

Abstract
Turner syndrome is a relatively common chromosomal abnormality presenting as primary amenorrhoea in gynaecological and endocrine clinics, caused by complete or partial X monosomy in some or all cells. Mayer-Rokitansky-Kuster-Hauser syndrome is another common cause of primary amenorrhoea characterised by Mullerian agenesis of varying degrees. We report a case of an 18-year-old girl, who presented with primary amenorrhoea, absence of secondary sexual characteristics and short stature. Hormonal profile confirms hypergonadotrophic hypogonadism. Karyotyping was consistent with Turner syndrome (45,XO). In addition, radiological imaging of the pelvis showed the absence of both ovaries as well as the uterus, cervix and vagina. This patient had therefore presented with two different syndromes as the cause of her primary amenorrhoea, which is extremely rare in a single patient. Moreover, oestrogen replacement therapy will trigger the development of secondary sexual characteristic and promote bone growth, but induction of menstruation and fertility is impossible.
Keywords: reproductive medicine; congenital disorders; endocrine system; obstetrics, gynaecology and fertility
Background
Primary amenorrhoea is defined as the absence of menstruation in women aged 14 years without the development of secondary sexual characteristics or a female who never menstruated by the age of 16 years, with the presence of secondary sexual characteristics.
Turner syndrome is the most common chromosomal abnormality, presenting as primary amenorrhoea. It affects approximately 1 in 2500 live-born females. Turner syndrome is usually characterised by the presence of typical physical features in a phenotypic female with complete or partial absence of X chromosome, or other X chromosome abnormalities, with or without cell line mosaicism.1 A patient can present with primary amenorrhoea and the absence of the development of secondary sexual characteristics, or normal secondary sexual characteristics but primary amenorrhoea or even beginning of menstrual bleeding.2 The development of Mullerian structures, that is, uterus, cervix, vagina and fallopian tubes, are typically normal in Turner syndrome.
On the contrary, Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome is characterised by an absent or hypoplastic uterus, cervix and upper two-thirds of the vagina in genetically and phenotypically normal female with the incidence of 1 in 5000 newborns. A patient with MRKH has normal secondary sexual characteristics because of normal functioning ovaries. There are two major types of MRKH syndrome. Type 1 is characterised by congenital aplasia of the uterus and upper two-thirds of the vagina. Type 2 also incorporates extragenital/extra-Müllerian malformations, including vertebral, cardiac, urological (upper tract) and otological anomalies. Type 2 includes MURCS association (Mullerian duct aplasia, renal dysplasia, cervical somite anomalies) and includes renal (unilateral agenesis, ectopic kidney or horseshoe kidney), vertebral skeletal (Klippel-Feil anomaly, fused vertebrae mainly cervical and scoliosis), hearing defects due to middle ear abnormalities and rarely cardiac and digital anomalies.
We report a case of primary amenorrhoea diagnosed to have Turner syndrome (45,XO), who also had absent uterus, cervix and vagina. These features raise the possibility of coexistent MRKH syndrome in the same patient, which is extremely rare.3 The incidence of having both abnormalities simultaneously in a patient is 1 in 1 500 000.4
Case presentation
An 18-year-old Asian girl, presented with primary amenorrhoea, absence of secondary sexual characteristics and short stature. She was born out of non-consanguineous marriage, at complete term by caesarian section. At birth, she was kept in special care for 3 days due to meconium aspiration. Maternal age at her birth was 21 years. There was no antenatal history of any viral or bacterial infection or chronic illness during gestation. Her developmental milestones were normal except stunted growth as reported by her mother (as she could not provide us with her birth and developmental records). There were no dysmorphic features on face, neck, trunk or extremities, during newborn, infant and childhood periods. Moreover, she never had any skeletal abnormalities. She has one elder and one younger brother, both were healthy. There was no family history of any genetic disease. On examination at present, her vitals were normal, height 138 cm, weight 41 kg, body mass index 21.5 kg/m2 (25th percentile for Turner syndrome girls) and mid-parental height was 152.5 cm. Her breast and pubic hair development was at Tanner stage 1. However, no features of broad chest, widely space nipple, web neck and wide carrying angle at the elbow were present. Other systemic examinations including neurological, cardiovascular, chest and abdominal examinations were unremarkable.
Investigations
Hormonal profile confirmed hypergonadotropic hypogonadism as shown in table 1.
Table 1.
