

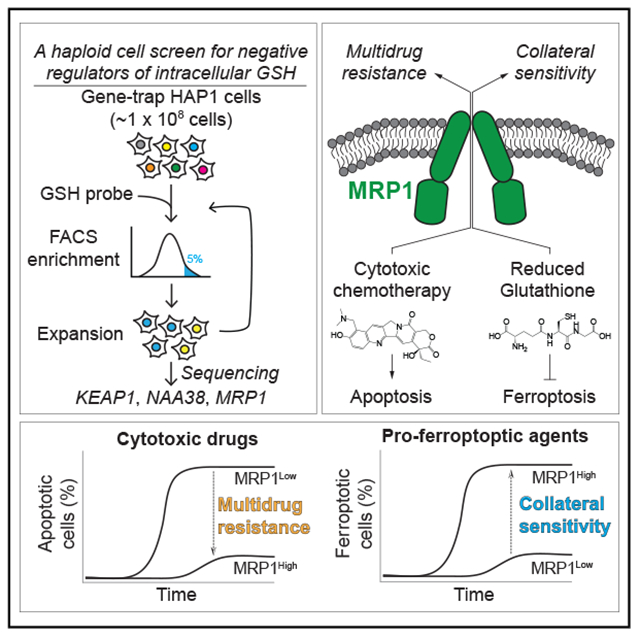
SUMMARY
The tripeptide glutathione suppresses the iron-dependent, non-apoptotic cell death process of ferroptosis. How glutathione abundance is regulated in the cell and how this regulation alters ferroptosis sensitivity is poorly understood. Using genome-wide human haploid genetic screening technology coupled to fluorescence-activated cell sorting (FACS), we directly identify genes that regulate intracellular glutathione abundance and characterize their role in ferroptosis regulation. Disruption of the ATP binding cassette (ABC)-family transporter multidrug resistance protein 1 (MRP1) prevents glutathione efflux from the cell and strongly inhibits ferroptosis. High levels of MRP1 expression decrease sensitivity to certain pro-apoptotic chemotherapeutic drugs, while collaterally sensitizing to all tested pro-ferroptotic agents. By contrast, disruption of KEAP1 and NAA38, leading to the stabilization of the transcription factor NRF2, increases glutathione levels but only weakly protects from ferroptosis. This is due in part to concomitant NRF2-mediated upregulation of MRP1. These results pinpoint glutathione efflux as an unanticipated regulator of ferroptosis sensitivity.
Graphical Abstract
In Brief
Glutathione suppresses the non-apoptotic cell death process of ferroptosis. Using genome-wide human haploid cell mutagenesis and FACS-based detection, Cao et al. identify negative regulators of intracellular glutathione abundance that affect glutathione efflux and NRF2 protein levels, altering ferroptosis sensitivity.
INTRODUCTION
Cellular metabolism supports the growth, proliferation, and survival of all cells and is altered in diseases such as cancer (Beadle and Tatum, 1941; DeBerardinis and Thompson, 2012; Vander Heiden and DeBerardinis, 2017). The intracellular abundance of specific metabolites is regulated by the expression and activity levels of enzymes and transport proteins (Ducker and Rabinowitz, 2017; Metallo and Vander Heiden, 2013). Genome-wide gene deletion analysis in the single-celled eukaryote Saccharomyces cerevisiae has identified hundreds of genes that can regulate the abundance of individual metabolites (Cooper et al., 2010; Mülleder et al., 2016). Human haploid cell genetic screening technology has recently been developed and applied to identify regulators of viral entry, cell death, and other processes (Carette et al., 2011a, 2011b; Dixon et al., 2015; Dovey et al., 2018). We envisioned that this technology could be combined with a metabolite-specific fluorescent reporter and fluorescence-activated cell sorting (FACS) to identify genes that regulate metabolite abundance in human cells. As proof-of-concept, we focused in this work on genes regulating the abundance of glutathione, an essential intracellular thiol-containing tripeptide.
Glutathione functions as an electron donor or acceptor by cycling between reduced (GSH) and oxidized (GSSG) forms and is important for xenobiotic detoxification, protein folding, antioxidant defense, and other processes (Deponte, 2013). As such, glutathione is especially important for the growth and survival of many cancer cells in vitro and in vivo (Harris et al., 2015; Lien et al., 2016; Piskounova et al., 2015). When intracellular GSH levels drop below a critical threshold, the GSH-dependent lipid hydroperoxidase glutathione peroxidase 4 (GPX4) cannot function, which can lead to a fatal buildup of lipid reactive oxygen species (ROS) and cell death via the iron-dependent, non-apoptotic process of ferroptosis (Dixon et al., 2012; Ingold et al., 2018; Yang et al., 2014). De novo GSH synthesis requires cysteine, which is typically found outside cells in the oxidized form as cystine. Small molecule inhibitors of cystine import via the cystine/glutamate antiporter system xc−, such as erastin, cause GSH depletion, lipid ROS accumulation, and ferroptosis induction (Dixon et al., 2012, 2014). Whether inhibition of de novo GSH synthesis alone accounts for the rapid induction of ferroptosis following system xc− inhibition, or whether other mechanisms contribute to GSH depletion is unclear.
Here, using genome-wide human haploid cell genetic screening, we identify negative regulators of intracellular glutathione levels that also alter ferroptosis sensitivity, including multidrug resistance protein 1 (MRP1), whose disruption reduces glutathione efflux from the cell (Cole, 2014a). High levels of MRP1-mediated glutathione efflux promote multidrug resistance and collaterally sensitize cancer cells to ferroptosis-inducing agents. Increased expression of the NRF2 antioxidant transcription factor can also elevate intracellular glutathione but has weak effects on ferroptosis sensitivity, in part because NRF2 upregulates MRP1 expression and therefore simultaneously increases both GSH synthesis and efflux.
RESULTS
A Genome-wide Screen for Negative Regulators of Intracellular GSH Abundance
We sought to identify genes that regulate glutathione abundance in human HAP1 haploid cells using the GSH probe monochlorobimane (MCB) (Figure S1A) and FACS technology. In HAP1 cells, the levels of intracellular GSH detected with MCB using flow cytometry correlated closely with the levels of total glutathione (GSH + GSSG) detected using a traditional biochemical method, Ellman’s reagent (Figures S1B and S1C). Thus, most glutathione within HAP1 cells is in the reduced form and susceptible to MCB labeling.
To identify negative regulators of glutathione abundance, a starting pool of ~100 million randomly mutagenized HAP1 cells was labeled with MCB and those with the highest (top 5%) MCB signal were isolated using FACS. These cells were expanded in culture for 3 days, and the same FACS-based selection process was repeated a second time. This isolated population was expanded in culture for 5 days and then the sites of gene-trap insertion were determined by deep sequencing (Figure 1A). Using a stringent statistical threshold (false-discovery rate [FDR]-corrected p < 0.001), we identified five candidate genes that were significantly enriched for independent gene-trap insertions over the control (unsorted) population: KEAP1 (p = 4.6 × 10−7), ABCC1 (p = 1 × 10−6), GSTO1 (p = 8.9 × 10−4), SETD5 (p = 1.8 × 10−3), and NAA38 (p = 3 × 10−3) Figures 1B and S1D). KEAP1 (kelch-like ECH associated protein 1), ABCC1 (encoding MRP1), and GSTO1 (glutathione S-transferase omega 1) were previously linked to glutathione metabolism: KEAP1 negatively regulates the accumulation of the “antioxidant” transcription factor nuclear factor erythroid 2-like 2 (NFE2L2, NRF2); MRP1 effluxes GSH, GSSG, and glutathione S-conjugates from the cell; and GSTO1 regulates protein glutathionylation (Cole and Deeley, 2006; Cole et al., 1990; Itoh et al., 1999; Marchan et al., 2008; Menon and Board, 2013; Sayin et al., 2017). SETD5 and NAA38, encoding a putative methyltransferase and a component of the NatC N-terminal acetylation complex, respectively, had not previously been associated with the regulation of glutathione abundance.
Figure 1. A Genetic Screen Identifies Negative Regulators of Intracellular GSH Abundance.
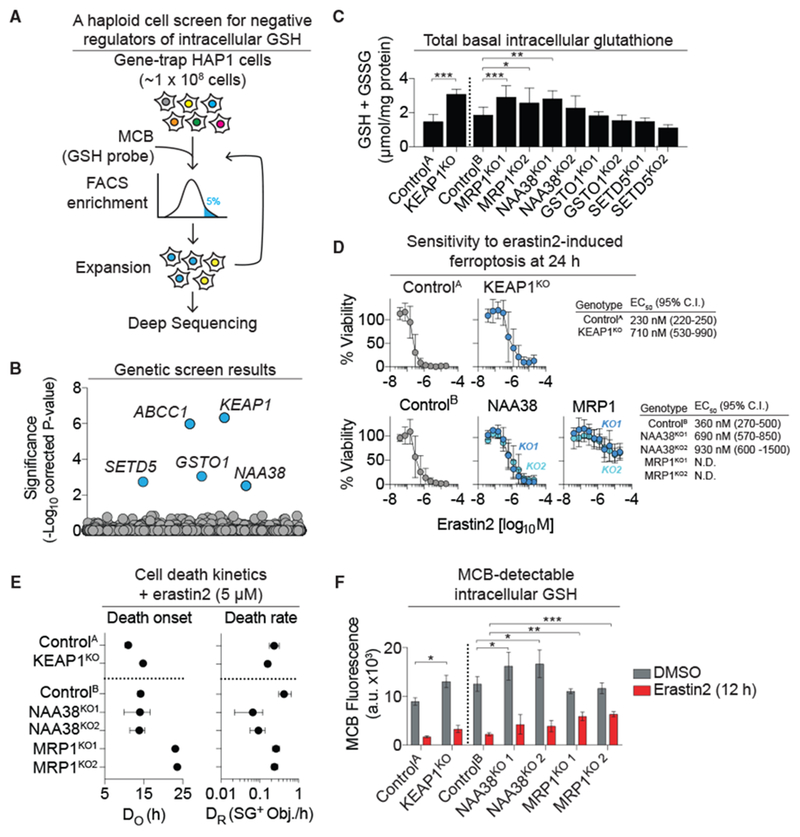
(A) Overview of the human haploid cell genetic screen for negative regulators of intracellular reduced glutathione (GSH). MCB, monochlorobimane.
(B) Gene-level enrichment summary plot for the screen in (A).
(C) Total basal intracellular glutathione levels determined using Ellman’s reagent in unmodified (ControlA/B) and CRISPR-Cas9 gene-disrupted (KO) HAP1 cell lines.
(D) Erastin2 potency determined using PrestoBlue. Cell viability is normalized to DMSO-treated controls (100%). N.D., not determinable.
(E) Population cell death kinetics determined from the analysis of dead cell counts (SYTOX Green positive [SG+] objects) over time followed by curve fitting and parameter value extraction. DO, death onset; DR, maximal death rate.
(F) MCB-detectable GSH levels ± erastin2 (5 μM) determined by flow cytometry.
Data in (C)–(F) were from three independent experiments and represent means ± SDs (C and F) or means ± 95% confidence intervals (D and E). In (C) and (F), data were analyzed using one-way ANOVA, with *p < 0.05, **p < 0.01, and ***p < 0.001.
To validate our primary screening results, we examined total glutathione (GSH + GSSG) levels using Ellman’s reagent and a microplate-based detection assay. A CRISPR-Cas9 gene-disrupted (“KO”) cell line lacking KEAP1 expression (i.e., KEAP1KO) and its paired control (ControlA) were obtained commercially. Separately, we generated two independent clonal gene-disrupted cell lines targeting the ABCC1/MRP1, NAA38, GSTO1, and SETD5 genes, using CRISPR-Cas9 technology. We also isolated an independent control cell line (ControlB) that underwent the CRISPR protocol but was unmodified. Consistent with the results obtained in the primary screen, intracellular total glutathione (GSH + GSSG) levels were significantly elevated in KEAP1KO, NAA38KO1, and both MRP1KO1/2 cell lines relative to the respective controls (Figure 1C; note that NAA38KO2 just missed the cutoff for statistical significance).
We unexpectedly found that total glutathione levels were not elevated in GSTO1KO1/2 or SETD5KO1/2 cells relative to ControlB cells (Figure 1C). It is possible that GSTO1 and SETD5 were false-positives in the original screen, or that the impact of these two genes on GSH levels may only be apparent in response to the stress of flow cytometry (Llufrio et al., 2018). Regardless, KEAP1, NAA38, and ABCC1/MRP1 were confirmed as negative regulators of intracellular GSH levels in HAP1 cells.
