

Abstract
One drug, two diseases is a rare and economical therapeutic strategy that is highly desirable in the pharmaceutical industry. We previously reported a 21-amino acid peptide named beta-structured inhibitor for neurodegenerative diseases (BIND) that can effectively inhibit expanded CAG trinucleotide toxicity in polyglutamine (polyQ) diseases. Here we report that BIND also effectively inhibits GGGGCC repeat-mediated neurodegeneration in vitro and in vivo. When fused with a cell-penetrating peptide derived from the transactivator of transcription (TAT) protein of the HIV, TAT-BIND reduces cell death, formation of GGGGCC RNA foci, and levels of poly-GR, poly-GA, and poly-GP dipeptide proteins in cell models of C9ORF72-associated amyotrophic lateral sclerosis and frontotemporal dementia (C9ALS-FTD). We showed that TAT-BIND disrupts the interaction between GGGGCC RNA and nucleolin protein, restores rRNA maturation, and inhibits mislocalization of nucleolin and B23, which eventually suppresses nucleolar stress in C9ALS-FTD. In a Drosophila model of C9ALS-FTD, TAT-BIND suppresses retinal degeneration, rescues climbing ability, and extends the lifespan of flies. In contrast, TAT-BIND has no effect on UAS-poly-glycine-arginine (poly-GR)100-expressing flies, which generate only poly-GR protein toxicity, indicating BIND ameliorates toxicity in C9ALS-FTD models via a r(GGGGCC)exp-dependent inhibitory mechanism. Our findings demonstrated that, apart from being a potential therapeutic for polyQ diseases, BIND is also a potent peptidylic inhibitor that suppresses expanded GGGGCC RNA-mediated neurodegeneration, highlighting its potential application in C9ALS-FTD treatment.
Keywords: G4C2, amyotrophic lateral sclerosis, frontotemporal dementia, cell-penetrating peptide, peptide inhibitor, C9ORF72
Graphical Abstract
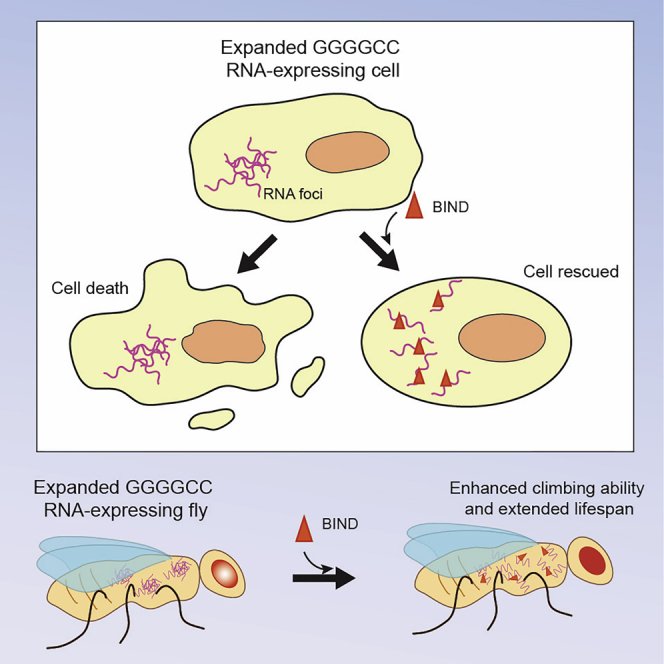
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are devastating neurodegenerative diseases with no effective treatment. The hexanucleotide (HRE) GGGGCC repeat expansion in the non-coding region of the C9ORF72 gene has been reported to contribute to up to 40% of ALS and FTD cases.1, 2 This group of diseases is commonly referred to as C9ALS-FTD. Both the sense and antisense RNAs transcribed from the GGGGCC expansion have been reported to cause nuclear RNA foci formation3, 4 and sequester diverse RNA-binding proteins, impairing the RNA-processing machinery.5, 6, 7, 8 The repeat-associated non-ATG (RAN)-translated dipeptide repeat (DPR) proteins,4, 9, 10 including poly-glycine-alanine (GA), poly-glycine-arginine (GR), poly-proline-arginine (PR), poly-proline-alanine (PA), and poly-glycine-proline (GP), are all reported to be toxic,11, 12, 13, 14, 15 while the arginine-containing poly-GR and poly-PR proteins are found to be particularly toxic.11, 12 Thus, GGGGCC repeat-containing RNAs and their respective protein products are both toxic species, and they both contribute to the pathogenesis of C9ALS-FTD.
A pathogenic feature of C9ALS-FTD is the notable buildup of nucleolar stress in affected cells,16, 17, 18 where nucleolar stress is a cellular response during the disruption of ribosome biogenesis and/or the malfunctioning of ribosomes.19 Failure in rRNA transcription20 and processing21, 22 hinders ribosome biogenesis,23, 24 upregulates cellular p53 expression,19 and eventually leads to apoptosis.25 Nucleolar stress is widely reported in neurodegenerative diseases, including polyglutamine (polyQ) diseases,26, 27, 28, 29, 30 Parkinson’s disease,31, 32, 33 and C9ALS-FTD.16, 17 The r(GGGGCC)exp has been reported to sequester nucleolin (NCL) proteins, obstruct the maturation of rRNA, and finally induce nucleolar stress.16 The poly-GR and poly-PR proteins have also been demonstrated to cause the translocation of the key nucleolar component nucleophosmin (B23) and NCL, leading to nucleolar stress and cell death.17
We recently reported the activity of a therapeutic peptide inhibitor candidate against CAG RNA toxicity named beta-structured inhibitor for neurodegenerative diseases (BIND).34 The BIND sequence is derived from the RNA recognition motif (RRM) 2 of NCL protein. By fusing with the cell-penetrating peptide (CPP), a sequence derived from the transactivator of transcription (TAT) protein of HIV-1, to BIND, the TAT-BIND peptide was capable of inhibiting NCL-expanded CAG RNA interaction and suppressing nucleolar stress in polyQ diseases.34 Here we report that TAT-BIND, apart from being a potent suppressor of expanded CAG RNA toxicity, is also a potent suppressor of expanded GGGGCC RNA-mediated toxicity. TAT-BIND reduced GGGGCC-induced cell death, RNA foci formation, poly-GP, poly-GR, and poly-GA dipeptide protein levels, and nucleolar stress in cell models of C9ALS-FTD. When applied to in vivo disease models, TAT-BIND effectively suppressed neurodegeneration in a GGGGCC repeat length-dependent manner. Not only did our findings open up a new potential therapeutic treatment for C9ALS-FTD, our results also presented a rare and economical one drug, two diseases strategy that is highly desirable for further therapeutic development.
Results
TAT-BIND Suppresses pAG3-(GGGGCC)66-Induced Cell Death
The central region of the NCL protein contains four RRMs.35 Based on sequence and structural analysis of the four individual RRMs, we predicted the potential RNA-binding region on each RRM, and we generated synthetic peptides derived from each of these regions in an attempt to identify sequences that could suppress r(GGGGCC)exp-mediated defects (Table 1). One of the peptides, RRM2-P1, has been designated previously as BIND, as it showed inhibitory activity against expanded CAG RNA-induced toxicity in our earlier work.34 To facilitate cellular uptake, we fused an 11-residue-long CPP (YGRKKRRQRRR) derived from residues 47–57 of the TAT protein from the HIV to the N termini of all the peptides.30, 34, 36, 37, 38 TAT peptide was selected because it exhibits little cytotoxic effect.39, 40 The pAG3-(GGGGCC)2/66 construct is a set of well-defined constructs; pAG3-(GGGGCC)66 expresses both expanded GGGGCC repeat RNA and RAN-translated DPR proteins.41, 42
Table 1.