Hormonal profile of the patient
| Test | Result | Reference range |
| FSH | 124 mIU/mL | Follicular phase: 3–14.4 |
| LH | 29.01 mIU/mL | 1.1–11.6 |
| TSH | 4.43 uIU/mL | 04–4.0 |
| FT3 | 169.9 ng/dL | 84–172 |
| FT4 | 11.28 mcg/dL | 4.5–12.5 |
| Prolactin | 14.21 ng/mL | 4.79–23.30 |
FSH, follicular-stimulating hormone; FT3, free tri-iodothyronine; FT4, free thyroxine; LH, luteinising hormone; TSH, thyroid-stimulating hormone.
Other haematological and biochemical tests were normal. Karyotyping was consistent with Turner syndrome (45,XO) (figure 1). Radiologically estimated bone age was equal to chronological age (figure 2). Moreover, there were no skeletal features associated with Turner syndrome reported in these images. In addition, ultrasound pelvis did not show uterus and ovaries. So MRI (figure 3) pelvis was done that confirmed the absence of uterus, cervix, vagina and both ovaries.
Figure 1.
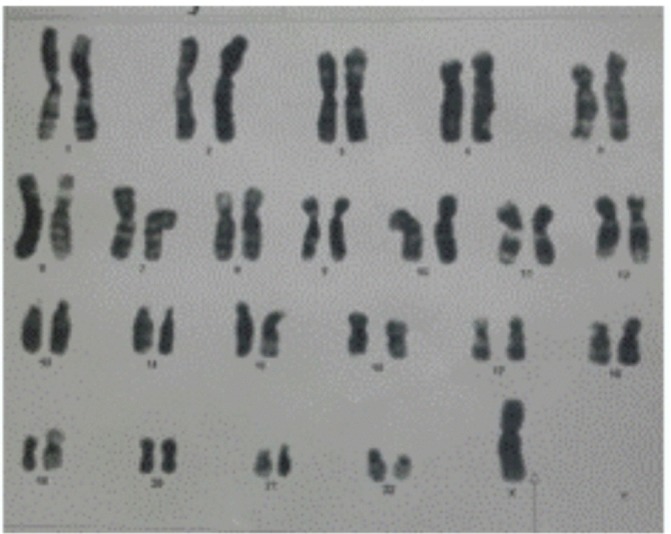
Karyotyping showing chromosomal pattern of TS (45,XO).
Figure 2.
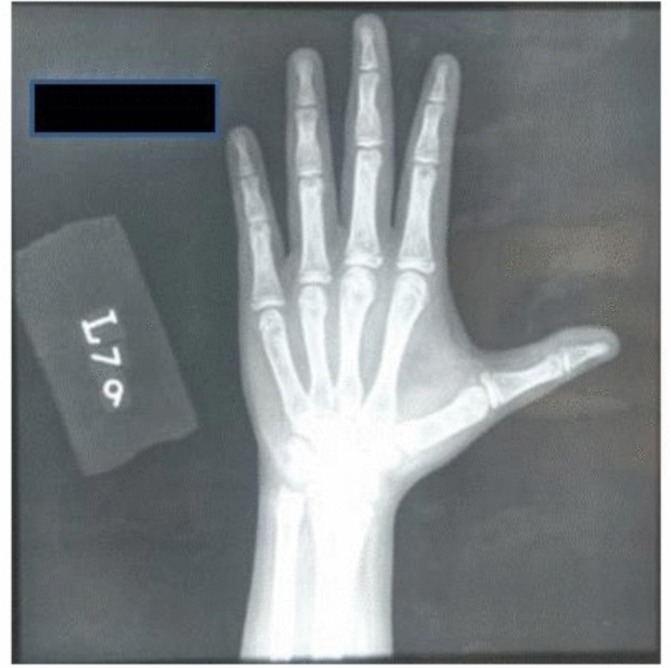
X-ray AP view of the left hand for bone age. AP, anteroposterior.
Figure 3.
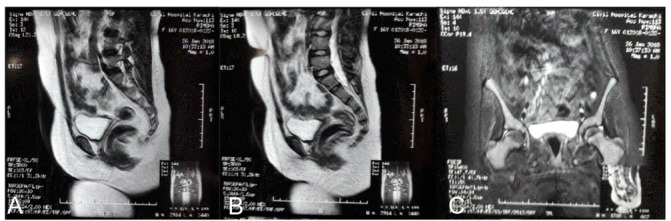
(A and B) Mid sagittal views of T2-weighted MRI with contrast showing the absence uterine tissue. (C) Coronal T2-weighted MRI with contrast showing the absence of uterine and ovarian tissue.