Regulators of Total Glutathione Levels Affect Ferroptosis Sensitivity
Intracellular GSH inhibits ferroptosis (Dixon et al., 2014; Yang et al., 2014). We therefore hypothesized that disruption of KEAP1, NAA38, and ABCC1/MRP1, leading to increased intracellular GSH, would reduce ferroptosis sensitivity. We confirmed that HAP1 cells were sensitive to erastin2, a potent system xc− inhibitor and ferroptosis-inducing agent (Dixon et al., 2014), and that erastin2-induced cell death was inhibited by co-treatment with the lipophilic radical-trapping antioxidant ferrostatin-1 or the iron chelator deferoxamine (DFO), but not the pan-caspase inhibitor Q-VD-OPh (Figure S2A). Both KEAP1KO and NAA38KO1/2 cells were less sensitive to erastin2-induced ferroptosis, but with only modest (~2- to 3-fold) shifts in potency relative to their respective control cell lines (Figures 1D and S2B). Compared to these cell lines, MRP1KO1/2 cells were far more resistant to erastin2 treatment, such that it was not possible to compute an EC50 value at 24 h (Figures 1D and S2B). The potency of erastin2 was not significantly altered in either GSTO1KO1 or SETD5KO1 cells relative to ControlB (Figure S2C), consistent with the observation that intracellular glutathione levels did not differ in these cells compared to the control cell line (Figure 1C).
We recently demonstrated that the initial onset of cell death and the maximal rate of cell death within a population can vary substantially between lethal stimuli (Forcina et al., 2017). These kinetic parameters can be determined using time-lapse imaging and curve fitting to the observed counts of live and/or dead cells within a population of cells over time. In the context of erastin2-induced ferroptosis, the onset of cell death correlates with the oxidation of the lipid peroxide-sensitive probe C11 BODIPY 581/591 (C11) specifically at the plasma membrane, linking the onset of cell death to a key terminal marker of the ferroptotic process (Figures S2D–S2F) (Magtanong et al., 2019). We therefore examined changes in cell death kinetics in response to erastin2 treatment in our control and gene-disrupted cell lines. HAP1 ControlA/B, KEAP1KO, NAA38KO1/2, and MRP1KO1/2 cells were treated with erastin2, and cell death within the population was monitored over time by counting the number of SYTOX Green+ (SG+) cells every 4 h for 48 h. Counts of SG+ objects over time for each cell line were used to compute the timing of cell death onset (DO) and maximal cell death rate (DR) within each population in response to erastin2. KEAP1KO cells treated with erastin2 (5 μM) exhibited a slight delay in the timing of cell death onset and a reduction in the maximal rate of cell death compared to ControlA cells, while NAA38KO1/2 cells had no change in the timing of cell death onset and a pronounced reduction in the maximal rate of cell death compared to ControlB (Figures 1E and S2G). The timing of cell death onset in MRP1KO1/2 cells was significantly delayed relative to ControlB cells (23 versus 14 h); however, once cell death was initiated, the maximal rate did not differ (Figures 1E and S2G).
We next examined whether these differences in cell death kinetics correlated with changes in GSH abundance basally or in response to erastin2. To do so, we measured MCB-detectable GSH pools in KEAP1KO, NAA38KO1/2, and MRP1KO1/2 cells treated with vehicle (DMSO) or erastin2 (5 μM) for 12 h. KEAP1KO and NAA38KO1/2 cells exhibited a basal increase in intracellular MCB-detectable GSH levels relative to control cells, but MCB-detectable GSH was depleted to a similar extent in these mutants relative to their respective controls following 12 h of erastin2 treatment (Figure 1F). Basal MCB-detectable GSH levels were not elevated in MRP1KO1/2 cells relative to ControlB cells, but erastin2 treatment caused significantly less apparent GSH depletion (Figure 1F). We infer that these cells are protected against ferroptosis due to increased retention of intracellular glutathione. Of note, GPX4 expression was similar in ControlA/B, KEAP1KO, NAA38KO1/2, and MRP1KO1/2 cell lines, demonstrating that the loss of these proteins did not result in compensatory changes in the expression of this key anti-ferroptotic protein (Figure S2H).
Basal MCB-detectable GSH was not increased in MRP1KO1/2 cells in these experiments, despite being isolated in the primary screen using this reagent and despite higher total (GSH + GSSG) levels detected using Ellman’s reagent (Figure 1C). One possibility was that the effects of MRP1 deletion on MCB-detectable GSH was amplified by the prolonged (~1 h) incubation of cells under detached conditions on ice during the genome-wide FACS sort, compared to the more rapid analysis of cells in the small-scale experiments (<5 min). Alternatively, or in parallel, some portion of the basal intracellular GSH pool may normally be inaccessible to MCB in cells lacking MRP1, at least under the conditions examined here in the small-scale experiments. Consistent with this latter possibility, when we examined GSH levels using a recently developed ratiometric GSH probe that spontaneously reacts with GSH (RealThiol [RT]) (Jiang et al., 2017), we observed that MRP1KO1/2 cells exhibited higher basal GSH levels compared to ControlB cells (Figure S2I). Thus, by two independent measures, loss of MRP1 increased the basal abundance of intracellular GSH within the cell through a mechanism that appeared to be distinct from that resulting from the loss of KEAP1 or NAA38, as inferred from the kinetic analysis of cell death over time.
MRP1-Dependent Glutathione Efflux Promotes Ferroptosis
MRP1 can efflux glutathione and glutathione S conjugates from the cell (Cole, 2014a), and we examined whether MRP1 could perform this function in HAP1 cells. HAP1 MRP1KO1 cells were reconstituted via lentiviral transduction with an empty vector control, wild-type MRP1 or MRP1K322L, a point mutant that is defective in the export of glutathione S-conjugates (Haimeur et al., 2002; Maeno et al., 2009) (Figure 2A). Relative to ControlB cells, MRP1KO1 cells reconstituted with empty vector or MRP1K322L had elevated levels of total (GSH + GSSG) intracellular glutathione, while total glutathione levels in MRP1KO1 cells reconstituted with wild-type MRP1 were similar to those observed in ControlB cells (Figure 2B). Glutathione efflux can be stimulated by placing cells in Dulbecco’s PBS (DPBS) (Sagara et al., 1996). In response to DPBS, MRP1KO1 cells reconstituted with empty vector or MRP1K322L effluxed less total glutathione than ControlB cells and MRP1KO1 cells reconstituted with wild-type MRP1 (Figure 2C). Thus, MRP1 can efflux glutathione from HAP1 cells.
Figure 2. MRP1 Expression Regulates Basal Glutathione Levels and Ferroptosis Sensitivity.
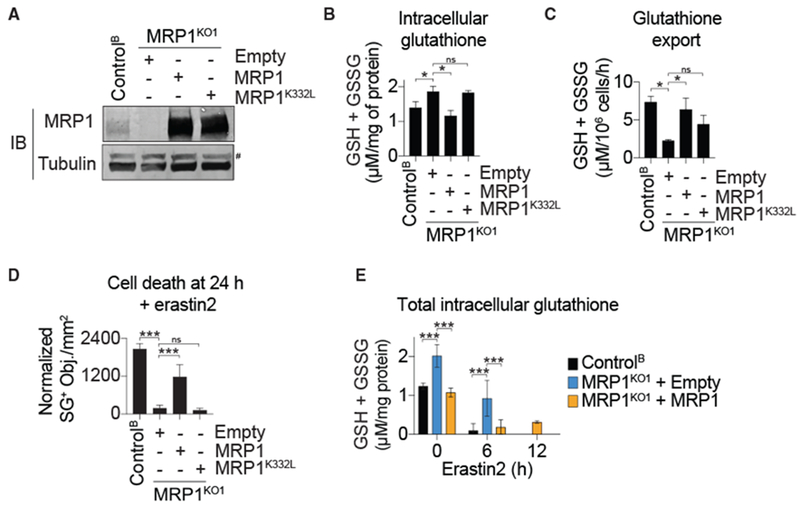
(A) MRP1 protein levels in HAP1 Control and MRP1KO cells stably complemented with wild-type MRP1, the glutathione-export defective MRP1K332L mutant, or empty vector control (Empty). #, non-specific band.
(B) Total intracellular glutathione determined using Ellman’s reagent.
(C) DPBS-stimulated glutathione export measured using Ellman’s reagent.
(D) Dead cell (SG+) counts following erastin2 treatment (5 μM).
(E) Intracellular glutathione over time following the addition of erastin2 (5 μM).
Data in (B)–(E) represent means ± SDs from three independent experiments. Data were analyzed using one-way ANOVA, with *p < 0.05 and ***p < 0.001; ns, not significant.
We next examined whether MRP1-mediated glutathione efflux promoted ferroptosis. Ferroptosis-resistant MRP1KO1 cells reconstituted with wild-type MRP1 but not MRP1K322L were resensitized to erastin2-induced cell death (Figures 2D and S3A). Moreover, MRP1KO1 cells reconstituted with empty vector exhibited greater total glutathione (GSH + GSSG) retention over time following erastin2 (5 μM) treatment compared to both Control and MRP1KO1 cells reconstituted with wild-type MRP1 (Figure 2E). Thus, MRP1-mediated glutathione efflux appeared to promote ferroptosis sensitivity in HAP1 cells.
To investigate further how MRP1-mediated glutathione efflux modulates ferroptosis sensitivity and intracellular glutathione levels, we transduced H1299 non-small-cell lung carcinoma (NSCLC) and U-2 OS osteosarcoma cells with a control (empty vector) lentivirus or lentivirus directing the expression of MRP1 or MRP1K332L (Figure 3A). Consistent with results obtained in HAP1 cells, overexpression of wild-type MRP1 but not MRP1K332L reduced intracellular total glutathione (GSH + GSSG) levels and increased DPBS-stimulated total glutathione export relative to empty vector control cell lines, with stronger effects in H1299 than U-2 OS cells (Figures 3A and 3B). In both cell lines, overexpression of wild-type MRP1 but not MRP1K332L accelerated the onset of ferroptosis induced by erastin2 treatment or cystine deprivation (Figure 3C). Cell death in both empty vector control and MRP1-overexpressing cells was completely suppressed by ferrostatin-1 (Fer-1) and DFO, but not by the pan-caspase inhibitor Q-VD-OPh, confirming that MRP1 expression accelerated the induction of ferroptosis and not a different mode of cell death (Figures 3D, S3B, and S3C).
Figure 3. MRP1 Overexpression Sensitizes to Ferroptosis.
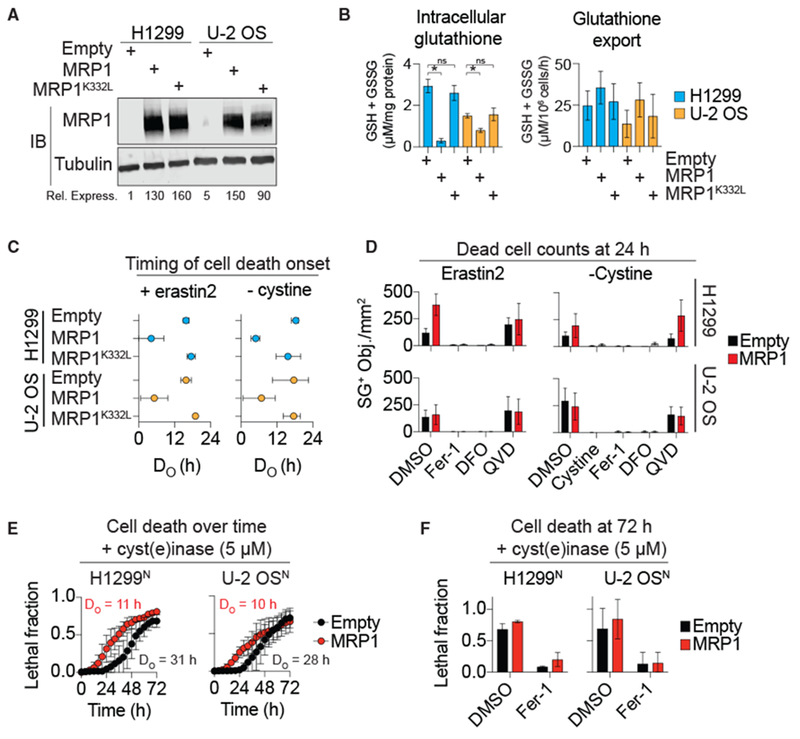
(A) MRP1 protein levels in H1299 or U-2 OS cells stably overexpressing empty vector control (Empty), wild-type MRP1, or MRP1 K332L. Rel. Express., quantification of the relative expression level of MRP1, normalized to tubulin. MRP1 levels in H1299 Empty cells set equal to 1.
