

Summary
The TAM receptor, Axl, has been implicated as a candidate entry receptor for Zika virus (ZIKV) infection but has been shown as inessential for virus infection in mice. To probe the role of Axl in murine ZIKV infection, we developed a mouse model lacking the Axl receptor and the interferon alpha/beta receptor (Ifnar−/−Axl−/−), conferring susceptibility to ZIKV. This model validated that Axl is not required for murine ZIKV infection and that mice lacking Axl are resistant to ZIKV pathogenesis. This resistance correlates to lower pro-interleukin-1β production and less apoptosis in microglia of ZIKV-infected mice. This apoptosis occurs through both intrinsic (caspase 9) and extrinsic (caspase 8) manners, and is age dependent, as younger Axl-deficient mice are susceptible to ZIKV pathogenesis. These findings suggest that Axl plays an important role in pathogenesis in the brain during ZIKV infection and indicates a potential role for Axl inhibitors as therapeutics during viral infection.
Subject Areas: Biological Sciences, Neurotoxicology, Virology
Graphical Abstract
Highlights
-
•
IFNAR−/−Axl−/− mice show Axl unnecessary for Zika virus replication in mice
-
•
Mice lacking Axl receptor are significantly resistant to Zika virus neuropathogenesis
-
•
IFNAR−/−Axl−/− mice have less ZIKV-driven caspase-dependent apoptosis in brain
-
•
Axl deficient mice have fewer apoptotic microglia after ZIKV infection
Biological Sciences; Neurotoxicology; Virology
Introduction
First discovered in Africa in 1947 (Dick, 1952, Dick et al., 1952), Zika virus (ZIKV) is a positive-sense enveloped flavivirus that is primarily transmitted by the Aedes aegypti mosquito (Li et al., 2012). Unlike other flaviviruses, ZIKV is capable infecting the male reproductive organs, leading to testicular atrophy (Govero et al., 2016, Ma et al., 2016, Uraki et al., 2017) and sexual transmission (Hastings and Fikrig, 2017). Most infections with this virus are asymptomatic (Duffy et al., 2009), and the majority of symptomatic infections result in a mild and self-limiting febrile illness (Simpson, 1964, Bearcroft, 1956). Rare cases of more severe illness have been reported including Guillain-Barré syndrome (GBS), marked by subacute flaccid paralysis (Oehler et al., 2014, Ioos et al., 2014) in infected adults, and infection of pregnant women has been associated with severe birth defects, including congenital malformations and severe birth defects in newborns (World Health Organization, 2016, Ventura et al., 2016, Schuler-Faccini et al., 2016).
Upon ZIKV infection, ZIKV is present in a variety of tissues and body fluids including the central nervous system (Tang et al., 2016), saliva (Musso et al., 2015), blood (Musso et al., 2016), urine (Zhang et al., 2016), and semen (Atkinson et al., 2016), many of which are unique among flaviviruses. Similar to other flaviviruses, ZIKV targets dendritic cells and macrophages in the skin and other tissues for replication (Wu et al., 2000, Jurado et al., 2016, Hamel et al., 2015), and replication of the virus in the testes (Govero et al., 2016, Ma et al., 2016, Uraki et al., 2017) and brain (Li et al., 2016a, Meertens et al., 2017) results in apoptosis of important cell types driving pathogenesis. This difference in tissue tropism for ZIKV compared with related flaviviruses has led to significant efforts to identify the entry receptor for this virus. One of the leading candidate proteins implicated as facilitating viral entry is a member of the TAM family of receptor tyrosine kinases, Axl (Hamel et al., 2015, Liu et al., 2016, Retallack et al., 2016, Meertens et al., 2017, Savidis et al., 2016). However, work from this group (Hastings et al., 2017) and others (Wang et al., 2017) has shown that Axl is dispensable in a murine model of ZIKV infection, and genetic ablation of Axl in human neural progenitor cells and cerebral organoids does not prevent ZIKV infection (Wells et al., 2016).
ZIKV infects several cell types that express high levels of Axl (Lemke and Burstyn-Cohen, 2010, Nowakowski et al., 2016, Ma et al., 2016, Tabata et al., 2016, Rothlin et al., 2015), and signaling of this protein contributes to infection of astrocytes by downregulating type I interferon (IFN) signaling (Chen et al., 2018). Axl is a member of the TAM family of tyrosine kinase receptors. These receptors bind the ligands, Gas6 and Protein S, which recognize phosphatidylserine present on enveloped viruses and dying cells (Shimojima et al., 2007, Lemke and Burstyn-Cohen, 2010). Type I IFN signaling upregulates TAM receptors, which are part of a negative feedback loop for inflammatory responses and inhibits the Toll-like receptor pathway (Rothlin et al., 2007, Carrera Silva et al., 2013). In dendritic cells, this inhibition is dependent on a physical interaction with the type I IFN receptor (Ifnar) (Rothlin et al., 2007). In addition, these receptors contribute to the clearance of apoptotic cells and the differentiation of natural killer cells (Bosurgi et al., 2013, Caraux et al., 2006a, Caraux et al., 2006b, Paolino et al., 2014). ZIKV infection is controlled by type I IFN signaling (Lazear et al., 2016) and is capable of using its NS5 protein to degrade human STAT2 and inhibit this signaling, but not mouse STAT2 (Grant et al., 2016), requiring the use of immune-deficient mice for assessment of infection in the mouse model. To further assess the role of Axl, we generated an Ifnar/Axl double knockout mouse, which is susceptible to infection, and tested ZIKV replication and pathogenesis in this mouse model.
