide repeat, repeat protein, protein–protein interaction, therapeutics, biologics
Introduction
Scaffold proteins can be given new functionalities by grafting of binding peptides onto an alpha‐helix or a loop. In a similar manner to chemical stapling methods, the scaffold constrains the binding epitope in its bioactive conformation and endows it with enhanced proteolytic stability.1, 2, 3, 4, 5, 6, 7, 8, 9 Helix grafting has been particularly successful, but there are challenges in the grafting of peptides into protein loops. The difficulty arises in part because it can be hard to predict loop conformations, and there may be a large entropic cost upon binding of the grafted peptide.10 There are alternative methods for creating proteins with new high‐affinity binding functions including experimental approaches, such as surface display by directed evolution, and computational design and combinations thereof.3, 11, 12, 13, 14 However, where there is a known binding epitope comprising a short linear motif, a simple “cut‐and‐paste” strategy may be sufficient, and indeed even relatively long binding peptides of around 40 amino acids have successfully been grafted in this way.8, 15
Tetratricopeptide repeat (TPR) proteins are a class of repeat proteins that are common in nature.16, 17 They are characterized by repeating units of 34 amino acids, which form two antiparallel helices joined by a turn region. A consensus‐designed tetratricopeptide repeat protein (CTPR) was first developed by Main et al. by alignment of proteins from a nonredundant database in order to determine the optimal sequence of the CTPR and resulted in proteins with very high stability and without need of disulphide bonds.18 Grove et al. used a modified CTPR sequence with two mutations, D18Q and E19K, and Cortajarena et al. later described how the E19K mutation promoted charge–charge interactions and increased the intrinsic stability of each repeat.19, 20 TPR proteins can utilize varied modes of binding to their partner proteins but the most well‐characterized involves the groove formed by 2–3 repeats binding to a short (~ 5 residue) negatively charged peptide.21, 22, 23 Exploiting this binding mode, Cortajarena et al. grafted the Hsp90‐binding residues from the natural TPR protein Hop onto a three‐repeat CTPR (CTPR3) to create a functional TPR module.24 They subsequently used both rational design approaches and library screening to make novel functional CTPR proteins.25, 26 In all cases, the affinities observed for this binding mode are fairly weak (low‐ to mid‐micromolar), presumably because the interfaces involved have relatively small areas.
The high thermodynamic stabilities of CTPRs combined with their modular nature means that even gross modifications are still likely to produce proteins with high stabilities, and indeed we recently showed that CTPR proteins can tolerate extension of the inter‐repeat loop by up to 25 residues.27 Here, we take advantage of this malleability to describe a new strategy for engineering high‐affinity binding functions into CTPR proteins by grafting functional peptides into the inter‐repeat loop. As proof of concept, we use a short peptide sequence derived from the protein Nrf2 (nuclear factor erythroid‐2‐related factor) that is a substrate of the E3 ubiquitin ligase Keap1 (Kelch‐like ECH‐associated protein 1). Nrf2 is a transcription factor that modulates the body's response to oxidative stress. Keap1 is known to target Nrf2 for ubiquitination leading to degradation by the proteasome. Keap1 binds to Nrf2 through a hinge‐and‐latch mechanism in which a single Nrf2 molecule binds to the two Kelch domains of the Keap1 dimer via two different binding motifs on Nrf2.28 The ETGE motif (residues 77–82) initiates binding and acts as the hinge, allowing the binding of 100‐fold weaker DLG motif “latch” (residues 27–32).29, 30 The ETGE motif of Nrf2 binds to Keap1 in a beta‐turn conformation with the side chains of E79 and E82 forming hydrogen bonds to Keap1 [Fig. 1(B)].31 Further contacts with Keap1 are made through four carbonyl groups and one amide group from the Keap1 peptide backbone.32 Additionally, L75, L84 and D77 have been suggested to stabilise the beta‐turn structure. The binding of isolated, unconstrained peptides based on the ETGE motif of Nrf2 to Keap1 is well‐characterized through biophysical, crystallographic and cell‐based assays.31, 33, 34, 35, 36, 37 Inhibiting this interaction has therapeutic potential for a range of diseases, such as the chemoprevention of cancer, diabetes Alzheimer's disease, and chronic obstructive pulmonary disease.38, 39
Figure 1.