Sequence of TAT and the Respective NCL RRM-Derived Peptide
| Peptide | Sequence | Derived from |
|---|---|---|
| TAT | YGRKKRRQRRR | HIV-1 virus transactivator |
| TAT-RRM1-P1 | YGRKKRRQRRRTGTNRKFGYVD | RRM1 of NCL |
| TAT-RRM2-P1 | YGRKKRRQRRRAEIRLVSKDGKSKGIAYIEFK | RRM2 of NCL |
| TAT-RRM3-P1 | YGRKKRRQRRRTFIKVPQNQNGKSKGYAF | RRM3 of NCL |
| TAT-RRM4-P1 | YGRKKRRQRRRRARIVTDRETGSSKGFGFVD | RRM4 of NCL |
We employed the lactate dehydrogenase (LDH) assay to detect cell death in our cell model,43 and we confirmed that (GGGGCC)66 expression caused significant cell death in SK-N-MC cells (Figures 1A–1D). Our results showed that the application of TAT-RRM2-P1, hereafter referred to as TAT-BIND, suppressed (GGGGCC)66-induced cell death in a dose-dependent manner (Figure 1B). The application of 1 μM TAT-BIND on cells suppressed approximately 25% of the relative cell death induced by (GGGGCC)66 expression, whereas the 10- and 20-μM treatments nearly completely suppressed cell death in our cell model (Figure 1B). On the other hand, a slight but significant suppression of cell death was observed when 0.1 or 1 μM TAT-RRM3-P1 was applied (Figure 1C). However, the suppressive effect of TAT-RRM3-P1 diminished when higher concentrations of peptide were used (Figure 1C). We suspect this might be the result of the peptide forming soluble aggregates due to its higher content of hydrogen bond donors and acceptors. Alternatively, it could be caused by toxicity induced by the TAT-RRM3-P1 peptide itself. Considering the significant dose-dependent inhibitory effect of TAT-BIND, we selected it for further investigation.
Figure 1.

TAT-RRM2-P1 (TAT-BIND) Significantly Suppressed Cell Death Induced by pAG3-(GGGGCC)66 in SK-N-MC Cells
(A) TAT-RRM1-P1 treatment did not alter pAG3-(GGGGCC)66-induced cell death. (B) TAT-RRM2-P1 suppressed pAG3-(GGGGCC)66-induced cell death in a dose-dependent manner. (C) Low concentrations, but not high concentrations, of TAT-RRM3-P1 slightly suppressed pAG3-(GGGGCC)66-induced cell death. (D) TAT-RRM4-P1 treatment did not alter pAG3-(GGGGCC)66-induced cell death. 1 μg pAg3-(GGGGCC)2/66 plasmid was used to transfect SK-N-MC cells, followed by application of the respective TAT peptides (0.1, 1, 10, and 20 μM). LDH enzyme activity in the cell culture medium was measured 48 h after treatment. The results of the experimental groups were normalized to the untransfected controls. (E) Application of TAT-BIND or the scrambled control TAT-BIND-S didn’t elicit any observable cytotoxicity to pAG3-(GGGGCC)2/66-expressing cells. TAT-BIND, but not TAT-BIND-S, could suppress pAG3-(GGGGCC)66-induced cell death. (F) Half-maximum inhibitory concentration (IC50) of TAT-BIND on the inhibition of pAG3-(GGGGCC)66-induced cell death. The IC50 value represents the concentration of TAT-BIND that decreased LDH enzyme activity by 50% relative to LDH enzyme activity in the untreated control group. The experiments were repeated at least thrice, and data were plotted as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001; NS, no significance.
To test the specificity of the peptide, we generated a control peptide TAT-BIND-S containing a scrambled amino acid sequence of BIND. We found that 10 μM TAT-BIND exerted no observable toxicity on control cells expressing (GGGGCC)2, but it completely suppressed cell death in (GGGGCC)66-expressing cells (Figure 1E). By contrast, the scrambled TAT-BIND-S had no effect on cells expressing (GGGGCC)2/66 (Figure 1E), indicating that the cell death-suppressing effect of TAT-BIND is dependent on its amino acid sequence. The calculated half-maximum inhibitory concentration (IC50) of TAT-BIND in inhibiting (GGGGCC)66-induced cell death is 103.9 ± 24.6 nM (Figure 1F).
TAT-BIND Suppresses the Formation of GGGGCC RNA Foci and Expression of DPR Proteins
Ribonuclear foci, which contain aggregates of expanded GGGGCC RNA, are frequently found in the neurons of patients with C9ALS-FTD,44, 45, 46 and they are key indicators of RNA toxicity.42, 47, 48 To determine whether TAT-BIND could inhibit the formation of GGGGCC RNA foci, we expressed (GGGGCC)2/66 in SK-N-MC cells, and we treated the cells with either TAT-BIND or TAT-BIND-S. We observed that TAT-BIND effectively suppressed foci formation in (GGGGCC)66-expressing cells, whereas TAT-BIND-S showed no modifying effect (Figures 2A and 2B). These findings suggested that TAT-BIND mitigates the RNA toxicity caused by expanded GGGGCC. We further demonstrated that TAT-BIND did not alter the (GGGGCC)66 RNA level in (GGGGCC)66-expressing cells (Figure S1), which excluded the involvement of a toxic event led by a reduction in C9ORF72 expression.
Figure 2.

TAT-BIND Suppressed GGGGCC RNA Foci Formation and RAN Translation in pAg3-(GGGGCC)66-Expressing SK-N-MC Cells
(A) TAT-BIND suppressed GGGGCC RNA foci formation in pAg3-(GGGGCC)66-expressing SK-N-MC cells. In situ hybridization was performed to detect GGGGCC RNA foci (red) by using a TYE563-labeled LNA probe. Nuclei (blue) were stained by Hoechst 33332. The scale bar represents 10 μm. (B) Quantification of the percentage of cells containing RNA foci after transfection. (C) TAT-BIND suppressed poly-GR protein expression in pAg3-(GGGGCC)66-expressing cells. (D) Statistical analysis of band intensity (poly-GR/GAPDH) of (C). (E) TAT-BIND suppressed poly-GA protein expression in pAg3-(GGGGCC)66-expressing cells. (F) Statistical analysis of band intensity (poly-GA/GAPDH) of (E). (G) TAT-BIND treatment suppressed poly-GP protein expression in pAg3-(GGGGCC)66-expressing cells. (H) Statistical analysis of band intensity (poly-GP/GAPDH) of (G). 1 μg pAg3-(GGGGCC)66 plasmid was used to transfect SK-N-MC cells, followed by application of 10 μM of each peptide. Cells were collected and lysed for western blot analysis 48 h after treatment. GAPDH was used as the loading control. Only representative blots are shown. All experiments were repeated at least thrice with consistent results. Data were plotted as mean ± SEM. ***p < 0.001 and ****p < 0.0001; NS, no significance.
Ribonuclear foci are common signs, but not necessarily the main source of toxicity in patients with C9ALS-FTD. In fact, DPR proteins resulting from the RAN translation was also suggested as the major determinant of toxicity and degeneration in this disease.11, 49 In particular, three different types of DPR proteins, the poly-GR, poly-GA, and poly-GP, could be produced by RAN translation of GGGGCC repeats.15 We sought to determine whether TAT-BIND has any effect on the formation of DPR proteins. Our western blot results showed that TAT-BIND, unlike its scrambled counterpart, significantly decreased the protein levels of poly-GR, poly-GA, and poly-GP DPR proteins (Figures 2C–2H), indicating that the peptide could also inhibit RAN translation of GGGGCC repeats.
TAT-BIND Relieves GGGGCC-Expressing Cells from Nucleolar Stress
Since both GGGGCC RNA foci and DPR proteins have been shown to contribute to nucleolar stress in C9ALS-FTD, we speculated that TAT-BIND could relieve the nucleolar stress in cells expressing GGGGCC repeats. A reduction in rRNA maturation is considered as an indicator of nucleolar stress in C9ALS-FTD.16 We expressed (GGGGCC)66 in SK-N-MC cells, and we performed real-time PCR analysis on 45S pre-rRNA (pre-45 s rRNA), 28 s, 18 s, and 5.8 s rRNA. A general reduction in rRNA maturation was observed in these cells. Upon TAT-BIND treatment, (GGGGCC)66-expressing cells showed a significant restoration of pre-45 s, 18 s, and 5.8 s rRNA expression (Figure 3A). No such effect was observed in the TAT-BIND-S-treated cells. A similar trend was also observed for 28 s rRNA expression (Figure 3A).
Figure 3.