Results
The TAM Receptor, Axl, Is Not Required for Replication of ZIKV In Vivo but Is Involved in Viral Pathogenesis
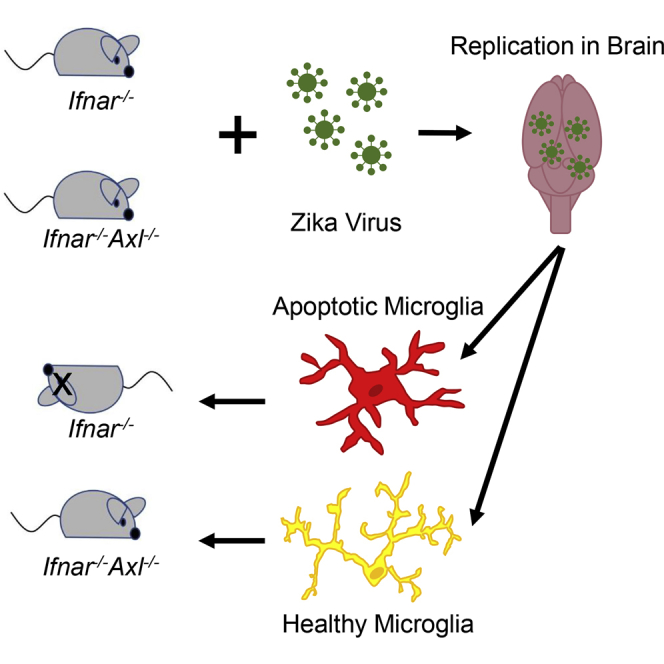
To probe the function of the TAM receptor Axl in a holistic murine infection model lacking this protein (Ifnar−/−Axl−/−), we bred together two existing mouse models. The first, an IFN-αβ receptor knockout (Ifnar−/−), will render this model susceptible to ZIKV infection, and the second, an Axl knockout (Axl−/−), will allow us to probe the role of Axl in ZIKV pathogenesis and replication in specific tissues (Figure 1A). To determine if Axl is involved in replication of ZIKV in this model, we subcutaneously inoculated Ifnar1−/−Axl−/− and Ifnar1−/− mice and show that Axl is not required for ZIKV replication in the blood at days 2, 4, and 6 as measured by qRT-PCR (Figure 1B), in the brain at day 6 as measured by qRT-PCR (Figure 1C), or in plaque assay (Figure 1D).
Figure 1.

The TAM Receptor Axl Is Not Required for Replication of ZIKV In Vivo but Is Involved in Viral Pathogenesis
(A–H) (A–F) Six-week old or (G and H) three-week old Ifnar1−/− or Ifnar1−/−Axl−/− mice were subcutaneously infected via footpad injection with 105 plaque-forming unit Cambodian strain of ZIKV. (A) Diagram of Axl knockout mouse model describing the purpose of gene ablation. (B) Whole blood was collected at days 2, 4, and 6 post-ZIKV infection from both groups and analyzed by qRT-PCR. (C and D) At day 6, when mice begin to show viral pathogenesis, mice were sacrificed, brains were collected, and ZIKV levels were analyzed using (C) qRT-PCR and (D) plaque assay. (E and G) Both groups were weighed daily for two weeks or until sacrificed due to severe pathogenesis, and data were graphed as a percentage of original body. (F and H) Mice were also monitored for survival for two weeks after ZIKV infection. ZIKV RNA levels were normalized to mouse β-actin (ACTB) RNA levels. (n = 8–10/group for each genotype). Significance was tested by (B) two-way ANOVA with a post-hoc Tukey test, (C and D) Student's t test, or (F and H) log rank (Mantel-Cox) test. Error bars represent SEM.
Interestingly, in mice lacking Axl expression ZIKV pathogenesis is significantly decreased, with infected animals showing a slight decrease in weight around days 6–8 (Figure 1E) but most recovering fully, whereas the virus is 100% lethal in mice expressing Axl (Figure 1F). The pathogenesis in this model appeared to be driven by replication of virus in the brain, as the mice that died had hindlimb paralysis and unsteadiness on their feet. This phenotype appears to be age dependent, as Axl knockout weanling mice (3 weeks old) show no difference in ZIKV pathogenesis compared with those expressing Axl (Figures 1G and 1H). These data indicate that although ZIKV does not require Axl to replicate in the mouse model, this protein is important for driving severe disease in vivo.