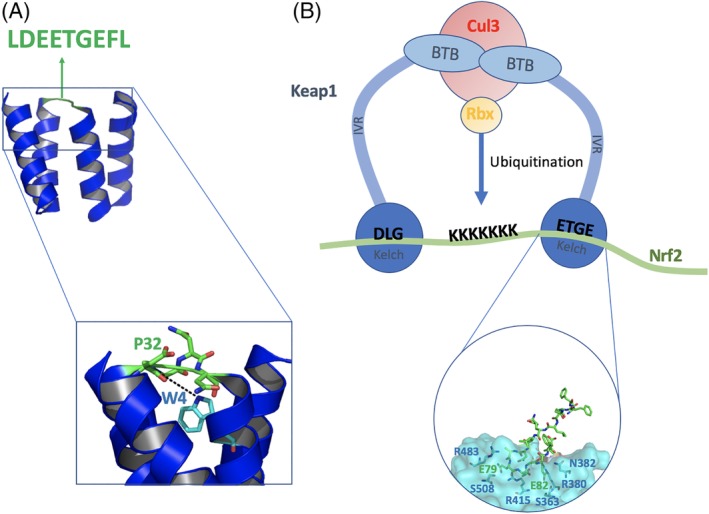
Schematics showing the proteins used in this study and the peptide grafting process. (A) The Nrf2 peptide (green) is grafted into the inter‐repeat loop of the CTPR2 protein (blue) PDB ID 1NA0. (B) Schematic representation of the structure of the Keap1‐Nrf2 complex. The BTB, IVR, and Kelch domains of the substrate‐recognition subunit, Keap1, assemble with Cullin 3 (Cul3) subunit and Rbx subunit to form the active E3 ubiquitin ligase. The ETGE motif of the substrate Nrf2 (residues 77–82, blue) adopts a β‐turn conformation. E79 of Nrf2 forms hydrogen bonds to R415, R483, and S508 of Keap1 whereas E82 hydrogen bonds to S363, N382, and R380 PDB ID 2FLU.48
The CTPR proteins are small, monomeric, and very stable without need of disulphide bonds. They are easy to produce recombinantly in high yields, facilitated further by the fact that a small‐scale expression/purification protocol is usually sufficient to produce milligrams quantities of high purity protein without need of multiple purification steps, and therefore many proteins can be made in parallel for testing.40 Our results indicate that the CTPR proteins could provide a scaffold with which to screen binding sequences before going on to producing the optimized sequence in the form of constrained chemically synthesized peptides such as those used for therapeutic purposes, as is costly and laborious to produce large numbers of such peptides themselves. Most importantly, whereas new binding functions engineered into TPR proteins to date have all had micromolar affinities for their targets, our approach can yield orders of magnitude higher affinities in the nanomolar range.
Results and discussion
Protein design
The CTPR sequence used previously by Grove et al. was chosen due to its high stability and solubility despite the lack of capping repeats that are often used for designed repeat proteins.19, 20 A scaffold without caps will be useful in further work intended to introduce additional functionalisation by using more repeats in a modular fashion. We grafted a Keap1‐binding peptide derived from Nrf2 into this CTPR scaffold. We chose the ETGE motif because it has been shown to have high affinity for Keap1 even without any chemical or protein constraints, and peptides based on this motif have already been shown to be amenable to grafting onto proteins.41, 42
The inter‐repeat loop of the CTPR scaffold comprises four amino acids (DPNN, residues 31–34 of the repeat). The “Consensus Nrf2 CTPR2” protein was designed with the Nrf2 peptide inserted into the loop of CTPR2 with a “DPNN” sequence flanking it on both sides repeated so as to preserve the chemical environment of the residues adjacent to the loop (Table 1).43 Additionally, P32 is also an important residue, as it promotes the turn conformation and provides backbone hydrogen bonding to W4 of the next repeat.18
Table 1.
Design of the Keap1‐binding loops grafted into the CTPR scaffold
| CTPR | Loop sequence |
|---|---|
| Consensus Nrf2 CTPR2 | DPNNLDEETGEFLDPNN |
| Flexible Nrf2 CTPR2 | DPNNGGLDEETGEFLGGDPNN |
| Charge Nrf2 CTPR2 | DPDNLDEETGEFLDPDN |
| CIDER NRf2 CTPR2 | DPNNLDEETGEFLDPRN |
The sequence of the “native” CTPR loop is in bold. The sequence corresponding to the Nrf2 peptide is shown in italics. The other residues, relating to the design strategy, are shown in plain font.