Differential diagnosis
MRKH syndrome
Androgen insensitivity syndrome (AIS).
Müllerian-inhibiting substance deficiency.
Turner’s syndrome
5-Alpha reductase deficiency.
17 Alpha-hydroxylase deficiency with 46XX.
Constitutional delay in growth and puberty.
Secondary hypogonadism.
Treatment
Growth hormone (GH) therapy should be initiated as early as possible (around 4–6 years of age, and preferably before 12–13 years) at a dose of 45–50 µg/kg/day, and adjusted according to patient’s growth response and insulin-like growth factor-1 level up to 68 µg/kg/day.5 As our patient presented in her second decade of life and her bone age was equal to the chronological age, GH therapy was considered to be less likely effective.
Ideally, oestrogen replacement should be started not later than 15 years and not before 12 years of age, when growth is the priority unless height has been maximised. Low-dose oestrogen 0.25–0.5 mg micronised estradiol or its equivalent should be given, and increased gradually up to 2 mg over a period of 2 years. Preferably, a transdermal route should be used, but due to the lack of availability, we pursued oral treatment. The addition of progesterone is delayed for at least 2 years or until breakthrough bleeding occurs to permit normal breast and uterine development and to mimic age-specific physiological pattern as closely as possible.5 Oestrogen replacement therapy is continued until the age of menopause to maintain feminisation and to prevent osteoporosis. This patient has been started on oral conjugated oestrogen 0.3 mg daily. However, the absence of uterus in our patient exclude the administration of progesterone.
The aim of treatment in the patient with Mullerian agenesis is the surgical creation of neo-vagina. Our plan is to follow the patient after a period of oestrogen replacement and then refer for the surgical procedure. Moreover, we had also referred the patient to a psychiatrist to cover the aspects of social and personal issues related to her diagnosis.
Outcome and follow-up
We planned to follow our patient after 6 months of oestrogen replacement and undergo a repeat MRI pelvis to review for the presence of any uterine tissue. Moreover, we had also advised her for cardiac and other Turner syndrome-associated health issues as per the clinical practice guidelines for the care of girls and women with Turner syndrome.5 Unfortunately, our patient has lost to follow-up due to her poor financial status and non-affordability of various investigations.
Discussion
During the first 6 weeks of development, the male and female fetuses are indistinct and exhibit both mesonephric and paramesonephric ducts. The presence of Y chromosome is associated with the formation of Mullerian inhibitory factor (MIF). The absence of MIF in female advances the development of paired Mullerian ducts from the paramesonephric system along with a recession of mesonephric ducts. Any interference in this process can lead to aplasia or hypoplasia of uterus, cervix and vagina. These features are described in the MRKH syndrome, perhaps related to the arrest of development between 8 and 12 weeks of gestation, and with clinical manifestation of primary amenorrhoea. As oestrogen is not obligatory for Mullerian duct development, the recession of Wolffian ducts, the development of gonads and external genitalia with secondary sexual characteristics are of the normal female. Diagnosis is based on physical examination and radiological imaging. The potential cause of MRKH is not very clear, however, familial case studies have supported the genetic basis as well as chromosomal aberrations.2 6
On the contrary, gonadal dysgenesis in the form of primary amenorrhoea and absent secondary sexual characteristic is the most common presentation described in Turner syndrome. This may originate from an error early in primordial follicle synthesis or in the differentiation of ovaries. There is no relationship with any risk factor, maternal age or familial inheritance.
Short stature is another most common presenting feature of Turner syndrome, which is predicted to be due to haploinsufficiency of short stature homeobox-containing gene (SHOX) located within Xp terminal, pseudoautosomal region of X-chromosome, resulting in average adult stature 20 cm shorter than their target height.
In addition, there is a high risk of atherosclerosis, hypertension and type 2 diabetes mellitus. Diabetes is usually mild and responsive to single drug therapy and lifestyle modification. Among the skeletal deformities, there may be a positive metacarpal sign; however, Madelung deformity of the wrist joint is infrequent.
It is hard to presume a theory for co-existence of MRKH syndrome and Turner syndrome simultaneously in an individual presenting with primary amenorrhoea. About 30 case reports have been published since 1976 with these two rare association of syndromes, summarised in table 2.