Axl Expression Is Important for IL-1β Expression during ZIKV Infection
To attempt to determine the mechanism underlying the increased pathogenesis of ZIKV in Axl-competent mice, we collected brains from Ifnar1−/− and Ifnar1−/−Axl−/− mice and isolated RNA to determine the expression of important inflammatory cytokines during viral infection. Using qRT-PCR, we show that the levels of transforming growth factor-β, tumor necrosis factor-α, IFN-γ, and IL-6 are not different between Axl−/− and Axl wild-type mice (Figures 2B–2E), but that Ifnar1−/−Axl−/− mice had significantly lower pro-IL-1β levels when compared with Ifnar1−/− mice (Figure 2A). Interestingly, no differences in the expression of inflammatory cytokines were observed between 3-week-old IFNAR−/−Axl−/− mice and Axl-competent mice of the same age during ZIKV infection (Figure S1).
Figure 2.

Axl Expression Is Important for IL-1β Expression during ZIKV Infection
Six-week old Ifnar1−/− or Ifnar1−/−Axl−/− mice were subcutaneously infected via footpad injection with 105 plaque-forming unit Cambodian strain of ZIKV.
(A–E) At day 6, when mice begin to show viral pathogenesis, they were sacrificed, brains were collected and (A) pro-IL-1β, (B) TGF-β, (C) TNF-α, (D) IFN-γ, and (E) IL-6 levels were analyzed using qRT-PCR. ZIKV RNA levels were normalized to mouse β-actin (ACTB) RNA levels. Data are expressed as a percentage of the average expression in the Ifnar1−/− group. Significance was tested using Student's t test (n = 8–10/group for each genotype).
To determine if this increased inflammatory response in 6-week-old Axl knockout mice is a result of enhanced inflammatory cell recruitment or proliferation in the brains of ZIKV-infected mice, we examined the number of immune cells in the brain at day 6 of ZIKV infection using flow cytometry (Figure S2). There is a robust expansion or infiltration of monocytes (Figure 3A), neutrophils (Figure 3B), macrophages (Figure 3C), dendritic cells (Figure 3D), microglia (Figure 3E), and both CD4+ (Figure 3G) and CD8+ (Figure 3H) T cells in infected mice, whereas there are no significant differences between Ifnar1−/− and Ifnar1−/−Axl−/− mice (Figure 3). Intriguingly, there was a trend toward slightly fewer CD11c+ activated microglia in Ifnar1−/−Axl−/− mice (Figure 3F), but this difference was not significant. IL-1β is highly expressed during apoptosis, so we hypothesized that Axl could instead be playing a role in driving apoptosis during ZIKV infection.
Figure 3.

Axl Expression Does Not Drive Differences in Immune Cell Infiltration during ZIKV Infection
Six-week old Ifnar1−/− or Ifnar1−/−Axl−/− mice were subcutaneously infected via footpad injection with 105 plaque-forming unit Cambodian strain of ZIKV.
(A–H) At day 6, when mice begin to show viral pathogenesis, mice were sacrificed, brains were collected, and (A) monocytes, (B) neutrophils, (C) macrophages, (D) dendritic cells, (E) microglia, (F) activated microglia, (G) CD4+ T cells, and (H) CD8+ T cells were analyzed using flow cytometry. Data are expressed as total number of cells present in the brain. Significance was tested by two-way ANOVA with a post-hoc Tukey test. No significant differences were detected among any groups. Error bars represent SEM (n = 8/group for each infected group and n = 2/group for each uninfected group).
Axl Drives Apoptosis in Microglia in the Brains of ZIKV-Infected Mice
To examine if Axl plays a role in driving apoptosis in the 6-week-old animals, we dissected brains from ZIKV-infected Ifnar1−/− or Ifnar1−/−Axl−/− mice at day 6, and performed a TUNEL assay, which selectively marks cells with breaks in DNA indicating apoptosis, on fixed brain sections. It is evident that Axl-competent mice have markedly increased apoptosis across several regions of the brain, including the ventral striatum (Figure 4A), the cerebellum (Figure 4B), and the hippocampus (Figure 4C).
Figure 4.

Axl Drives Apoptosis in the Brain of ZIKV-Infected Mice
Six-week old Ifnar1−/− or Ifnar1−/−Axl−/− mice were subcutaneously infected via footpad injection with 105 plaque-forming unit Cambodian strain of ZIKV.
(A–C) At day 6, when mice begin to show viral pathogenesis, mice were sacrificed, brains were collected, fixed in buffered formalin, and prepared for histology. Slides were stained using the TUNEL assay, and the entire brain was imaged at 20×. Shown are (A) the ventral striatum, (B) the cerebellum, and (C) the hippocampus (n = 4/group for each genotype). Scale bar, 500 μM in main image and 100 μM in inset image.
By co-staining these tissue sections with TUNEL and NeuN (neurons, Figure 5A), glial fibrillary acidic protein (GFAP) (astrocytes, Figure 5B), or Iba1 (microglia, Figure 5C), we can clearly show that microglia represent the main apoptotic cell type present in the brain. As these cells are capable of phagocytosing dead cells, we cannot rule out the possibility that some of the microglial cells appear TUNEL positive owing to phagocytosis of other cell types; however, the majority of these cells show TUNEL staining in the nucleus (co-localizing with DAPI staining) and not in the cytoplasm (Figure 5C), which indicates that these cells are undergoing apoptosis themselves. When TUNEL+ microglia were quantified, significantly more TUNEL+ microglia were observed in the brains of Axl-competent mice compared with Axl−/− mice (Figure 5D), further suggesting a role of Axl in driving apoptosis in these cells.