Peptide grafting onto protein scaffolds is more likely to be successful when there is a good geometrical match between the peptide and the scaffold. This requirement can be hard to meet in addition to finding a scaffold that is amenable to grafting without destabilization. The rigid CTPR, like other scaffolds, means that the grafted peptide may not be able to adopt the correct conformation. Therefore, we proposed the insertion of a flexible “GG” linker both before and after the grafted peptide in order to overcome this potential problem (named “Flexible Nrf2 CTPR2”; Table 1). We hypothesize that this may reduce conformational strain on the grafted peptide, allowing it to more easily adopt a bioactive conformation. Flexible loop regions are often observed in nature to modulate binding, and they may also help to prevent steric clashes between the protein scaffold and the target protein.44
The Keap1‐ETGE interface involves multiple electrostatic interactions, and many studies have focused on optimizing electrostatics so as to optimize binding affinities.28, 45, 46 We hypothesized that the mutation of asparagine to aspartic acid in the amino acids both preceding and proceeding the grafted Nrf2 peptide could help increase the favorable electrostatic interactions with residues on Keap1, thereby enhancing the binding affinity. This approach is seen in Charge Nrf2 CTPR2 design (Table 1).
CIDER is a program developed by Pappu et al. that predicts the structure of intrinsically disordered proteins.47 If we make the assumption that loop regions are, for the most part, disordered, we can use the programme as an aid to predict how certain mutations might move the peptide from region 1 (swollen coils) to region 3 (coils, hairpins, and chimeras), which may allow the grafted Nrf2 peptide to adopt its bioactive β‐turn conformation and thus lead to a higher affinity of binding. This design is referred to as CIDER Nrf2 CTPR2 (Table 1).
Secondary structure content and thermal stability
The proteins expressed in Escherichia coli in the soluble fraction with high yields of 1–2 mg from 90 mL of culture. Samples were pure as judged by mass spectrometry. Circular dichroism (CD) spectra showed the proteins to be folded [Fig. 2(B)] and to have high degrees of α‐helicity. The very high stability of the parent sequence (CTPR2) means that, although there is a significant decrease in thermal stability upon introduction of the grafted peptide sequences, the proteins still have high thermal stabilities with melting temperatures around 70°C [Fig. 2(C), Table 2]. The melting temperature was found to be relatively insensitive to the nature and length of the grafted peptide. All proteins were found to refold after thermal denaturation, further demonstrating the stability of the CTPR scaffold [Fig. 2(A)].
Figure 2.
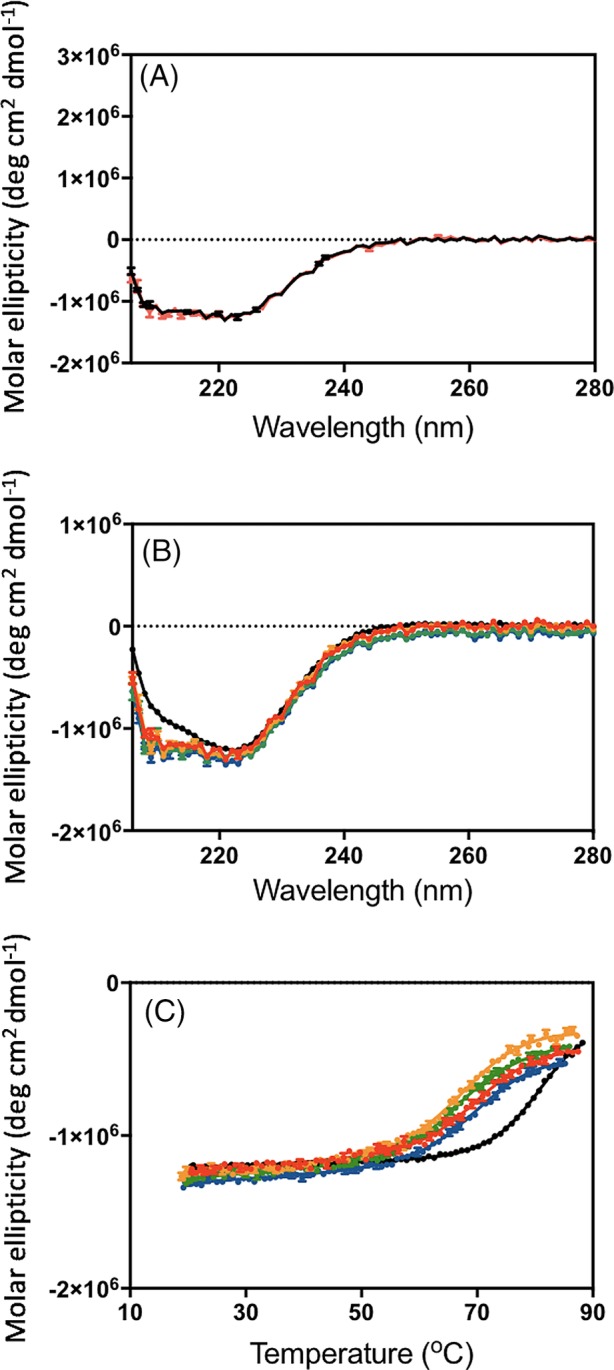
CD analysis of the Nrf2 CTPR2 proteins. (A) CD spectrum of consensus Nrf2 CTPR2 before (black) and after (red) thermal denaturation. (B) CD spectra of CTPR2 (black), consensus Nrf2 CTPR2 (red), flexible Nrf2 CTPR2 (orange), charge Nrf2 CTPR2 (green) and CIDER Nrf2 CTPR2 (blue). (C) Thermal denaturation curves monitored by CD. CTPR2 (black), consensus Nrf2 CTPR2 (red), flexible Nrf2 CTPR2 (orange), charge Nrf2 CTPR2 (green), and CIDER Nrf2 CTPR2 (blue).