Table 2.
Literature review of published cases showing co-existence of Turner syndrome and MRKH syndrome
| S.No. | Author (year) | Age of presentation (years) | Karyotype | Ovaries | Uterus | Fallopian tubes | Consanguinity | Other abnormalities |
| 1 | Elamparidhi et al
(2017)13 |
17 | 45,XO | Agenetic | Absent | NA | Absent | Horseshoe kidney Short 4th metacarpal |
| 2 | Białka et al (2016)14 | 17 | 46,X/X(q10) | Dysgenetic | Hypoplastic | NR | Absent | NA |
| 3 | Bhandari and Chaudhary (2017)15 | 17 | 46,XX | Agenetic | Absent | NR | Absent | NA |
| 4 | Meena et al 16 | 15 | 45,XO/46,XX | Agenetic | Absent | NA | Absent | No |
| 5 | De Chavez et al (2014)17 | 18 | 45,X | Agenetic | Absent | NA | NA | --- |
| 6 | Kebaili et al (2013)18 | 21 | 46,XX | Agenetic | Absent | Absent | Absent | No malformation |
| 7 | Vaddadi et al (2013)3 | 35 | 45,X | Agenetic | Absent | NA | Present | Primary hypothyroidism |
| 8 | Shah et al (2013)7 | 21 | 46,XX | Agenetic | Absent | Absent | Absent | ------- |
| 9 | Bousfiha et al (2010)19 | 19 | 46,XX | Dysgenetic | Absent | Absent | Absent | None |
| 10 | Tatar et al (2009)2 | 2 sisters (34 and 23) | 46,XX | Agenetic | Hypoplastic | Hypoplastic | Present | Partial alopecia, mental retardation, microcephaly, kyphosis, sensorineural deafness in one of them |
| 11 | Zaman and Nisar (2009)8 | 2 sisters (22 and 13 | 46,XX | Dysgenetic | One absent the other, rudimentary | Hypoplastic | Present | Hypoplastic vagina, alopecia totalis |
| 12 | Güven et al (2008)20 | 17 | 45,X/46,X delX (p11.21) | Agenetic | Absent | NR | Absent | Short stature, bone age was 12 |
| 13 | Marcial SG(2008)4 | NA | 45,X6
(46,X,i(X) (q10)16) |
NA | Absent | NA | Present | No abnormality |
| 14 | Kumar et al (2007)21 | 18 | 46,XX | Rt. side agenetic | Absent | NR | Absent | Solitary malrotated pelvic kidney with PUJ obstruction |
| 15 | Colombani et al (2007)9 | 15 | 46,XX | Dysgenetic | Absent | N | Present | Autoimmune thyroiditis with secondary hypothyroidism |
| 16 | Marrakchi et al (2004)22 | 19 | 46,XX | Dysgenetic | Absent | N | Absent | None |
| 17 | Plevraki et al (2004)23 | 6 patients | 46,XX with testis-specific protein 1-Y linked gene (in patients 1 and 4) | Patient 1: left side, agenetic Patient 6: agenetic | Patient 1: hypoplastic uterus with symmetrical uterine buds, with no endometrium Patient 6: uterus, symmetrical hypoplastic | Patient 1: left fallopian tube, absent Patient 6: both fallopian tubes were symmetric, but hypoplastic | Absent | Patient 1: short fourth metacarpal Patient 6: bifid first sacral vertebra, lumbar scoliosis |
| 18 | Kaya et al (2003)24 | 17 | 46,XX | Left Agenetic | Absent | Right normal left hypoplastic | Absent | Right kidney malrotated |
| 19 | Aydos et al (2003)25 | 19 | 46,X, del(X)(Pter>q 22) | Agenetic | Rudimentary | NR | Absent | Mild torticollis, cutis marmotara, hallux valgus |
| 20 | Mégarbané et al (2003)10 | 2 sisters | 46,XX | Dysgenetic | Hypoplastic | Hypoplastic | Present | Microcephaly, flat occiput, partial alopecia |
| 21 | Gorgojo et al (2002)12 | 17 | 46,XX | Agenetic | Absent | Absent | Absent | Single pelvic kidney, primary subclinical hypothyroidism |
| 22 | Ting and Chang (2002)26 | 22 | 45,X/46,X, del(X) (p22.22) | Dysgenetic | Absent | Rudimentary | Absent | Scoliosis of thoracic spine |
| 23 | Güitrón-Cantú et al (1995)27 | 19 | 45,X/46,Xdic(X) | Agenetic | Absent | N | Absent | None |
| 24 | Oyer et al (1994)28 | Neonate | 46,XX | Agenetic | Defects in Mullerian derivatives | NA | Absent | Diaphragmatic hernia, bicuspid aortic valve |
| 25 | Aughton (1993)29 | NA | 46,XX | Dysgenetic | Absent | Absent | Absent | The girl’s mother and maternal grandmother have low galactose-1-phosphate uridyl transferase activities and heterozygous for classic galactosaemia |