Figure 5.

ZIKV Induces Apoptosis in Microglia
Six-week old Ifnar1−/− or Ifnar1−/−Axl−/− mice were subcutaneously infected via footpad injection with 105 plaque-forming unit Cambodian strain of ZIKV.
(A–C) At day 6, when mice begin to show viral pathogenesis, mice were sacrificed, brains were collected, fixed in buffered formalin, and prepared for histology. Slides were stained using the TUNEL assay and antibodies toward (A) NeuN (neurons), (B) GFAP (astrocytes), or (C) Iba1 (microglia) and imaged at 20×. Insets are digitally enlarged by 3× to show individual cells. Scale bar is 100 μM in main image. Shown is representative image from Ifnar1−/− mice.
(D) Percent microglia that were TUNEL+ were quantified and graphed from two to three random fields in the brains of each genotype. Significance was tested by two-way ANOVA. Error bars represent SEM (n = 4/group for each genotype).
The canonical apoptosis pathway is caspase 3 dependent and can be either extrinsic through death receptor (like tumor necrosis factor-related apoptosis-inducing ligand receptor) signaling and caspase 8, or intrinsic through mitochondrial-associated Bcl-2 homology proteins and caspase 9. Downstream of both these pathways is cleavage of poly (ADP-ribose) polymerase (PARP), which is responsible for repairing DNA. Six-week-old Axl-deficient mice have significantly lower levels of cleaved PARP shown by western blot analysis of whole ZIKV-infected brain homogenates of ZIKV-infected mice, and there was significantly higher caspase 8 and cleaved caspase 9, and a trend toward higher caspase 3, cleaved caspase 3, and caspase 9, in the Ifnar1−/− mice when compared with the Axl-deficient mice, which indicates that the observed apoptosis is induced both through the intrinsic and extrinsic pathways (Figures 6 and S3). Caspase 12 is involved in a less common apoptotic pathway, and whereas less caspase 12 expression was seen in the Axl-deficient group (Figure 6H), no change in cleaved caspase 12 was observed (Figure 6I). This suggests that Axl signals through a caspase-dependent PARP-dependent manner leading to apoptosis in microglia.
Figure 6.

ZIKV Induces Apoptosis by a PARP-Dependent Caspase-Dependent Mechanism
Six-week old Ifnar1−/− or Ifnar1−/−Axl−/− mice were subcutaneously infected via footpad injection with 105 plaque-forming unit Cambodian strain of ZIKV.
(A–J) At day 6, when mice begin to show viral pathogenesis, they were sacrificed, brains were collected in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitor, and a western blot was performed. Membrane was probed with antibodies toward proteins involved in apoptosis. Protein expressions of (A) PARP, (B) cleaved PARP, (C) caspase 3, (D) cleaved caspase 3, (E) caspase 8, (F) caspase 9, (G) cleaved caspase 9, (H) caspase 12, and (I) cleaved caspase 12 were quantified using ImageJ. Data shown are representative of at least two independent experiments. Significance was tested with two-tailed Mann-Whitney U test. Error bars represent SEM (n = 4/group for each genotype).
Discussion
Axl has been shown to be involved in the immune response during ZIKV infection and has been implicated as an entry factor for specific cell types. To further examine the role of Axl in replication and pathogenesis in mice, we generated a mouse lacking Axl that was also deficient in the Ifnar protein, rendering these animals highly susceptible to ZIKV infection. Using this model, we demonstrate that although, similar to our previous work, ZIKV replication levels are unaffected by the lack of Axl, the loss of this molecule protected mice from severe viral pathogenesis in the brain. This protection coincided with significantly lower inflammatory pro-IL-1β expression and a decrease in apoptosis, specifically in glial cells, in the brains of Axl-deficient mice. These data suggest that Axl present on microglia is involved in facilitating ZIKV pathogenesis by inducing apoptosis in these cells.
Small-molecule inhibition of Axl in human cell lines (Hamel et al., 2015, Liu et al., 2016, Retallack et al., 2016, Meertens et al., 2017, Savidis et al., 2016), small interfering RNA knockdown of Axl human alveolar basal epithelial carcinoma cells (A549) (Hamel et al., 2015), and CRISPR knockout of Axl in human cervical adenocarcinoma cells (HeLa) (Savidis et al., 2016), a human glioblastoma line (U87) (Retallack et al., 2016), a human microglial cell line (CHME3) (Meertens et al., 2017), and human embryonic kidney cells (293T) (Liu et al., 2016) has been shown to block or reduce ZIKV infection, but Axl is not necessary for replication of ZIKV in the mouse model (Hastings et al., 2017, Wang et al., 2017). Intriguingly, our group has shown that murine Axl is capable of acting as an entry factor by using a transcomplementation assay (Hastings et al., 2017). This raises the possibility that this protein is important for entry into specific cell types, but these cell types are not a major contributor to overall ZIKV replication in the mouse model. Meertens et al. demonstrated that Axl can mediate ZIKV infection of astrocytes and microglia in vitro and that the subsequent signaling in these cells through Axl dampens type I IFN signaling and increases viral replication in glial cells. As the mouse model that we generated is deficient in Ifnar, any phenotype observed in these mice is independent of Axl's role in the regulation of type I IFNs. Therefore the lack of difference in viral titer suggests that if Axl is indeed involved in entry to these cells, these cells do not represent a major source of viral replication in the mouse model. The data presented here are consistent with Axl facilitating entry into microglia and indicate that the inflammatory response by these cells is critical for the pathogenesis of ZIKV in the brain.