TAT-BIND Treatment Suppressed Nucleolar Stress in pAg3-(GGGGCC)66-Expressing SK-N-MC Cells
(A) TAT-BIND rescued impaired rRNA transcription in pAg3-(GGGGCC)66-expressing cells. (B) TAT-BIND inhibited the mislocalization of NCL proteins in pAg3-(GGGGCC)66-expressing cells. (C) Statistical analysis of the nuclear NCL fold change of (A). (D) TAT-BIND inhibited the translocation of B23 protein from the nucleolus to the nucleoplasm in pAg3-(GGGGCC)66-expressing cells. (E) Statistical analysis of nuclear B23 fold change of (C). (F) TAT-BIND disrupted the interaction between expanded GGGGCC RNA and NCL. For (A), total RNA was extracted followed by reverse transcription. Real-time PCR was employed to determine the expression level of corresponding rRNA. Actin was used for normalization. Fold change of gene expression level was compared to untransfected cells. For (B)–(E), the cells were subjected to immunofluorescence using anti-NCL or anti-B23 antibody (red). Nuclei were stained with Hoechst 33343 (blue). The heatmap of NCL or B23 intensities highlighted the differences of protein localization among the cells. The scale bars indicate 10 μm. The pixel area of the NCL relative to the area of the nucleus was calculated and normalized to that of the untransfected control. 150–300 cells were measured for each condition. For (F), cells expressing pAG3-(GGGGCC)2/66 RNA were co-transfected with either the pcDNA3.1(+)-TAT-BIND-myc or pcDNA3.1(+)-TAT-BIND-S-myc construct. Endogenous NCL protein was immunoprecipitated with anti-NCL antibody, followed by RT-PCR to determine the presence of GGGGCC2/66 RNA. “+” indicates the presence of anti-NCL antibody in the immunoprecipitants, while “−” indicates that anti-NCL antibody was not included. All experiments were repeated at least thrice with consistent results. Data were plotted as mean ± SEM. *p < 0.05, **p < 0.01, and ****p < 0.0001; NS, no significance.
During nucleolar stress, the cell nucleolus rapidly loses its compact organization, and several nucleolar proteins, including NCL and nucleophosmin (B23), mislocalize to the nucleoplasm.16, 17, 50, 51 To investigate if TAT-BIND could restore the nucleolar organization in pAG3-(GGGGCC)66-transfected cells, we performed immunofluorescence to monitor the localization of the NCL and B23 nucleolar marker proteins. As expected, both NCL and B23 were largely mislocalized to the nucleoplasm in SK-N-MC cells transfected with pAG3-(GGGGCC)66 (Figures 3B–3E). In contrast, the nucleolar localization pattern of both nucleolar marker proteins was significantly restored upon TAT-BIND treatment (Figures 3B–3E). Such restoration was not observed in cells treated with TAT-BIND-S (Figures 3B–3E). These findings strongly indicated that TAT-BIND could relieve (GGGGCC)66-expressing cells from nucleolar stress.
Recruitment of NCL to expanded GGGGCC RNA represents one of the culprits that induce nucleolar stress.16 We therefore reasoned that TAT-BIND relieved the nucleolar stress in (GGGGCC)66-expressing by directly inhibiting the interaction between NCL and GGGGCC RNA. We performed protein-RNA interaction assay to test this. Briefly, SK-N-MC cells were transfected with either pAG3-(GGGGCC)2 or pAG3-(GGGGCC)66 in the presence or absence of TAT-BIND. After crosslinking, the RNA-protein complexes were immunoprecipitated using anti-NCL antibody followed by RT-PCR to determine the levels of (GGGGCC)2/66 RNA in the samples (Figure 3F). Consistent with previous findings, we observed an interaction between GGGGCC RNA and NCL proteins in (GGGGCC)66-expressing, but not (GGGGCC)2-expressing, cells. Co-expression of TAT-BIND, but not the TAT-BIND-S control, significantly reduced the amount of GGGGCC RNA immunoprecipitated together with NCL (Figure 3F). This suggests that TAT-BIND suppresses C9ALS-FTD-associated nucleolar stress by targeting the interaction between NCL and expanded GGGGCC RNA.
TAT-BIND Suppresses Expanded GGGGCC-Induced Neurodegeneration In Vivo
In Drosophila, the expression of (GGGGCC)36 under the gmr-GAL4 driver causes retinal degeneration in adult flies.11 We employed this Drosophila model of C9ALS-FTD, which expressed the expanded GGGGCC repeat RNA and DPR proteins,11 to examine the effect of TAT-BIND on suppressing C9ALS-FTD-associated neurodegeneration in vivo. Animals that expressed the expanded (GGGGCC)36 or the unexpanded (GGGGCC)3 using gmr-GAL4 were fed with 100 μM TAT-BIND or TAT-BIND-S at the third instar larval stage. Retinal degeneration was observed in flies expressing the expanded (GGGGCC)36, but not in flies expressing the unexpanded (GGGGCC)3 (Figures 4A and 4B). The (GGGGCC)36 retinal degeneration was ameliorated when animals were fed with TAT-BIND, but not in the ones fed with TAT-BIND-S (Figures 4A and 4B). This indicates that TAT-BIND mitigates expanded GGGGCC-associated neurodegeneration in vivo.
Figure 4.

TAT-BIND Inhibited Eye Degeneration, Delayed the Onset of Climbing Defects, and Extended the Lifespan of UAS-(GGGGCC)36 Flies
(A) TAT-BIND treatment inhibited the eye degeneration of UAS-(GGGGCC)36 flies. The flies were treated with 100 μM TAT-BIND or TAT-BIND-S for the external eye assay. Images of 1-day-old adult flies were taken. Genotypes were w; GMR-GAL4/UAS-(GGGGCC)3 and w; GMR-GAL4/UAS-(GGGGCC)36. The experiments were repeated thrice, and at least 30 flies were captured for scoring. (B) Statistical analysis of the scar formation of (A). (C) TAT-BIND treatment rescued the climbing defect of UAS-(GGGGCC)36 flies at 10 days post-eclosion (dpe). (D) TAT-BIND treatment extended the lifespan of UAS-(GGGGCC)36 flies. (E) Area under curve (AUC) analysis of (D). For the climbing ability and lifespan assays, flies at 2 dpe were fed with food containing different drug combinations, including vehicle control (ethanol), mifepristone (RU486, 200 μM), RU486 (200 μM) plus TAT-BIND (50 μM), and RU486 (200 μM) plus TAT-BIND-S (50 μM). Transgene expression was induced with mifepristone (RU486, 200 μM). The climbing ability assay was repeated six times, and at least 90 flies per treatment were scored. The lifespan assay was repeated at least six times, and more than 100 flies per treatment were recorded. Genotypes used in (C)–(E) were w; UAS-(GGGGCC)3/+; elavGS-GAL4/+ and w; UAS-(GGGGCC)36/+; elavGS-GAL4/+. Data were expressed as mean ± SEM. **p < 0.01, ***p < 0.001, and ****p < 0.0001; NS, no significance.
The gmr-GAL4 driver is expressed throughout development in Drosophila. However, C9ALS-FTD are adult-onset diseases. To circumvent the manifestation of developmental effects, we utilized the inducible pan-neuronal elav-GeneSwitch driver (elavGS-GAL4) to restrict the expression of (GGGGCC)36 to only adult neurons. We continuously (every 3 days) fed the animals with 200 μM mifepristone (RU486) starting from 2 days post-eclosion (dpe) to induce (GGGGCC)36 expression, and we allowed neurodegeneration to gradually occur. It was reported that the expression of (GGGGCC)36 using elavGS-GAL4 causes defective locomotor activity and shortened lifespan.11 We performed the established climbing assay on 10-dpe flies to measure the locomotor activity of the (GGGGCC)36-expressing animals,52 and we assessed the modifying effect of TAT-BIND on (GGGGCC)36 flies. Our results showed that 50 μM TAT-BIND significantly rescued the climbing behavior of the (GGGGCC)36-expressing flies, whereas TAT-BIND-S exerted no effect (Figure 4C). The severity of GGGGCC-induced neurodegeneration is age dependent.11 Thus, to gain insight into the effect of TAT-BIND on older animals with more severe phenotypes, we repeated this experiment on 15-, 20-, and 25-dpe files. The percentage of (GGGGCC)36-expressing flies that failed to climb significantly increased with age (Figures S2A–S2C). Nonetheless, TAT-BIND treatment was still able to significantly rescue their climbing behavior in 25-dpe flies (Figure S2C).
In addition to the climbing assay, we also employed the established lifespan assay to determine the effects of TAT-BIND on the longevity of (GGGGCC)36-expressing flies.12 Flies that pan-neuronally expressed (GGGGCC)36 at the adult stage had a significantly shorter lifespan than that of the control flies without the induced (GGGGCC)36 expression (Figures 4D and 4E). TAT-BIND treatment significantly rescued the reduced lifespan of (GGGGCC)36-expressing flies, whereas TAT-BIND-S showed no such effect (Figures 4D and 4E). Both TAT-BIND and TAT-BIND-S had no impact on the longevity of the control (GGGGCC)3-expressing flies (Figures S2D and S2E). The results of the retinal degeneration assay, climbing assay, and lifespan assay above provided consistent evidence that TAT-BIND ameliorates (GGGGCC)36-induced neurodegeneration in vivo.