| 26 | Alper et al (1985)30 | 16 | NA | Dysgenetic | Absent | NA | Absent | Normal vagina |
| 27 | Al-Awadi et al (1985)11 | 2 sisters (18 and 16) |
46,XX | One agenetic, other dysgenetic | Hypoplastic | One absent, other hypoplastic | Present | Partial alopecia |
| 28 | De Leon et al (1984)31 | NA | 46,X,i(Xq) | Agenetic | Absent | NR | Absent | Subtle features of Turner syndrome |
| 29 | Levinson et al 32 | 17 | 46,XX | Agenetic | Absent | Absent | Absent | Absent vagina, double ureter on left |
Courtesy, Meena A, Daga MK, Dixit R. Unusual association of Turner syndrome and Mayer-Rokitansky-Kuster-Hauser syndrome. BMJ case reports. 2016; 2016. doi: 10.1136/bcr-2015–2 12 634. PubMed PMID: 27207981; PubMed Central PMCID: PMC4885519.
n, normal; NA, not available; NR,not reported; PUJ, pelvi ureteral junction.
Although the literature shows that these two syndromes are being reported for the last four decades, however, no link between the two syndromes has ever been identified to date. One of the case reports was in French language, so it was not possible for us to analyse and include in the literature review. All patients except one presented in adult age with primary amenorrhoea and underdeveloped secondary sexual characteristics. In eight cases,2–4 7–11 there was a history of consanguinity. Five cases have a positive family history of Mullerian and gonadal agenesis. Out of 30 cases, 16 patients had normal (46,XX) karyotype, while others had microdeletion or mosaicism, and only 4 had classic 45,XO karyotype. Among all cases, only three cases had primary hypothyroidism.3 9 12 In the light of these cases, it is important to consider the diagnosis of Turner syndrome in all cases with short stature and primary amenorrhoea. Besides, the radiological investigation should always be taken into account apart from hormonal and chromosomal analyses. First, to confirm the presence or absence ovarian tissue in a 45,XO individual, and second to identify Mullerian structures, as this will guide treatment plan and assess in prognostication not only in Turner syndrome (more favourable from a fertility point of view) but also in MRKH syndrome. Moreover, these investigations are also important to exclude other differential diagnoses (gonadal dysgenesis and AIS).
All individuals with Turner syndrome require monitoring and screening for auditory disturbances, thyroid dysfunction, hypertension, diabetes and dyslipidaemia annually.5 In our society as well as in general, the reproductive ability of a female is taken as an extreme social stigma, because of which recognition of cases like our patient is very important who is a teenager. Therefore, these individuals need a highly skilled multidisciplinary team effort for addressing these important issues in their lives. Hence, endocrinologist (paediatric and adult), geneticist, psychiatrist, psychologist, sociologist, cardiologist and gynaecologist need to come together on the same platform for individual case management and care.
Learning points.
Turner syndrome is an important diagnosis to consider in all females with primary amenorrhoea.
Absence of Mullerian structures should prompt one to consider Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome in any genotypic female.
Association of Turner syndrome with MRKH is a rare entity with no specific cause, but it is increasingly reported and need awareness among healthcare providers.
One of the main issues for the girls with MRKH is the impossibility to get pregnant, which differentiate them from the girls with the Turner syndrome only, who variably have some chances.5
Footnotes
Contributors: ZK: conceived, designed, did data interpretation and review of case report. TJ: did acquisition of the data, manuscript writing and editing.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Obtained.