The apoptosis observed in our histology (TUNEL) and western blot (cleaved PARP) experiments appears to be Axl dependent. Canonical apoptosis is induced through an extrinsic pathway, dependent on death ligands or receptors and caspase 8, or an intrinsic pathway, dependent on mitochondrial changes and caspase 9 (Elmore, 2007). To determine if this phenomenon is due to an increase in components of canonical apoptosis pathways, we performed western blot analysis of various proteins involved in intrinsic and extrinsic apoptosis. Interestingly, Axl-deficient mice had slightly lower levels of these proteins when compared with Axl-competent mice. Axl expression appears to increase both intrinsic and extrinsic apoptosis during ZIKV infection, and it is possible that the difference in apoptosis is caused by a difference in viral entry and infection in a specific cell type, perhaps glial cells, or the mechanistic role of Axl in specific functions of microglia.
ZIKV is capable of targeting and replicating in both developing neural stem cells and to a lesser extent in mature neurons (Nowakowski et al., 2016, Tang et al., 2016, Onorati et al., 2016). In addition, the symptoms of GBS that have been associated with acute ZIKV infection are likely related to infection or autoimmune-mediated death of neurons or glial cells (do Rosario et al., 2016, Siu et al., 2016). Susceptible knockout mice infected with ZIKV are marked by acute and severe neuropathology and hindlimb paralysis (Li et al., 2016b), and the data here indicate that this pathogenesis is related to Axl expression and apoptosis of microglia. Interestingly, microglia have been shown to undergo a process termed phagoptosis, in which they become activated and phagocytose live neurons, or other cells (Brown and Neher, 2014). This process is directly related to the expression of TAM receptors including Axl (Fourgeaud et al., 2016). It is possible that the neuropathogenesis observed during ZIKV infection could be related to the phagoptosis of ZIKV-infected neurons by activated microglia.
It is also possible that the increase in pathogenesis observed in the presence of Axl is related to uncontrolled inflammatory responses. Neuropathogenesis in Japanese encephalitis virus, another flavivirus, has been attributed to inflammation in the brain, including excessive IL-1β expression (Myint et al., 2014), and expression of this inflammatory cytokine is also responsible for pathogenesis in the brains of patients infected with HIV (Yang et al., 2010) and Alzheimer disease (Shaftel et al., 2007). Counterintuitively, Axl kinase signaling has been shown to suppress inflammation and IL-1β expression (Ganesh et al., 2012, Han et al., 2016), but we hypothesize that the difference observed in inflammation and pathogenesis in our experiments is due to decreased infection of, or phagocytosis of virus and virally infected cells by, microglia lacking Axl. This would be consistent with studies showing that entry via clathrin-mediated endocytosis of ZIKV into glial cells is decreased when Axl is blocked (Meertens et al., 2017).
Taken together, these data suggest an important role for Axl in the pathogenesis of ZIKV in the brain. In addition, using a susceptible mouse model lacking Ifnar we further corroborate our previous work showing that Axl is not necessary for ZIKV infection. The similarity between ZIKV levels in wild-type and Axl knockout mice indicate that, whereas Axl is important for entry into cell types, like microglia, that are crucial for viral neuropathogenesis, it is dispensable for entry and replication into cell types that are major producers of virus in vivo. Therefore pharmacological agents that have been developed to target Axl as therapeutics for ZIKV could represent an effective strategy for combatting ZIKV pathogenesis.
Limitations of Study
These experiments were performed in IFNAR knockout mice, which allows us to not only infect with ZIKV and observe IFN-independent effects of Axl but also reduce the physiological relevance of this model. We have not fully characterized the phenotype of microglia, astrocytes, and neurons in the IFNAR knockout model, and it is possible that some other virally independent phenotype could drive the striking difference in survival that we observed. It would also be particularly interesting to generate microglia-specific (or other cell-type-specific) knockouts of Axl to observe if these effects are driven by Axl on these cells or on others. This study uses the pre-pandemic Cambodian strain of ZIKV, and it is possible that this phenotype would change with a pandemic strain. We did infect these mice with a pandemic Mexican strain of virus (MEX2-81), but we did not observe the same severe pathogenesis in IFNAR knockout mice.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to acknowledge Akiko Iwasaki and Carla Rothlin, along with their respective laboratories, for providing mice, expert advice, and the use of equipment for these experiments. We would also like to acknowledge the animal care technicians who maintained our mouse colonies. E.F. is an HHMI investigator, and funds from HHMI were used to perform these experiments.