(B) Total intracellular glutathione and DPBS-stimulated glutathione export determined using Ellman’s reagent. Data were analyzed using a one-way ANOVA with *p < 0.05; ns, not significant.
(C) The timing of population cell death onset (DO) determined by counting SYTOX Green+ dead cells over time followed by curve fitting and parameter value extraction. Data represent means ± 95% confidence intervals.
(D) SYTOX Green+ dead cells with erastin2 (5 μM) or cystine-free medium ± ferrostatin-1 (Fer-1,2 μM), deferoxamine (DFO) (100 μM), Q-VD-OPh (QVD, 25 μM), or cystine (200 μM).
(E) Population cell death (lethal fraction) over time determined using the STACK method.
(F) Population cell death at 72 h determined using STACK. Fer-1 (2 μM).
Data in (B) and (D)–(F) represent means ± SDs for three independent experiments.
New agents capable of inducing ferroptosis in vivo have recently been developed, including the engineered cystine/cysteine-degrading enzyme cyst(e)inase (Cramer et al., 2017). With a view to future in vivo studies, we examined whether MRP1 expression increased sensitivity to cyst(e)inase-induced cell death. For these experiments, we examined cell death in H1299 and U-2 OS cell lines expressing the live cell marker nuclear-mKate2 (denoted by a superscript “N“). This enabled us to use the sensitive scalable time-lapse analysis of cell death kinetics (STACK) approach, in which the fraction of dead cells in the total population (i.e., lethal fraction) is determined by the direct observation of live and dead cells over time (Forcina et al., 2017). Consistent with expectations, overexpression of MRP1 accelerated the onset of cyst(e)inase-induced cell death compared to empty vector controls by >15 h in both H1299 and U-2 OS cells (Figure 3E). This acceleration of cell death onset was not due to the induction of a different mode of cell death, as in both MRP1-overexpressing and empty vector control cell lines cyst(e)inase-induced cell death was suppressed by co-treatment with Fer-1 (Figure 3F). Thus, MRP1-mediated glutathione efflux promotes ferroptosis in response to various ferroptosis-inducing conditions.
MRP1 Expression Collaterally Sensitizes Cells to Ferroptosis
Genetic alterations that decrease sensitivity to one lethal agent while simultaneously increasing sensitivity to another are said to cause collateral sensitization (Lorendeau et al., 2017; Szybalski and Bryson, 1952). We predicted that MRP1, which can efflux a number of conventional chemotherapeutics in a glutathione-dependent manner (Cole, 2014a; Franco and Cidlowski, 2006; Hammond et al., 2007; Trompier et al., 2004), would simultaneously collaterally sensitize cells to ferroptosis-inducing agents. Consistent with this prediction, overexpression of MRP1 reduced the sensitivity of both H1299 and U-2 OS cells to the lethal effects of the MRP1 substrates vincristine and doxorubicin and increased sensitivity to the lethal effects of the system xc− inhibitor erastin2, the GPX4 inhibitors RSL3 and ML162, and the de novo GSH synthesis inhibitor buthionine sulfoximine (BSO), in some cases by >100-fold (Figure 4A; Table 1). The lethal potencies of the proteasome inhibitor bortezomib and the sarco/endoplasmic reticulum Ca2+-ATPase pump inhibitor thapsigargin were not altered by MRP1 overexpression, suggesting that this transporter does not non-specifically alter cell death sensitivity in these cells (Figure 4A; Table 1).
Figure 4. MRP1 Expression Collaterally Sensitizes to Ferroptosis.
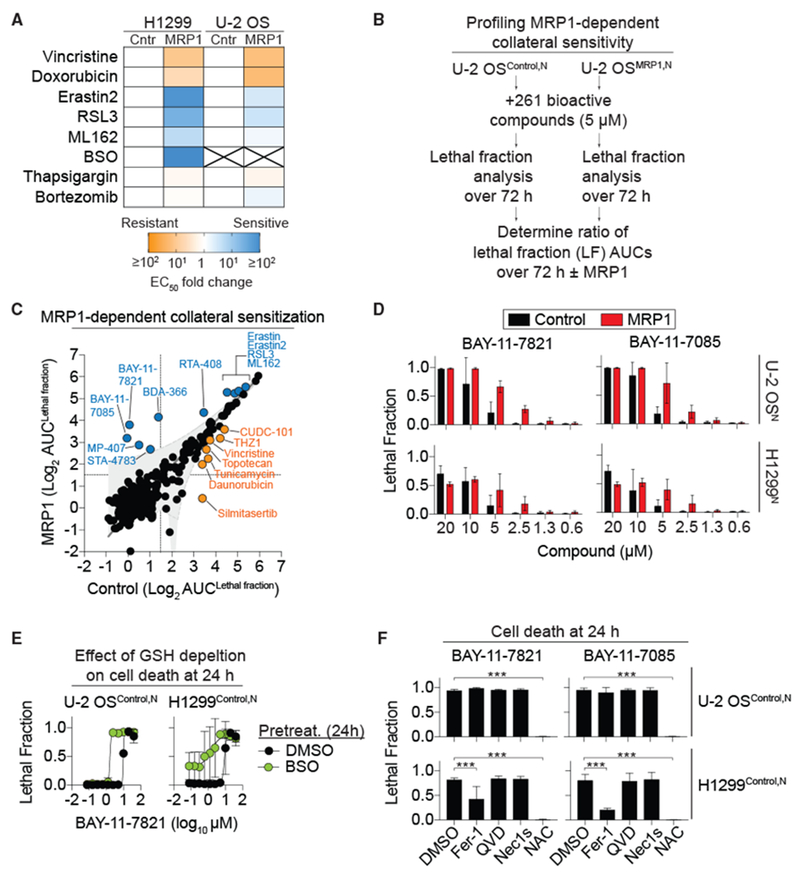
(A) Summary of lethal compound EC50 fold changes for control (Cntr) and MRP1-overexpressing (MRP1) cells. X, EC50 values could not be computed. See also Table 1.
(B) Outline of the collateral sensitivity profiling experiment in Control and MRP1-overexpressing cells in response to 261 bioactive compounds using STACK.
(C) Results of the comparative analysis of cell death ± MRP1. Each dot represents a single compound. MRP1 overexpression had no effect (black), reduced sensitivity (yellow), or increased sensitivity (blue) to compound-induced cell death.
(D) Cell death at 48 h in Control and MRP1-overexpressing cells determined using STACK.
(E) BAY-11-7821-induced cell death determined using STACK ± buthionine sulfoximine (BSO) pretreatment (200 μM, 24 h).
(F) Cell death determined using STACK. Conditions were: BAY-11-7821 or BAY-11-7085 (both 20 μM) ± Fer-1 (2 μM), Q-VD-OPh (25 μM), Nec-1 s (1 μM), or N-acetylcysteine (NAC, 1 mM). Data were analyzed by one-way ANOVA with ***p < 0.001.
Data in (D)–(F) represent means ± SDs for three independent experiments.
Table 1.
Lethal Compound Potency in Control and MRP1-Overexpressing Cells
| Compound | H1299Control | H1299MRP1 | U-2 OSControl | U-2 OSMRP1 |
|---|---|---|---|---|
| Vincristine | 2.8 (1.9–4.2) nM | 32 (19–54) nM | 3.3 (1.4–8.2) nM | 53 (18–65) nM |
| Doxorubicin | 0.2 (0.1–0.3) μM | 1.0 (0.8–1.4) μM | 0.2 (0.1–0.3) μM | 3.7 (1.9–13) μM |
| Erastin2 | 80 (65–105) nM | 1.1 (0.1–12) nM | 130 (100–170) nM | 47 (20–77) nM |
| RSL3 | 140 (54–530) nM | 0.055 (0.015–0.19) nM | 5.9 (4.8–7.2) μM | 1.8 (0.8–42) μM |
| ML162 | 140 (46–870) nM | 0.3 (0.1–0.6) nM | 4.9 (4.4–5.5) μM | 3.4 (2.6–4.6) μM |
| BSO | >20 mM | 83 (67–100) nM | N.D. | N.D. |
| Thapsigargin | 3.7 (3.4–4.1) nM | 5.4 (2.4–11) nM | 1.4 (0.9–2.0) nM | 2.3 (0.4–4.1) nM |
| Bortezomib | 21 (12–47) nM | 25 (16–43) nM | 12 (6.9–27) nM | 7.4 (4.6–14) nM |
For vincristine, doxorubicin, erastin2, RSL3, ML162, BSO, and thapsigargin treatments, cell viability was assayed using PrestoBlue. For bortezomib treatment, cell viability was assayed by counting of SYTOX Green+ dead cells. Erastin2, RSL3, ML162, and BSO effects were assayed after 48 h of treatment; doxorubicin and thapsigargin were assayed after 72 h of treatment. Vincristine was assayed after 48 or 72 h of treatment in H1299 and U-2 OS cells, respectively. Data represent means and 95% confidence intervals (in brackets). N.D., not determined.
MRP1 expression has been linked to enhanced sensitivity to a number of agents that disrupt intracellular thiol metabolism (Lorendeau et al., 2017). It has also been suggested that MRP1-mediated GSH efflux can contribute to apoptosis under certain conditions (Hammond et al., 2007). These findings led us to examine more broadly how MRP1 expression affected sensitivity to compound-induced cell death. Using STACK, we profiled cell death over time in empty vector (Control) and MRP1-overexpressing U-2 OSN cells treated with 261 structurally diverse bioactive compounds, each tested at a fixed concentration of 5 μM. Lethal fraction scores for each compound over 72 h were summarized as area under the curve (AUC) values and were averaged across three independent experiments, from which we pinpointed compounds whose lethality differed significantly as a function of MRP1 expression (see Method Details) (Figure 4C). The lethality of 244 of 261 compounds (93%) was not significantly altered by MRP1 expression, suggesting that in these cells, MRP1 expression had little effect on cell death induced by most compounds. Seven compounds were less lethal to MRP1-overexpressing cells compared to empty vector Control cells, including vincristine, CUDC-101, THZ1, and silmitasertib. Ten compounds were more lethal to MRP1-overexpressing cells compared to empty vector Control cells, including erastin, erastin2, RSL3, and ML162. Thus, MRP1 expression confers resistance to a small number of compounds while specifically collaterally sensitizing cells to canonical ferroptosis-inducing compounds and a select number of additional agents.
In addition to known ferroptosis-inducing compounds, our profiling identified two structurally related sulfonyl acrylonitriles, BAY-11-7821 and BAY-11-7085 (BAY compounds), that were more lethal to MRP1-overexpressing cells than Control cells (Figures 4C and 4D). BAY compounds are cysteine reactive and can form covalent adducts with a number of proteins in the cell (Pierce et al., 1997; Strickson et al., 2013; Yu et al., 2018). There is some evidence that these compounds may induce ferroptosis (Chang et al., 2018). Sensitivity to BAY-11–7821 was enhanced by pretreatment with the de novo GSH biosynthesis inhibitor BSO, suggesting that the lethality of this compound is opposed by GSH (Figure 4E). However, unlike erastin2, cell death induced by BAY-11-7821 and BAY-11-7085 was only partially (H1299N) or not at all (U-2 OSN) suppressed by co-treatment with the canonical ferroptosis inhibitors DFO or Fer-1 (Figures 4F and S4A). Oxidation of the lipid ROS-sensitive probe C11 at the plasma membrane is highly correlated with the induction of ferroptosis (Magtanong et al., 2019). We observed plasma membrane C11 oxidation in H1299 cells but not U-2 OS cells (Figure S4B), which is consistent with the inhibitor studies, suggesting that BAY compounds can induce a degree of ferroptosis in H1299 but not U-2 OS cells. These findings suggest that while cell death is enhanced by GSH depletion, BAY compounds kill cells mostly through a non-apoptotic cell death mechanism that is distinct from ferroptosis.