Table 2.
Circular dichroism (CD) and fluorescence polarization (FP) data for the thermal stability and Keap1‐binding affinities of the designed TPR proteins
| CTPR | Tm ± SE (°C) | K i ± SE (nM) |
|---|---|---|
| CTPR2 | >80 | ‐ |
| Consensus Nrf2 CTPR2 | 70.0 ± 1.3 | 282 ± 64 |
| Flexible Nrf2 CTPR2 | 68.0 ± 1.0 | 109 ± 18 |
| Charge Nrf2 CTPR2 | 65.6 ± 0.9 | 817 ± 75 |
| CIDER NRf2 CTPR2 | 70.6 ± 1.5 | 103 ± 15 |
Binding of grafted CTPR proteins to Keap1
First, we used FP to measure the K D of a FITC‐labeled Nrf2 peptide (FITC‐β‐Ala‐DEETGEF‐OH) for the Keap1 Kelch domain (referred to subsequently simply as Keap1). The value obtained was 238 ± 29 nM [Fig. 3(A)], which is consistent with previously published data. 31 Next, we used competition FP to determine the Keap1‐binding affinities of the Nrf2‐grafted CTPR proteins [Fig. 3(B)]. We found that, relative to the isolated Nrf2 peptide, small increases in affinity could be achieved by grafting the peptide into the CTPR scaffold with a flexible linker (Flexible Nrf2 CTPR2) and with the CIDER‐designed sequence (CIDER Nrf2 CTPR2). The binding affinity of Flexible Nrf2 CTPR2 was verified using ITC, and a K D of 74.6 ± 17 nM was obtained [Fig. 3(C)].
Figure 3.
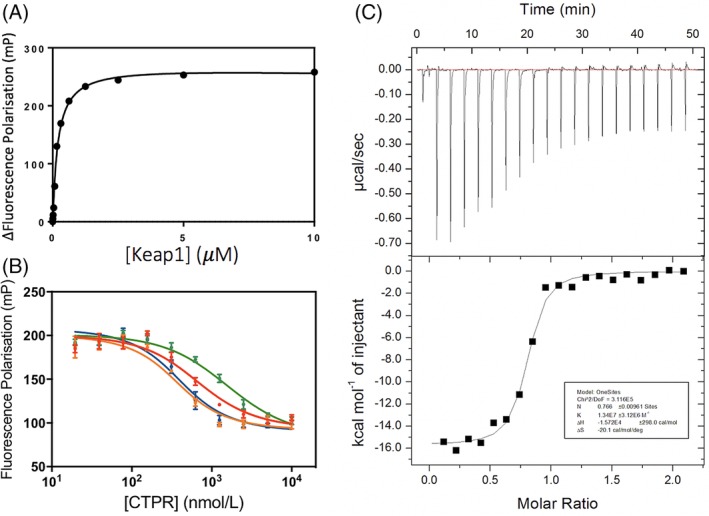
Binding of the Nrf2 CTPR2 proteins to Keap1 monitored by FP and ITC. (A) FP of Keap1 binding to FITC‐β‐Ala‐DEETGEF‐OH peptide. (B) Competition FP of consensus Nrf2 CTPR2 (red), flexible Nrf2 CTPR2 (orange), charge Nrf2 CTPR2 (green), and CIDER Nrf2 CTPR2 (blue) with the preformed complex of Keap1 and Fl‐β‐Ala‐DEETGEF‐OH. (C) ITC of flexible Nrf2 CTPR2 and Keap1.
It is interesting that modest (two‐fold) improvements in binding affinity are observed, since the Nrf2 peptide sequence already makes intramolecular interactions through residues L76 and L84 and so it was not clear whether changing the residues flanking these leucines would have any further impact on the binding affinity. We hypothesize that the higher binding affinities of the Flexible Nrf2 CTPR2 and CIDER Nrf2 CTPR compared with the original Nrf2 CTPR arise because these designs enable the grafted peptide to adopt a bioactive conformation. We note that the two approaches yield almost identical binding affinities, suggesting that we may be close to the maximum affinity that can be achieved using this Nrf2 sequence.