Author Contributions
Conceptualization, A.K.H. and J.H.; Methodology, A.K.H., K.H., R.U., and J.H.; Investigation, A.K.H., K.H., R.U., J.H., H.G., K.D., and E.W.; Validation, A.K.H.; Writing – Original Draft, A.K.H.; Writing – Review & Editing, A.K.H., K.H., R.U., J.H., H.G., K.D., E.W., and E.F.; Funding Acquisition, E.F.; Resources, E.F.; Supervision, A.K.H. and E.F.
Declaration of Interests
The authors declare no competing interests.
Published: March 29, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.03.003.
Supplemental Information
References
- Atkinson B., Hearn P., Afrough B., Lumley S., Carter D., Aarons E.J., Simpson A.J., Brooks T.J., Hewson R. Detection of zika virus in semen. Emerg. Infect. Dis. 2016;22:940. doi: 10.3201/eid2205.160107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearcroft W.G. Zika virus infection experimentally induced in a human volunteer. Trans. R. Soc. Trop. Med. Hyg. 1956;50:442–448. [PubMed] [Google Scholar]
- Bosurgi L., Bernink J.H., Delgado Cuevas V., Gagliani N., Joannas L., Schmid E.T., Booth C.J., Ghosh S., Rothlin C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. U S A. 2013;110:13091–13096. doi: 10.1073/pnas.1302507110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G.C., Neher J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014;15:209. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- Caraux A., Kim N., Bell S.E., Zompi S., Ranson T., Lesjean-Pottier S., Garcia-Ojeda M.E., Turner M., Colucci F. Phospholipase C-gamma2 is essential for NK cell cytotoxicity and innate immunity to malignant and virally infected cells. Blood. 2006;107:994–1002. doi: 10.1182/blood-2005-06-2428. [DOI] [PubMed] [Google Scholar]
- Caraux A., Lu Q., Fernandez N., Riou S., di Santo J.P., Raulet D.H., Lemke G., Roth C. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nat. Immunol. 2006;7:747–754. doi: 10.1038/ni1353. [DOI] [PubMed] [Google Scholar]
- Carrera Silva E.A., Chan P.Y., Joannas L., Errasti A.E., Gagliani N., Bosurgi L., Jabbour M., Perry A., Smith-Chakmakova F., Mucida D. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity. 2013;39:160–170. doi: 10.1016/j.immuni.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Yang Y.-F., Yang Y., Zou P., Chen J., He Y., Shui S.-L., Cui Y.-R., Bai R., Liang Y.-J. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018;3:302–309. doi: 10.1038/s41564-017-0092-4. [DOI] [PubMed] [Google Scholar]
- Dick G.W. Zika virus. II. Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952;46:521–534. doi: 10.1016/0035-9203(52)90043-6. [DOI] [PubMed] [Google Scholar]
- Dick G.W., Kitchen S.F., Haddow A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952;46:509–520. doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- do Rosario M.S., de Jesus P.A., Vasilakis N., Farias D.S., Novaes M.A., Rodrigues S.G., Martins L.C., Vasconcelos P.F., Ko A.I., Alcantara L.C., de Siqueira I.C. Guillain-barre syndrome after zika virus infection in Brazil. Am. J. Trop. Med. Hyg. 2016;95:1157–1160. doi: 10.4269/ajtmh.16-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy M.R., Chen T.H., Hancock W.T., Powers A.M., Kool J.L., Lanciotti R.S., Pretrick M., Marfel M., Holzbauer S., Dubray C. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009;360:2536–2543. doi: 10.1056/NEJMoa0805715. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L., Través P.G., Tufail Y., Leal-Bailey H., Lew E.D., Burrola P.G., Callaway P., Zagórska A., Rothlin C.V., Nimmerjahn A., Lemke G. TAM receptors regulate multiple features of microglial physiology. Nature. 2016;532:240. doi: 10.1038/nature17630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh K., Das A., Dickerson R., Khanna S., Parinandi N.L., Gordillo G.M., Sen C.K., Roy S. Prostaglandin E(2) induces oncostatin M expression in human chronic wound macrophages through Axl receptor tyrosine kinase pathway. J. Immunol. 2012;189:2563–2573. doi: 10.4049/jimmunol.1102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govero J., Esakky P., Scheaffer S.M., Fernandez E., Drury A., Platt D.J., Gorman M.J., Richner J.M., Caine E.A., Salazar V. Zika virus infection damages the testes in mice. Nature. 2016;540:438–442. doi: 10.1038/nature20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant A., Ponia S.S., Tripathi S., Balasubramaniam V., Miorin L., Sourisseau M., Schwarz M.C., Sánchez-Seco M.P., Evans M.J., Best S.M., García-Sastre A. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe. 2016;19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R., Dejarnac O., Wichit S., Ekchariyawat P., Neyret A., Luplertlop N., Perera-Lecoin M., Surasombatpattana P., Talignani L., Thomas F. Biology of zika virus infection in human skin cells. J. Virol. 2015;89:8880–8896. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Bae J., Choi C.Y., Choi S.P., Kang H.S., Jo E.K., Park J., Lee Y.S., Moon H.S., Park C.G. Autophagy induced by AXL receptor tyrosine kinase alleviates acute liver injury via inhibition of NLRP3 inflammasome activation in mice. Autophagy. 2016;12:2326–2343. doi: 10.1080/15548627.2016.1235124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings A.K., Fikrig E. Zika virus and Sexual transmission: a new route of transmission for mosquito-borne flaviviruses. Yale J. Biol. Med. 2017;90:325–330. [PMC free article] [PubMed] [Google Scholar]