BAY compound lethality was not suppressed in either cell line by the pan-caspase inhibitor Q-VD-OPh or the receptor interacting protein kinase 1 inhibitor Nec-1 s, indicating that these compounds were unlikely to induce apoptosis or necroptosis. BAY compound-induced cell death was completely suppressed by the thiol N-acetylcysteine (NAC) (Figures 4F and S4A). NAC can form covalent adducts with, and thereby inactivate, BAY compounds (Strickson et al., 2013), possibly accounting for how they suppress BAY-induced cell death. We posit that MRP1-mediated glutathione efflux sensitizes to BAY compound-induced cell death by limiting thiol-mediated inactivation of these cysteine-reactive compounds.
NAA38 Controls NRF2 Stability and Function
ABCC1/MRP1 expression can be positively regulated by the antioxidant transcription factor NRF2 (Ji et al., 2013). KEAP1KO HAP1 cells exhibited a small but significant increase in MRP1 protein levels compared to ControlA cells (Figure 5A). We observed a similar small but significant increase in MRP1 levels in NAA38KO1/2 cells relative to ControlB cells (Figure 5A). Moreover, both KEAP1KO and NAA38KO1/2 cells had higher levels of NRF2 protein and increased expression of the NRF2 target genes NQO1, SLC7A11, and glutamate-cysteine ligase modifier subunit (GCLM) relative to their respective ControlA/B cell lines (Figures 5A and 5B). By contrast, NRF2 protein and NQO1, SLC7A11, and GCLM mRNA levels were not increased in MRP1KO1/2 cells relative to ControlB cells (Figures 5A and 5B). These results suggest that NAA38 disruption altered ferroptosis sensitivity through effects on NRF2 stability and the expression of glutathione biosynthetic genes.
Figure 5. Deletion of KEAP1 and NAA38 Stabilize NRF2 and Activate the NRF2 Pathway While Deletion of MRP1 Does Not.
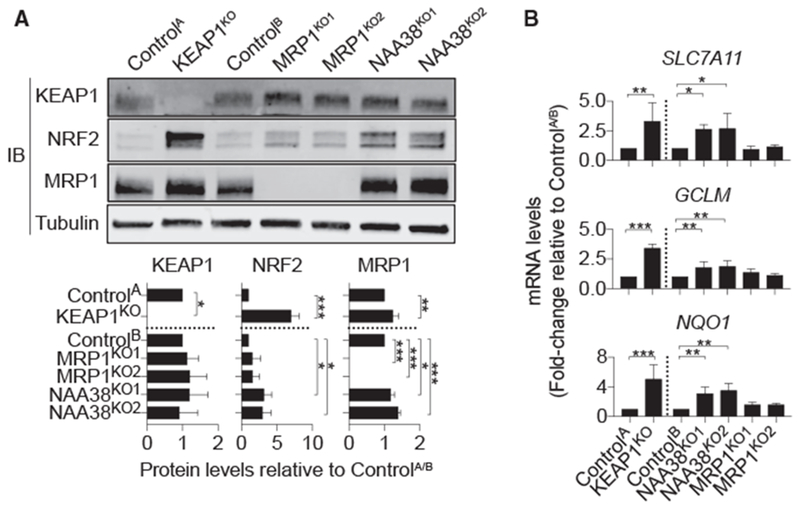
(A) Kelch-like ECH associated protein 1 (KEAP1), MRP1, and NRF2 protein levels in HAP1 Control and gene-disrupted cells. Quantification (mean intensity ± SD) is from three independent blots.
(B) Relative expression of NRF2 target genes in control and gene-disrupted cell lines.
Data are means ± SDs from three independent experiments. Data were analyzed using one-way ANOVA, with *p < 0.05 and ***p < 0.001; ns = not significant.
NRF2-Dependent MRP1 Regulation Balances Glutathione Synthesis with Efflux
NRF2 stabilization is reported to suppress ferroptosis (Fan et al., 2017; Roh et al., 2017; Sun et al., 2016). We were therefore surprised that NRF2 stabilization in HAP1 cells, due to disruption of KEAP1 or NAA38, only weakly inhibited ferroptosis (Figures 1D, 1E, and 5A). By contrast, disruption of MRP1, which had no effect on NRF2 expression, inhibited ferroptosis more potently (Figures 1D, 1E, and 5A). This led us to hypothesize that glutathione efflux via MRP1 limited the ability of NRF2 to protect cells from ferroptosis, despite increasing GSH synthesis.
To begin exploring this hypothesis, we first examined GCLM and MRP1 protein expression and glutathione levels in human tumor cell lines with high and low basal NRF2 expression. A549 NSCLC and T98G glioblastoma cells express high levels of NRF2 due to the inactivation of KEAP1 (Ma et al., 2012). By contrast, H1299 NSCLC, U-2 OS osteosarcoma, HT-1080 fibrosarcoma, and HAP1 cells express low basal NRF2 levels (Figure 6A). High NRF2 expression correlated with increased GCLM and MRP1 protein levels, as expected (Figure 6B). Basal total intracellular glutathione levels (GSH + GSSG) were also positively correlated with NRF2 and GCLM levels (Figure 6C). High levels of MRP1 expression were also positively correlated with total intracellular glutathione, indicating that NRF2 can significantly increase basal intracellular glutathione levels despite enhanced MRP1 expression (Figure 6C).
Figure 6. NRF2 and MRP1 Regulate Intracellular Glutathione Levels and Ferroptosis Sensitivity in Distinct Ways.
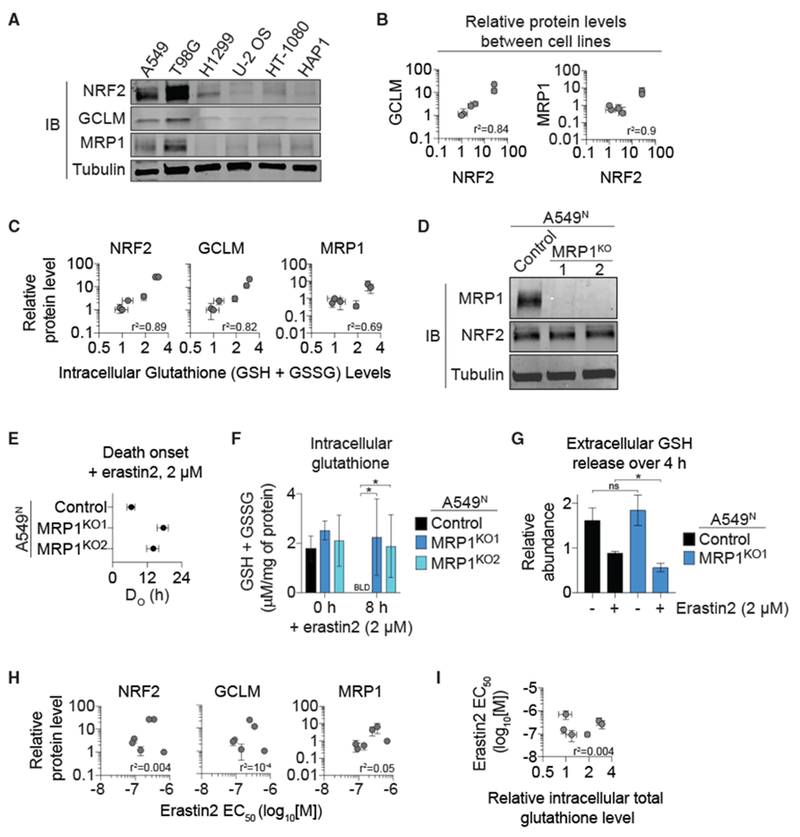
(A) NRF2, GCLM, and MRP1 protein levels in six cancer cell lines of diverse tissue origin.
(B) Pearson correlation of NRF2 protein levels to GCLM and MRP1 protein levels in the cell lines from (A). Protein expression was determined from three independent experiments and the levels of each protein normalized to those observed in HAP1 cells, which was set equal to 1.
(C) Pearson correlation of NRF2, GCLM, and MRP1 protein expression to intracellular glutathione levels in these cell lines.
(D) Immunoblotting of lysates from A549N Control and MRP1KO1/2 cells using antibodies against MRP1, NRF2, and α-tubulin.
(E) The timing of population cell death onset (DO) determined in A549N Control and MRP1KO1/2 cells using STACK.
(F) Intracellular glutathione levels determined using Ellman’s reagent. BLD, below the limit of detection.
(G) Extracellular GSH release determined by stable isotope tracing and mass spectrometry.
(H) Pearson correlation between NRF2, GCLM, or MRP1 protein levels and erastin2 potency in the six cell lines from (A).
(I) Pearson correlation between erastin2 potency and intracellular glutathione levels in the six cell lines from (A).
Data are means ± 95% confidence intervals (B, C, E, H, and I) or means ± SDs (F and G) from three independent experiments. Data in (F) and (G) were analyzed using one-way ANOVA, with *p < 0.05; ns, not significant.
Given these results, we examined in A549 cells whether MRP1 disruption affected ferroptosis sensitivity and glutathione metabolism when challenged with the deprivation of the rate-limiting GSH biosynthetic precursor cysteine. Using CRISPR-Cas9 technology, we generated A549N cell lines lacking MRP1 expression (Figure 6D). Compared to an unmodified Control cell line, MRP1KO1/2 cells were less sensitive to the induction of erastin2-induced ferroptosis, and this correlated with the greater retention of total intracellular glutathione (GSH + GSSG) over time and less GSH efflux into the growth medium, as detected using stable isotope tracing and mass spectrometry (Figures 6E-6G). GSSG was not detected in the medium from either Control or MRP1KO1 cells in this experiment, suggesting that it may not be a transport substrate in these cells (data not shown). Of note, basal GSH efflux was not altered in the absence of MRP1, suggesting that other transporters may also contribute to this process in parallel to MRP1.
Thus, while increased NRF2 expression is associated with the accumulation of high basal intracellular glutathione levels, it is also associated with increased MRP1-driven GSH efflux, limiting the ability of cells to retain this metabolite when de novo synthesis is halted. Given this observation, we hypothesized that the expression of NRF2 protein and NRF2 target gene products, as well as basal intracellular glutathione levels, would be poor predictors of ferroptosis sensitivity. Consistent with this hypothesis, sensitivity to erastin2-induced ferroptosis was not significantly correlated with NRF2, GCLM, or MRP1 protein levels across the panel of six cell lines examined here (Figure 6H). Furthermore, the potency of erastin2 was not correlated with basal intracellular glutathione levels in these cells (Figure 6I). Thus, NRF2 pathway activity and intracellular total glutathione levels are poor predictors of ferroptotic sensitivity, possibly in part due to the pro-ferroptotic effect of MRP1-mediated glutathione efflux.
DISCUSSION
The regulation of metabolite abundance is poorly understood, especially in mammalian cells (Metallo and Vander Heiden, 2013). This work provides a proof-of-concept that fluorescent probes, FACS, and genome-wide haploid cell retroviral mutagenesis can be combined to directly identify regulators of metabolite abundance. Given the unbiased nature of this approach, it could be applied to any metabolite for which a suitable reporter is available. Our study also suggests the importance of confirming primary screening results using orthogonal assays. One limitation of the FACS-based genome-wide analysis is that it requires prolonged incubation of the large cell population under non-standard conditions followed by passage through the FACS instrument, which itself likely alters cellular metabolism (Llufrio et al., 2018). Importantly, the results obtained here using MCB to detect intracellular GSH in the genome-wide FACS screen were validated in follow-up experiments by traditional biochemical methods and an additional GSH-sensitive probe.
We identified three genes–KEAP1, NAA38, and ABCC1/MRP1–as negative regulators of glutathione abundance in human cells. Like KEAP1, NAA38 appears to regulate NRF2 stability, as NAA38 deletion increased NRF2 protein levels and the expression of NRF2 target genes. NAA38 is a component of the NatC N-terminal acetylation complex (Aksnes et al., 2015), which acetylates protein N-termini with a broad range of sequences (Van Damme et al., 2016). N-terminal acetylation can be a signal for proteasomal degradation via the Ac/N-end rule pathway (Drazic et al., 2016). Since KEAP1 protein levels were unchanged in NAA38KO1/2 cells relative to Control cells, it is unlikely that NAA38 affects NRF2 stability by reducing KEAP1 levels. An alternative possibility requiring further study is that NAA38-dependent NatC complex activity destabilizes NRF2 directly via N-terminal acetylation.