References
- 1. Bondy CA. Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab 2007;92:10–25. 10.1210/jc.2006-1374 [DOI] [PubMed] [Google Scholar]
- 2. Tatar A, Ocak Z, Tatar A, et al. Primary hypogonadism, partial alopecia, and Mullerian hypoplasia: report of a third family and review. Am J Med Genet A 2009;149A:501–4. 10.1002/ajmg.a.32645 [DOI] [PubMed] [Google Scholar]
- 3. Vaddadi S, Murthy RS, Rahul CH, et al. A rare case of Turner’s syndrome presenting with Mullerian agenesis. J Hum Reprod Sci 2013;6:277 10.4103/0974-1208.126313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sharon Gaila Marcial. A rare case primary amenorrhea in a patient with Turner Syndrome with concomitant Mayer-Rokitansky-Kuster-Hauser Syndrome. Philippine Journal of Reproductive Endocrinology and Infertility 2008;5:67–74. [Google Scholar]
- 5. Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol 2017;177:G1–G70. 10.1530/EJE-17-0430 [DOI] [PubMed] [Google Scholar]
- 6. Rall K, Eisenbeis S, Henninger V, et al. Typical and Atypical Associated Findings in a Group of 346 Patients with Mayer-Rokitansky-Kuester-Hauser Syndrome. J Pediatr Adolesc Gynecol 2015;28:362–8. 10.1016/j.jpag.2014.07.019 [DOI] [PubMed] [Google Scholar]
- 7. Shah VN, Ganatra PJ, Parikh R, et al. Coexistence of gonadal dysgenesis and Mayer-Rokitansky-Kuster-Hauser syndrome in 46, XX female: A case report and review of literature. Indian J Endocrinol Metab 2013;17(Suppl 1):274 10.4103/2230-8210.119605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zaman SM, Nisar M. Primary hypergonadotrophic hypogonadism, alopecia totalis, and müllerian hypoplasia: a clinical study. J Pak Med Assoc 2009;59:571–3. [PubMed] [Google Scholar]
- 9. Colombani M, Cau D, Sapin E, et al. Fourth case of uterine aplasia, ovarian dysgenesis, amenorrhea and impuberism: a variant of Mayer-Rokitansky-Kuster-Hauser syndrome. Acta Paediatr 2007;96:1371–71. 10.1111/j.1651-2227.2007.00404.x [DOI] [PubMed] [Google Scholar]
- 10. Mégarbané A, Gannage‐Yared M, Khalifé A, et al. Primary hypergonadotropic hypogonadism, partial alopecia, and Müllerian hypoplasia: report of a second family with additional findings. American Journal of Medical Genetics Part A 2003;119:214–7. [DOI] [PubMed] [Google Scholar]
- 11. Al-Awadi SA, Farag TI, Teebi AS, et al. Primary hypogonadism and partial alopecia in three sibs with müllerian hypoplasia in the affected females. Am J Med Genet 1985;22:619–22. 10.1002/ajmg.1320220322 [DOI] [PubMed] [Google Scholar]
- 12. Gorgojo JJ, Almodóvar F, López E, et al. Gonadal agenesis 46,XX associated with the atypical form of Rokitansky syndrome. Fertil Steril 2002;77:185–7. 10.1016/S0015-0282(01)02943-0 [DOI] [PubMed] [Google Scholar]
- 13. Elamparidhi P, Kumar RR, Sivaranjinie S, et al. Mullerian Agenesis Associated with Turner’s Syndrome. J Clin Diagn Res 2017;11:TD01 10.7860/JCDR/2017/23305.9157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Białka A, Gawlik A, Drosdzol-Cop A, et al. Coexistence of Mayer-Rokitansky-Küster-Hauser Syndrome and Turner Syndrome: A Case Report. J Pediatr Adolesc Gynecol 2016;29:e35–e38. 10.1016/j.jpag.2015.10.019 [DOI] [PubMed] [Google Scholar]
- 15. Bhandari B, Chaudhary BK. Gonadal dysgenesis and the Mayer-Rokitansky-Kuster-Hauser syndrome in a girl with a 46, XX karyotype: a case report. International Journal of Contemporary Pediatrics 2017;2:246–8. [Google Scholar]