The Suppression of Neurodegeneration by TAT-BIND Is Dependent on Expanded GGGGCC RNA
The suppression effect of TAT-BIND on neurodegeneration in vitro can be partially attributed to the reduction of DPR protein levels by TAT-BIND, which we showed in Figures 2C–2H. We next sought to determine whether TAT-BIND exerts its rescuing effect on reducing DPR protein production or suppressing DPR protein toxicity per se. We employed a GGGGCC RNA-only model, UAS-(GGGGCC)288-RO, and a poly-GR protein-only model, UAS-poly-GR100.11 The (GGGGCC)288-RO model carries stop codons in the sense strand to prevent RAN translation without affecting the tertiary structure of the RNA.11
To examine the suppression mechanism of TAT-BIND in vivo, we expressed (GGGGCC)288-RO using OK6-GAL4 to induce RNA foci in the salivary gland. Fluorescence in situ hybridization showed clear foci formation in the nuclei of salivary gland cells (Figures 5A and 5B). TAT-BIND, but not TAT-BIND-S, treatment significantly reduced the percentage of salivary gland cells containing nuclear RNA foci, indicating the suppression of GGGGCC RNA toxicity (Figures 5A and 5B). In poly-GR100 flies, the poly-GR protein is expressed using alternate codons, thus bypassing GGGGCC RAN translation. We found that the poly-GR100 flies showed climbing defects in 10- (Figure 5C) and 15-dpe (Figure S3) flies. Neither TAT-BIND nor TAT-BIND-S treatment showed any modifying effect on the poly-GR100 climbing phenotype (Figure 5C; Figure S3). In addition to climbing behavioral defects, poly-GR100 animals showed shortened lifespan (Figures 5D and 5E).11 We found that both TAT-BIND and TAT-BIND-S treatment showed no lifespan-extending effect on these flies (Figures 5D and 5E).
Figure 5.

TAT-BIND Inhibited Expanded GGGGCC-Associated Neurodegeneration in a Repeat RNA-Dependent Manner
(A) TAT-BIND suppressed GGGGCC RNA foci formation in UAS-(GGGGCC)288-RO flies. In situ hybridization was performed to detect GGGGCC RNA foci (red) by using a TYE563-labeled LNA probe. Nuclei (blue) were stained by Hoechst 33332. The scale bar represents 10 μm. (B) Quantification of the percentage of salivary gland cells containing nuclear RNA foci. An OK6-GAL4 driver was used to express 288 RNA-only (RO) GGGGCC repeats in Drosophila salivary glands at third instar larval stage. The genotype was w; OK6-GAL4-GFP/UAS-(GGGGCC)288-RO. At least 4 salivary glands with a total over 100 nuclei were calculated. (C) TAT-BIND treatment did not rescue the climbing defect of UAS-poly-GR100 flies at 15 dpe. (D) TAT-BIND treatment did not prolong the lifespan of UAS-poly-GR100 flies. (E) Area under curve (AUC) analysis of (D). For the climbing ability and lifespan assays, flies at 2 dpe were fed with food containing different drug combinations, including vehicle control (ethanol), mifepristone (RU486, 200 μM), RU486 (200 μM) plus TAT-BIND (50 μM), and RU486 (200 μM) plus TAT-BIND-S (50 μM). Transgene expression was induced with mifepristone (RU486, 200 μM). The climbing ability assay was repeated six times, and at least 90 flies per treatment were scored. The lifespan assay was repeated at least six times, and more than 100 flies per treatment were recorded. The genotype was w; UAS-poly-GR100/+; elavGS-GAL4/+. Data were expressed as mean ± SEM. *p < 0.05 and ****p < 0.0001; NS, no significance.
Structural Activity Relationship Study of BIND
Since the inhibitory activity of BIND is dependent on its primary structure, we next performed structure-activity relationship (SAR) study to identify the pharmacophores of BIND. We synthesized 19 TAT-BIND mutants (TAT-BIND-MT1–19), where each mutant contains a single alanine substitution on a different amino acid (Figure 6A). Similar to the TAT-BIND peptide, the mutant peptides TAT-BIND-MT1, -2, -3, -5, -8 to -11, -14, -15, and -19 suppressed cell death in (GGGGCC)66-expressing SK-N-MC cells. This result indicates that the residues at these positions were not essential for the bioactivity of TAT-BIND (Figure 6B).
Figure 6.

Calculated IC50 Detection and Structure-Activity Relationship Study of TAT-BIND
(A) Sequences of TAT-BIND mutants. TAT peptide (sequence in bold letters) was attached to the N terminus of each BIND mutant. Alanine substitutions for each mutant have been highlighted in red. (B) Structure-activity relationship studies of TAT-BIND. 1 μg pAg3-(GGGGCC)66 plasmid was used to transfect SK-N-MC cells followed by application of the respective TAT peptides (10 μM). LDH enzyme activity in the cell culture medium was measured 48 h after treatment. (C) Structure of BIND as predicted by PEP-FOLD server.73 Residues that are important for the suppression of both expanded CAG RNA- and expanded GGGGCC RNA-induced cytotoxicity are all located on the same side as strand β2 and colored yellow. Residues crucial for the suppression of expanded GGGGCC RNA-induced cytotoxicity only are colored green. Numbering is based on the amino acid sequence of BIND. (D) Schematic diagram illustrating mechanism of actions of BIND in suppressing neurodegeneration in C9ALS-FTD. The results of the experimental groups were normalized to the untransfected controls. The experiments were repeated at least thrice, and the data were plotted as mean ± SEM. ***p < 0.001 and ****p < 0.0001; NS, no significance.
By contrast, the cell death-suppressing effects of TAT-BIND-MT4, -6, -7, -12, -13, and -16 to -18 were significantly lower than that of TAT-BIND (Figure 6B), indicating that these residues were critical for TAT-BIND to suppress expanded GGGGCC-induced toxicity. Interestingly, while a few of these pharmacophores, namely, K13, G14, I18, E19, and F20, also play important roles in the inhibitory activity of TAT-BIND against expanded CAG RNA,34 the mutation of residues L5, S7, and K8 showed no involvement in the pharmacophore of CAG RNA toxicity. This suggests that TAT-BIND might employ different inhibitory mechanisms to prevent NCL-RNA interaction in the two different classes of toxic RNAs.
Discussion
ALS and FTD are two age-dependent multifactorial neurodegenerative disorders, which share many clinical, genetic, and pathological aspects.53 Mutations of the C9ORF72 gene is the most common cause of hereditary ALS-FTD.1 Accumulating evidence demonstrates that both the expanded GGGGCC repeat RNA and the RAN-translated DPR proteins contribute to the pathogenesis of C9ALS-FTD.4, 11, 13, 14, 15, 16, 42, 54 However, available therapeutics that can target these toxic species are limited. TAT-BIND, a pre-defined nucleolar stress inhibitor fused with the CPP TAT, was originally developed to neutralize expanded CAG-induced toxicity in polyQ diseases.13 Surprisingly, in this study, we discovered that TAT-BIND could also inhibit expanded GGGGCC-induced toxicity in C9ALS-FTD models. Although GGGGCC RNA induces toxicity via multiple pathways,11, 16, 55, 56, 57, 58, 59, 60 here we showed that TAT-BIND effectively suppressed GGGGCC RNA foci formation both in vitro (Figures 2A and 2B) and in vivo (Figures 4A and 4B). All of the five DPR proteins have been reported to contribute to neurodegeneration in C9ALS-FTD,11, 15, 17, 60 among which poly-GR and poly-PR proteins were emphasized to be extremely toxic.11, 17 Application of TAT-BIND significantly reduced poly-GA/GP/GR protein levels in (GGGGCC)66-expressing cells (Figures 2C–2H).
Nucleolar stress can be caused by toxic expanded repeat RNAs (polyQ diseases, myotonic dystrophy type 1, and C9ALS-FTD)16, 25, 26, 27, 28, 61 or aggregated proteins (Parkinson’s disease and C9ALS-FTD).17, 32, 33 Consistent with previous studies,16, 17 we confirmed an induction of nucleolar stress by reduced rRNA expression and mislocalization of NCL and B23 proteins in (GGGGCC)66-expressing cells (Figures 3A–3E). We previously showed that TAT-BIND is capable of mitigating these pathogenic defects in models of polyQ diseases.34 Here we found that TAT-BIND also ameliorates nucleolar stress-associated cellular defects in cell models of C9ALS-FTD (Figures 3A–3E). However, the potential inhibitory mechanism of TAT-BIND in C9ALS-FTD could be more sophisticated than in polyQ diseases.