- Hastings A.K., Yockey L.J., Jagger B.W., Hwang J., Uraki R., Gaitsch H.F., Parnell L.A., Cao B., Mysorekar I.U., Rothlin C.V. TAM receptors are not required for zika virus infection in mice. Cell Rep. 2017;19:558–568. doi: 10.1016/j.celrep.2017.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioos S., Mallet H.P., Leparc Goffart I., Gauthier V., Cardoso T., Herida M. Current Zika virus epidemiology and recent epidemics. Med. Mal. Infect. 2014;44:302–307. doi: 10.1016/j.medmal.2014.04.008. [DOI] [PubMed] [Google Scholar]
- Jurado K.A., Simoni M.K., Tang Z., Uraki R., Hwang J., Householder S., Wu M., Lindenbach B.D., Abrahams V.M., Guller S., Fikrig E. Zika virus productively infects primary human placenta-specific macrophages. JCI Insight. 2016;1:e88461. doi: 10.1172/jci.insight.88461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear H.M., Govero J., Smith A.M., Platt D.J., Fernandez E., Miner J.J., Diamond M.S. A mouse model of Zika virus pathogenesis. Cell Host Microbe. 2016;19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke G., Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann. N. Y. Acad. Sci. 2010;1209:23–29. doi: 10.1111/j.1749-6632.2010.05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Xu D., Ye Q., Hong S., Jiang Y., Liu X., Zhang N., Shi L., Qin C.F., Xu Z. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell. 2016;19:672. doi: 10.1016/j.stem.2016.10.017. [DOI] [PubMed] [Google Scholar]
- Li H., Saucedo-Cuevas L., Regla-Nava J.A., Chai G., Sheets N., Tang W., Terskikh A.V., Shresta S., Gleeson J.G. Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell. 2016;19:593–598. doi: 10.1016/j.stem.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.I., Wong P.S., Ng L.C., Tan C.H. Oral susceptibility of Singapore aedes (Stegomyia) aegypti (Linnaeus) to zika virus. PLoS Negl. Trop. Dis. 2012;6:e1792. doi: 10.1371/journal.pntd.0001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Delalio L.J., Isakson B.E., Wang T.T. AXL-mediated productive infection of human endothelial cells by zika virus. Circ. Res. 2016;119:1183–1189. doi: 10.1161/CIRCRESAHA.116.309866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W., Li S., Ma S., Jia L., Zhang F., Zhang Y., Zhang J., Wong G., Zhang S., Lu X. Zika virus causes testis damage and leads to male infertility in mice. Cell. 2016;167:1511–1524.e10. doi: 10.1016/j.cell.2016.11.016. [DOI] [PubMed] [Google Scholar]
- Meertens L., Labeau A., Dejarnac O., Cipriani S., Sinigaglia L., Bonnet-Madin L., le Charpentier T., Hafirassou M.L., Zamborlini A., Cao-Lormeau V.-M. Axl mediates Zika virus entry in human glial cells and modulates innate immune responses. Cell Rep. 2017;18:324–333. doi: 10.1016/j.celrep.2016.12.045. [DOI] [PubMed] [Google Scholar]
- Musso D., Roche C., Nhan T.X., Robin E., Teissier A., Cao-Lormeau V.M. Detection of Zika virus in saliva. J. Clin. Virol. 2015;68:53–55. doi: 10.1016/j.jcv.2015.04.021. [DOI] [PubMed] [Google Scholar]
- Musso D., Stramer S.L., Transfusion-Transmitted Diseases Committee. Busch M.P., International Society of Blood Transfusion Working Party on Transfusion-Transmitted Infectious Diseases Zika virus: a new challenge for blood transfusion. Lancet. 2016;387:1993–1994. doi: 10.1016/S0140-6736(16)30428-7. [DOI] [PubMed] [Google Scholar]
- Myint K.S.A., Kipar A., Jarman R.G., Gibbons R.V., Perng G.C., Flanagan B., Mongkolsirichaikul D., van Gessel Y., Solomon T. Neuropathogenesis of Japanese encephalitis in a primate model. PLoS Negl. Trop. Dis. 2014;8:e2980. doi: 10.1371/journal.pntd.0002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski T.J., Pollen A.A., di Lullo E., Sandoval-Espinosa C., Bershteyn M., Kriegstein A.R. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell. 2016;18:591–596. doi: 10.1016/j.stem.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehler E., Watrin L., Larre P., Leparc-Goffart I., Lastere S., Valour F., Baudouin L., Mallet H., Musso D., Ghawche F. Zika virus infection complicated by Guillain-Barre syndrome–case report, French Polynesia, December 2013. Euro Surveill. 2014;19:20720. doi: 10.2807/1560-7917.es2014.19.9.20720. [DOI] [PubMed] [Google Scholar]