Our results suggest a role for GSH efflux in the regulation of ferroptosis sensitivity. Unlike deletion of KEAP1 and NAA38, which enhance NRF2 protein levels and the expression of GSH biosynthetic genes, ABCC1/MRP1 deletion increases the size of the basal intracellular glutathione pool and/or slows the depletion of intracellular glutathione following the inhibition of de novo GSH synthesis. Either effect would enable the GSH-dependent pro-survival activity of GPX4 to be sustained over a longer period. Of note, the KM of MRP1 for GSH is relatively high (~10 mM; Cole, 2014a). It is therefore unlikely that MRP1 would efflux GSH from the cell once intracellular levels fell below a certain threshold. The depletion of intracellular glutathione to near-zero levels following erastin2 treatment, even in cells lacking MRP1, must therefore refect increased GSH catabolism or efflux from the cell through an alternative route. Regardless, pharmacological inhibition of MRP1-mediated glutathione efflux could be useful as a means to slow the onset of ferroptosis in the various pathological cell death scenarios in which this process has been observed (Cole, 2014b; Stockwell et al., 2017).
MRP1 can export certain chemotherapeutics, and high MRP1 expression in cancer cells confers a multidrug resistance phenotype (Cole et al., 1994). However, MRP1-mediated glutathione efflux can collaterally sensitize to compounds that target glutathione metabolism in various ways (Cole, 2014b; Cole et al., 1990; Lorendeau et al., 2017). In particular, we find that MRP1 expression can strongly collaterally sensitize cancer cells to all tested ferroptosis-inducing agents. Thus, a potential strategy to selectively kill cancer cells with high levels of MRP1 expression may be to target the ferroptosis pathway. Cancer cells with more mesenchymal, de-differentiated, or stem-like phenotypes can have lower intracellular glutathione levels, reduced sensitivity to targeted and cytotoxic chemotherapies, and increased sensitivity to ferroptosis (Hangauer et al., 2017; Tsoi et al., 2018; Viswanathan et al., 2017). A switch to higher expression of MRP1 could provide a unifying explanation for these observations.
KEAP1 mutation and/or increased NRF2 expression has been associated with the inhibition of ferroptosis, presumably due to the ability of this transcription factor to stimulate the expression of genes involved in GSH synthesis (e.g., GCLM) (Fan et al., 2017; Roh et al., 2017; Sun et al., 2016). However, NRF2 can also increase the expression of MRP1 and potentially other ABC family metabolite transporters that promote glutathione efflux, counterbalancing the protective effects of enhanced NRF2-driven GSH synthesis. Indeed, in HAP1 cells, the effects of NRF2 stabilization on ferroptosis sensitivity were modest, and no correlation was observed between NRF2 protein levels or basal intracellular glutathione levels and erastin2 potency. Consistent with this, mRNA levels for NFE2L2/NRF2 are a relatively weak predictor of sensitivity to ferroptosis-inducing compounds across hundreds of cancer cell lines cataloged in the Cancer Therapeutics Response Portal database (https://portals.broadinstitute.org/ctrp/) (Rees et al., 2016). Thus, while further investigation is required, high NRF2 expression does not appear to be an insurmountable barrier to the induction of ferroptosis, especially when the pro-ferroptotic effects of MRP1-mediated glutathione efflux are engaged.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Scott Dixon (sjdixon@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Culture Conditions
HAP1 (gender: male) cells were described previously (Carette et al., 2011b). A549 (gender: male), H1299 (gender: male), U-2 OS (gender: female), HT-1080 (gender: male) and T98G (gender: male) were purchased from ATCC. A KEAP1KO cell line (Cat# HZGHC003774c005) and a paired HAP1 control line (herein referred to as ControlA) were obtained from Horizon Discovery (Cambridge, UK). HT-1080N cells (i.e., HT-1080 cells stably expressing nuclear-localized mKate2) were described previously (Forcina et al., 2017). Clonal HAP1 cell lines harboring gene disruptions (‘KO’) of ABCC1/MRP1, GSTO1, NAA38 and SETD5, along with an unmodified Control cell line (herein referred to as ControlB), were generated from parental HAP1 cells as described below (see Table S1). Clonal A549 cell lines with disruption of ABCC1/MRP1, along with an unmodified control cell line, were generated from parental A549 cells in the same way. These cells were then transduced with a lentivirus expressing nuclear-localized mKate2 (Cat# 4627, Essen BioSciences, Ann Arbor, MI) and mKate2-positive cells were isolated using a BD FACSAria instrument (BD Biosciences). U-2 OS and H1299 cells overexpressing MRP1, MRP1K332L or empty vector (Control) were generated via lentiviral transduction, as described below. U-2 OSControl,N, U-2 OSMRP1,N, H1299Control,N and H1299MRP1,N cells were generated from these cells via transduction with a lentivirus directing the expression of nuclear-localized mKate2 (Cat# 4627, Essen BioSciences) followed by isolation of mKate2-positive cells using FACS. HAP1N cells were generated via transduction with a lentivirus directing the expression of nuclear-localized mKate2 (Cat# 4478, Essen BioSciences) followed by isolation of mKate2-positive cells using FACS.
All cell lines were cultured at 37°C and 5% CO2 in DMEM (Cat# 10003CV, Corning) supplemented with 10% fetal bovine serum (Cat# 26140-079, GIBCO) and 1% penicillin/streptomycin (P/S, Cat# 15070-063, Life Technologies). HT-1080 cells were also supplemented with 1× non-essential amino acids (NEAAs, Cat# 11140-050, GIBCO). HBSS (Cat# 14025-134) and trypsin (Cat# 25200114) were from GIBCO. Low cystine medium was constituted using DMEM (Cat# 17-204-CI, Thermo Fisher Scientific) supplemented with 10% FBS, 0.5 U/mL Pen/Strep, 30 mg/L L-methionine (Cat# M9625, Sigma Aldrich, St. Louis, MO) and 584 mg/L L-glutamine (Cat# G3126, Sigma Aldrich). For cell seeding of all experiments, cells were counted using a Cellometer Auto T4 Bright Field Cell Counter (Nexcelcom, Lawrence, MA).
Chemicals and Reagents
Erastin2 (compound 35MEW28 in (Dixon et al., 2014)) and ML162 were synthesized by Acme Bioscience (Palo Alto, CA, USA). Bortezomib (Cat# NC0587961), thapsigargin (Cat# T9033), N-Acetyl-L-Cysteine (Cat# A8199) and ferrostatin-1 (Cat# SML0583) were from Sigma-Aldrich. Buthionine sulfoximine (Cat# AC23552-0010), Q-VD-OPh (Cat# OPH00101M) and monochlorobimane (Cat# M-1381MP) were from Thermo Fisher. C11 BODIPY 581/591 (Cat# D3861) was from Molecular Probes (Eugene, OR) and dissolved in methanol. RSL3 (Cat # S8155), Doxorubicin (Cat# S1208), vincristine (Cat# S1241), BAY-11-7085 (Cat# S7352) and BAY-11-7821/7082 (Cat# S2913) were from Selleck Chemicals (Houston, TX). Nec-1s (Cat# 2263) was from BioVision (Milpitas, CA). Buthionine sulfoximine was dissolved directly into cell media. All other compounds were prepared as stock solutions in DMSO and stored prior to use at −20°C. Cyst(e)inase enzyme was purified as described and stored at −80°C until use (Cramer etal., 2017).
METHOD DETAILS
Flow Cytometry
Flow cytometry was performed at the Stanford Shared FACS facility. To perform the haploid genetic screens, we carried out FACS on a BD FACSAria instrument (BD Biosciences). Glutathione measurements using monochlorobimane or RealThiol were carried out on a customized Stanford and Cytek upgraded FACScan flow cytometer. Data were analyzed and assembled using FlowJo software (Becton, Dickinson & Company, Franklin Lakes, NJ).
Human Haploid Cell Genetic Screening
100 million human haploid HAP1 cells were randomly mutagenized with GFP gene trap retrovirus as described (Carette et al., 2011b). Cells were then labeled with 40 μM monochlorobimane (MCB) at a density of 2.5 × 106 cell/mL in warm complete medium for 20 min in a tissue culture incubator (37°C, 5% CO2) in the dark, followed by one wash with 50 mL cold HBSS (to stop the reaction) and centrifugation (200 × g, 4°C, 5 min). The pelleted cells were then re-suspended in 5 mL cold complete media and place on ice in the dark until fluorescence-activated cell sorting (FACS). HAP1 cells were sorted based on MCB-GSH signal using a 405-nm laser detected through a 450/50 nm filter, where the top 5% of the total cell population were isolated and grown for three days. This population was labeled with MCB and sorted a second time for the top 5% MCB-GSH signal and these cells were then expanded in culture for five days. 30 million cells from this expanded population were used for genomic DNA isolation using the QIAamp DNA Mini Kit (QIAGEN, Cat# 51306) according to manufacturer’s instructions. Gene trap insertion sites were determined by linear amplification of the genomic DNA flanking regions of the gene trap, sequenced using Genome Analyzer (Illumina, San Diego, CA), and aligned to the human genome. Next, inactivating insertion events in the selected sample dataset were compared with the unselected dataset to calculate an enrichment score for each gene. A P-value for enrichment (corrected for false discovery rate) relative to an unselected control population of cells was calculated using the one-sided Fisher exact test in R, as described (Carette et al., 2011b).
CRISPR/Cas9-Mediated Gene Disruption
CRISPR/Cas9-mediated gene disruption was performed in HAP1 and A549 cells using pSpCas9(BB)-2A-GFP (PX458; Addgene #48138). Specific sgRNA sequences for ABCC1, NAA38, GSTO and SETD5 were designed using http://zlab.bio/guide-design-resources (see Table S2 for sgRNA sequences) and cloned into PX458 as described (Ran et al., 2013). Following plasmid transfection with PolyJet DNA transfection reagent (Cat# SL100688, SignaGen Laboratories, Rockville, MD) for HAP1 or Lipofectamine LTX reagent (Cat# 15338-100, ThermoFisher) for A549, positively transfected cells were single-cell sorted based on the transiently expressed GFP marker using a BD Influx cell sorter into 96-well plates containing DMEM with 10% FBS for HAP1, or DMEM with 30% FBS for A549. Several weeks after sorting, the presence of single colonies were visually confirmed and mutant clones were expanded into T-75 flasks and frozen down. Mutant clones were confirmed by PCR sequencing from genomic DNA prior to phenotypic analysis (sequences listed in Table S1). In brief, genomic DNA was extracted from 0.5-1 × 106 cells using QuickExtract DNA Extraction Solution (Cat# QE09050, Epicenter, Madison, WI) per the manufacturer’s instructions. An approximate 1000 bp genomic region encompassing the CRISPR sgRNA target site was PCR-amplified and sequenced by Sanger sequencing (see Table S2 for amplification and sequencing primer sequences). Clones were selected in which frameshift or large indels occurred at the sgRNA target site.
Glutathione Measurements
For glutathione measurement using monochlorobimane (MCB), 1 million cells were labeled with 40 μM MCB in 1 mL of warm complete medium for 20 min in a tissue culture incubation (37°C, 5% CO2) in the dark. The reaction was terminated using 1 mL cold complete medium, followed by centrifugation (200 × g, 1 min, 22°C. The pelleted cells were then re-suspended in 0.5 mL cold complete medium, filtered through a single cell strainer into FACS tubes and place on ice in the dark until analysis by flow cytometry. The MCB-GSH signal was detected using a 405 nm laser through a 450/50-nm filter.
For glutathione measurements using RealThiol (RT), 1 million cells were labeled with 1 μM RT in 0.5 mL of warm complete medium for 20 min in the dark in a tissue culture incubator (37°C, 5% CO2). The pelleted cells were then re-suspended in 0.5 mL cold complete medium, filtered through a single cell strainer into FACS tubes and place on ice in the dark until analysis by flow cytometry. The RT-GSH signal was detected using a 405 nm laser through a 450/50-nm filter, and free RT probe signal was detected using a 488 nm laser through a 525/50-nm filter. Intracellular GSH levels calculated by taking the ratio of RT-GSH signal over free RT probe signal.