In our point of view, TAT-BIND could target at least one of the following mechanisms to suppress nucleolar stress and neurodegeneration in C9ALS-FTD: (1) through direct interaction with GGGGCC RNA to suppress the repeat RNA-induced nucleolar stress; (2) through inhibiting the expression of DPR proteins via direct TAT-BIND-GGGGCC RNA interaction to eventually suppress DPR protein-induced nucleolar stress; and/or (3) through suppressing the aggregation of DPR proteins and/or its subsequent cellular response to mitigate DPR protein-induced nucleolar stress. Since BIND is a short peptide derived from RRM2 of NCL, we speculate that it inhibits nucleolar stress through peptide-RNA interaction. Our results clearly revealed that TAT-BIND titrated NCL from binding to expanded GGGGCC RNA in vitro (Figure 3F) and it was not able to suppress neurodegeneration in flies expressing poly-GR protein in vivo via non-GGGGCC sequences (Figures 5C–5E; Figure S3). These collectively suggest that TAT-BIND acts on GGGGCC RNA to execute its therapeutic effects in C9ALS-FTD.
Similar to polyQ diseases, C9ALS-FTD is also an age-dependent neurodegenerative disease.62 The patients with C9ALS-FTD show progressive motor dysfunction after adult onset of the disease.62 Here we used Drosophila, a genetically tractable model, to mimick the onset and progression of C9ALS-FTD in human beings. Our results clearly demonstrated that continuous treatment of (GGGGCC)36-expressing flies with TAT-BIND inhibited the progression of climbing defect up to 25 dpe and, moreover, extended its lifespan from ∼33 to ∼50 days (nearly 50% extension). Although BIND inhibited neurodegeneration in both polyQ and C9ALS-FTD models, different SAR data implied a specific inhibitory mechanism in each disease scenario. In particular, residues K13, G14, I18, E19, and F20, which all lie on the same side as the strand of the predicted β-hairpin structure, are found to be crucial to the suppression of both expanded GGGGCC and CAG RNA-associated toxicity, whereas residues L5, S7, and K8 are unique to the former (Figures 6A and 6B).34 Since TAT-BIND also inhibits the binding of NCL to expanded-CAG RNA by directly interacting with the RNA, we speculated that the five residues located on the β-hairpin might be important for RNA binding in a non-sequence-specific manner. On the other hand, L5, S7, and K8 might specifically recognize the structure of the expanded GGGGCC RNA, contributing to the unique SAR study result. Further structural and biophysical investigations are required to elucidate the molecular basis of TAT-BIND inhibitory activity against the different expanded RNAs. Nevertheless, the SAR study results obtained thus far have provided insights into the interaction between NCL and expanded CAG RNA in polyQ diseases and GGGGCC RNA in C9ALS-FTD, respectively.
To date, antisense oligonucleotide (ASO) and small molecule therapy targeting GGGGCC RNA have shown promising effects in suppressing foci formation, DPR protein expression, and eventually neurodegeneration in C9ALS-FTD models.41, 48, 63, 64, 65 However, the administration of ASO therapeutics remains a big challenge and usually requires invasive delivery routes, such as intrathecal or intraventricular injections. In addition, ASO may be recognized by Toll-like receptors as foreign DNA and cause undesired effects on innate immunity.66 These challenges must be overcome before ASO therapeutics become clinically useful. On the other hand, RNA-targeting small molecule inhibitors pose different challenges, including specificity and selectivity, due to the highly anionic and dynamic nature of RNA.
Here we demonstrated, for the first time, that a peptidylic inhibitor can effectively inhibit r(GGGGCC)exp-initiated toxicities in a repeat RNA-dependent manner. Besides high specificity, peptide inhibitors usually have less chance of drug-drug interactions and, unlike small molecules, show less accumulation in tissues, thus reducing the risks of complications caused by the generation of intermediate metabolites. Although peptides are prone to proteolytic degradation and, therefore, native peptide therapeutics are limited by short plasma half-life and negligible oral bioavailability, recent advancements in synthetic strategies and medicinal chemistry techniques have improved candidates of peptide therapeutic to be more drug-like with favorable pharmaceutical properties.67 In fact, we have previously demonstrated that lipidation of a peptide inhibitor could significantly improve its pharmacokinetic profile and brain uptake in rats when administered intranasally, providing proof of concept that peptidyl inhibitors represent a promising class of therapeutic agents for the treatment of neurodegenerative diseases. Additionally, a CPP such as the TAT peptide has been shown to facilitate the transport of fusion cargos across the blood-brain barrier (BBB) and deliver them into nearly all tissues, including the mouse brain.68, 69, 70 The fusion of a CPP to BIND is thus a feasible strategy to enhance the drug ability of BIND as an alternative therapeutic for the treatment of C9ALS-FTD. Importantly, TAT-BIND exhibited no observable toxic effects in vitro and in vivo in our study. Further exploration of the fusion of different CNS-specific CPPs, such as Angiopep-2 or glutathione (GSH), will likely enhance the specificity of delivery of BIND and optimize it into a more promising drug candidate.71
Materials and Methods
Synthesis of Peptides
All peptides were purchased from GenScript. The sequences of TAT peptide, TAT-RRM1-P1, TAT-RRM2-P1 (TAT-BIND), TAT-RRM3-P1, and TAT-RRM4-P1, are shown in Table 1. The sequences of 19 TAT-BIND mutants are shown in Figure 2C. The amino acid sequence of BIND-S that served as a scrambled control of BIND in this study was GGEDIKSRVEAASILYFIKKK. The TAT CPP peptide was attached at the N terminus of the respective peptides. The purity of peptides used in cell experiments was over 90%. Desalted peptides were used in Drosophila feeding assays.
Cell Culture, Plasmid Construction, Plasmid Transfection, and Peptide Treatment
SK-N-MC cells were kindly provided by professor Dobrila D. Rudnicki (John Hopkins University, USA). They were cultured at 37°C with 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The pAg3-(GGGGCC)2/66 plasmids were kindly provided by professor Leonard Petrucelli (Mayo Clinic, USA). Construction of pcDNA3.1(+)-TAT-BIND-myc and pcDNA3.1(+)-TAT-BIND-S-myc was indicated previously.25 Transient transfection of plasmids to SK-N-MC cells was performed using Lipofectamine 2000 (Thermo Fisher Scientific). 10 μM of the respective peptide was added into culture well immediately after transfection unless otherwise stated.
LDH Cytotoxicity Assay and IC50 Determination
SK-N-MC cells were seeded on a 24-well plate at a density of 0.8 × 105, and the pAg3-(GGGGCC)2/66 construct was used to transfect the cells. LDH enzyme activity in the cell culture medium was measured 48 h post-drug treatment using the Cytotox 96 non-radioactive cytotoxicity assay (Promega).
For IC50 detection, various amounts of TAT-BIND (0.1, 0.5, 1, 10, 50, 100, and 500 nM and 1 and 10 μM) were added to individual culture wells after transfection. At 48 h post-treatment, LDH enzyme activity in the cell culture medium was measured as described before. Experimental groups were normalized to the untransfected control. After normalization, data were analyzed using the dose response-inhibition curve (nonlinear regression-variable slope) to determine the IC50 value (Prism 6 software, GraphPad).
RNA Fluorescence In Situ Hybridization of pAG3-(GGGGCC)2/66-Expressing Cells
In situ hybridization was carried out to evaluate the effect of TAT-BIND on GGGGCC RNA foci formation in pAG3-(CCCCGG)2/66-expressing SK-N-MC cells. In brief, 0.6 × 105 SK-N-MC cells were seeded and grown on glass coverslips in a 24-well plate. 10 μM TAT-BIND or TAT-BIND-S was added immediately after transfection of 1 μg pAg3-(GGGGCC)2/66 plasmid. At 48 h post-treatment, cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.5% Triton X-100 for 5 min at room temperature. Cells were then washed with PBS treated with diethylpyrocarbonate (DEPC-PBS) three times, and they were hybridized with 40 nM denatured TYE563-labeled locked nucleic acid (LNA) probe (5′-TYE563-CCCGGCCCCGGCCCC-3′; Exiqon) in hybridization buffer (50% formamide, 10% dextran sulfate, 2× saline-sodium citrate (SSC) buffer, and 50 mM sodium phosphate buffer) for 6 h at 65°C. After washing once with 0.1% Tween-20-2× SSC for 5 min at room temperature, cells were further washed with 0.1× SSC 3 times at 65°C for 10 min. Nuclei were counterstained with Hoechst 33342 (1 μg/mL, Thermo Fisher Scientific) prior to mounting on coverslips. Images were obtained on an Olympus FV1000 IX81-TIRF (total internal reflection fluorescence) confocal microscope.