- Onorati M., Li Z., Liu F., Sousa A.M.M., Nakagawa N., Li M., Dell'anno M.T., Gulden F.O., Pochareddy S., Tebbenkamp A.T.N. Zika virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial Glia. Cell Rep. 2016;16:2576–2592. doi: 10.1016/j.celrep.2016.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolino M., Choidas A., Wallner S., Pranjic B., Uribesalgo I., Loeser S., Jamieson A.M., Langdon W.Y., Ikeda F., Fededa J.P. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507:508–512. doi: 10.1038/nature12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retallack H., di Lullo E., Arias C., Knopp K.A., Laurie M.T., Sandoval-Espinosa C., Mancia Leon W.R., Krencik R., Ullian E.M. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. U S A. 2016;113:14408–14413. doi: 10.1073/pnas.1618029113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin C.V., Carrera-Silva E.A., Bosurgi L., Ghosh S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015;33:355–391. doi: 10.1146/annurev-immunol-032414-112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin C.V., Ghosh S., Zuniga E.I., Oldstone M.B., Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Savidis G., Mcdougall W.M., Meraner P., Perreira J.M., Portmann J.M., Trincucci G., John S.P., Aker A.M., Renzette N., Robbins D.R. Identification of zika virus and dengue virus dependency factors using functional genomics. Cell Rep. 2016;16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Schuler-Faccini L., Ribeiro E.M., Feitosa I.M., Horovitz D.D., Cavalcanti D.P., Pessoa A., Doriqui M.J., Neri J.I., Neto J.M., Wanderley H.Y., Brazilian Medical Genetics Society–Zika Embryopathy Task Force Possible association between Zika virus infection and microcephaly - Brazil, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016;65:59–62. doi: 10.15585/mmwr.mm6503e2. [DOI] [PubMed] [Google Scholar]
- Shaftel S.S., Kyrkanides S., Olschowka J.A., Miller J.N., Johnson R.E., O'banion M.K. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest. 2007;117:1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima M., Ikeda Y., Kawaoka Y. The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 2007;196(Suppl 2):S259–S263. doi: 10.1086/520594. [DOI] [PubMed] [Google Scholar]
- Simpson D.I. Zika virus infection in man. Trans. R. Soc. Trop. Med. Hyg. 1964;58:335–338. [PubMed] [Google Scholar]
- Siu R., Bukhari W., Todd A., Gunn W., Huang Q.S., Timmings P. Acute zika infection with concurrent onset of Guillain-barre syndrome. Neurology. 2016;87:1623–1624. doi: 10.1212/WNL.0000000000003038. [DOI] [PubMed] [Google Scholar]
- Tabata T., Petitt M., Puerta-Guardo H., Michlmayr D., Wang C., Fang-Hoover J., Harris E., Pereira L. Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe. 2016;20:155–166. doi: 10.1016/j.chom.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H., Hammack C., Ogden S.C., Wen Z., Qian X., Li Y., Yao B., Shin J., Zhang F., Lee E.M. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uraki R., Hwang J., Jurado K.A., Householder S., Yockey L.J., Hastings A.K., Homer R.J., Iwasaki A., Fikrig E. Zika virus causes testicular atrophy. Sci. Adv. 2017;3:e1602899. doi: 10.1126/sciadv.1602899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura C.V., Maia M., Bravo-Filho V., Gois A.L., Belfort R., Jr. Zika virus in Brazil and macular atrophy in a child with microcephaly. Lancet. 2016;387:228. doi: 10.1016/S0140-6736(16)00006-4. [DOI] [PubMed] [Google Scholar]
- Wang Z.Y., Wang Z., Zhen Z.D., Feng K.H., Guo J., Gao N., Fan D.Y., Han D.S., Wang P.G., An J. Axl is not an indispensable factor for Zika virus infection in mice. J. Gen. Virol. 2017;98:2061–2068. doi: 10.1099/jgv.0.000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells M.F., Salick M.R., Wiskow O., Ho D.J., Worringer K.A., Ihry R.J., Kommineni S., Bilican B., Klim J.R., Hill E.J. Genetic ablation of <em>AXL</em> does not protect human neural progenitor cells and cerebral organoids from zika virus infection. Cell Stem Cell. 2016;19:703–708. doi: 10.1016/j.stem.2016.11.011. [DOI] [PubMed] [Google Scholar]
- World Health Organization, 2016. Zika Situation Report.
- Wu S.J., Grouard-Vogel G., Sun W., Mascola J.R., Brachtel E., Putvatana R., Louder M.K., Filgueira L., Marovich M.A., Wong H.K. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 2000;6:816–820. doi: 10.1038/77553. [DOI] [PubMed] [Google Scholar]
- Yang Y., Wu J., Lu Y. Mechanism of HIV-1-TAT induction of interleukin-1beta from human monocytes: Involvement of the phospholipase C/protein kinase C signaling cascade. J. Med. Virol. 2010;82:735–746. doi: 10.1002/jmv.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.C., Li X.F., Deng Y.Q., Tong Y.G., Qin C.F. Excretion of infectious Zika virus in urine. Lancet Infect. Dis. 2016;16:641–642. doi: 10.1016/S1473-3099(16)30070-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.