Future studies will focus on testing different types of flanking sequences in combination with different binding peptides. This approach will be particularly important for those binding epitopes that have a poor structural match to the native tight‐turn conformation of the CTPR loop, as the flanking regions may be able to either provide constraint or be flexible enough to allow the peptide to adopt its bioactive conformation. The low affinity of Charge Nrf2 CTPR2 for Keap1 could be explained if the introduction of the N33D mutation induces structural changes in the loop that distort the binding epitope away from its bioactive conformation. These studies will allow us to understand the relationship between the length/sequence composition of the grafted peptide and its binding affinity within the context of the CTPR scaffold. In terms of the maximum peptide length, we have found that we can successfully graft binding peptides of up to 15 residues onto the inter‐repeat loop, and we have also shown that the loop can be extended by up to 40 residues without disrupting the CTPR structure (27; unpublished results).
The small size of these CTPR2 proteins (at 11.5 KDa), their amenability to peptide grafting without disrupting the structure or drastically reducing the overall stability, and their capacity to display peptides with nanomolar affinities for their targets, could make them potential candidates for future biotherapeutics. This study provides new strategies for peptide grafting into scaffolds without the need for extensive computational design or directed evolution experiments and introduces a new scaffold for peptide grafting.
Materials and methods
Protein expression constructs
The CTPR constructs were cloned from gBlock oligos purchased from Integrated DNA Technologies and inserted into the PRSet B multicloning site of the vector using restriction digestion‐ligation cloning between with BamHI and HindIII restriction enzymes (ThermoFisher Scientific, Waltham, MA) and Bioline quick stick ligase according to the manufacturer's protocol. The Keap1 construct has an N‐terminal His‐tag and the Human Keap1 Kelch domain (residues 321–624) with a TEV cleavage site in a pNIC28‐Bsa4 vector. This was a kind gift from Alex Bullock (Structural Genomics Consortium, Oxford).
Peptide synthesis
FITC‐β‐Ala‐DEETGEF‐OH was synthesized by Cambridge Peptides Ltd and provided at a purity of >90%.
Protein expression and purification
CTPR2 constructs were transformed into E. coli Lemo21 cells, apart from CTPR2, which was transformed into C41 cells. Colonies were individually selected and grown in 15 mL 2xYT media for approx. 16 hours at 37°C until an OD of 0.8 was reached, and then induced with 0.5 mM IPTG and grown for 24 hr at 20°C. The cells were then pelleted and purified according to the protocol published by Perez‐Riba et al.40 Samples were pure as judged by mass spectrometry.
The Keap1 Kelch domain expression plasmid was transformed into C41 cells and grown at 37°C until O.D. of 0.8 was reached. Cells were then induced with 0.5 mM IPTG at 20°C overnight. Cells were pelleted at 5000 RPM for 5 min. Cells were resuspended in 35 mL of 50 mM Tris–HCl pH 8, 150 mM NaCl, 2 mM DTT with a Sigmafast protease tablet (EDTA‐free) and then lysed using an Emulsiflex C‐5 homogenizer using three runs. The lysed cells were centrifuged at 17000 RPM for 45 min. The cleared lysates were incubated with 4 mL Ni‐NTA beads for 1 hr at 4°C. The beads were then washed from using 3 × 50 mL washes with 50 mM Tris‐HCl pH 8, 150 mM NaCl, 2 mM DTT and eluted with 10 mL 50 mM Tris‐HCl pH 8, 150 mM NaCl, 300 mM Imidazole, 2 mM DTT. The mixture was then filtered through a 0.22 μm syringe and further purified by gel filtration with a HiLoad 26/60 Superdex 75 column in 50 mM Tris‐HCl pH 8, 150 mM NaCl, 2 mM DTT.
CD spectroscopy and thermal unfolding experiments
Secondary structure content was assessed using CD. The spectra of 10 μM CTPR proteins in 50 mM Tris‐HCl pH 8.5, 150 mM NaCl was measured using an Applied Photophysics Chirascan CD spectrophotometer with a 1 mm cuvette between wavelengths of 206 nm and 280 nm at 20°C at 1 nm increments at a rate of 0.5 s/reading with three repeats being taken. The thermal stability of the proteins was determined by measuring the molar ellipticity of the solution at 222 nm upon heating the sample to 94°C at a rate of 0.5°C per minute with five repeats of the readings being taken. The solution was then cooled to 20°C, and the CD spectrum was re‐measured. The data were analyzed using GraphPad Prism 6 software and melting temperatures determined using a sigmoidal sloppy Boltzmann equation. The reported standard error is the fitting error.