For biochemical determination of glutathione levels using Ellman’s reagent, the day before the experiment, HAP1 cells were seeded at 500,000 cells/well, while H1299, U-2 OS or A549N cells were seeded at 250,000 cells/well in a 6-well plate (Cat# 07-200-83, Corning). The next day, following any treatments, cells were washed once with HBSS and collected into MES buffer with 1 mM EDTA using a cell scraper, followed by brief sonication and centrifugation (16,110 × g, 4°C, 15 min). Supernatants were collected and protein was quantified by Bradford assay using the Bio-Rad Protein Assay reagent (Cat# 5000002, Bio-Rad, Hercules, CA). The samples were deproteinated with equal volume of 12.5 M metaphosphoric acid, and total glutathione was measured using the Glutathione Assay Kit per manufacturer’s instructions (Cat# 703002, Cayman Chemical, Ann Arbor, MI). Assay readings were performed on a Synergy Neo2 reader (BioTek, Winooski, VT). Results were normalized to total protein concentration for each sample.
Stimulated Glutathione Export Assay
Cells were seeded (500,000 cells/well for HAP1, 250,000 cells/well for H1299, U-2 OS or A549N cells) in a 6-well plate 24 h prior to the experiment. The next day, cells were washed 2× with DPBS (Cat# 14287-080, GIBCO) with 1× Pen/Strep and then incubated in 1 mL of DPBS medium for 2 h (37°C, 5% CO2). The DPBS medium was then collected, lyophilized overnight using a VirTis Sentry 2.0 (SP Industries Inc., Warminster, PA), and resuspended in 100 μL MES buffer provided from the Glutathione Assay kit (Cayman Chemical). Total glutathione was measured using the Glutathione Assay kit per manufacturer’s instructions. Values were normalized to total cell count, determined using a Cellometer Auto T4 Bright Field Cell Counter (Nexcelcom).
Cell Viability Analysis using PrestoBlue
Cells were seeded (4000 cells/well for HAP1 cells, or 1500 cells/well for H1299 or U-2 OS cells) into 384-well plates (Cat# 07-201-013, Corning) 24 h prior to the experiment. The next day, cells were treated with various doses of drug as indicated. Cell viability was assayed using PrestoBlue (Life Technologies, Cat# A13262) at the indicated time after drug treatment. 10% final (v/v) PrestoBlue reagent was added to the existing treatment medium with mixing and incubated for 30 min (37°C, 5% CO2). PrestoBlue signal was measured using a Synergy Neo2 reader (BioTek Instruments) at ex/em of 530/590 nm. Background fluorescence from medium-only controls with 10% final PrestoBlue was subtracted from all values, and samples were normalized to an internal control treated with DMSO that was set to 100% viability. EC50 was calculated using standard sigmoidal four-point logistic regression with Prism 7.0 (GraphPad, La Jolla, CA, USA).
Imaging Analysis of Cell Death and Cell Death Kinetics
In some experiments, cell death within the population was assessed by counting dead cells. HAP1 cells were seeded in 6-well (500,000 cells/well) or 384-well (4000 cells/well) plates 24 h prior to the start of treatment. The next day, equivalent cell seeding between wells was confirmed by visual inspection and the medium was replaced with fresh medium containing lethal compounds together with the normally membrane-impermeant DNA intercalating dye SYTOX Green (SG) (Life Technologies, Cat# S7020) at a final concentration of 20 nM. Cells were imaged using an IncuCyte live cell analysis system (Essen BioScience) either at a fixed time point (e.g., 24 h), or every 4 h over 48 h for kinetic experiments. SG positive (SG+) objects (i.e., dead cells) were counted within the population at each time point using the IncuCyte ZOOM Live-Cell Analysis System software as described (Forcina et al., 2017). Counting routines were empirically optimized for each cell line using the Zoom software package (V2016A/B) and training data from DMSO and lethal compound-treated samples. HAP1 cells populations were analyzed using a routine with the following settings (in parentheses) to count SG+ objects (Parameter adaption, threshold adjustment: 2; Edge split on; Filter area min 50 μm2, maximum 500 μm2; Filter mean intensity min: 38 AU). Dead cell SG positivity can be transient due to disintegration of nuclear DNA within long-dead cell corpses (Forcina et al., 2017). Thus, the maximum SG count within the population at each time point in the series was determined and the maximum number observed at any time point was used for all subsequent calculations. In some experiments, cell death was expressed as the number of SG+ (i.e., dead) cells within the population at a single time point. In some experiments we determined cell death kinetics using counts of SG+ objects within the population over time, essentially as described (Forcina et al., 2017). Briefly, the timing of cell death onset within the population is a value termed DO. The maximal rate of cell death within the population is a value termed DR. DO and DR are parameter values extracted from curves fits to counts of SG+ objects over time within the population. Curve fits were computed using Prism 7.0 using the ‘plateau followed by one-phase association’ function. Within the Prism output, DO corresponds to the best-fit value for the X0 parameter, and DR corresponds to the best-fit for the K parameter.
In some experiments, both live and dead cells were counted. For these experiments we employed cells stably expressing nuclear-localized mKate2, denoted by a superscript ‘N’. Lethal fraction scores at each time point were computed from counts of live and dead cells, as described (Forcina et al., 2017). Briefly, lethal fraction scores represent the fraction of dead (SG+) cells over total cells (mKate2+ + SG+) cells within the population at a given time point. Lethal fraction scoring accounts for the loss of SG signal from long-dead cell corpses by taking the maximum number of SG+ objects observed within the population up to a given time point as the dead cell count, as described above, and in (Forcina et al., 2017). In some experiments, the death onset (DO) within the population was computed from lethal fraction scores overtime as described (Forcina et al., 2017). Briefly, the computed lethal fraction scores over time were fit using Prism 7.0 with the ‘plateau followed by one-phase association’ function. Within the Prism output, DO corresponds to the best-fit value for the X0 parameter, and DR corresponds to the best-fit for the K parameter.
Thus, HAP1N cells were seeded in 6-well (500,000 cells/well) or 384-well (4000 cells/well) plates 24 h prior to the start of treatment. HAP1n populations were analyzed using a routine with the following settings (in parentheses) to count mKate2+ objects (Parameter adaption, threshold adjustment: 2; Edge split on) and SG+ objects (Parameter adaption, threshold adjustment: 2; Edge split on; Filter area min 50 μm2, maximum 500 μm2; Filter mean intensity min: 38 AU). U-2 OSN and A549N populations were analyzed using a routine with the following settings (in parentheses) to count mKate2+ objects (Parameter adaption, threshold adjustment: 2.5; Edge split on; Edge sensitivity −2; Filter area min 50 μm2, maximum 8100 μm2) and SG+ objects (Parameter adaption, threshold adjustment: 10; Edge split on; Filter area min 20 μm2, maximum 750 μm2). H1299N populations were analyzed using a routine with the following settings (in parentheses) to count mKate2+ objects (Parameter adaption, threshold adjustment: 6; Edge split on; Edge sensitivity −10) and SG+ objects (Parameter adaption, threshold adjustment: 2; Edge split on; Edge sensitivity −38). In some experiments, we captured phase contrast images and measured confluence using the phase contrast image analyzer (segmentation adjustment, background: 0.5). mKate2+ and SG+ counts, expressed as objects per mm2, and confluence measures, expressed as an overall percentage (0%-100%), were exported to Excel (Microsoft) for further processing.
Cell death and cell death kinetics were also determined in nuclear-localized mKate2-expressing U-2 OSN, H1299N or A549N cells. These cells were seeded in 384-well plates 24 h prior to the initiation of treatment at a density of 1500 cells/well. The next day, the medium was replaced with fresh medium containing lethal compounds or conditions (e.g., −cystine, +cyst(e)inase) together with SG (20 nM). For cell death experiments with cystine-free media and BSO, cells were washed three times with HBSS before adding the indicated treatments along with SYTOX Green. In all cases, cells were imaged using an IncuCyte live cell analysis system every 4 h for up to 72 h and counts of both dead (SG+) and live (mKate2+) cells determined at each time point and lethal fraction scores and cell death kinetic parameter values determined as described above.
Western Blotting
1 million cells were lysed in a modified RIPA buffer (50 mM Tris pH 7.5, 150mM NaCl, 0.5% NP-40, 0.1% Sodium deoxycolate, 1% Triton X-100, 0.1% SDS, 2 mM EDTA) with complete protease inhibitor cocktail (Cat# P8340, Sigma) on ice for 20 min. Followed by brief sonication and centrifugation at 16,110 × g at 4°C for 15 min, the clarified lysates were quantified using BCA Protein Assay Kit (Cat # 23225, Thermo Fisher). 40 μg of lysates were prepared using Laemmli buffer (Cat# 1610737, Bio-Rad) containing beta-mercaptoethanol by incubating at room temperature for 15 min, then the samples were subjected to SDS-PAGE using pre-cast 4%-15% polyacrylamide gels (Cat# 4561084, Bio-Rad) and transferred onto nitrocellulous membranes using the iBlot Dry Blotting System (Cat# IB21001, Thermo Fisher). Membranes were probed with primary antibodies against MRP1 (Cat#ab3368, Abcam, 1:500 dilution), KEAP1 (Cat# sc-365626, Santa Cruz, 1:500 dilution), NRF2 (Cat# ab137550, Abcam, 1:1000 dilution), GCLM (Cat# NBP1-33405, Novus Biologicals), and alpha-tubulin (Cat# MS581P1, Fisher Scientific) in Odyssey Buffer (Cat# 927-40100, LI-COR Biotechnology, Lincoln, NE) (60 rpm, 4°C, overnight). After washing three times in TBST, membranes were incubated with the respective secondary antibodies (Cat# 926-68024, Donkey anti-goat-680; Cat# 926-32214; Donkey anti-goat-800; Cat# 926-68023, Donkey anti-rabbit-680; Cat# 926-32213, Donkey anti-rabbit-800; Cat# 926-68022, Donkey anti-mouse-680; Cat# 926-32212, Donkey anti-mouse-800; all at 1:15,000 dilution; LI-COR) in Odyssey Buffer (40 min, 60 rpm, RT) The membranes were washed three times in TBST and imaged using a LI-COR Odyssey CLx imager. Western blot band quantification was done using the LI-COR ImageStudio program. KEAP1, MRP1, NRF2 and GCLM signals were normalized to the alpha-tubulin loading control for each sample. Relative protein expression was determined by dividing the normalized value determined for each sample by the control samples.
RT-qPCR
Cells were washed once in PBS and harvested following trypsinization and centrifugation. The pelleted cells (with media removed) were frozen immediately on dry ice for at least 15 min. RNA was extracted using a QIAGEN QIAshredder extraction column (Cat# 79654) and RNeasy Plus RNA Extraction Kit (Cat# 74134). cDNA was generated using the TaqMan Reverse Transcriptase Kit according to the manufacturer’s instructions (Cat# N8080234, Applied Biosystems/Thermo Fisher Scientific, Foster City, CA). See Table S2 for qPCR primer sequences. Quantitative PCR reactions were prepared with SYBR Green Master Mix (Cat# 4367659, Life Technologies) and run on an Applied Biosystems QuantStudio 3 real-time PCR machine (Thermo Fisher). Relative transcript levels were calculated using the ΔΔCT method and normalized to the ACTB gene as described (Tarangelo et al., 2018).
Lipid ROS Imaging
Imaging and quantification of C11 BODIPY 581/591 (C11) (Cat# D3861, Molecular Probes) were performed as described previously (Magtanong et al., 2019) with the following modifications. For the C11 time course, the day before the experiment, 150,000 HT-1080 cells/well were seeded into 6-well dishes with one 22 mm2 glass coverslip in each well. The next day, cells were treated with erastin2 (1 μM) for 0 (i.e., no treatment), 4 or 8 h. At the end of the time course, the treatment medium was removed and cells were washed once with HBSS. HT-1080 cells were then labeled with 1 mL C11 BODIPY 581/591 (5 μM) dissolved in HBSS. Cells were incubated at 37°C for 10 min. After 10 min, the label mixture was removed and 1 mL of fresh HBSS was added to the cells. The coverslip was removed from the well and inverted onto a glass microscope slide onto which 25 μL of fresh HBSS had been applied. Cells were imaged using a Zeiss Axio Observer microscope with a confocal spinning-disk head (Yokogawa, Tokyo, Japan), PlanApoChromat 63 X /1.4 NA oil immersion objective, and a Cascade II:512 electron-multiplying (EM) CCD camera (Photometrics, Tucson, AZ). Imaging was performed on two independent biological replicates per treatment. Images were processed in ImageJ 1.48v and quantified as described previously (Magtanong et al., 2019). A similar imaging approach was used to treat and image H1299N and U-2 OSN cells with the following modifications. Cells were treated with DMSO (vehicle), BAY-11-7821 (20 μM) or ML162 (5 μM) for 4 h before C11 labeling. For labeling, a mixture of Concanavalin A-Alexa Fluor 350 (25 μg/mL) and C11 BODIPY 581/591 (5 μM) was made in HBSS. Cells were incubated in the labeling mixture at 37°C for 10 min prior to mounting onto microscope slides.