RNA Fluorescence In Situ Hybridization of Salivary Gland of UAS-(GGGGCC)288-RO-Expressing Flies
The salivary gland of third instar larvae with corresponding genotype was dissected as previously reported11 and fixed with 4% paraformaldehyde for 10 min. After fixation, samples were washed 3 times in PBS-T (PBS containing 0.3% Tween), followed by dehydration in methanol at −20°C for at least 12 h. After that, a methanol gradient (90%, 75%, 50%, and 25%) was used for re-hydration and washed three times in 1× DEPC-PBS. Samples were then treated with 0.5 μg/mL proteinase K in 50 mM Tris and 50 mM EDTA buffer for 1 h at room temperature, and they were washed 1 time with 1× DEPC-PBS and 2× SSC buffer. Pre-hybridization was performed in 50% formamide and 2× SSC buffer for 30 min at 80°C. Samples were hybridized with 400 nM denatured TYE563-labeled LNA probe (5′-TYE563-CCCGGCCCCGGCCCC-3′; Exiqon) in hybridization buffer (50% formamide, 0.02% BSA, 10% dextran sulfate, 2× SSC, 1 mg/mL salmon sperm, 1.6 mM vanadyl ribonucleoside, and 50 mM sodium phosphate buffer) for 3 h at 80°C. After hybridization, samples were washed with 50% formamide, 0.2× SSC three times, followed by a brief wash in 1× DEPC PBS. Nuclei were counterstained with Hoechst 33342 (1 μg/mL, Thermo Fisher Scientific) prior to mounting on coverslips. Images were obtained on an Olympus FV1000 IX81-TIRF confocal microscope.
Western Blotting
All protein samples were resolved on 15% SDS-PAGE and detected using the following antibodies: anti-C9orf72 poly (GR) (Cosmo Bio; 1:1,000) for poly-GR proteins, anti-C9orf72 poly (GA) (Cosmo Bio; 1:1,000) for poly-GA proteins, and anti-C9orf72 poly (GP) (Cosmo Bio; 1:1,000) for poly-GP proteins. GAPDH was detected by 6C5 (Thermo Fisher Scientific; 1:2,000). Each experiment was repeated at least three times and comparable results were obtained.
Immunostaining
SK-N-MC cells were seeded and grown on glass coverslips at a density of 0.6 × 105 in a 24-well plate. At 48 h post-treatment, cells were fixed with 4% paraformaldehyde followed by three times wash with 1× PBS. Cells were then permeabilized in 0.5% Triton X-100 for 10 min at room temperature followed by three washes with 1× PBS. After the washing step, cells were blocked with 5% BSA in 1× PBS for 1 h at room temperature. The NCL (1:500, Abcam) or B23 (1:500, Abcam) antibody was then applied with 5% BSA (1:500) for 2 h at room temperature. Once the primary antibody was removed, cells were washed three times with 1× PBS and incubated with an anti-rabbit Cy3 or an anti-mouse Cy3 secondary antibody (1:400, Jackson Labs) for 1 h at room temperature. After washing with 1× PBS, nuclei were counterstained with Hoechst 33342 (1 μg/mL, Thermo Fisher Scientific) prior to mounting on coverslips. Images were obtained on an Olympus FV1000 IX81-TIRF confocal microscope. A single focal plan was obtained through the center of the nucleus. The method of quantification of NCL and B23 nuclear area fold change was described previously.16 To quantify both dispersed NCL and dense nucleolar NCL, a threshold setting in ImageJ ranging from 25 to 100 was used to measure the pixel area of NCL relative to the area of the nucleus outlined by the Hoechst staining. Over 150 cells were measured per treatment and data were normalized to the untransfected control.
RNA Extraction, Reverse Transcription, and PCR
RNA was extracted from cells by TRIzol reagent (Thermo Fisher Scientific), and 1 μg purified RNA was then used for reverse transcription using the ImPromII Reverse Transcription System (Promega). Random hexamer (Roche) was used as the primer in reverse transcription. Real-time PCR was performed on a Bio-Rad CFX96 Real-time PCR system, and data were analyzed as previously described.29 The following primers were used: human pre-45 s rRNA-F, 5′-GAACGGTGGTGTGTCGTT-3′; human pre-45 s rRNA-R, 5′-GCGTCTCGTCTCGTCTCACT-3′; human 28 s rRNA-F, 5′-AGAGGTAAACGGGTGGGGTC-3′; human 28 s rRNA-R, 5′-GGGGTCGGGAGGAACGG-3′; human 18 s rRNA-F, 5′-GATGGTAGTCGCCGTGCC-3′; human 18 s rRNA-R, 5′-GCCTGCTGCCTTCCTTGG-3′; human 5.8 s rRNA-F, 5′-ACTCGGCTCGTGCGTC-3′; human 5.8 s rRNA-R, 5′-GCGACGCTCAGACAGG-3′; human GGGGCC RNA-F, 5′-AGTACTCGCTGAGGGTG-3′; human GGGGCC RNA-R, 5′-TAGCGCGCGACTCCTGAG-3′; Human actin-F, 5′-TGTGCAAGGCCGGTT TCG C-3′; and human actin-R, 5′-CGACACGCAGCTCATTGTAG-3′.
Protein-RNA Interaction
Cells were transfected with 2 μg pAG3-(GGGGCC)2/66 simultaneously with 2 μg pcDNA3.1(+)-TAT-BIND-myc or pcDNA3.1(+)-TAT-BIND-S-myc construct. At 48 h after transfection, cells were fixed with 1% formaldehyde for 10 min at 37°C. Ice-cold 1× PBS was used to wash cells twice. Binding buffer (10 mM HEPES [pH 7.5], 10% glycerol, 5 mM MgCl2, 142.5 mM KCl, 1 mM EDTA, and 1% Triton X-100) supplemented with protease inhibitor cocktail (Sigma-Aldrich) and 40 U RNAsin (Promega) was then added to the samples. Sonication was applied to lyse the cells (Duty Cycle 30, Output Control 3, Timer 20 s, Sonifier 450, Branson Ultrasonics), and the samples were incubated at 4°C for 1 h with rotation. The samples were centrifuged at 4°C for 30 min at 16,100 × g. 10% supernatant was saved as input. 40 μL protein G agarose beads, together with anti-NCL antibody (1:200; ab22758, Abcam), was quickly added to the remaining samples, and the mixture was incubated at 4°C overnight with gentle rotation. For no antibody control, the same amount of protein G agarose beads was added to the reaction followed by gentle rotation at 4°C overnight. The protein-RNA complex was collected by centrifuging at 4°C for 10 s at 16,100 × g, followed by washing with binding buffer three times. 50 μL binding buffer was used to resuspend the protein-RNA complex, and the reaction was incubated for 45 min at 65°C to reverse cross-linking. 20 μL suspension was subject to immunoblot analysis. The primary antibody used was anti-NCL (ab22758, 1:1,000) from Abcam. The secondary antibody used was goat-anti-rabbit (11-035-045, 1:5,000) from Jackson ImmunoResearch Laboratories. The remaining 30 μL suspension, as well as the input, was subject to RNA extraction and subsequent RT-PCR. GGGGCC cDNA was amplified using the following primers: GGGGCC-F, 5′-AGTACTCGCTGAGGGTG-3′; and GGGGCC-R, 5′-TAGCGCGCGACTCCTGAG-3′. TAT-BIND-myc or TAT-BIND-S-myc cDNA was amplified using the following primers: TAT-F, 5′-ATGTATGGCCGCAAAAAAC-3′; and myc-R, 5′-CAGATCCTCTTCTGAGATGAG-3′.
Peptide Feeding, External Eye Assay, Climbing Ability Assay, and Lifespan Analysis
Flies were raised at 25°C on cornmeal medium supplemented with dry yeast. Eye images of 1-day-old adult UAS-(GGGGCC)3 or UAS-(GGGGCC)36 flies were captured using a SPOT Insight charge coupled device (CCD) camera (Diagnostic Instruments) on an Olympus SZX-12 stereomicroscope.