Fluorescence polarization assays
A Fluorescence Polarisation assay based on the assay developed by Hancock et al. was employed using a 384‐well black opaque optiplate microplates with a total volume of 40 μL in order to minimise protein usage.31 1 nM FITC‐beta‐Ala‐DEETGEF‐OH peptide in 50 mM Tris‐HCl 150 mM NaCl pH 8.5 was incubated with a serial dilution of Keap1 protein for 30 minutes. The experiment was repeated three times, and the data were fitted in GraphPad Prism 6.0 to a one‐site binding model using the following equation:
where AF is the polarisation fraction, B is the maximum specific binding, [P] is the protein concentration in M, K D is the midpoint concentration where half the maximum signal is reached, N s is the gradient of the non‐specific binding, and C is the background polarisation fraction.
1 nM FITC‐beta‐Ala‐DEETGEF‐OH tracer and 237.5 nM Keap1 protein in 50 mM Tris‐HCl 150 mM NaCl pH 8.5 was then incubated with a serial dilution of CTPR protein for 30 minutes using a 384‐well black opaque optiplate microplates with a total volume of 40 μL. The experiment was repeated three times, and the data were fitted in GraphPad Prism 6.0.
Isothermal titration calorimetry
A Microcal iTC200 isothermal titration calorimeter was used. Proteins were dialysed overnight into 50 mM Tris‐HCl pH 8.5, 150 mM NaCl 0.5 mM TCEP at 4°C. 100 μM Keap1 was titrated into a 10 μM Flexible Nrf2 CTPR2 solution over 20 injections of 2 μL, with an initial delay of 60 s, a reference power of 5 μcal/s, an injection duration of 0.8 s, 150 s spacing and a stirring speed of 750 RPM. The experiment was repeated with a titration of 100 μM Keap1 into buffer and the previous data were then subtracted from this background data. The data were analyzed using Origin 7.0 and fitted using a one‐site binding model.
Cider plots
The sequences “DPNNLDEETGEFLDPNN” and “DPNNLDEETGEFLDPRN” were input into the CIDER server and found to predict negatively charged strong polyelectrolytes and strong polyampholytes respectively.47
Declaration of Interest
Authors AP and LSI are inventors on patent application PCT/EP2018/068580. Authors AP and LSI are founders of PolyProX Therapeutics Limited (company number 11664980).
Acknowledgments
L.S.I. acknowledges the support of a Senior Fellowship from the UK Medical Research Foundation and a CRUK Pioneer Award. S. K. M. acknowledges a BBSRC Doctoral Training Scholarship (BB/J014540/1) and a Society of Chemical Industry (SCI) Messel Scholarship. A.P was supported by a BBSRC Doctoral Training Scholarship and an Oliver Gatty Studentship. We thank Jane Clarke for useful discussions and Alex Bullock for the gift of the Keap1 expression plasmid.
Brief summary: Finding suitable scaffolds for biotherapeutics and research tools that are amenable to extensive protein engineering without detriment to structure and stability presents a major challenge, as many modified proteins are not stable. Here we present a consensus‐designed protein (CTPR) that, due to its very high stability, can tolerate grafting of a functional peptide into its inter‐repeat protein loop, imparting nanomolar affinity for a target.