- 16. Meena A, Daga MK, Dixit R. Unusual association of Turner syndrome and Mayer-Rokitansky-Küster-Hauser syndrome. BMJ Case Rep 2016;2016:bcr2015212634 10.1136/bcr-2015-212634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maria Delina E DC, Capco-Dichoso M, Opulencia MR. Double burden: A rare case of Turner’s syndrome with concomitant Mayer-Rokitanski-Kuster-Hauser syndrome. Philippine Journal of Obstetrics and Gynecology 2014;38:31–7. [Google Scholar]
- 18. Kebaili S, Chaabane K, Mnif MF, et al. Gonadal dysgenesis and the Mayer-Rokitansky-Kuster-Hauser Syndrome in a girl with a 46, XX karyotype: A case report and review of literature. Indian J Endocrinol Metab 2013;17:505 10.4103/2230-8210.111663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bousfiha N, Errarhay S, Saadi H, et al. Gonadal Dysgenesis 46, XX Associated with Mayer-Rokitansky-Kuster-Hauser Syndrome: One Case Report. Obstet Gynecol Int 2010;2010:1–3. 10.1155/2010/847370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Güven A, Kara N, Sağlam Y, et al. The Mayer–Rokitansky–Kuster–Hauser and gonadal dysgenesis anomaly in a girl with 45, X/46, X, del (X)(p11. 21). American Journal of Medical Genetics Part A 2008;146:128–31. [DOI] [PubMed] [Google Scholar]
- 21. Kumar A, Mishra S, Dogra PN. Management of an unusual case of atypical Mayer–Rokitansky–Kuster–Hauser syndrome, with unilateral gonadal agenesis, solitary ectopic pelvic kidney, and pelviureteric junction obstruction. Int Urogynecol J 2007;18:823–5. 10.1007/s00192-006-0238-z [DOI] [PubMed] [Google Scholar]
- 22. Gonadal dysgenesis associated with Mayer-Rokitansky-Küster-Hauser syndrome: a case report. Annales d’endocrinologie 2004. [DOI] [PubMed] [Google Scholar]
- 23. Plevraki E, Kita M, Goulis DG, et al. Bilateral ovarian agenesis and the presence of the testis-specific protein 1-Y-linked gene: two new features of Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril 2004;81:689–92. 10.1016/j.fertnstert.2003.07.029 [DOI] [PubMed] [Google Scholar]
- 24. Kaya H, Sezik M, Ozkaya O, et al. Mayer-Rokitansky-Küster-Hauser syndrome associated with unilateral gonadal agenesis. A case report. J Reprod Med 2003;48:902–4. [PubMed] [Google Scholar]
- 25. Aydos S, Tükün A, Bökesoy I. Gonadal dysgenesis and the Mayer-Rokitansky-Kuster-Hauser syndrome in a girl with 46,X,del(X)(pter-->q22:). Arch Gynecol Obstet 2003;267:173–4. 10.1007/s00404-001-0274-3 [DOI] [PubMed] [Google Scholar]
- 26. Ting TC, Chang SP. Coexistence of gonadal dysgenesis and Mullerian agenesis with two mosaic cell lines 45,X/46,X,del(X)(p22.2). Zhonghua Yi Xue Za Zhi 2002;65:450–2. [PubMed] [Google Scholar]
- 27. Güitrón-Cantú A, López-Vera E, Forsbach-Sánchez G, et al. Gonadal dysgenesis and Rokitansky syndrome. A case report. J Reprod Med 1999;44:891–3. [PubMed] [Google Scholar]
- 28. Oyer CE, Ramos D, Shoji T, et al. 46,XX gonadal agenesis in a neonate with multiple congenital anomalies: case report and review of the literature. Pediatr Pathol 1994;14:967–72. 10.3109/15513819409037693 [DOI] [PubMed] [Google Scholar]
- 29. Aughton DJ. Müllerian duct abnormalities and galactosaemia heterozygosity: report of a family. Clin Dysmorphol 1993;2:55–61. [PubMed] [Google Scholar]
- 30. Alper MM, Garner PR, Spence JE. Coexistence of gonadal dysgenesis and uterine aplasia. A case report. J Reprod Med 1985;30:232–4. [PubMed] [Google Scholar]
- 31. De Leon FD, Hersh JH, Sanfilippo JS, et al. Gonadal and müllerian duct agenesis in a girl with 46,X,i(Xq). Obstet Gynecol 1984;63(3 Suppl):81S–3. [PubMed] [Google Scholar]
- 32. Levinson G, Zárate A, Guzmán-Toledano R, et al. An XX female with sexual infantilism, absent gonads, and lack of Müllerian ducts. J Med Genet 1976;13:68–9. 10.1136/jmg.13.1.68 [DOI] [PMC free article] [PubMed] [Google Scholar]