For lifespan and climbing ability assays, flies of 2 dpe were continuously (every 3 days) fed with food containing different drug combinations, including vehicle control (ethanol), Mifepristone (RU486, 200 μM), RU486 (200 μM) plus TAT-BIND (50 μM), and RU486 (200 μM) plus TAT-BIND-S (50 μM). Mifepristone (RU486, 200 μM) was used to induce transgene expression. For climbing ability assay, 10–15 flies were allocated to each experimental vial (total of 90–100 flies per condition) and monitored every 5 days. Fly climbing ability was analyzed by negative geotaxis. Groups of ∼15 flies were anesthetized and placed in a vertical plastic column. After 1-h recovery, flies were banged to the bottom, and then they were scored for climbing ability as the percentage of flies remaining at the bottom (<2 cm) at 25 s. Three trials were performed at 3-min intervals in each experiment. For lifespan analysis, 100–120 flies were tested per treatment as previously described.72
Statistical Analyses
Data were analyzed by one-way ANOVA followed by post hoc Tukey test (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
Author Contributions
Q.Z., A.C.K., K.-F.L., J.C.K.N., and H.Y.E.C. conceived the study and designed the experiments. Q.Z., Y.A., and Z.S.C. performed the experiments. Q.Z., A.C.K., K.-F.L., J.C.K.N., and H.Y.E.C. analyzed the data and wrote the manuscript. All authors contributed to discussions and reviewed the manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Acknowledgments
This work was supported by the ANR/RGC Joint Research Scheme (A-CUHK401/14) and Collaborative Research Fund (CUHK1/CRF/13G) of the Hong Kong Research Grants Council, CUHK Lui Che Woo Institute of Innovative Medicine BRAIN Initiative (8303404), CUHK Group Research Scheme (3110102), CUHK Vice-Chancellor’s One-Off Discretionary Fund (VCF2014011), CUHK One-off Funding for Joint Lab/Research Collaboration (3132980), CUHK Gerald Choa Neuroscience Centre (7105306), CUHK Faculty of Science Strategic Development Fund, and donations from the Hong Kong Spinocerebellar Ataxia Association (6903291). Z.S.C. is supported by the Postdoctoral Fellowship in Clinical Neurosciences of The Chinese University of Hong Kong.
Footnotes
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.omtn.2019.02.015.
Contributor Information
Jacky Chi Ki Ngo, Email: jackyngo@cuhk.edu.hk.
Ho Yin Edwin Chan, Email: hyechan@cuhk.edu.hk.
Supplemental Information
References
- 1.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., ITALSGEN Consortium A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu J., Liu J., Li L., Gagnon K.T., Corey D.R. Engineering Duplex RNAs for Challenging Targets: Recognition of GGGGCC/CCCCGG Repeats at the ALS/FTD C9orf72 Locus. Chem. Biol. 2015;22:1505–1511. doi: 10.1016/j.chembiol.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zu T., Liu Y., Bañez-Coronel M., Reid T., Pletnikova O., Lewis J., Miller T.M., Harms M.B., Falchook A.E., Subramony S.H. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA. 2013;110:E4968–E4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gendron T.F., Belzil V.V., Zhang Y.J., Petrucelli L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014;127:359–376. doi: 10.1007/s00401-013-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper-Knock J., Walsh M.J., Higginbottom A., Robin Highley J., Dickman M.J., Edbauer D., Ince P.G., Wharton S.B., Wilson S.A., Kirby J. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014;137:2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper-Knock J., Higginbottom A., Stopford M.J., Highley J.R., Ince P.G., Wharton S.B., Pickering-Brown S., Kirby J., Hautbergue G.M., Shaw P.J. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 2015;130:63–75. doi: 10.1007/s00401-015-1429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee Y.B., Chen H.J., Peres J.N., Gomez-Deza J., Attig J., Stalekar M., Troakes C., Nishimura A.L., Scotter E.L., Vance C. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackenzie I.R., Frick P., Grässer F.A., Gendron T.F., Petrucelli L., Cashman N.R., Edbauer D., Kremmer E., Prudlo J., Troost D., Neumann M. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015;130:845–861. doi: 10.1007/s00401-015-1476-2. [DOI] [PubMed] [Google Scholar]
- 10.Mori K., Weng S.M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 11.Mizielinska S., Grönke S., Niccoli T., Ridler C.E., Clayton E.L., Devoy A., Moens T., Norona F.E., Woollacott I.O.C., Pietrzyk J. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wen X., Tan W., Westergard T., Krishnamurthy K., Markandaiah S.S., Shi Y., Lin S., Shneider N.A., Monaghan J., Pandey U.B. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y.J., Gendron T.F., Grima J.C., Sasaguri H., Jansen-West K., Xu Y.F., Katzman R.B., Gass J., Murray M.E., Shinohara M. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 2016;19:668–677. doi: 10.1038/nn.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y.J., Jansen-West K., Xu Y.F., Gendron T.F., Bieniek K.F., Lin W.L., Sasaguri H., Caulfield T., Hubbard J., Daughrity L. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128:505–524. doi: 10.1007/s00401-014-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee Y.B., Baskaran P., Gomez-Deza J., Chen H.J., Nishimura A.L., Smith B.N., Troakes C., Adachi Y., Stepto A., Petrucelli L. C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum. Mol. Genet. 2017;26:4765–4777. doi: 10.1093/hmg/ddx350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haeusler A.R., Donnelly C.J., Periz G., Simko E.A., Shaw P.G., Kim M.S., Maragakis N.J., Troncoso J.C., Pandey A., Sattler R. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao Z., Wang H., Xia Q., Li K., Li K., Jiang X., Xu G., Wang G., Ying Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 2015;24:2426–2441. doi: 10.1093/hmg/ddv005. [DOI] [PubMed] [Google Scholar]
- 18.Freibaum B.D., Lu Y., Lopez-Gonzalez R., Kim N.C., Almeida S., Lee K.H., Badders N., Valentine M., Miller B.L., Wong P.C. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.James A., Wang Y., Raje H., Rosby R., DiMario P. Nucleolar stress with and without p53. Nucleus. 2014;5:402–426. doi: 10.4161/nucl.32235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y., Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16:369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ginisty H., Amalric F., Bouvet P. Nucleolin functions in the first step of ribosomal RNA processing. EMBO J. 1998;17:1476–1486. doi: 10.1093/emboj/17.5.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burger K., Mühl B., Kellner M., Rohrmoser M., Gruber-Eber A., Windhager L., Friedel C.C., Dölken L., Eick D. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 2013;10:1623–1630. doi: 10.4161/rna.26214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouvet P., Diaz J.J., Kindbeiter K., Madjar J.J., Amalric F. Nucleolin interacts with several ribosomal proteins through its RGG domain. J. Biol. Chem. 1998;273:19025–19029. doi: 10.1074/jbc.273.30.19025. [DOI] [PubMed] [Google Scholar]
- 24.Rickards B., Flint S.J., Cole M.D., LeRoy G. Nucleolin is required for RNA polymerase I transcription in vivo. Mol. Cell. Biol. 2007;27:937–948. doi: 10.1128/MCB.01584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong C.H., Nguyen L., Peh J., Luu L.M., Sanchez J.S., Richardson S.L., Tuccinardi T., Tsoi H., Chan W.Y., Chan H.Y. Targeting toxic RNAs that cause myotonic dystrophy type 1 (DM1) with a bisamidinium inhibitor. J. Am. Chem. Soc. 2014;136:6355–6361. doi: 10.1021/ja5012146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan H.Y. RNA-mediated pathogenic mechanisms in polyglutamine diseases and amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014;8:431. doi: 10.3389/fncel.2014.00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsoi H., Chan H.Y. Expression of expanded CAG transcripts triggers nucleolar stress in Huntington’s disease. Cerebellum. 2013;12:310–312. doi: 10.1007/s12311-012-0447-6. [DOI] [PubMed] [Google Scholar]
- 28.Tsoi H., Chan H.Y. Roles of the nucleolus in the CAG RNA-mediated toxicity. Biochim. Biophys. Acta. 2014;1842:779–784. doi: 10.1016/j.bbadis.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 29.Tsoi H., Lau T.C., Tsang S.Y., Lau K.F., Chan H.Y. CAG expansion induces nucleolar stress in polyglutamine diseases. Proc. Natl. Acad. Sci. USA. 2012;109:13428–13433. doi: 10.1073/pnas.1204089109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q., Tsoi H., Peng S., Li P.P., Lau K.F., Rudnicki D.D., Ngo J.C., Chan H.Y. Assessing a peptidylic inhibitor-based therapeutic approach that simultaneously suppresses polyglutamine RNA- and protein-mediated toxicities in patient cells and Drosophila. Dis. Model. Mech. 2016;9:321–334. doi: 10.1242/dmm.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang H., Shin J.H. Repression of rRNA transcription by PARIS contributes to Parkinson’s disease. Neurobiol. Dis. 2015;73:220–228. doi: 10.1016/j.nbd.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Evsyukov V., Domanskyi A., Bierhoff H., Gispert S., Mustafa R., Schlaudraff F., Liss B., Parlato R. Genetic mutations linked to Parkinson’s disease differentially control nucleolar activity in pre-symptomatic mouse models. Dis. Model. Mech. 2017;10:633–643. doi: 10.1242/dmm.028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parlato R., Liss B. How Parkinson’s disease meets nucleolar stress. Biochim. Biophys. Acta. 2014;1842:791–797. doi: 10.1016/j.bbadis.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Q., Chen Z.S., An Y., Liu H., Hou Y., Li W., Lau K.F., Koon A.C., Ngo J.C.K., Chan H.Y.E. A peptidylic inhibitor for neutralizing expanded CAG RNA-induced nucleolar stress in polyglutamine diseases. RNA. 2018;24:486–498. doi: 10.1261/rna.062703.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginisty H., Sicard H., Roger B., Bouvet P. Structure and functions of nucleolin. J. Cell Sci. 1999;112:761–772. doi: 10.1242/jcs.112.6.761. [DOI] [PubMed] [Google Scholar]