References
- 1. Sormanni P, Aprile FA, Vendruscolo M (2015) Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc Natl Acad Sci U S A 112:9902–9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chin JW, Schepartz A (2001) Design and evolution of a miniature Bcl‐2 binding protein. Angew Chem Int Ed Engl 40:3806–3809. [DOI] [PubMed] [Google Scholar]
- 3. Azoitei ML, Ban YE, Julien JP, Bryson S, Schroeter A, Kalyuzhniy O, Porter JR, Adachi Y, Baker D, Pai EF, Schief WR (2012) Computational design of high‐affinity epitope scaffolds by backbone grafting of a linear epitope. J Mol Biol 415:175–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sia SK, Kim PS (2003) Protein grafting of an HIV‐1‐inhibiting epitope. Proc Natl Acad Sci U S A 100:9756–9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsomaia N (2015) Peptide therapeutics: targeting the undruggable space. Eur J Med Chem 94:459–470. [DOI] [PubMed] [Google Scholar]
- 6. Montclare JK, Schepartz A (2003) Miniature homeodomains: high specificity without an N‐terminal arm. J Am Chem Soc 125:3416–3417. [DOI] [PubMed] [Google Scholar]
- 7. Chin JW, Grotzfeld RM, Fabian MA, Schepartz A (2001) Methodology for optimizing functional miniature proteins based on avian pancreatic polypeptide using phage display. Bioorg Med Chem Lett 11:1501–1505. [DOI] [PubMed] [Google Scholar]
- 8. Rossmann M, J Greive S, Moschetti T, Dinan M, Hyvönen M (2017) Development of a multipurpose scaffold for the display of peptide loops. Protein Eng Des Sel 30:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu W, Lau YH, Fischer G, Tan YS, Chattopadhyay A, de la Roche M, Hyvönen M, Verma C, Spring DR, Itzhaki LS (2017) Macrocyclized extended peptides: inhibiting the substrate‐recognition domain of tankyrase. J Am Chem Soc 139:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Der BS, Kuhlman B (2013) Strategies to control the binding mode of de novo designed protein interactions. Curr Opin Struct Biol 23:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gilbreth RN, Koide S (2012) Structural insights for engineering binding proteins based on non‐antibody scaffolds. Curr Opin Struct Biol 22:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Škrlec K, Štrukelj B, Berlec A (2015) Non‐immunoglobulin scaffolds: a focus on their targets. Trends Biotechnol 33:408–418. [DOI] [PubMed] [Google Scholar]
- 13. Tlatli R, Nozach H, Collet G, Beau F, Vera L, Stura E, Dive V, Cuniasse P (2013) Grafting of functional motifs onto protein scaffolds identified by PDB screening: an efficient route to design optimizable protein binders. FEBS J 280:139–159. [DOI] [PubMed] [Google Scholar]
- 14. Owens B (2017) Faster, deeper, smaller‐the rise of antibody‐like scaffolds. Nat Biotechnol 35:602–603. [DOI] [PubMed] [Google Scholar]
- 15. Stadler LK, Hoffmann T, Tomlinson DC, Song Q, Lee T, Busby M, Nyathi Y, Gendra E, Tiede C, Flanagan K, Cockell SJ, Wipat A, Harwood C, Wagner SD, Knowles MA, Davis JJ, Keegan N, Ferrigno PK (2011) Structure‐function studies of an engineered scaffold protein derived from Stefin a. II: development and applications of the SQT variant. Protein Eng Des Sel 24:751–763. [DOI] [PubMed] [Google Scholar]
- 16. D'Andrea LD, Regan L (2003) TPR proteins: the versatile helix. Trends Biochem Sci 28:655–662. [DOI] [PubMed] [Google Scholar]
- 17. Javadi Y, Itzhaki LS (2013) Tandem‐repeat proteins: regularity plus modularity equals design‐ability. Curr Opin Struct Biol 23:622–631. [DOI] [PubMed] [Google Scholar]
- 18. Main ER, Xiong Y, Cocco MJ, D'Andrea L, Regan L (2003) Design of stable alpha‐helical arrays from an idealized TPR motif. Structure 11:497–508. [DOI] [PubMed] [Google Scholar]
- 19. Cortajarena AL, Mochrie SG, Regan L (2011) Modulating repeat protein stability: the effect of individual helix stability on the collective behavior of the ensemble. Protein Sci 20:1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grove TZ, Osuji CO, Forster JD, Dufresne ER, Regan L (2010) Stimuli‐responsive smart gels realized via modular protein design. J Am Chem Soc 132:14024–14026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Allan RK, Ratajczak T (2011) Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones 16:353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taylor P, Dornan J, Carrello A, Minchin RF, Ratajczak T, Walkinshaw MD (2001) Two structures of cyclophilin 40: folding and fidelity in the TPR domains. Structure 9:431–438. [DOI] [PubMed] [Google Scholar]
- 23. Brinker A, Scheufler C, Von Der Mulbe F, Fleckenstein B, Herrmann C, Jung G, Moarefi I, Hartl FU (2002) Ligand discrimination by TPR domains. Relevance and selectivity of EEVD‐recognition in Hsp70 x hop x Hsp90 complexes. J Biol Chem 277:19265–19275. [DOI] [PubMed] [Google Scholar]
- 24. Cortajarena AL, Yi F, Regan L (2008) Designed TPR modules as novel anticancer agents. ACS Chem Biol 3:161–166. [DOI] [PubMed] [Google Scholar]