MRP1 Gene Expression
The cDNA of human ABCC1 (NM_004996) was subcloned from pShuttle-GATEWAY-ABCC1 with STOP (Cat# GC-Z4479, GeneCopoeia, Rockville, MD) into the lentiviral expression plasmid, pLenti-CMV-Puro DEST (Addgene plasmid #17452, from Eric Campeau and Paul Kaufman) using Gateway LR Clonase II Enzyme Mix (Cat# 11791-020, Life Technologies). The cDNA of ABCC1K332L was subcloned from pCDNA3.1-ABCC1K332L into pLenti-CMV-Puro DEST plasmid via Gibson Assembly (Cat# E5510S, New England Biolabs, Ipswich, MA). All plasmids were verified by Sanger sequencing. Lentiviruses were generated in HEK293T cells using a 3rd generation lentivirus packaging system. In brief, cells were transfected with 1 μg pLentiCMV DEST plasmid + 0.25 mg of each of three 3rd generation lentiviral packaging plasmids (pMDLg/pRRE, pRSV-Rev and pMD2.G, Addgene plasmids #12251, #12253 and #12259, respectively, from Didier Trono) using PolyJet DNA Transfection Reagent (SignaGen Laboratories, Cat# SL100688 Rockville, MD) as per the manufacturer’s instructions. Lentivirus was harvested twice (three and four days post-transfection), filtered through a 0.45 μM PVDF filter (Cat# SLHV033RS, Merck Millipore, Burlington, MA) and stored at −80°C until use. For infections, 0.5 mL of freshly-thawed virus soup was mixed with polybrene (Cat# H9268, Sigma-Aldrich) to a final concentration of 8 μg/mL and used to infect HAP1 MRP1KO1, U-2 OS and H1299 cell lines. Stably transduced cell populations were selected with 1 μg/mL puromycin (Cat# A11138-03, Life Technologies) for three days.
Stable Isotope Tracing of Glutathione Efflux
A549N cells were seeded at 125,000 cells/well in a 6-well plate. The next day, the growth medium was replaced with medium reconstituted with 13C5-L-glutamine (Cat# 605166, Sigma Aldrich) for incorporation into glutathione via de novo synthesis. After 24 h, cells were washed twice with HBSS and replaced in 0.5 mL of fresh 13C5-L-glutamine-containing media + acivicin (100 μM, Cat# SML0312, Sigma Aldrich) ± erastin2 (2 μM) for 4 h. 13C5-glutamine is deaminated to make 13C5-glutamate, which can then be incorporated into GSH via de novo synthesis, yielding M+5 GSH. Note: acivicin was included to inhibit gamma-glutamyl transpeptidase-mediated extracellular GSH degradation. After 4 h, the spent medium was collected and 10 μL of medium was combined with 40 μL of 100% methanol (pre-chilled −80°C) on ice, vortexed twice for 1 min, and centrifuged (20,000 × g, 4°C, 10 min). Supernatants were then transferred to liquid chromatography vials for HPLC injection. For normalization purposes, the remaining cells were trypsinized and cell number was quantified using a Cellometer Auto T4 Bright Field Cell Counter. Liquid chromatography coupled to High Resolution Mass Spectrometry (LC-HRMS) was carried out using a Q Exactive Plus MS (Thermo Scientific) as described (Liberti et al., 2017; Liu et al., 2014). HPLC injection volume was 5 μL. Peak alignment and detection were conducted using Sieve 2.0 software (Thermo Scientific) according to the manufacturer’s guidelines. M+5 GSH intensities were normalized to total cell number.
MRP1 Collateral Sensitivity Screen
A bioactive compound library (Cat# L2000) was obtained from Selleck Chemicals (Houston, TX), stored, re-formatted and screened as described (Magtanong et al., 2019) with the following modifications. The day before screen, U-2 OSControl,N or U-2 OSMRP1,N cells were seeded (1,500 cells/well) into 384-well plates (1 plate per cell line) at a final volume of 40 μL. The next day, compounds were added from freshly thawed library master stock plates to a final concentration of 5 μM in each well, using a Versette liquid handler equipped with a 384-channel pipetting head. Plates were imaged immediately and every 4 h thereafter for a total of 72 h, on the Essen IncuCyte Zoom. Counts of SG+ and mKate2+ objects/mm2 were obtained and lethal fraction scores calculated as described (Forcina et al., 2017). The area under the curve (AUC) of the lethal fraction scores (AUCLethal fraction) over 72 h was computed using the ‘area under curve’ function in Prism 7.0 with default settings. The profiling experiment was performed three times on different days and an average AUCLethal fraction score for each condition was computed and converted to a log2 value, and values for each compound tested in U-2 OSControl,N cells was plotted against the corresponding values obtained from U-2 OSMRP1,N cells. A linear regression line with 95% confidence interval was calculated with the average AUCLethal fraction scores from all compounds using Prism 7.0 (the gray area in Figure 4C). Bioactive compounds with average AUCLethal fraction scores outside of the 95% confidence interval for this linear regression line, and also three standard deviations away from vehicle (DMSO)-treated cells (dotted-line on either the X- or Y- axis), were identified as being differentially-sensitive to MRP1 expression.
QUANTIFICATION AND STATISTICAL ANALYSIS
Except when noted, all data represent mean ± standard deviation (SD) from three independent experiments performed on separate days. Lethal fraction scores were calculated using Microsoft Excel 14.6.0 (Microsoft Corporation, Redmond, WA). Flow cytometry data were processed using FlowJo 10.1r5 (FlowJo LLC, Ashland, OR). Confocal images were processed and quantified in ImageJ 1.48v (U.S. National Institutes of Health, Bethesda, MD). Figures were compiled using Adobe Illustrator (Adobe Systems, San Jose, CA). STACK analysis, graphing and all statistical analyses were performed using Prism 7.0 (GraphPad Software, La Jolla, CA). Pearson r coefficients of determination (r2) are reported. One-way ANOVAs were analyzed using Sidak post-tests.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-MRP1 | Abcam | Cat# ab3368; RRID: AB_303746 |
| Rabbit polyclonal anti-NRF2 | Abcam | Cat# ab137550; RRID: AB_2687540 |
| Mouse monoclonal anti-KEAP1 | Sigma-Aldrich | Cat# K2769; RRID: AB_10602870 |
| Rabbit polyclonal anti-GCLM | Novus Biologicals | Cat# NBP1-33405; RRID: AB_2107841 |
| Mouse monoclonal anti-Tubulin alpha, clone DM1A | Fisher Scientific | Cat# MS581P1; RRID: AB_144075 |
| IRDye 680LT Goat anti-Rat IgG | LI-COR | Cat# 925-68076 |
| IRDye 800CW Goat anti-Rat IgG | LI-COR | Cat# 925-32219 |
| IRDye 800CW Donkey anti-Rabbit IgG | LI-COR | Cat# 926-32213 |
| IRDye 800CW Donkey anti-Mouse IgG | LI-COR | Cat# 926-32212 |
| IRDye 680LT Donkey anti-Rabbit IgG | LI-COR | Cat# 926-68023 |
| IRDye 680LT Donkey anti-Mouse IgG | LI-COR | Cat# 926-68022 |
| Bacterial and Virus Strains | ||
| NucLight Red lentivirus reagent (EF1a, Puromycin) Nuclear-localized mKate2 (Nuc::mKate2) |
Essen BioSciences | Cat# 4265 |
| NucLight Red lentivirus reagent (EF1a, Bleomycin) Nuclear-localized mKate2 (Nuc::mKate2) |
Essen BioSciences | Cat# 4627 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SYTOX Green | Life Technologies | Cat# S7020 |
| DFO mesylate | Cayman Chemical | Cat# 14595; CAS: 138-14-7 |
| Cyst(e)inase | Cramer et al., 2017 | N/A |
| Erastin2 (Compound 35MEW28 in Dixon et al., 2014) | Dixon et al., 2014 | N/A |
| Buthionine sulfoximine | Fisher Scientific | Cat# AC23552-0010; CAS: 83730-53-4 |
| Q-VD-OPh | Fisher Scientific | Cat# OPH00101M |
| Puromycin | Life Technologies | Cat# A11138-03 |
| SYTOX Green | Life Technologies | Cat# S7020 |
| Lipofectamine LTX Reagent with PLUS Reagent | Life Technologies | Cat# 15338-100 |
| C11 BODIPY 581/591 (4,4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diaza-s-indacene-3-undecanoic acid | Molecular Probes | Cat# D3861 |
| Concanavalin A-Alexa Fluor 350 | Molecular Probes | Cat# C11254 |
| 261 member bioactive chemical library | Selleck Chemicals | Cat# L2000 |
| BAY-11-7821/7082 | Selleck Chemicals | Cat# S2913; CAS: 19542-67-7 |
| BAY-11-7085 | Selleck Chemicals | Cat# S7352; CAS: 196309-76-9 |
| RSL3 | Selleck Chemicals | Cat# S8155; CAS: 1219810-16-8 |
| Doxorubicin hydrochloride | Selleck Chemicals | Cat# S1208; CAS: 25316-40-9 |
| Vincristine sulfate | Selleck Chemicals | Cat# S1241; CAS: 2068-78-2 |
| Bortezomib | Sigma-Aldrich | Cat# NC0587961; CAS: 179324-69-7 |
| N-Acetyl-L-Cysteine | Sigma-Aldrich | Cat# A8199; CAS: 616-91-1 |
| Thapsigargin | Sigma-Aldrich | Cat# T9033; CAS: 67526-95-8 |
| Ferrostatin-1 | Sigma-Aldrich | Cat# SML0583; CAS: 347174-05-4 |
| Polybrene | Sigma-Aldrich | Cat# H9268; CAS: 28728-55-4 |
| Acivicin | Sigma-Aldrich | Cat# SML0312; CAS: 42228-92-2 |
| 13C5-L-glutamine | Sigma-Aldrich | Cat# 605166; CAS: 184161-19-1 |
| PolyJet In Vitro DNA Transfection Reagent | SigmaGen Laboratories | Cat# SL100688 |
| Monochlorobimane | Thermo Fisher | Cat# M-1381MP; CAS: 76421-73-3 |
| Critical Commercial Assays | ||
| QuickExtract DNA Extraction Solution | Epicenter | Cat# QE09050 |
| QIAshredder RNA Extraction Column Kit | QIAGEN | Cat# 79654 |
| RNeasy Plus RNA Extraction Kit | QIAGEN | Cat# 74134 |
| TaqMan Reverse Transcriptase Kit | TaqMan | Cat# N8080234 |
| Cayman Glutathione Kit | Cayman Chemical | Cat# 703002 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| SYBR Green Master Mix | Life Technologies | Cat# 4367659 |
| Gateway LR Clonase II Enzyme Mix | Thermo Fisher | Cat# 11791-020 |
| BCA Protein Assay Kit | Thermo Fisher | Cat# 23225 |
| PrestoBlue Cell Viability Reagent | Thermo Fisher | Cat# A13261 |
| Bradford Assay Kit | Bio-Rad | Cat# 5000002 |
| Experimental Models: Cell Lines | ||
| Human: HAP1 | Carette et al., 2011b | N/A |
| Human: HAP1N | This paper | N/A |
| Human: HAP1 ControlA | Horizon Discovery | Cat# C631 |
| Human: HAP1 KEAP1KO | Horizon Discovery | Cat# HZGHC003774c005 |
| Human: HAP1 ControlB | This paper | N/A |
| Human: HAP1 MRP1KO1 | This paper | N/A |
| Human: HAP1 MRP1KO2 | This paper | N/A |
| Human: HAP1 MRP1KO1 + CMV-empty | This paper | N/A |
| Human: HAP1 MRP1KO1 + CMV-MRP1 | This paper | N/A |
| Human: HAP1 MRP1KO1 + CMV-MRP11K332L | This paper | N/A |
| Human: HAP1 NAA38KO1 | This paper | N/A |
| Human: HAP1 NAA38KO2 | This paper | N/A |
| Human: HAP1 GSTOKO1 | This paper | N/A |
| Human: HAP1 GSTOKO2 | This paper | N/A |
| Human: HAP1 SETD5KO1 | This paper | N/A |
| Human: HAP1 SETD5KO2 | This paper | N/A |
| Human: A549 | ATCC | Cat# CCL-185; RRID: CVCL_0023 |
| Human: A549N | Forcina et al., 2017 | N/A |
| Human: A549N MRP1KO1 | This paper | N/A |
| Human: A549N MRP1KO2 | This paper | N/A |
| Human: HT-1080 | ATCC | Cat# CCL-121; RRID: CVCL_0317 |
| Human: HT-1080N | Forcina et al., 2017 | N/A |
| Human: U-2 OS | ATCC | Cat# HTB-96; RRID: CVCL_0042 |
| Human: U-2 OSN | Forcina et al., 2017 | N/A |
| Human: T98GN | Forcina et al., 2017 | N/A |
| Human: NCI-H1299 (called H1299) | ATCC | Cat# CRL-5803; RRID: CVCL_0060 |
| Human: H1299N | This paper | N/A |
| Human: U-2 OS + CMV-empty | This paper | N/A |
| Human: U-2 OS + CMV-MRP1K332L | This paper | N/A |
| Human: U-2 OS + CMV-MRP1K332L | This paper | N/A |
| Human: U-2 OSN + CMV-empty | This paper | N/A |
| Human: U-2 OSN + CMV-MRP1 | This paper | N/A |
| Human: H1299 + CMV-empty | This paper | N/A |
| Human: H1299 + CMV-MRP1 | This paper | N/A |
| Human: H1299 + CMV-MRP1K332L | This paper | N/A |
| Human: H1299N + CMV-empty | This paper | N/A |
| Human: H1299N + CMV-MRP1 | This paper | N/A |
| Oligonucleotides | ||
| See Table S2. | N/A | |
| Recombinant DNA | ||
| pSpCas9 (BB)-2A-GFP-sgRNA-p53 | This paper | N/A |
| pLenti-CMV-Puro-MRP1 | This paper | N/A |
| pLenti-CMV-Puro-MRP1K332L | This paper | N/A |
| Other Plasmids | ||
| pSpCas9 (BB)-2A-GFP (PX458) | Addgene | Cat# 48138; RRID: Addgene_48138 |
| pLenti-CMV-Puro DEST (w118-1) | Addgene | Cat# 17452; RRID: Addgene_17452 |
| pShuttle-GATEWAY-ABCC1 with STOP | GeneCopoeia | Cat# GC-Z4479 |
| pCDNA3.1-ABCC1K332L | Dr. Susan Cole (Queen’s University) | N/A |
| Software and Algorithms | ||
| Prism 7.0 | GraphPad Software | N/A |
| Microsoft Excel | Microsoft | N/A |
| ImageJ 1.48v | Rasband, W.S. | https://imagej.nih.gov/ij/ |
| FlowJo 10.1r5 | Becton, Dickinson & Company | N/A |
Highlights.