- 36.Frankel A.D., Pabo C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 37.Green M., Loewenstein P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Dis. Markers. 1990;8:34–35. [PubMed] [Google Scholar]
- 38.Rizzuti M., Nizzardo M., Zanetta C., Ramirez A., Corti S. Therapeutic applications of the cell-penetrating HIV-1 Tat peptide. Drug Discov. Today. 2015;20:76–85. doi: 10.1016/j.drudis.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 39.El-Andaloussi S., Järver P., Johansson H.J., Langel U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study. Biochem. J. 2007;407:285–292. doi: 10.1042/BJ20070507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saar K., Lindgren M., Hansen M., Eiríksdóttir E., Jiang Y., Rosenthal-Aizman K., Sassian M., Langel U. Cell-penetrating peptides: a comparative membrane toxicity study. Anal. Biochem. 2005;345:55–65. doi: 10.1016/j.ab.2005.07.033. [DOI] [PubMed] [Google Scholar]
- 41.Su Z., Zhang Y., Gendron T.F., Bauer P.O., Chew J., Yang W.Y., Fostvedt E., Jansen-West K., Belzil V.V., Desaro P. Discovery of a Biomarker and Lead Small Molecules to Target r(GGGGCC)-Associated Defects in c9FTD/ALS. Neuron. 2014;84:239. doi: 10.1016/j.neuron.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 42.Gendron T.F., Bieniek K.F., Zhang Y.J., Jansen-West K., Ash P.E., Caulfield T., Daughrity L., Dunmore J.H., Castanedes-Casey M., Chew J. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bañez-Coronel M., Porta S., Kagerbauer B., Mateu-Huertas E., Pantano L., Ferrer I., Guzmán M., Estivill X., Martí E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012;8:e1002481. doi: 10.1371/journal.pgen.1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conlon E.G., Lu L., Sharma A., Yamazaki T., Tang T., Shneider N.A., Manley J.L. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife. 2016;5:e17820. doi: 10.7554/eLife.17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang J., Zhu Q., Gendron T.F., Saberi S., McAlonis-Downes M., Seelman A., Stauffer J.E., Jafar-Nejad P., Drenner K., Schulte D. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 2016;90:535–550. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gitler A.D., Tsuiji H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 2016;1647:19–29. doi: 10.1016/j.brainres.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mizielinska S., Lashley T., Norona F.E., Clayton E.L., Ridler C.E., Fratta P., Isaacs A.M. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126:845–857. doi: 10.1007/s00401-013-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lagier-Tourenne C., Baughn M., Rigo F., Sun S., Liu P., Li H.R., Jiang J., Watt A.T., Chun S., Katz M. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA. 2013;110:E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran H., Almeida S., Moore J., Gendron T.F., Chalasani U., Lu Y., Du X., Nickerson J.A., Petrucelli L., Weng Z., Gao F.B. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron. 2015;87:1207–1214. doi: 10.1016/j.neuron.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubbi C.P., Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003;22:6068–6077. doi: 10.1093/emboj/cdg579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meng L., Yasumoto H., Tsai R.Y. Multiple controls regulate nucleostemin partitioning between nucleolus and nucleoplasm. J. Cell Sci. 2006;119:5124–5136. doi: 10.1242/jcs.03292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li L.B., Yu Z., Teng X., Bonini N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107–1111. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo W., Fumagalli L., Prior R., Van Den Bosch L. Current Advances and Limitations in Modeling ALS/FTD in a Dish Using Induced Pluripotent Stem Cells. Front. Neurosci. 2017;11:671. doi: 10.3389/fnins.2017.00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freibaum B.D., Taylor J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017;10:35. doi: 10.3389/fnmol.2017.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moens T.G., Mizielinska S., Niccoli T., Mitchell J.S., Thoeng A., Ridler C.E., Grönke S., Esser J., Heslegrave A., Zetterberg H. Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta. Neuropathol. 2018;135:445–457. doi: 10.1007/s00401-017-1798-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Donnelly C.J., Zhang P.W., Pham J.T., Haeusler A.R., Mistry N.A., Vidensky S., Daley E.L., Poth E.M., Hoover B., Fines D.M. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orr H.T. Toxic RNA as a driver of disease in a common form of ALS and dementia. Proc. Natl. Acad. Sci. USA. 2013;110:7533–7534. doi: 10.1073/pnas.1305239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang K., Donnelly C.J., Haeusler A.R., Grima J.C., Machamer J.B., Steinwald P., Daley E.L., Miller S.J., Cunningham K.M., Vidensky S. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang K., Grima J.C., Rothstein J.D., Lloyd T.E. Nucleocytoplasmic transport in C9orf72-mediated ALS/FTD. Nucleus. 2016;7:132–137. doi: 10.1080/19491034.2016.1172152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamakawa M., Ito D., Honda T., Kubo K., Noda M., Nakajima K., Suzuki N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet. 2015;24:1630–1645. doi: 10.1093/hmg/ddu576. [DOI] [PubMed] [Google Scholar]
- 61.Mykowska A., Sobczak K., Wojciechowska M., Kozlowski P., Krzyzosiak W.J. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011;39:8938–8951. doi: 10.1093/nar/gkr608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Achi E.Y., Rudnicki S.A. ALS and Frontotemporal Dysfunction: A Review. Neurol. Res. Int. 2012;2012:806306. doi: 10.1155/2012/806306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simone R., Balendra R., Moens T.G., Preza E., Wilson K.M., Heslegrave A., Woodling N.S., Niccoli T., Gilbert-Jaramillo J., Abdelkarim S. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol. Med. 2018;10:22–31. doi: 10.15252/emmm.201707850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sareen D., O’Rourke J.G., Meera P., Muhammad A.K., Grant S., Simpkinson M., Bell S., Carmona S., Ornelas L., Sahabian A. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schludi M.H., Edbauer D. Targeting RNA G-quadruplexes as new treatment strategy for C9orf72 ALS/FTD. EMBO Mol. Med. 2018;10:4–6. doi: 10.15252/emmm.201708572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frazier K.S. Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol. 2015;43:78–89. doi: 10.1177/0192623314551840. [DOI] [PubMed] [Google Scholar]
- 67.Goldflam M., Ullman C.G. Recent Advances Toward the Discovery of Drug-Like Peptides De novo. Front Chem. 2015;3:69. doi: 10.3389/fchem.2015.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cao G., Pei W., Ge H., Liang Q., Luo Y., Sharp F.R., Lu A., Ran R., Graham S.H., Chen J. In Vivo Delivery of a Bcl-xL Fusion Protein Containing the TAT Protein Transduction Domain Protects against Ischemic Brain Injury and Neuronal Apoptosis. J. Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwarze S.R., Ho A., Vocero-Akbani A., Dowdy S.F. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 70.Stalmans S., Bracke N., Wynendaele E., Gevaert B., Peremans K., Burvenich C., Polis I., De Spiegeleer B. Cell-Penetrating Peptides Selectively Cross the Blood-Brain Barrier In Vivo. PLoS ONE. 2015;10:e0139652. doi: 10.1371/journal.pone.0139652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oller-Salvia B., Sánchez-Navarro M., Giralt E., Teixidó M. Blood-brain barrier shuttle peptides: an emerging paradigm for brain delivery. Chem. Soc. Rev. 2016;45:4690–4707. doi: 10.1039/c6cs00076b. [DOI] [PubMed] [Google Scholar]
- 72.Linford N.J., Bilgir C., Ro J., Pletcher S.D. Measurement of lifespan in Drosophila melanogaster. J. Vis. Exp. 2013;(71):50068. doi: 10.3791/50068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maupetit J., Derreumaux P., Tufféry P. A fast method for large-scale de novo peptide and miniprotein structure prediction. J. Comput. Chem. 2010;31:726–738. doi: 10.1002/jcc.21365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.