- 25. Jackrel ME, Valverde R, Regan L (2009) Redesign of a protein‐peptide interaction: characterization and applications. Protein Sci 18:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cortajarena AL, Liu TY, Hochstrasser M, Regan L (2010) Designed proteins to modulate cellular networks. ACS Chem Biol 5:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perez‐Riba A, Lowe AR, Main ERG, Itzhaki LS (2018) Context‐dependent energetics of loop extensions in a family of tandem‐repeat proteins. Biophys J 114:2552–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Canning P, Sorrell FJ, Bullock AN (2015) Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med 88:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tong KI, Kobayashi A, Katsuoka F, Yamamoto M (2006) Two‐site substrate recognition model for the Keap1‐Nrf2 system: a hinge and latch mechanism. Biol Chem 387:1311–1320. [DOI] [PubMed] [Google Scholar]
- 30. Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M (2006) Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two‐site molecular recognition model. Mol Cell Biol 26:2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hancock R, Bertrand HC, Tsujita T, Naz S, El‐Bakry A, Laoruchupong J, Hayes JD, Wells G (2012) Peptide inhibitors of the Keap1‐Nrf2 protein‐protein interaction. Free Radic Biol Med 52:444–451. [DOI] [PubMed] [Google Scholar]
- 32. Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M (2006) Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell 21:689–700. [DOI] [PubMed] [Google Scholar]
- 33. Hancock R, Schaap M, Pfister H, Wells G (2013) Peptide inhibitors of the Keap1‐Nrf2 protein‐protein interaction with improved binding and cellular activity. Org Biomol Chem 11:3553–3557. [DOI] [PubMed] [Google Scholar]
- 34. Schaap M, Hancock R, Wilderspin A, Wells G (2013) Development of a steady‐state FRET‐based assay to identify inhibitors of the Keap1‐Nrf2 protein‐protein interaction. Protein Sci 22:1812–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu MC, Jiao Q, Liu T, Tan SJ, Zhou HS, You QD, Jiang ZY (2018) Discovery of a head‐to‐tail cyclic peptide as the Keap1‐Nrf2 protein‐protein interaction inhibitor with high cell potency. Eur J Med Chem 143:1578–1589. [DOI] [PubMed] [Google Scholar]
- 36. Lu M‐C, Chen Z‐Y, Wang Y‐L, Jiang Y‐L, Yuan Z‐W, You Q‐D, Jiang Z‐Y (2015) Binding thermodynamics and kinetics guided optimization of potent Keap1‐Nrf2 peptide inhibitors. RSC Advan 5:85983–85987. [Google Scholar]
- 37. Steel R, Cowan J, Payerne E, O'Connell MA, Searcey M (2012) Anti‐inflammatory effect of a cell‐penetrating peptide targeting the Nrf2/Keap1 interaction. ACS Med Chem Lett 3:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu MC, Ji JA, Jiang ZY, You QD (2016) The Keap1‐Nrf2‐ARE pathway as a potential preventive and therapeutic target: an update. Med Res Rev 36:924–963. [DOI] [PubMed] [Google Scholar]
- 39. Copple IM (2012) The Keap1‐Nrf2 cell defense pathway: a promising therapeutic target? Adv Pharmacol 63:43–79. [DOI] [PubMed] [Google Scholar]
- 40. Perez‐Riba A, Itzhaki LS (2017) A method for rapid high‐throughput biophysical analysis of proteins. Sci Rep 7:9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guntas G, Lewis SM, Mulvaney KM, Cloer EW, Tripathy A, Lane TR, Major MB, Kuhlman B (2016) Engineering a genetically encoded competitive inhibitor of the KEAP1‐NRF2 interaction via structure‐based design and phage display. Protein Eng Des Sel 29:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu X, Taylor RD, Griffin L, Coker SF, Adams R, Ceska T, Shi J, Lawson AD, Baker T (2017) Computational design of an epitope‐specific Keap1 binding antibody using hotspot residues grafting and CDR loop swapping. Sci Rep 7:41306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Phillips JJ, Javadi Y, Millership C, Main ER (2012) Modulation of the multistate folding of designed TPR proteins through intrinsic and extrinsic factors. Protein Sci 21:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kempner ES (1993) Movable lobes and flexible loops in proteins. Structural deformations that control biochemical activity. FEBS Lett 326:4–10. [DOI] [PubMed] [Google Scholar]
- 45. Lee LP, Tidor B (2001) Optimization of binding electrostatics: charge complementarity in the barnase‐barstar protein complex. Protein Sci 10:362–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang T, Tomic S, Gabdoulline RR, Wade RC (2004) How optimal are the binding energetics of barnase and barstar? Biophys J 87:1618–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holehouse AS, Das RK, Ahad JN, Richardson MO, Pappu RV (2017) CIDER: resources to analyze sequence‐ensemble relationships of intrinsically disordered proteins. Biophys J 112:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M (2006) Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J 25:3605–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]