FACS-based screening can identify genetic regulators of glutathione abundance
MRP1 effluxes glutathione, which accelerates ferroptosis
NAA38 and KEAP1 regulate expression of NRF2 and also MRP1
High MRP1 expression promotes collateral sensitivity to ferroptosis-inducing agents
ACKNOWLEDGMENTS
We thank Giovanni Forcina for assistance with compound profiling, Pin-Joe Ko for help with the figures, and Tim Stearns for help with microscopy. This work was supported by a Stanford ChEM-H Postdocs at the Interface grant (to J.Y.C. and C.M.D.) and the NIH (R01GM115622, R01CA207701, and R21CA213535, to J.W.; R01CA193256, to J.W.L.; 1R01CA189623 to E.S.; and 1R01GM122923, to S.J.D.). M.A.R. is supported by the American Cancer Society (131615-PF-17-210-01-TBE). S.J.D. is supported by a Damon Run-yon-Rachleff Innovation Award.
Footnotes
DECLARATION OF INTERESTS
S.J.D. is a member of the scientific advisory board of Ferro Therapeutics. E.S. is an inventor of intellectual property related to this work and has an equity interest in Aeglea BioTherapeutics.
REFERENCES
- Aksnes H, Hole K, and Arnesen T (2015). Molecular, cellular, and physiological significance of N-terminal acetylation. Int. Rev. Cell Mol. Biol 316, 267–305. [DOI] [PubMed] [Google Scholar]
- Beadle GW, and Tatum EL (1941). Genetic Control of Biochemical Reactions in Neurospora. Proc. Natl. Acad. Sci. USA 27, 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C, Bell G, Yuan B, Muellner MK, Nijman SM, et al. (2011a). Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat. Biotechnol 29, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. (2011b). Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L-C, Chiang S-K, Chen S-E, Yu Y-L, Chou R-H, and Chang W-C (2018). Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 416, 124–137. [DOI] [PubMed] [Google Scholar]
- Cole SPC. (2014a). Multidrug resistance protein 1 (MRP1, ABCC1), a “multi-tasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem 289, 30880–30888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SPC (2014b). Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu. Rev. Pharmacol. Toxicol 54, 95–117. [DOI] [PubMed] [Google Scholar]
- Cole SPC, and Deeley RG (2006).Transport ofglutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci 27, 438–446. [DOI] [PubMed] [Google Scholar]
- Cole SPC, Downes HF, Mirski SE, and Clements DJ (1990). Alterations in glutathione and glutathione-related enzymes in a multidrug-resistant small cell lung cancer cell line. Mol. Pharmacol 37, 192–197. [PubMed] [Google Scholar]
- Cole SPC, Sparks KE, Fraser K, Loe DW, Grant CE, Wilson GM, and Deeley RG (1994). Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 54, 5902–5910. [PubMed] [Google Scholar]
- Cooper SJ, Finney GL, Brown SL, Nelson SK, Hesselberth J, Mac-Coss MJ, and Fields S (2010). High-throughput profiling of amino acids in strains of the Saccharomyces cerevisiae deletion collection. Genome Res 20, 1288–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S, et al. (2017). Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med 23, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Thompson CB (2012). Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deponte M (2013). Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 1830, 3217–3266. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, and Stockwell BR (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015). Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol 10, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey CM, Diep J, Clarke BP, Hale AT, McNamara DE, Guo H, Brown NW Jr., Cao JY, Grace CR, Gough PJ, et al. (2018). MLKL Requires the Inositol Phosphate Code to Execute Necroptosis. Mol. Cell 70, 936–948.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazic A, Myklebust LM, Ree R, and Arnesen T (2016). The world of protein acetylation. Biochim. Biophys. Acta 1864, 1372–1401. [DOI] [PubMed] [Google Scholar]
- Ducker GS, and Rabinowitz JD (2017). One-Carbon Metabolism in Health and Disease. Cell Metab. 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Wirth A-K, Chen D, Wruck CJ, Rauh M, Buchfelder M, and Savaskan N (2017). Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcina GC, Conlon M, Wells A, Cao JY, and Dixon SJ (2017). Systematic Quantification of Population Cell Death Kinetics in Mammalian Cells. Cell Syst. 4, 600–610.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, and Cidlowski JA (2006). SLCO/OATP-like transport of glutathione in FasL-induced apoptosis: glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J. Biol. Chem 281, 29542–29557. [DOI] [PubMed] [Google Scholar]
- Haimeur A, Deeley RG, and Cole SPC (2002). Charged amino acids in the sixth transmembrane helix of multidrug resistance protein 1 (MRP1/ABCC1) are critical determinants of transport activity. J. Biol. Chem 277, 41326–41333. [DOI] [PubMed] [Google Scholar]
- Hammond CL, Marchan R, Krance SM, and Ballatori N (2007). Glutathione export during apoptosis requires functional multidrug resistance-associated proteins. J. Biol. Chem 282, 14337–14347. [DOI] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. (2015). Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222. [DOI] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422.e21. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, and Yamamoto M (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji L, Li H, Gao P, Shang G, Zhang DD, Zhang N, and Jiang T (2013). Nrf2 pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PLoS One 8, e63404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Chen J, Bajić A, Zhang C, Song X, Carroll SL, Cai Z-L, Tang M, Xue M, Cheng N, et al. (2017). Quantitative real-time imaging of glutathione. Nat. Commun 8, 16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, et al. (2017). A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 26, 648–659.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien EC, Lyssiotis CA, Juvekar A, Hu H, Asara JM, Cantley LC, and Toker A (2016). Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol 18, 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, and Locasale JW (2014). Development and quantitative evaluation of a high-resolution metabolomics technology. Anal. Chem 86, 2175–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llufrio EM, Wang L, Naser FJ, and Patti GJ (2018). Sorting cells alters their redox state and cellular metabolome. Redox Biol. 16, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorendeau D, Dury L, Nasr R, Boumendjel A, Teodori E, Gutschow M, Falson P, Di Pietro A, and Baubichon-Cortay H (2017). MRP1-dependent collateral sensitivity of multidrug-resistant cancer cells: Identifying selective modulators inducing cellular glutathione depletion. Curr. Med. Chem 24, 1186–1213. [DOI] [PubMed] [Google Scholar]
- Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, Mehta M, Cheung KL, Ganesan S, Kong A-NT, et al. (2012). PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol 32, 1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeno K, Nakajima A, Conseil G, Rothnie A, Deeley RG, and Cole SPC (2009). Molecular basis for reduced estrone sulfate transport and altered modulator sensitivity of transmembrane helix (TM) 6 and TM17 mutants of multidrug resistance protein 1 (ABCC1). Drug Metab. Dispos 37, 1411–1420. [DOI] [PubMed] [Google Scholar]
- Magtanong L, Ko P-J, To M, Yinuo Cao J, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol Published online January 24, 2019. 10.1016/j.chembiol.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchan R, Hammond CL, and Ballatori N (2008). Multidrug resistance-associated protein 1 as a major mediator of basal and apoptotic glutathione release. Biochim. Biophys. Acta 1778, 2413–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon D, and Board PG (2013). A role for glutathione transferase Omega 1 (GSTO1-1) in the glutathionylation cycle. J. Biol. Chem 288, 25769–25779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, and Vander Heiden MG (2013). Understanding metabolic regulation and its influence on cell physiology. Mol. Cell 49, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mülleder M, Calvani E, Alam MT, Wang RK, Eckerstorfer F, Zelezniak, , and Ralser M (2016). Functional Metabolomics Describes the Yeast Biosynthetic Regulome. Cell 167, 553–565.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, and Gerritsen ME (1997). Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem 272, 21096–21103. [DOI] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, and Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, et al. (2016). Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol 12, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh J-L, Kim EH, Jang H, and Shin D (2017). Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 11, 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara J, Makino N, and Bannai S (1996). Glutathione efflux from cultured astrocytes. J. Neurochem 66, 1876–1881. [DOI] [PubMed] [Google Scholar]
- Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM, et al. (2017).Activation ofthe NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 6, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickson S, Campbell DG, Emmerich CH, Knebel A, Plater L, Ritorto MS, Shpiro N, and Cohen P (2013). The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem. J 451, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D (2016). Activation ofthe p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szybalski W, and Bryson V (1952). Genetic studies on microbial cross resistance to toxic agents. I. Cross resistance of Escherichia coli to fifteen antibiotics. J. Bacteriol 64, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, and Dixon SJ (2018). p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 22, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompier D, Chang X-B, Barattin R, du Moulinet D’Hardemare A, Di Pietro A, and Baubichon-Cortay H (2004). Verapamil and its derivative trigger apoptosis through glutathione extrusion by multidrug resistance protein MRP1. Cancer Res. 64, 4950–4956. [DOI] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Kalvik TV, Starheim KK, Jonckheere V, Myklebust LM, Menschaert G, Varhaug JE, Gevaert K, and Arnesen T (2016). A Role for Human N-alpha Acetyltransferase 30 (Naa30) in Maintaining Mitochondrial Integrity. Mol. Cell. Proteomics 15, 3361–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C-H, Chou C-C, Lee D-Y, Khoo K-H, and Chang G-D (2018). Target identification reveals protein arginine methyltransferase 1 is a potential target of phenyl vinyl sulfone and its derivatives. Biosci. Rep 38, BSR20171717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.